

Abstract
Introduction
Smith-Lemli-Opitz Syndrome (SLOS) is a malformation syndrome inherited in an autosomal recessive fashion. It is due to a metabolic defect in the conversion of 7-dehydrocholesterol to cholesterol, which leads to an accumulation of 7-dehydrocholesterol and frequently a deficiency of cholesterol. The syndrome is characterized by typical dysmorphic facial features, multiple malformations, and intellectual disability.
Areas covered
In this paper we provide an overview of the clinical phenotype and discuss how the manifestations of the syndrome vary depending on the age of the patients. We then explore the underlying biochemical defect and pathophysiological alterations that may contribute to the many disease manifestations. Subsequently we explore the epidemiology and succinctly discuss population genetics as they relate to SLOS. The next section presents the diagnostic possibilities. Thereafter, the treatment and management as is standard of care are presented.
Expert opinion
Even though the knowledge of the underlying molecular mutations and the biochemical alterations is being rapidly accumulated, there is currently no efficacious therapy addressing neurological dysfunction. We discuss the difficulty of treating this disorder, which manifests as a combination of a malformation syndrome and an inborn error of metabolism. A very important factor in developing new therapies is the need to rigorously establish efficacy in controlled trials.
1. Introduction
Smith-Lemli-Opitz syndrome (SLOS, OMIM #270400) is an autosomal recessive, multiple malformation/cognitive impairment syndrome resulting from an underlying inborn error of metabolism, specifically a deficiency of 7-dehydrocholesterol reductase (DHCR7) activity. SLOS was first described by Drs. Smith, Lemli and Opitz [1] in 1964 in a case series of three male pediatric patients with small stature, failure to thrive, severe feeding disorder, hypospadias and a characteristic facial appearance that includes micrognathia, broad alveolar ridges, anteverted nares, and microcephaly. In 1993, Irons et al. [2,3] described an abnormal plasma sterol profile consisting of low cholesterol and elevated 7-dehydrocholesterol (7-DHC) in a subject with SLOS. This finding strongly suggested that mutations of DHCR7, the gene encoding the enzyme DHCR7 likely were the cause of SLOS. In 1998, DHCR7 was cloned and subsequently three groups independently identified DHCR7 mutations in SLOS patients [4–7]. The advent of biochemical and molecular testing lead to a significant expansion of the SLOS phenotype which ranges from severely affected individuals with multiple malformations and high mortality to a mild disorder including behavioral abnormalities and learning impairments. SLOS is a panethnic disorder; however, the carrier frequency appears to be higher in Caucasians, especially of Northern European heritage where it approaches 1–2 % [8–10]. Currently the mainstay of therapy is oral cholesterol supplementation. Even though this intervention has shown some somatic benefit, new therapies are needed to address the neurological deficits.
2. Clinical phenotype
2.1 General overview
The SLOS phenotype is extremely broad and ranges from severely affected infants with multiple major congenital malformations who die in the perinatal period to mildly affected individuals with minor physical anomalies and a behavioral phenotype that frequently includes autistic traits [11]. SLOS severity is a continuum and cases at the severe end of the spectrum correspond to what was previously recognized as SLOS type II. The SLOS phenotypic spectrum likely merges with normal. A severity scale first developed by Bialer [12] then modified by Kelley and Hennekam [13] can be used to clinically classify patients. This rating scale is based on the degree of malformations present in ten developmental domains. The major limitation of this scale is that it does not account for behavioral and cognitive deficits.
2.2 Dysmorphic features
Although the SLOS phenotype is variable, the facial features tend to be readily recognizable (Figure 1A-F). Typical craniofacial features include microcephaly with bitemporal narrowing, a short upturned nose with anteverted nares, unilateral or bilateral ptosis, epicanthus, and retrognathia [1,14]. The lateral view frequently shows flat facial profile and low set ears. Postaxial polydactyly of hands (Figure 2A) or feet can be found in more severe cases, and cutaneous 2,3 syndactyly of the toes (Figure 2B) is usually noted, although its very high frequency may be due to ascertainment bias. Oral features can include dental crowding, broad alveolar ridges, cleft palate, and bifid uvula (Figure 2C). Cleft lip is uncommon. The classical facial appearance changes over time with some features becoming less apparent with age. Adult individuals may look older than their chronological age [14]. The nose often appears more bulbous and the lips fuller than in children, making the typical gestalt less apparent. However, a recent anthropometric study that evaluated 42 individuals with SLOS demonstrated that standardized measurements of the craniofacial patterning were consistent from infancy to adulthood [15].
Figure 1. SLOS craniofacial phenotype.
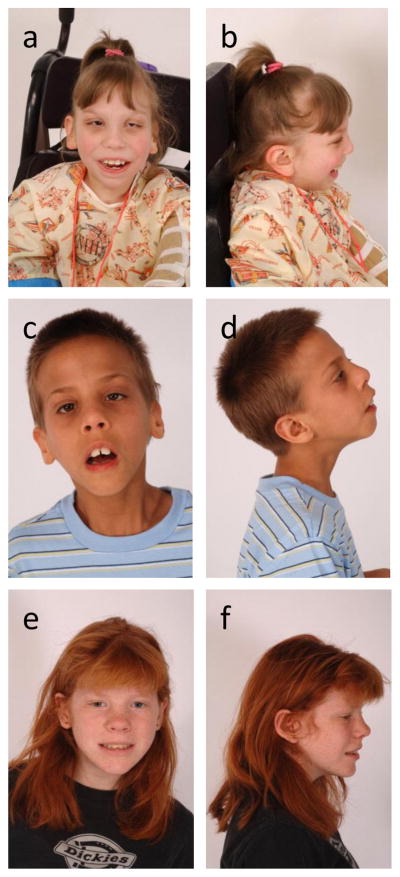
Portrait and profile views of severe (1a, 1b), classical (1c, 1d), and mild disease (1e, 1f). Characteristic craniofacial features include microcephaly, bitemporal narrowing, midface hypoplasia, small upturned nose, ptosis, strabismus, a flat facial profile and micrognathia.
Figure 2. SLOS malformations.

Limb anomalies include postaxial polydactyly of the hands (2a) or feet, a short posteriorly placed thumb (2a), and syndactyly of the 2nd and 3rd toe (2b). A bifid uvula (2c) is the mildest form of a midline cleft palate. Permission for the publication of photographs was obtained from guardians.
2.3 Manifestations in the fetal period
Although most cases of SLOS are not diagnosed during prenatal ultrasound evaluations, in many pregnancies there appear to be findings that would support additional evaluation to exclude SLOS. A recent case series of 10 individuals diagnosed with SLOS during the fetal period described by Quélin [16] shows that among the 10 cases, 4/8 had increased nuchal translucency on screening, 7/10 had intrauterine growth restriction (IUGR) determined by prenatal ultrasound, 7/10 had oligo- or anhydramnios noted, 5/10 had polydactyly in upper or lower extremity noted on ultrasound, 9/10 had a heart defect (7 atrioventricular canal defects, 1 ventricular septal defect, 1 pulmonary valve dysplasia). In addition, various other malformations were noted on prenatal ultrasound including agenesis of the corpus callosum, microcephaly, abnormal gyration of hemispheres, cleft lip, cleft palate, sexual ambiguity with male karyotype, bilateral agenesis of the kidneys. On autopsy these abnormalities were confirmed and moreover all fetuses had 2,3 toe syndactyly and facial dysmorphism consistent with SLOS. It was further noted that 8/10 had lobation anomalies of the lung, and 1/10 had lung hypoplasia secondary to renal hypoplasia. A previous case series describes the antenatal manifestations of 30 cases, of which 10 were diagnosed antenatally and 20 were diagnosed at or after birth [17]. When considering the 20 cases diagnosed after birth, the findings on prenatal ultrasound were milder with 7 cases having isolated IUGR, and six cases having IUGR associated with another abnormal finding, such as nuchal edema, urinary tract dilatation, ventricular septal defect and aortic hypoplasia, or polydactyly. Two additional cases had isolated cardiac defects and 5 cases had no abnormalities noted on antenatal ultrasounds.
2.4 Manifestations of the neonatal period and infancy
The SLOS phenotype is readily apparent in most cases at birth. Malformations identified at birth include postaxial polydactyly, syndactyly of the second and third toes, cleft palate or bifid uvula. The hands often show a single palmar crease and a proximally placed thumb. Poor neonatal growth, poor suck, formula intolerance, significant gastrointestinal reflux and constipation frequently contribute to a diagnosis of failure to thrive. However, even with the provision of adequate calories, growth typically remains poor during the neonatal period. The lack of adequate feeding often requires gastric tube placement, and a continuous feeding schedule is frequently required to provide sufficient calories. Because growth does not respond normally to caloric intake, a healthy infant and slow and steady weight gain are the goals, rather than trying to obtain a normal growth pattern. If an infant does not adequately feed, it is unlikely that one can temporize with nasogastric tube feedings. The feeding issues can be further complicated by the development of liver disease, pyloric stenosis, or by the presence of malformations, such as colonic aganglionosis or malrotation. In addition, infants often manifest irritability, hypersensitivity to the environment and may develop feeding aversion. Genital malformations range from chordee or hypospadias to sex reversal with a male karyotype and female appearing or ambiguous genitalia with bilateral cryptorchidism [12,18]. In severely affected newborns, electrolyte abnormalities and hypoglycemia due to adrenal insufficiency have been reported and may contribute to the increased morbidity and mortality [19,20]. No evidence of adrenal insufficiency was found in a cohort of mild to moderately affected individuals [21]. Likewise severely affected individuals may show signs of bile acid deficiency as neonates, but no deficiency was noted in mildly affected patients [22]. A variable extent of liver disease and structural abnormalities may be seen in SLOS [13]. Rossi et al. [23] suggest that hepatic dysfunction is underappreciated and reported hepatic dysfunction in 16% of SLOS cases. Prenatal cataracts, most often not impairing vision, have been described in about one eighth of cases [24]. Postnatal development of cataracts has also been described [25]. Strabismus is not uncommon. Imaging of the central nervous system often shows malformations, which range from septum pellucidum abnormalities and agenesis of the corpus callosum to holoprosencephaly in severe cases [26,27].
2.5 Manifestations in childhood
During childhood, behavior, feeding issues, and gastrointestinal symptoms remain major concerns. Abnormal gastrointestinal mobility manifesting with recurrent vomiting and reflux, in combination with very selective food intake due to oral sensitivity, with avoidance of specific tastes and textures, likely contributes to poor growth [28]. Specific growth charts have been developed, and individuals should be plotted on these in addition to the regular World Health Organization/Centers for Disease Control and Prevention standardized pediatric growth charts [29]. Constipation can become a severe problem further exacerbating abdominal symptoms. An increased rate of otitis media, frequent skin infections and respiratory infections can be seen in childhood without evidence of an underlying immune deficiency [13]. The initial hypotonia of infancy slowly improves and often progresses to increased tone, especially in the extremities, which may lead to contractures. Individuals suffer from an increased sensitivity to light, which manifests as photosensitivity reaction of the skin and significant photophobia [30]. Autistic traits, in addition to mild to moderate intellectual disability contribute to the typical behavioral phenotype [31,32]. Abnormal sleep patterns with difficulty settling, frequent night awakening, and early morning awakenings are often noted by parents [33]. A correlation between the severity of the cholesterol synthesis defect and the biological genesis of challenging behavior has been described [34].
2.6 Adult manifestations
The adult phenotype is characterized by shorter than average stature. Food aversions and gastrointestinal symptoms may persist. The psychiatric manifestations often include depression and anxiety. The degree of intellectual disability varies, most often mild to moderate intellectual disability are seen, although both borderline intellectual disability and individuals with normal intelligence have been reported [35]. After menarche a gynecologist should be consulted regularly. The hormonal changes may lead to worsening behavior with increased aggression and self-injury related to the menstrual cycle. Fertility should be assumed, and a pregnancy with good outcome has been reported in a mildly affected individual [35]. Even though no other pregnancies have been reported, an adequate form of birth control is recommended. Anecdotally, hormonal based birth control can improve irritability associated with menses. Currently, there are no systematic studies of adult morbidity and mortality.
3. Pathogenesis
3.1 Biochemical abnormality/molecular abnormality
The underlying biochemical defect is due to decreased reduction of the C(7–8) double bond of 7-DHC to yield cholesterol in the final step of the Kandutsch-Russel synthetic cholesterol pathway [2,3] (Figure 3). This enzymatic step is catalyzed by DHCR7. Deficient DHCR7 activity results in elevated levels of 7-DHC and its isomer 8-dehydrocholesterol (8-DHC), and decreased levels of cholesterol. Serum cholesterol is often low, although it can be in the normal range.
Figure 3.

The ultimate step of cholesterol biosynthesis from 7-DHC to cholesterol including down-stream pathways that cholesterol and its precursors are utilized in.
DHCR7 was cloned and mutations initially identified in 1998 [4–7]. The gene maps to chromosome 11q12-13 and consists of 9 coding exons. DHCR7 is an integral membrane protein; however, its membrane topology has not been experimentally determined. Based on in silico modeling, Waterham and Wanders [36] proposed six transmembrane domains and Fitzky [4] proposed nine. We found that the Protter predictive algorithm [37] predicts a protein with seven transmembrane domains (Figure 4). All of these topology models should be viewed as hypothetical until experimental data is available. Since the initial sequencing of DHCR7, over 150 mutations and multiple benign variants have been reported [38] (see Figure 4 and Table 1). A database of DHCR7 mutations has been established [39]. The most frequent mutation in the United States (US) population c.964-1G>C (IVS8-1G>C) is a splice acceptor mutation which results in an insertion of 134 base pairs. When ascertained in SLOS patients, the c.964-1G>C mutation accounts for approximately a third of the mutant alleles. It is likely underreported, due to the fact that in the homozygous state, a severe, often prenatal lethal, phenotype is present [40,41]. The twelve most frequent mutations, c.964-1G4C (30%), p.T93M (10%), p.W151X (6%), p.R404C (5%), and p.V326L (5%), R352W (3%), E448K (3%), G410S (2%), R242C (2%), S169L (2%), F302L (1%), and R242H (1%) account for approximately two thirds of all mutant alleles found in DHCR7 [42]. More recently, a number of whole exon deletions have been found to be causative for SLOS [43,44]. Many cases are due to one frequent mutation in combination with a unique or low frequency mutation. Even though an effort has been made to find a correlation between the severity of SLOS and the genotype, the correlations are not strong enough to allow for counseling in individual cases. Generally, it can be stated that an individual with two null mutations or with mutations in putative loop 8 or 9, in the 9 transmembrane model, will have a more severe phenotype [38]. Patients with two missense mutations seem to be more mildly affected. For the majority of individuals who harbor a combination of a nonsense and missense mutation, phenotype-genotype correlation is relatively poor. Modifier genes likely influence the SLOS phenotype. Maternal ApoE and ABCA1 genotypes, perhaps by altering maternoplacental cholesterol transfer, appear to modulate SLOS phenotypic severity [45,46].
Figure 4.

Putative membrane structure of DHCR7 with location of mutations. Accompanying table (see supplemental materials Table 1) specifies the mutations and if they have been clinically identified or are predicted to be pathogenic utilizing PolyPhen-2 [115].
3.2 Pathophysiology
Cholesterol is one of the most ubiquitous lipids in the body and has multiple physiological functions. Cholesterol is found in all cells as a critical component of cellular membranes. In cellular membranes it has a major influence on fluidity and intracellular signaling [47]. Cholesterol is the precursor for steroid hormones, bile acids, and oxysterols. 7-dehydrocholesterol is the precursor for vitamin D. Cholesterol also plays a major role in embryonic development in that it is involved in hedgehog signaling [48]. In SLOS one typically observes both a cholesterol deficiency and an elevation of dehydrocholesterol levels. Thus one needs to consider whether the observed pathology is due to cholesterol deficiency, a toxic effect of dehydrocholesterol, or a combination of these two factors.
A number of studies have demonstrated that dehydrocholesterol combined with cholesterol deficiency clearly alters membrane properties. Boeze-Battaglia et al. [49] were able to determine an altered ratio of sterols and fatty acids, and changes in rigidity in membranes isolated from cells treated with AY9944, an inhibitor of DHCR7. Gondre- Lewis et al. [50] demonstrated that cholesterol derivatives incorporated into the cell membrane caused fewer granules and morphologically aberrant granules to be produced. Haldar et al. [51] showed that incorporation of 7-DHC or other cholesterol precursors significantly alters the electrostatic properties of biological membranes, a finding that implicates the possibility of changes in the activity of ion-dependent ATPases and ion channels. Transmembrane signaling is also perturbed. DHCs are incorporated into the lipid raft domains of mast cells in a mouse model of SLOS and decrease the stability of the lipid rafts, leading to an increase in degranulation of mast cells and to an enhanced cytokine response [52]. Altered sterol composition in SLOS fibroblast lead to decreased cavolein levels in the cell membrane, and therefore, altered signaling [53]. In an in-vitro model of SLOS utilizing a pharmacological DHCR7 inhibitor, the lipid raft protein composition was shown to be changed [54]. Paila et al. [55] showed that the serotonin 1A receptor, that requires cholesterol for functioning, exhibits reduced ligand binding, and consequently, reduced signaling efficiency, when elevated levels of 7-DHC and reduced cholesterol levels were present. Serotonin signaling may be altered in SLOS patients. Analysis of cerebral spinal fluid from 21 SLOS patients demonstrated decreased levels of 5-hydroxyindoleacetic acid, a serotonin metabolite [56]. Although the effects on any given cellular signaling process may be relatively minor, the potential additive impact of a general alteration in cellular signaling pathways may have a major impact on cellular function.
DHC, like cholesterol, can serve as a precursor molecule for sterol metabolites. Both 7- and 8-DHC can enter the steroid biosynthetic pathway. DHC derivatives of pregnanolone and allopregnanolone have been identified in SLOS patients [57]. To date, the biological activity of these DHC-derived steroids has not been defined. Theoretically, these derivatives could have agonistic or antagonistic hormonal effects via multiple steroid hormone receptors. Synthesis of DHC bile acids [58,59] and enzymatically produced oxysterols [60–62] have been described. Again, the biological activity of DHC-derived oxysterols and bile acids has not been systematically studied. In most cases, the level of cholesterol-derived sterol metabolites also has not been systematically evaluated in SLOS patients, thus the degree to which cholesterol deficiency might contribute to impaired steroid hormone, bile acid or oxysterol function is not known.
In addition to enzymatically produced oxysterols, cholesterol can undergo nonenzymatic oxidation leading to production of biologically active oxysterols [63,64]. 7-DHC is extremely sensitive to oxidation [65]. The extreme sensitivity of 7-DHC to non-enzymatic oxidation combined with the potential toxicity of the resulting oxidative metabolites provides the rational for a controlled trial of antioxidants in SLOS [63,66]. For example, in a pharmacologically induced SLOS rat model the retinas were found to be light-sensitive and showed histological signs of retinal degeneration, a development that could be prevented by administration of a synthetic antioxidant prior to light exposure [67].
Cholesterol deficiency during development may impair proper signaling by hedgehog family members. Cholesterol is necessary for the normal maturation and signaling of hedgehog, a family of proteins that play critical roles in pattern formation during embryonic development [48,68]. Impaired hedgehog signaling plausibly underlies some of the malformations found in SLOS such as holoprosencephaly/agenesis of the corpus callosum and postaxial polydactyly [68].
4. Epidemiological aspects
Although most frequently described in individuals of European descent, SLOS cases have been reported in individuals of African, Arabic and Japanese heritage [69–74]. Witsch-Baumgartner and coworkers [8,75] have characterized the distribution of DHCR7 mutations in European populations. The common splice acceptor mutation c.964-1G>C arose in the British Isles and decreases in frequency as one progresses eastward across Europe. In contrast p.W151X and p.V326L appear to have arisen in Northeastern Europe and p.T93M seems to have originated in the Mediterranean area. A missense mutation, p.R352Q appears to be the most frequent mutation in Japanese SLOS patients [74]. Based on biochemical testing, Kelley [76] reported an incidence of 1/50,000 in the US. Recently Cross et al. [9] analyzed exome sequence from 17,836 alleles and estimated a carrier frequency of 1.01%, corresponding to a predicted SLOS incidence of approximately 1/39,000. Discrepancies between projected cases and actual clinical incidence are likely due to high intrauterine mortality of severe cases and perhaps underdiagnoses of mild cases. Unless region or ethnic group specific carrier frequencies have been determined, for genetic counseling purposes, a carrier frequency of 1–2% in Caucasians is a conservative and reasonable estimate.
5. Diagnosis
5.1 Postnatal diagnosis
Upon clinical suspicion the recommended screening for SLOS is measurement of plasma 7-DHC via gas chromatography- mass spectrometry. Values above 2 mcg/mL are considered abnormal. Plasma cholesterol is typically reduced, but may be normal. Most clinical laboratories measure cholesterol using a cholesterol oxidase-based test, which is not specific for cholesterol. Thus, the measured value will include cholesterol precursors (such as 7- and 8-DHC) as well as cholesterol, and can therefore be falsely elevated. A normal plasma cholesterol level does not exclude SLOS. Plasma 7-DHC is a sensitive marker for detection of SLOS, but false positive elevations have been noted with psychoactive medications including aripiprazole and trazodone, which are inhibitors of DHCR7 [13,77]. Other psychoactive agents, such as haloperidol, can lead to increases in 7-DHC levels by increasing cholesterol synthesis [78]. Other conditions that increase cholesterol synthesis, such as bile acid loss after ileal resection, can also result in increased 7-DHC levels, leading to diagnostic difficulties. In these cases molecular testing of DHCR7 is recommended. Sequence analysis can detect up to 96% of mutations. If no mutation or only one mutation is found, a quantitative test such as quantitative PCR, multiple probe amplification analysis, or chromosomal microarray with coverage of DHCR7 and the flanking region is recommended, since multiple exon deletions have been described [43,44]. If molecular analysis does not identify pathogenic mutations and the potential confounder leading to a possible false positive biochemical test cannot be removed, then the sterol profile of fibroblasts or lymphoblasts grown in cholesterol depleted media can be used to establish a diagnosis (CAW and FDP unpublished observation). Carriers cannot be identified via biochemical assays, since their biochemical profile overlaps normal subjects’ profile [79]. If the primary proband’s mutations are known, carriers can be identified through molecular analysis.
5.2 Prenatal diagnosis
If intrauterine growth restriction or multiple congenital anomalies are noted during fetal ultrasound, it is possible to test either tissue obtained during chorionic villus sample or amniotic fluid obtained during amniocentesis for an abnormal level of 7-DHC via gas-chromatography- mass spectrometry [80]. A large multi-center trial showed that abnormal sterols can be screened for in maternal urine. The measurement of 7-dehydropregnanetriol to pregnanetriol ratio was found to be a reliable non-invasive diagnostic test in the second trimester [81]. Sometimes prenatal testing for SLOS is pursued after unusually low levels of unconjugated estriol are noted on second trimester maternal serum screening test. Since fetoplacental synthesis of unconjugated estriol requires cholesterol as a precursor, pregnancies affected by SLOS may have low unconjugated estriol levels, while the other two markers, alpha-fetoprotein and human chorionic gonadotropin stay relatively unaffected. Palomaki et al. [82] describe an algorithm that can be used to assess risk of SLOS in an abnormal screening result.
5.3 Future diagnostic trends
Currently there are about 20 different commercially available panels that include massively parallel sequencing techniques and include the DHCR7 gene in their analysis [83]. The clinical indications range from autism or developmental delay to cholestatic disease and epilepsy. It is unclear if this development will further broaden the phenotype with new characterizations of atypical or hypomorphic presentations. In addition, more and more disorders are being included in newborn screening panels in an effort to ease the diagnostic journey and make disorders amenable to presymptomatic therapies, as they become available. There has been an effort to develop techniques to analyze 7-DHC in dried blood spots [84,85]. However, one of the main problems remains that 7-DHC is not as stable as other diagnostic compounds on dried blood spots, so that pretreated filter paper may be needed [86].
6. Treatment and management
6.1 Pharmacological management
6.1.1 Cholesterol supplementation
The majority of SLOS patients are treated with oral cholesterol supplementation via administration of food-based cholesterol (such as egg yolks), crystalline cholesterol suspension in oil or in Ora Plus® (a colloidal aqueous solution used in medication preparations), or a microencapsulated cholesterol powder. The cholesterol preparations must be compounded at a pharmacy. The microencapsulated cholesterol is currently available as Cholextra® [87] or as SLOesterol® [88] in the US. Since the absorption of cholesterol is presumed to be different depending on the actual formulation, different doses are recommended depending on the mode of administration [89]. Cholesterol supplementation leads to improved cholesterol levels through increased absorption in the diet [90]. Chan et al. showed that the fractional cholesterol synthesis rate was lower on a high cholesterol diet in comparison to a low cholesterol diet in individuals with SLOS [91]. In addition, the higher cholesterol levels provide a feedback inhibition for endogenous synthesis, so that less 7-DHC is produced. Case series have reported improvement in symptoms, including growth, susceptibility to infection and behavior [92–94]. Improvements in skin photosensitivity have been quantified [95]. However, a double blinded, cross-over study could find no difference in short term behavior when patients were on cholesterol supplementation or on placebo [96]. A number of factors likely limit the efficacy of dietary cholesterol synthesis. In vitro studies indicate that SLOS cells have impaired intracellular cholesterol transport, suggesting a possible defect in the ability of SLOS cells to properly utilize exogenous cholesterol [97]. In addition, dietary cholesterol does not cross the blood-brain-barrier to any significant extent and the central nervous system is dependent upon endogenous synthesis [98].
6.1.2 Bile acid replacement
In severe cases of SLOS there may be cholestatic liver disease in infancy [99]. In these cases bile acid replacement can be considered. Bile acid replacement was also tried in combination with cholesterol supplementation as a means to increase intestinal cholesterol absorption, but no difference was found in plasma cholesterol levels compared to patients only receiving cholesterol [93]. There is no evidence of bile acid production deficit in mild to moderate phenotypes.
6.1.3 Fresh frozen plasma
When an SLOS patient experiences acute severe illness or undergoes a major surgical procedure, one can consider the use of fresh frozen plasma (FFP). FFP contains cholesterol rich lipoproteins, such as LDL. This intervention has not been validated with clinical trials, but there is anecdotal evidence supporting this therapy in acutely ill or severely stressed patients [13,100,101]. There is also a report of FFP being used in the antenatal setting via fetal intravenous and fetal intraperitoneal transfusion [102].
6.1.4 Stress steroid dosing
Even though moderate to mild cases do not show evidence of adrenal insufficiency [21], in severely affected individuals adrenal insufficiency and the need of stress steroid replacement should be considered if the clinical picture is indicative of adrenal insufficiency.
6.1.5 Simvastatin
Several case reports and a case series suggest that adding HMG-CoA reductase inhibitors may benefit patients by improving the cholesterol profile [103–105]. Jira et al. [103] studied the effects of simvastatin on two patients and reported a significant increase in serum cholesterol with concomitant reduction in cholesterol precursors. Shortly after this, Starck et al. [106] also attempted treatment with simvastatin in two patients with SLOS, and noted some decrease in dehydrocholesterol, but no notable improvement of cholesterol levels. One of these patients experienced liver dysfunction which required stopping simvastatin treatment. The retrospective study [104], which included 14 SLOS patients receiving simvastatin, showed that the improvement in cholesterol/dehydrocholesterol ratio was due to decrease in 7- and 8- DHC, without any notable increase in cholesterol levels. Chan et al. [91] showed that adding simvastatin to a high cholesterol diet in three patients further decreased their 7-DHC levels while maintaining their total cholesterol levels.
6.2 Clinical management
6.2.1 Health supervision and general management
Management includes regular physical examinations with special attention paid to growth parameters such as weight, height and head circumference, which ideally should be plotted on SLOS specific charts in addition to standard CDC or WHO charts [29]. In phenotypically female patients with manifestations of a severe phenotype, a karyotype to exclude sex reversal is recommended, and if necessary, subsequent removal of intraabdominal testes. Genital malformations such as hypospadias or cryptorchidism may need surgical repair. Cleft palate may need surgical intervention. Limb malformations, such as polydactyly, may need repair to attain optimal functioning.
6.2.2 Gastrointestinal management
Treatment of the growth failure, due to dysphagia, reflux, oromotor dysfunction, and feeding aversion involves speech and swallowing therapy, nutritional support, and placement of a gastrostomy tube to meet nutritional needs, if necessary. Gastrointestinal manifestations such as pyloric stenosis and colonic aganglionosis need surgical intervention.
6.2.3 Neurological and psychiatric management
A developmental pediatric specialist should be involved in care to assess developmental delay, the appearance of autistic features, and provide timely interventions, such as physical therapy or occupational therapy. Communication can be facilitated by using sign language, picture based communication devices, or computer based communication devices. A pediatric neurologist should follow the patients regularly and assess hypotonia, hypertonia, the presence of seizures, and symptoms of autism spectrum disorder or attention deficit hyperactivity disorder. If independent ambulation is reached, ankle-foot orthoses are often needed due to poor tone and ensuing instability. If spasticity occurs, botulinum toxin injections or tendon release procedures may be necessary. Behavioral problems such as self-injurious behavior, compulsion, obsessions and anxiety may require psychiatric evaluation and medications in childhood. Affect dysregulation can be noted in infancy and childhood and may manifest with depressive or psychotic symptoms in adults necessitating psychiatric care. If pharmacological intervention is needed care should be taken to try to avoid psychoactive medications that interfere with DHCR7 metabolism (haloperidol, trazodone, and aripiprazole). Serum 7- DHC levels and clinical status should be monitored frequently to avoid negative consequences of therapy. Sleep disturbances may need referral to a sleep specialist for further evaluation and management.
6.2.4 Ophthalmological management
A baseline ophthalmologic assessment is needed for assessment of cataracts, strabismus, and other ocular malformations, such as retinal dysplasia or optic nerve atrophy [107]. Some ocular manifestations, such as strabismus, cataracts, and ptosis, may require surgical intervention.
6.2.5 Cardiological management
A baseline cardiological examination including echocardiography and electrocardiogram is recommended to assess for congenital malformations. SLOS patients should be monitored for the development of hypertonia and treated if necessary. The decision, how to approach cardiac anomalies, should be made on an individual basis with the input of a pediatric cardiologist, a neonatologist and the parents, and depends on the severity of the cardiac malformation in combination with the overall health of the patient.
6.3 Genetic counseling
All families should be referred to genetic counseling services. Carrier detection using molecular techniques should be reserved to adults or minors who are pregnant or considering a pregnancy, according to the current published recommendations [108]. The recurrence risk of SLOS is 25 % with each conception. In high risk pregnancies based on a positive family history, the diagnosis can be made by measuring the 7-DHC level of amniotic fluid collected at fifteen to eighteen weeks gestation or in tissue obtained by chorionic villus sampling at ten to twelve weeks gestation [109]. Preimplantation genetic testing may be an option for affected families if the pathogenic mutations are known.
7. Expert Opinion
SLOS is a unique disorder in that it is both a malformation syndrome and an inborn error of metabolism. Thus, the clinical problems found in SLOS may arise from developmental defects or be a consequence of altered sterol biochemistry. Developmental defects arise in utero and thus are not likely to be amendable to therapeutic intervention in the majority of cases since malformations associated with disturbed pattern formation occur prior to recognition of pregnancy. However, potentially one could consider early interventions in “at risk” pregnancies. During normal development the vast majority of fetal cholesterol is synthesized by the fetus itself and the placenta is considered to be a barrier to maternal cholesterol transport; however, some data suggest that modulation of maternal factors influencing cholesterol metabolism and homeostasis could potentially impact clinical severity. For example, Witsch-Baumgartner et al. [46] showed that a more severe SLOS phenotype is associated with maternal heterozygosity for an ApoE2 allele. ApoE2 binding to LDL receptor is not as efficient as with other ApoE alleles. The paternal ApoE phenotype has no impact on severity of SLOS in the offspring. Lindegaard et al. [110] demonstrated that maternal cholesterol transport, although still very limited, can be modulated by treatment with LXR agonists. These data suggest that one could theoretically modulate maternal transfer of cholesterol to the developing fetus and, if done early enough in pregnancy, could potentially decrease developmental defects. However, a major limitation of this approach is that one would also need to determine how to increase cholesterol transport across the fetal blood-brain-barrier. Work by Tint et al. [111] utilizing an SLOS mouse model has shown that the blood-brain- barrier becomes impermanent to peripheral cholesterol early in development of the central nervous system. Although early developmental defects that give rise to structural abnormalities might be prevented, the biochemical abnormalities would persist in the CNS once the blood-brain-barrier precludes transfer of cholesterol to the developing central nervous system.
The issue of blood-brain-barrier impermanence to cholesterol transport is a major limiting factor in the current treatment of SLOS behavioral and cognitive issues. Although multiple case reports indicate subjective and anecdotal improvement in behavior after SLOS patients are started on dietary cholesterol supplementation, a direct effect of dietary cholesterol on CNS function is not biologically plausible due to the presence of the blood-brain-barrier. Although indirect mechanisms, such as alteration of neuroactive steroids, can be postulated, it is critical to demonstrate that the reported beneficial effect of dietary cholesterol on behavior is real. To date this has not been demonstrated in a controlled study. In fact, the only reported placebo-controlled trial of dietary cholesterol supplementation identified no effect of dietary cholesterol supplementation on aberrant behavior [96]. Without controlled evidence that dietary cholesterol supplementation has a demonstrable effect on behavior, it is unlikely that hypothetical indirect mechanisms, which could give insight into directed therapy, will be explored in detail.
One possible solution to circumventing the impermeable blood-brain-barrier limiting dietary cholesterol supplementation could be using an approach focused on increasing expression of the endogenous enzyme. Most SLOS patients with a mild to classical phenotype express a DHCR7 allele with residual enzymatic function. Increased expression of DHCR7 mutant alleles or stabilization of the mutant DHCR7 protein may have therapeutic potential. In fact, simvastatin was found to lead to an increase in cholesterol levels by increasing expression of mutant DHCR7 proteins with residual enzymatic activity [112,113]. High-throughput drug screens evaluating drugs that have the potential to stabilize proteins and could be repurposed for SLOS need to be conducted. Initial experiments in a mouse model have explored the potential of gene therapy using adeno-associated virus vectors [114].
The observation that 7-DHC is extremely sensitive to oxidation and the oxidative metabolites are potentially toxic suggests that antioxidant therapy directed at reducing 7-DHC derived metabolites may play a role in the treatment of SLOS. In fact, a recent preclinical trial utilizing human SLOS fibroblasts showed that an antioxidant mixture consisting of vitamin A, coenzyme Q10, vitamin C and vitamin E improved the biochemical phenotype [66]. There was even some evidence that when the antioxidant mixture was fed to pregnant mice the oxysterols measurement in the pups’ brain were lowered. However, like dietary cholesterol supplementation, one needs to be very skeptical of subjective, anecdotal reports of efficacy in SLOS patients. It is absolutely critical that this potential treatment modality be evaluated in a placebo-controlled trial.
Supplementary Material
Acknowledgments
J.L.C. is co-supervised in her studies by Professor Frances Platt, University of Oxford, and is a recipient of the Wellcome Trust/NIH Studentship. FDP, CAW and SB are supported by the intramural research program of the Eunice Kennedy ShriverNational Institute of Child Health and Human Development.
References
- 1*.Smith DW, Lemli L, Opitz JM. A newly recognized syndrome of multiple congenital anomalies. J Pediatr. 1964;64:210–7. doi: 10.1016/s0022-3476(64)80264-x. first description. [DOI] [PubMed] [Google Scholar]
- 2.Irons M, Elias ER, Salen G, Tint GS, Batta AK. Defective cholesterol biosynthesis in Smith-Lemli-Opitz syndrome. Lancet. 1993;341(8857):1414. doi: 10.1016/0140-6736(93)90983-n. [DOI] [PubMed] [Google Scholar]
- 3.Tint GS, Irons M, Elias ER, Batta AK, Frieden R, Chen TS, et al. Defective cholesterol biosynthesis associated with the Smith-Lemli-Opitz syndrome. N Engl J Med. 1994;330(2):107–13. doi: 10.1056/NEJM199401133300205. [DOI] [PubMed] [Google Scholar]
- 4.Fitzky BU, Witsch-Baumgartner M, Erdel M, Lee JN, Paik YK, Glossmann H, et al. Mutations in the Delta7-sterol reductase gene in patients with the Smith-Lemli-Opitz syndrome. Proc Natl Acad Sci U S A. 1998;95(14):8181–6. doi: 10.1073/pnas.95.14.8181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Waterham HR, Wijburg FA, Hennekam RC, Vreken P, Poll-The BT, Dorland L, et al. Smith-Lemli-Opitz syndrome is caused by mutations in the 7-dehydrocholesterol reductase gene. Am J Hum Genet. 1998;63(2):329–38. doi: 10.1086/301982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wassif CA, Maslen C, Kachilele-Linjewile S, Lin D, Linck LM, Connor WE, et al. Mutations in the human sterol delta7-reductase gene at 11q12-13 cause Smith-Lemli-Opitz syndrome. Am J Hum Genet. 1998;63(1):55–62. doi: 10.1086/301936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moebius FF, Fitzky BU, Lee JN, Paik YK, Glossmann H. Molecular cloning and expression of the human delta7-sterol reductase. Proc Natl Acad Sci U S A. 1998;95(4):1899–902. doi: 10.1073/pnas.95.4.1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Witsch-Baumgartner M, Schwentner I, Gruber M, Benlian P, Bertranpetit J, Bieth E, et al. Age and origin of major Smith-Lemli-Opitz syndrome (SLOS) mutations in European populations. J Med Genet. 2008;45(4):200–9. doi: 10.1136/jmg.2007.053520. [DOI] [PubMed] [Google Scholar]
- 9.Cross JL, Iben J, Simpson CL, Thurm A, Swedo S, Tierney E, et al. Determination of the allelic frequency in Smith-Lemli-Opitz syndrome by analysis of massively parallel sequencing data sets. Clin Genet. 2014 doi: 10.1111/cge.12425. [epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nowaczyk MJ, Waye JS, Douketis JD. DHCR7 mutation carrier rates and prevalence of the RSH/Smith-Lemli-Opitz syndrome: where are the patients? Am J Med Genet A. 2006;140(19):2057–62. doi: 10.1002/ajmg.a.31413. [DOI] [PubMed] [Google Scholar]
- 11.Langius FA, Waterham HR, Romeijn GJ, Oostheim W, de Barse MM, Dorland L, et al. Identification of three patients with a very mild form of Smith-Lemli-Opitz syndrome. Am J Med Genet A. 2003;122A(1):24–9. doi: 10.1002/ajmg.a.20207. [DOI] [PubMed] [Google Scholar]
- 12.Bialer MG, Penchaszadeh VB, Kahn E, Libes R, Krigsman G, Lesser ML. Female external genitalia and mullerian duct derivatives in a 46,XY infant with the smith-lemli-Opitz syndrome. Am J Med Genet. 1987;28(3):723–31. doi: 10.1002/ajmg.1320280320. [DOI] [PubMed] [Google Scholar]
- 13.Kelley RI, Hennekam RC. The Smith-Lemli-Opitz syndrome. J Med Genet. 2000;37(5):321–35. doi: 10.1136/jmg.37.5.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14**.Nowaczyk MJM, Irons MB. Smith Lemli Opitz syndrome: Phenotype, natural history, and epidemiology. American Journal of Medical Genetics Part C: Seminars in Medical Genetics. 2012;160C(4):250–62. doi: 10.1002/ajmg.c.31343. very thorough clinical review, part of an issue dedicated to SLOS. [DOI] [PubMed] [Google Scholar]
- 15.Nowaczyk MJ, Tan M, Hamid JS, Allanson JE. Smith-Lemli-Opitz syndrome: Objective assessment of facial phenotype. Am J Med Genet A. 2012;158A(5):1020–8. doi: 10.1002/ajmg.a.35285. [DOI] [PubMed] [Google Scholar]
- 16.Quelin C, Loget P, Verloes A, Bazin A, Bessieres B, Laquerriere A, et al. Phenotypic spectrum of fetal Smith-Lemli-Opitz syndrome. Eur J Med Genet. 2012;55(2):81–90. doi: 10.1016/j.ejmg.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 17.Goldenberg A, Wolf C, Chevy F, Benachi A, Dumez Y, Munnich A, et al. Antenatal manifestations of Smith-Lemli-Opitz (RSH) syndrome: a retrospective survey of 30 cases. Am J Med Genet A. 2004;124A(4):423–6. doi: 10.1002/ajmg.a.20448. [DOI] [PubMed] [Google Scholar]
- 18.Lachman MF, Wright Y, Whiteman DA, Herson V, Greenstein RM. Brief clinical report: a 46,XY phenotypic female with Smith-Lemli-Opitz syndrome. Clin Genet. 1991;39(2):136–41. doi: 10.1111/j.1399-0004.1991.tb03000.x. [DOI] [PubMed] [Google Scholar]
- 19.Andersson HC, Frentz J, Martinez JE, Tuck-Muller CM, Bellizaire J. Adrenal insufficiency in Smith-Lemli-Opitz syndrome. Am J Med Genet. 1999;82(5):382–4. [PubMed] [Google Scholar]
- 20.Chemaitilly W, Goldenberg A, Baujat G, Thibaud E, Cormier-Daire V, Abadie V. Adrenal insufficiency and abnormal genitalia in a 46XX female with Smith-Lemli-Opitz syndrome. Horm Res. 2003;59(5):254–6. doi: 10.1159/000070226. [DOI] [PubMed] [Google Scholar]
- 21.Bianconi SE, Conley SK, Keil MF, Sinaii N, Rother KI, Porter FD, et al. Adrenal function in Smith-Lemli-Opitz syndrome. Am J Med Genet A. 2011;155A(11):2732–8. doi: 10.1002/ajmg.a.34271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Steiner RD, Linck LM, Flavell DP, Lin DS, Connor WE. Sterol balance in the Smith-Lemli-Opitz syndrome. Reduction in whole body cholesterol synthesis and normal bile acid production. J Lipid Res. 2000;41(9):1437–47. [PubMed] [Google Scholar]
- 23.Rossi M, Vajro P, Iorio R, Battagliese A, Brunetti-Pierri N, Corso G, et al. Characterization of liver involvement in defects of cholesterol biosynthesis: long-term follow-up and review. Am J Med Genet A. 2005;132A(2):144–51. doi: 10.1002/ajmg.a.30426. [DOI] [PubMed] [Google Scholar]
- 24.Ryan AK, Bartlett K, Clayton P, Eaton S, Mills L, Donnai D, et al. Smith-Lemli-Opitz syndrome: a variable clinical and biochemical phenotype. J Med Genet. 1998;35(7):558–65. doi: 10.1136/jmg.35.7.558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goodwin H, Brooks BP, Porter FD. Acute postnatal cataract formation in Smith-Lemli-Opitz syndrome. Am J Med Genet A. 2008;146A(2):208–11. doi: 10.1002/ajmg.a.32084. [DOI] [PubMed] [Google Scholar]
- 26.Lee RW, Conley SK, Gropman A, Porter FD, Baker EH. Brain magnetic resonance imaging findings in Smith-Lemli-Opitz syndrome. Am J Med Genet A. 2013;161(10):2407–19. doi: 10.1002/ajmg.a.36096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weaver DD, Solomon BD, Akin-Samson K, Kelley RI, Muenke M. Cyclopia (synophthalmia) in Smith-Lemli-Opitz syndrome: First reported case and consideration of mechanism. Am J Med Genet C Semin Med Genet. 2010;154C(1):142–5. doi: 10.1002/ajmg.c.30241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Merkens MJ, Sinden NL, Brown CD, Merkens LS, Roullet JB, Nguyen T, et al. Feeding Impairments Associated with Plasma Sterols in Smith-Lemli-Opitz Syndrome. J Pediatr. 2014;165(4):836–41. doi: 10.1016/j.jpeds.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29**.Lee RW, McGready J, Conley SK, Yanjanin NM, Nowaczyk MJ, Porter FD. Growth charts for individuals with Smith-Lemli-Opitz syndrome. Am J Med Genet A. 2012;158A(11):2707–13. doi: 10.1002/ajmg.a.35376. SLOS specific growth charts for clinical use. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Anstey AV, Ryan A, Rhodes LE, Charman CR, Arlett CF, Tyrrell RM, et al. Characterization of photosensitivity in the Smith-Lemli-Opitz syndrome: a new congenital photosensitivity syndrome. Br J Dermatol. 1999;141(3):406–14. doi: 10.1046/j.1365-2133.1999.03032.x. [DOI] [PubMed] [Google Scholar]
- 31.Diaz-Stransky A, Tierney E. Cognitive and behavioral aspects of Smith-Lemli-Opitz syndrome. Am J Med Genet C Semin Med Genet. 2012;160C(4):295–300. doi: 10.1002/ajmg.c.31342. [DOI] [PubMed] [Google Scholar]
- 32.Bukelis I, Porter FD, Zimmerman AW, Tierney E. Smith-Lemli-Opitz syndrome and autism spectrum disorder. Am J Psychiatry. 2007;164(11):1655–61. doi: 10.1176/appi.ajp.2007.07020315. [DOI] [PubMed] [Google Scholar]
- 33.Zarowski M, Vendrame M, Irons M, Kothare SV. Prevalence of sleep problems in Smith-Lemli-Opitz syndrome. Am J Med Genet A. 2011;155A(7):1558–62. doi: 10.1002/ajmg.a.34021. [DOI] [PubMed] [Google Scholar]
- 34.Freeman KA, Eagle R, Merkens LS, Sikora D, Pettit-Kekel K, Nguyen-Driver M, et al. Challenging behavior in Smith-Lemli-Opitz syndrome: initial test of biobehavioral influences. Cogn Behav Neurol. 2013;26(1):23–9. doi: 10.1097/WNN.0b013e31828bf6d5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ellingson MS, Wick MJ, White WM, Raymond KM, Saenger AK, Pichurin PN, et al. Pregnancy in an individual with mild Smith-Lemli-Opitz syndrome. Clin Genet. 2014;85(5):495–7. doi: 10.1111/cge.12209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Waterham HR, Wanders RJ. Biochemical and genetic aspects of 7-dehydrocholesterol reductase and Smith-Lemli-Opitz syndrome. Biochim Biophys Acta. 2000;1529(1–3):340–56. doi: 10.1016/s1388-1981(00)00159-1. [DOI] [PubMed] [Google Scholar]
- 37.Omasits U, Ahrens CH, Muller S, Wollscheid B. Protter: interactive protein feature visualization and integration with experimental proteomic data. Bioinformatics. 2014;30(6):884–6. doi: 10.1093/bioinformatics/btt607. [DOI] [PubMed] [Google Scholar]
- 38**.Waterham HR, Hennekam RC. Mutational spectrum of Smith-Lemli-Opitz syndrome. Am J Med Genet C Semin Med Genet. 2012;160C(4):263–84. doi: 10.1002/ajmg.c.31346. most up to date review of clinically relevant mutations. [DOI] [PubMed] [Google Scholar]
- 39.Lanthaler B, Wieser S, Deutschmann A, Schossig A, Fauth C, Zschocke J, et al. Genotype-based databases for variants causing rare diseases. Gene. 2014;550(1):136–40. doi: 10.1016/j.gene.2014.08.016. [DOI] [PubMed] [Google Scholar]
- 40.Kelley RI, Herman GE. Inborn errors of sterol biosynthesis. Annu Rev Genomics Hum Genet. 2001;2:299–341. doi: 10.1146/annurev.genom.2.1.299. [DOI] [PubMed] [Google Scholar]
- 41.Opitz JM, Gilbert-Barness E, Ackerman J, Lowichik A. Cholesterol and development: the RSH (“Smith-Lemli-Opitz”) syndrome and related conditions. Pediatr Pathol Mol Med. 2002;21(2):153–81. doi: 10.1080/15227950252852078. [DOI] [PubMed] [Google Scholar]
- 42.Correa-Cerro LS, Porter FD. 3beta-hydroxysterol Delta7-reductase and the Smith-Lemli-Opitz syndrome. Mol Genet Metab. 2005;84(2):112–26. doi: 10.1016/j.ymgme.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 43.Aradhya S, Lewis R, Bonaga T, Nwokekeh N, Stafford A, Boggs B, et al. Exon-level array CGH in a large clinical cohort demonstrates increased sensitivity of diagnostic testing for Mendelian disorders. Genet Med. 2012;14(6):594–603. doi: 10.1038/gim.2011.65. [DOI] [PubMed] [Google Scholar]
- 44.Lanthaler B, Hinderhofer K, Maas B, Haas D, Sawyer H, Burton-Jones S, et al. Characterization of large deletions in the DHCR7 gene. Clin Genet. 2014 doi: 10.1111/cge.12454. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 45.Lanthaler B, Steichen-Gersdorf E, Kollerits B, Zschocke J, Witsch-Baumgartner M. Maternal ABCA1 genotype is associated with severity of Smith-Lemli-Opitz syndrome and with viability of patients homozygous for null mutations. Eur J Hum Genet. 2013;21(3):286–93. doi: 10.1038/ejhg.2012.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Witsch-Baumgartner M, Gruber M, Kraft HG, Rossi M, Clayton P, Giros M, et al. Maternal apo E genotype is a modifier of the Smith-Lemli-Opitz syndrome. J Med Genet. 2004;41(8):577–84. doi: 10.1136/jmg.2004.018085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hissa B, Pontes B, Roma PM, Alves AP, Rocha CD, Valverde TM, et al. Membrane cholesterol removal changes mechanical properties of cells and induces secretion of a specific pool of lysosomes. PLoS One. 2013;8(12):e82988. doi: 10.1371/journal.pone.0082988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Porter JA, Young KE, Beachy PA. Cholesterol modification of hedgehog signaling proteins in animal development. Science. 1996;274(5285):255–9. doi: 10.1126/science.274.5285.255. [DOI] [PubMed] [Google Scholar]
- 49.Boesze-Battaglia K, Damek-Poprawa M, Mitchell DC, Greeley L, Brush RS, Anderson RE, et al. Alteration of retinal rod outer segment membrane fluidity in a rat model of Smith-Lemli-Opitz syndrome. J Lipid Res. 2008;49(7):1488–99. doi: 10.1194/jlr.M800031-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gondre-Lewis MC, Petrache HI, Wassif CA, Harries D, Parsegian A, Porter FD, et al. Abnormal sterols in cholesterol-deficiency diseases cause secretory granule malformation and decreased membrane curvature. J Cell Sci. 2006;119(Pt 9):1876–85. doi: 10.1242/jcs.02906. [DOI] [PubMed] [Google Scholar]
- 51.Haldar S, Kanaparthi RK, Samanta A, Chattopadhyay A. Differential effect of cholesterol and its biosynthetic precursors on membrane dipole potential. Biophys J. 2012;102(7):1561–9. doi: 10.1016/j.bpj.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kovarova M, Wassif CA, Odom S, Liao K, Porter FD, Rivera J. Cholesterol deficiency in a mouse model of Smith-Lemli-Opitz syndrome reveals increased mast cell responsiveness. J Exp Med. 2006;203(5):1161–71. doi: 10.1084/jem.20051701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ren G, Jacob RF, Kaulin Y, Dimuzio P, Xie Y, Mason RP, et al. Alterations in membrane caveolae and BKCa channel activity in skin fibroblasts in Smith-Lemli-Opitz syndrome. Mol Genet Metab. 2011;104(3):346–55. doi: 10.1016/j.ymgme.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Keller RK, Arnold TP, Fliesler SJ. Formation of 7-dehydrocholesterol-containing membrane rafts in vitro and in vivo, with relevance to the Smith-Lemli-Opitz syndrome. J Lipid Res. 2004;45(2):347–55. doi: 10.1194/jlr.M300232-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Paila YD, Murty MR, Vairamani M, Chattopadhyay A. Signaling by the human serotonin(1A) receptor is impaired in cellular model of Smith-Lemli-Opitz Syndrome. Biochim Biophys Acta. 2008;1778(6):1508–16. doi: 10.1016/j.bbamem.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 56.Sparks SE, Wassif CA, Goodwin H, Conley SK, Lanham DC, Kratz LE, et al. Decreased cerebral spinal fluid neurotransmitter levels in Smith-Lemli-Opitz syndrome. J Inherit Metab Dis. 2014;37(3):415–20. doi: 10.1007/s10545-013-9672-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marcos J, Guo LW, Wilson WK, Porter FD, Shackleton C. The implications of 7-dehydrosterol-7-reductase deficiency (Smith-Lemli-Opitz syndrome) to neurosteroid production. Steroids. 2004;69(1):51–60. doi: 10.1016/j.steroids.2003.09.013. [DOI] [PubMed] [Google Scholar]
- 58.Nittono H, Takei H, Unno A, Kimura A, Shimizu T, Kurosawa T, et al. Diagnostic determination system for high-risk screening for inborn errors of bile acid metabolism based on an analysis of urinary bile acids using gas chromatography-mass spectrometry: results for 10 years in Japan. Pediatr Int. 2009;51(4):535–43. doi: 10.1111/j.1442-200X.2008.02799.x. [DOI] [PubMed] [Google Scholar]
- 59.Natowicz MR, Evans JE. Abnormal bile acids in the Smith-Lemli-Opitz syndrome. Am J Med Genet. 1994;50(4):364–7. doi: 10.1002/ajmg.1320500413. [DOI] [PubMed] [Google Scholar]
- 60.Meljon A, Watson GL, Wang Y, Shackleton CH, Griffiths WJ. Analysis by liquid chromatography-mass spectrometry of sterols and oxysterols in brain of the newborn Dhcr7(Delta3-5/T93M) mouse: a model of Smith-Lemli-Opitz syndrome. Biochem Pharmacol. 2013;86(1):43–55. doi: 10.1016/j.bcp.2013.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wassif CA, Yu J, Cui J, Porter FD, Javitt NB. 27-Hydroxylation of 7- and 8-dehydrocholesterol in Smith-Lemli-Opitz syndrome: a novel metabolic pathway. Steroids. 2003;68(6):497–502. doi: 10.1016/s0039-128x(03)00090-4. [DOI] [PubMed] [Google Scholar]
- 62.Goyal S, Xiao Y, Porter NA, Xu L, Guengerich FP. Oxidation of 7-dehydrocholesterol and desmosterol by human cytochrome P450 46A1. J Lipid Res. 2014;55(9):1933–43. doi: 10.1194/jlr.M051508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Korade Z, Xu L, Shelton R, Porter NA. Biological activities of 7-dehydrocholesterol-derived oxysterols: implications for Smith-Lemli-Opitz syndrome. J Lipid Res. 2010;51(11):3259–69. doi: 10.1194/jlr.M009365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xu L, Korade Z, Rosado DA, Jr, Mirnics K, Porter NA. Metabolism of oxysterols derived from nonenzymatic oxidation of 7-dehydrocholesterol in cells. J Lipid Res. 2013;54(4):1135–43. doi: 10.1194/jlr.M035733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xu L, Porter NA. Free Radical Oxidation of Cholesterol and Its Precursors: Implications in Cholesterol Biosynthesis Disorders. Free Radic Res. 2014:1–30. doi: 10.3109/10715762.2014.985219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66**.Korade Z, Xu L, Harrison FE, Ahsen R, Hart SE, Folkes OM, et al. Antioxidant supplementation ameliorates molecular deficits in Smith-Lemli-Opitz syndrome. Biol Psychiatry. 2014;75(3):215–22. doi: 10.1016/j.biopsych.2013.06.013. recent study describing therapeutic effects of antioxidant supplementation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vaughan DK, Peachey NS, Richards MJ, Buchan B, Fliesler SJ. Light-induced exacerbation of retinal degeneration in a rat model of Smith-Lemli-Opitz syndrome. Exp Eye Res. 2006;82(3):496–504. doi: 10.1016/j.exer.2005.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cooper MK, Wassif CA, Krakowiak PA, Taipale J, Gong R, Kelley RI, et al. A defective response to Hedgehog signaling in disorders of cholesterol biosynthesis. Nat Genet. 2003;33(4):508–13. doi: 10.1038/ng1134. [DOI] [PubMed] [Google Scholar]
- 69.Hanissian AS, Summitt RL. Smith-Lemli-Opitz syndrome in a negro child. J Pediatr. 1969;74(2):303–5. doi: 10.1016/s0022-3476(69)80082-x. [DOI] [PubMed] [Google Scholar]
- 70.Nezarati MM, Loeffler J, Yoon G, MacLaren L, Fung E, Snyder F, et al. Novel mutation in the Delta-sterol reductase gene in three Lebanese sibs with Smith-Lemli-Opitz (RSH) syndrome. Am J Med Genet. 2002;110(2):103–8. doi: 10.1002/ajmg.10367. [DOI] [PubMed] [Google Scholar]
- 71.Tsukahara M, Fujisawa K, Yamamoto K, Hasui M, Saito C, Yamamaka T, et al. Smith-Lemli-Opitz syndrome in Japan. Am J Med Genet. 1998;75(1):118–9. [PubMed] [Google Scholar]
- 72.Al-Owain M, Imtiaz F, Shuaib T, Edrees A, Al-Amoudi M, Sakati N, et al. Smith-Lemli-Opitz syndrome among Arabs. Clin Genet. 2012;82(2):165–72. doi: 10.1111/j.1399-0004.2011.01742.x. [DOI] [PubMed] [Google Scholar]
- 73.Wright BS, Nwokoro NA, Wassif CA, Porter FD, Waye JS, Eng B, et al. Carrier frequency of the RSH/Smith-Lemli-Opitz IVS8-1G>C mutation in African Americans. Am J Med Genet A. 2003;120A(1):139–41. doi: 10.1002/ajmg.a.10207. [DOI] [PubMed] [Google Scholar]
- 74.Matsumoto Y, Morishima K, Honda A, Watabe S, Yamamoto M, Hara M, et al. R352Q mutation of the DHCR7 gene is common among Japanese Smith-Lemli-Opitz syndrome patients. J Hum Genet. 2005;50(7):353–6. doi: 10.1007/s10038-005-0267-3. [DOI] [PubMed] [Google Scholar]
- 75*.Witsch-Baumgartner M, Loffler J, Utermann G. Mutations in the human DHCR7 gene. Hum Mutat. 2001;17(3):172–82. doi: 10.1002/humu.2. information for mutation database. [DOI] [PubMed] [Google Scholar]
- 76.Kelley RI. A new face for an old syndrome. Am J Med Genet. 1997;68(3):251–6. doi: 10.1002/(sici)1096-8628(19970131)68:3<251::aid-ajmg1>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 77.Hall P, Michels V, Gavrilov D, Matern D, Oglesbee D, Raymond K, et al. Aripiprazole and trazodone cause elevations of 7-dehydrocholesterol in the absence of Smith-Lemli-Opitz Syndrome. Mol Genet Metab. 2013;110(1–2):176–8. doi: 10.1016/j.ymgme.2013.04.004. [DOI] [PubMed] [Google Scholar]
- 78.Porter FD. Smith-Lemli-Opitz syndrome: pathogenesis, diagnosis and management. Eur J Hum Genet. 2008;16(5):535–41. doi: 10.1038/ejhg.2008.10. [DOI] [PubMed] [Google Scholar]
- 79**.Nowaczyk MJM. Smith-Lemli-Opitz Syndrome. In: Pagon RA, Adam MP, Ardinger HH, Bird TD, Dolan CR, Fong CT, et al., editors. GeneReviews(R) Seattle WA: University of Washington, Seattle; 1993. regularly updated clinical overview with emphasis on testing strategies and molecular background. [PubMed] [Google Scholar]
- 80.McGaughran JM, Clayton PT, Mills KA, Rimmer S, Moore L, Donnai D. Prenatal diagnosis of Smith-Lemli-Opitz syndrome. Am J Med Genet. 1995;56(3):269–71. doi: 10.1002/ajmg.1320560306. [DOI] [PubMed] [Google Scholar]
- 81.Shackleton CH, Marcos J, Palomaki GE, Craig WY, Kelley RI, Kratz LE, et al. Dehydrosteroid measurements in maternal urine or serum for the prenatal diagnosis of Smith-Lemli-Opitz syndrome (SLOS) Am J Med Genet A. 2007;143A(18):2129–36. doi: 10.1002/ajmg.a.31901. [DOI] [PubMed] [Google Scholar]
- 82.Palomaki GE, Bradley LA, Knight GJ, Craig WY, Haddow JE. Assigning risk for Smith-Lemli-Opitz syndrome as part of 2nd trimester screening for Down’s syndrome. J Med Screen. 2002;9(1):43–4. doi: 10.1136/jms.9.1.43. [DOI] [PubMed] [Google Scholar]
- 83.GeneTests. 2015 [cited 2015 01/10/2015] [Available from: https://www.genetests.org/
- 84.Gelzo M, Clericuzio S, Barone R, D’Apolito O, Dello Russo A, Corso G. A routine method for cholesterol and 7-dehydrocholesterol analysis in dried blood spot by GC-FID to diagnose the Smith-Lemli-Opitz syndrome. J Chromatogr B Analyt Technol Biomed Life Sci. 2012;907:154–8. doi: 10.1016/j.jchromb.2012.08.025. [DOI] [PubMed] [Google Scholar]
- 85.Zimmerman PA, Hercules DM, Naylor EW. Direct analysis of filter paper blood specimens for identification of Smith-Lemli-Opitz syndrome using time-of-flight secondary ion mass spectrometry. Am J Med Genet. 1997;68(3):300–4. doi: 10.1002/(sici)1096-8628(19970131)68:3<300::aid-ajmg10>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 86.Gelzo M, Dello Russo A, Corso G. Stability study of dehydrocholesterols in dried spot of blood from patients with Smith-Lemli-Opitz syndrome, using filter-paper treated with butylated hydroxytoluene. Clin Chim Acta. 2012;413(3–4):525–6. doi: 10.1016/j.cca.2011.11.008. [DOI] [PubMed] [Google Scholar]
- 87.Solace Nutrition Cholextra Tm. Solace Nutrition; 2015. [cited 2015 01/15/2015] [Available from: http://www.solacenutrition.com/products/cholextra/cholextra.php. [Google Scholar]
- 88.MetaGenes Product SloEsterol Tm. [cited 2015 1/15/2015] MetaGenes; 2012. [Available from: http://metagenes.co/main/product/sloesterol. [Google Scholar]
- 89.Lin DS, Steiner RD, Flavell DP, Connor WE. Intestinal absorption of cholesterol by patients with Smith-Lemli-Opitz syndrome. Pediatr Res. 2005;57(6):765–70. doi: 10.1203/01.PDR.0000157723.98422.B5. [DOI] [PubMed] [Google Scholar]
- 90.Linck LM, Lin DS, Flavell D, Connor WE, Steiner RD. Cholesterol supplementation with egg yolk increases plasma cholesterol and decreases plasma 7-dehydrocholesterol in Smith-Lemli-Opitz syndrome. Am J Med Genet. 2000;93(5):360–5. doi: 10.1002/1096-8628(20000828)93:5<360::aid-ajmg4>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 91.Chan YM, Merkens LS, Connor WE, Roullet JB, Penfield JA, Jordan JM, et al. Effects of dietary cholesterol and simvastatin on cholesterol synthesis in Smith-Lemli-Opitz syndrome. Pediatr Res. 2009;65(6):681–5. doi: 10.1203/PDR.0b013e31819ea4eb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nwokoro NA, Mulvihill JJ. Cholesterol and bile acid replacement therapy in children and adults with Smith-Lemli-Opitz (SLO/RSH) syndrome. Am J Med Genet. 1997;68(3):315–21. doi: 10.1002/(sici)1096-8628(19970131)68:3<315::aid-ajmg13>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 93.Irons M, Elias ER, Abuelo D, Bull MJ, Greene CL, Johnson VP, et al. Treatment of Smith-Lemli-Opitz syndrome: results of a multicenter trial. Am J Med Genet. 1997;68(3):311–4. [PubMed] [Google Scholar]
- 94.Elias ER, Irons MB, Hurley AD, Tint GS, Salen G. Clinical effects of cholesterol supplementation in six patients with the Smith-Lemli-Opitz syndrome (SLOS) Am J Med Genet. 1997;68(3):305–10. doi: 10.1002/(sici)1096-8628(19970131)68:3<305::aid-ajmg11>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 95.Azurdia RM, Anstey AV, Rhodes LE. Cholesterol supplementation objectively reduces photosensitivity in the Smith-Lemli-Opitz syndrome. Br J Dermatol. 2001;144(1):143–5. doi: 10.1046/j.1365-2133.2001.03964.x. [DOI] [PubMed] [Google Scholar]
- 96.Tierney E, Conley SK, Goodwin H, Porter FD. Analysis of short-term behavioral effects of dietary cholesterol supplementation in Smith-Lemli-Opitz syndrome. Am J Med Genet A. 2010;152A(1):91–5. doi: 10.1002/ajmg.a.33148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wassif CA, Vied D, Tsokos M, Connor WE, Steiner RD, Porter FD. Cholesterol storage defect in RSH/Smith-Lemli-Opitz syndrome fibroblasts. Mol Genet Metab. 2002;75(4):325–34. doi: 10.1016/S1096-7192(02)00010-0. [DOI] [PubMed] [Google Scholar]
- 98.Dietschy JM. Central nervous system: cholesterol turnover, brain development and neurodegeneration. Biol Chem. 2009;390(4):287–93. doi: 10.1515/BC.2009.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ko JS, Choi BS, Seo JK, Shin JY, Chae JH, Kang GH, et al. A novel DHCR7 mutation in a Smith-Lemli-Opitz syndrome infant presenting with neonatal cholestasis. J Korean Med Sci. 2010;25(1):159–62. doi: 10.3346/jkms.2010.25.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Boctor FN, Wilkerson ML. Fresh frozen plasma as a source of cholesterol for newborn with smith-lemli-opitz syndrome associated with defective cholesterol synthesis. Ann Clin Lab Sci. 2014;44(3):332–3. [PubMed] [Google Scholar]
- 101.Krakowiak PA, Nwokoro NA, Wassif CA, Battaile KP, Nowaczyk MJ, Connor WE, et al. Mutation analysis and description of sixteen RSH/Smith-Lemli-Opitz syndrome patients: polymerase chain reaction-based assays to simplify genotyping. Am J Med Genet. 2000;94(3):214–27. [PubMed] [Google Scholar]
- 102.Irons MB, Nores J, Stewart TL, Craigo SD, Bianchi DW, D’Alton ME, et al. Antenatal therapy of Smith-Lemli-Opitz syndrome. Fetal Diagn Ther. 1999;14(3):133–7. doi: 10.1159/000020906. [DOI] [PubMed] [Google Scholar]
- 103.Jira PE, Wevers RA, de Jong J, Rubio-Gozalbo E, Janssen-Zijlstra FS, van Heyst AF, et al. Simvastatin. A new therapeutic approach for Smith-Lemli-Opitz syndrome. J Lipid Res. 2000;41(8):1339–46. [PubMed] [Google Scholar]
- 104.Haas D, Garbade SF, Vohwinkel C, Muschol N, Trefz FK, Penzien JM, et al. Effects of cholesterol and simvastatin treatment in patients with Smith-Lemli-Opitz syndrome (SLOS) J Inherit Metab Dis. 2007;30(3):375–87. doi: 10.1007/s10545-007-0537-7. [DOI] [PubMed] [Google Scholar]
- 105.Szabo GP, Olah AV, Kozak L, Balogh E, Nagy A, Blahakova I, et al. A patient with Smith-Lemli-Opitz syndrome: novel mutation of the DHCR7 gene and effects of therapy with simvastatin and cholesterol supplement. Eur J Pediatr. 2010;169(1):121–3. doi: 10.1007/s00431-009-0987-z. [DOI] [PubMed] [Google Scholar]
- 106.Starck L, Lovgren-Sandblom A, Bjorkhem I. Simvastatin treatment in the SLO syndrome: a safe approach? Am J Med Genet. 2002;113(2):183–9. doi: 10.1002/ajmg.10722. [DOI] [PubMed] [Google Scholar]
- 107.Atchaneeyasakul LO, Linck LM, Connor WE, Weleber RG, Steiner RD. Eye findings in 8 children and a spontaneously aborted fetus with RSH/Smith-Lemli-Opitz syndrome. Am J Med Genet. 1998;80(5):501–5. doi: 10.1002/(sici)1096-8628(19981228)80:5<501::aid-ajmg12>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 108.Ross LF, Saal HM, David KL, Anderson RR. Technical report: Ethical and policy issues in genetic testing and screening of children. Genet Med. 2013;15(3):234–45. doi: 10.1038/gim.2012.176. [DOI] [PubMed] [Google Scholar]
- 109.Kelley RI. Diagnosis of Smith-Lemli-Opitz syndrome by gas chromatography/mass spectrometry of 7-dehydrocholesterol in plasma, amniotic fluid and cultured skin fibroblasts. Clin Chim Acta. 1995;236(1):45–58. doi: 10.1016/0009-8981(95)06038-4. [DOI] [PubMed] [Google Scholar]
- 110.Lindegaard ML, Wassif CA, Vaisman B, Amar M, Wasmuth EV, Shamburek R, et al. Characterization of placental cholesterol transport: ABCA1 is a potential target for in utero therapy of Smith-Lemli-Opitz syndrome. Hum Mol Genet. 2008;17(23):3806–13. doi: 10.1093/hmg/ddn278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Tint GS, Yu H, Shang Q, Xu G, Patel SB. The use of the Dhcr7 knockout mouse to accurately determine the origin of fetal sterols. J Lipid Res. 2006;47(7):1535–41. doi: 10.1194/jlr.M600141-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Correa-Cerro LS, Wassif CA, Kratz L, Miller GF, Munasinghe JP, Grinberg A, et al. Development and characterization of a hypomorphic Smith-Lemli-Opitz syndrome mouse model and efficacy of simvastatin therapy. Hum Mol Genet. 2006;15(6):839–51. doi: 10.1093/hmg/ddl003. [DOI] [PubMed] [Google Scholar]
- 113.Wassif CA, Krakowiak PA, Wright BS, Gewandter JS, Sterner AL, Javitt N, et al. Residual cholesterol synthesis and simvastatin induction of cholesterol synthesis in Smith-Lemli-Opitz syndrome fibroblasts. Mol Genet Metab. 2005;85(2):96–107. doi: 10.1016/j.ymgme.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 114.Ying L, Matabosch X, Serra M, Watson B, Shackleton C, Watson G. Biochemical and Physiological Improvement in a Mouse Model of Smith-Lemli-Opitz Syndrome (SLOS) Following Gene Transfer with AAV Vectors. Mol Genet Metab Rep. 2014;1:103–13. doi: 10.1016/j.ymgmr.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–9. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.