

INTRODUCTION
Despite tremendous advances in basic and clinical oncology, the prognosis for most patients with neoplastic diseases is still dismal. Therefore, new therapeutic strategies must be sought. A novel drug for the treatment of malignant diseases should have a mechanism of action different from those known for established oncological therapeutics. In addition, it is desirable to attempt to relate the antitumor activity of a candidate compound with molecular effects on proteins relevant for the pathogenesis of malignant diseases.
Geldanamycin (GA; NSC 122750) has the potential to fulfill the aforementioned criteria. It was first purified in 1970 from the broth of Streptomyces hygroscopicus var geldanus var nova (DeBoer et al 1970). As a benzoquinone ansamycin (BA), it consists of a quinone ring and a hydrophobic ansa bridge (Fig 1; Rinehart and Shield 1976). The DNA sequences responsible for GA biosynthesis have been previously characterized (Allen and Ritchie 1994). The antineoplastic effect of GA was already noted in its first description (DeBoer et al 1970).
Fig. 1.

Structural formula of geldanamycin
Molecular studies revealed the binding of GA to members of the heat shock protein 90 (Hsp90) family of molecular chaperones (Whitesell et al 1994). Interference with the function of these Hsps seems to be the major mechanism of action of GA.
This review focuses on oncological aspects and will not deal with other biological activities of GA, such as its virucidal (Li et al 1977) or ischemia protective (Conde et al 1997) effects.
MECHANISM OF ACTION
The use of various model systems generated data on the in vitro and in vivo activity against malignant tumors. Table 1 summarizes these results. In Table 2, studies are listed that were undertaken to define the mechanism of action of GA.
Table 1.
Reported antineoplastic activities of geldanamycin in preclinical models
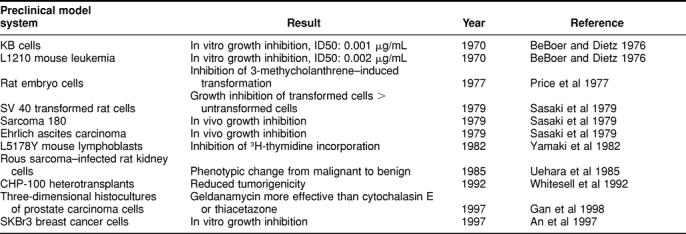
Table 2.
Postulated mechanisms of action

Three concepts are of particular importance: (1) formation of free radicals, (2) assumed inhibition of tyrosine kinases, and (3) binding to and interference with the function of members of the Hsp90 family of proteins.
Free radical formation
The fact that a quinone ring is part of the GA molecule led to the initiation of detailed studies on the generation of intracellular free radicals through redox cycling (Benchekroun et al 1994b). It was shown that the treatment of eukaryotic cells with GA indeed causes the formation of free radicals. However, this could only be observed at high concentrations of GA, eg, 100 μM, whereas the same investigators—as others later—found cytotoxic effects in the nanomolar range. It can be reasonably concluded that the antitumor activity of GA cannot be explained by the formation of free radicals.
Inhibition of tyrosine kinases
Since GA inhibited the tyrosine kinase activity of v-src, it was classified as a tyrosine kinase inhibitor (Yamaki et al 1995). The assumption was that BAs directly act on sulfhydryl groups of v-src (Uehara et al 1989). Yet it was known that the tyrosine kinase activity of v-src could only be influenced by herbimycin A—an analog of GA—in vivo but not in vitro (Uehara et al 1986). This suggested that the target molecule of GA is either downstream of v-src or an interacting protein. A detailed analysis of the course of GA effects showed that there is no correlation between its cytocidal activity and the late onset of inhibition of kinases of the src-family (Whitesell et al 1992).
Binding of GA to and interference with the function of members of the Hsp90 family of proteins
All these findings could not explain the multitude of effects of BAs. Finally, through chemical cross-linking of GA to Sepharose beads, it was possible to precipitate the target molecule of GA. Further analysis identified it as Hsp90 (Whitesell et al 1994). The discovery that GA binds to Hsp90 was a decisive breakthrough in understanding the mechanism of action of GA and structurally or functionally related substances. For the first time, a molecular chaperone was identified as the target molecule of an anticancer drug.
In transformed cells, v-src forms a heterocomplex with Hsp90 (Mimnaugh et al 1995). Thus, the described reversion of the malignant phenotype (Uehara et al 1986) could be attributed to GA-induced dissolution of the Hsp90–v-src heterocomplex (Whitesell et al 1994). Some experimental findings implicate phosphoserine and phosphothreonine contents of Hsp90 as crucial for the maintenance of the Hsp90–v-src complex (Mimnaugh et al 1995).
Further detailed studies revealed that GA binds to the amino terminus of Hsp90 (Grenert et al 1997). Crystallographic experiments confirmed this and found a pocketlike conformation of the binding site (Prodromou et al 1997; Stebbins et al 1997; Roe et al 1999). It was demonstrated that this region has an adenosine triphosphatase activity that is inhibited by GA through competitive binding with adenosine triphosphate (Grenert et al 1997; Obermann et al 1998).
The amino terminal end of Hsp90 was also described as being able to bind unfolded peptides and to prevent their aggregation in a GA-sensitive manner (Young et al 1997). The charged linker region connecting the amino and carboxy terminal domains of Hsp90 modulates the interaction of GA with its binding site (Scheibel et al 1999).
Further experiments showed that GA also binds to the glucose-regulated protein 94 (Grp94), another member of the Hsp90 family located in the endoplasmic reticulum (Chavany et al 1996; Schulte et al 1999).
ROLE OF Hsp90 IN CELLULAR PHYSIOLOGY
Hsp90 like other Hsps is named for its increased synthesis after heat shock that is contrary to the reduced synthesis of most cellular proteins under these conditions. Members of the Hsp90 family of molecular chaperones include Hsp90α (Hickey et al 1989), Hsp90β (Rebbe et al 1987), Grp94 (Little et al 1994), and Hsp75 (Chen et al 1996), also known as TNF receptor-associated protein-1 (TRAP-1) (Song et al 1995). So far no differential function has been assigned to the α and β subforms of Hsp90. Research on yeast cells shows that Hsp90 is essential (Picard et al 1990). Mutants with abrogated adenosine triphosphatase activity are not viable (Panaretou et al 1998). Since Hsp90 may represent 1% of the proteome even in the absence of heat shock, it must have functions beyond its role in the heat shock reaction. At least 3 functions of Hsp90 can be delineated. First, it is a molecular chaperone that prevents the aggregation and assists the refolding of proteins after various stresses, including heat shock (Nathan et al 1997). This finding is supported by experiments demonstrating the Hsp90-dependent renaturation of denatured enzymes (Bose et al 1996; Hartson et al 1996). In vitro, GA partially blocks protein renaturation (Schumacher et al 1996). Second, it exerts a “genomic buffer” function to suppress phenotypic traits that only become expressed after certain stresses (Rutherford and Lindquist 1998). Third, it stabilizes key regulatory proteins through the formation of heteroprotein complexes (Lindquist 1988) or keeps them in a defined functional state. Well-studied examples of the latter are the progesterone and the glucocorticosteroid receptors, which through interaction with Hsp90 and other accessory proteins are kept in a hormone-binding conformation (Sullivan and Toft 1993; Johnson and Toft 1995; Whitesell and Cook 1996). After hormone binding, Hsp90 is released from the complex.
Hsp90 is probably involved in either initiating or maintaining the transformed state: Hsp90 messenger RNA (mRNA) has been found to be overexpressed in human ovarian carcinoma cells, but not in normal ovarian cells or in those derived from benign ovarian tumors (Mileo et al 1990).
EFFECTS ON ONCOLOGICALLY IMPORTANT PROTEINS
There is evidence that Hsp90 interacts with several proteins important to oncology. Interactions have been described for cdk4 (Chen et al 1997), cyclin B1 and Cdc2 (Kim et al 1999), heterotrimeric Gα subunits (Busconi et al 2000), telomerase (Holt et al 1999), and epidermal growth factor (EGF) and platelet-derived growth factor receptors (Sakagami et al 1999). In addition, it may influence fusion proteins, which are present only in certain neoplasias as evidenced from its effect on the bcr-abl protein found in most chronic myelogenous leukemias and some acute lymphatic leukemias (An et al 1999).
At least 4 proteins of major interest for oncology are being influenced by BAs: mutant p53, erbB2, raf-1, and focal adhesion kinase. Most cellular proteins are not affected by treatment with GA, as can be deduced from the comparison of radioactive methionine-labeled cell lysates before and after treatment (Ochel et al 1999).
p53
As a transactivating protein, which acts as a tumor suppressor (Marx 1993), p53 is mutated or absent in approximately half of all human cancers and is the protein with the most commonly found molecular alteration in neoplasias (Nigro et al 1989; Levine 1992).
A role for Hsp90 in the pathophysiology of the malignant transformation was suggested by the fact that treatment of several breast cancer, prostate cancer, and leukemia cell lines with GA results in the destabilization of mutated p53 (Blagosklonny et al 1995). This was the first evidence of a specific pharmacological modulation of mutated p53. In this model system no influence on wildtype p53 levels was found, although in a different experimental setting accumulation of wildtype p53 accompanied by cell-cycle arrest was detected (McIlwrath et al 1996). These findings have been extended by studies using rabbit reticulocyte lysate and wheat germ extract. In these studies, the presence of Hsp90 was found to be necessary for p53 to acquire a mutated conformation (Blagosklonny et al 1996). Conversely, in vitro translation of mutated p53 in wheat germ extract, known to be functionally Hsp90 deficient, yielded p53 proteins only with wildtype but not with mutant conformation. This led to the hypothesis that mutant p53 stabilization, found in malignant cells, is not due to stabilization of the protein per se, but is caused by the formation of heterocomplexes of p53 with other proteins, such as Hsp90 (Blagosklonny 1997). The in vivo existence of a Hsp90–mutated p53 complex, which in some cases is located in the cytoplasm, has been demonstrated (Sepehrnia et al 1996). Further studies revealed that other chaperones, such as Hsp70 or Hop, can be coimmunoprecipitated with p53 and that these associations may be modulated by GA (Whitesell et al 1998). In some experiments, GA treatment induced the disruption of the Hsp90–mutated p53 complex (Whitesell et al 1998; Nagata et al 1999). In other studies, this was not found (Dasgupta and Momand 1997), pointing to different roles for this interaction in the pathophysiology of malignant diseases. All investigations showed a rapid destabilization of mutated p53.
It was already known that p53 may become ubiquitinated (Kubbutat and Vousden 1997). The ubiquitination increases with GA treatment (Nagata et al 1999), and this in turn stimulates p53 degradation by the proteasome (Whitesell et al 1997).
In summary, GA is the prototype of drugs that are able to selectively destabilize mutated p53. No restoration of wildtype transcriptional activity was detected under these conditions.
erbB2
The product of the ERBB2 gene, also known as NEU or HER-2, is also a target of GA. The gene codes for the mRNA of a transmembrane protein, which belongs to the EGF receptor family (Bargmann et al 1986). ErbB2 is a receptor subunit that homodimerizes or heterodimerizes with other members of the EGF-receptor family and thus creates receptors of different function and ligand affinity (Goldman et al 1990). The significance for oncology stems from the finding that overexpression of this protein mainly by means of gene amplification (King et al 1985) in breast or ovarian cancers (Slamon et al 1989) is associated with a dismal prognosis (Slamon et al 1987). Hence, therapeutic, pharmacological modulation of erbB2 is desirable.
The original observation that erbB2 overexpressing SKBr-3 breast cancer cells treated with BAs exhibit a rapid and profound reduction of erbB2 protein was attributed to enhanced degradation (Miller et al 1994a). ErbB2 mRNA levels were found to be unchanged. A first hint of the underlying molecular mechanism came from experiments with photoaffinity-labeled ansamycins, which were shown to bind to an approximately 100-kDa protein (Miller et al 1994b). Competition of binding with GA proved that this was the target molecule of the BAs. By coimmunoprecipitation and Western blotting experiments, it was identified as Grp94 (Chavany et al 1996). Grp94 is a homologue of Hsp90 that is located in the endoplasmic reticulum (Little et al 1994). Treatment with GA results in the disruption of the Grp94-erbB2 complex, followed by polyubiquitination of the transmembrane protein and proteasomal degradation (Chavany et al 1996; Mimnaugh et al 1996; Hartmann et al 1997). A relation between BA structure and their respective erbB2 depletion activity was described (Schnur et al 1995a). ErbB2 depletion by GA could also be demonstrated in vivo in heterotransplanted malignant tumors (Schnur et al 1995b).
raf-1
Raf-1 is part of a conserved signal transduction pathway that connects cytosolic and transmembrane tyrosine kinases with mitogen-activated protein kinases (Magnuson et al 1994). It is activated and recruited to the internal side of the plasma membrane through binding to the small, guanosine 5′-triphosphate binding protein ras (Marais et al 1995). Ras mutations leading to constitutive activation of this signal transduction pathway have been identified in different malignant tumors, but are especially prevalent in pancreatic cancers (Almoguera et al 1988). Raf-1 may also be oncogenically activated by mutation (Heidecker et al 1990). A physical interaction of raf-1 and Hsp90 in the form of a heterocomplex together with other accessory proteins was identified in vitro and in vivo (Stancato et al 1993). The treatment of MCF-7 breast cancer cells with GA results in the rapid destabilization of raf-1, mediated by the proteasome (Schulte et al 1997), and the dissolution of the association of raf-1 with ras (Schulte et al 1995). Upstream and downstream effectors of this signal transduction pathway are not affected by GA (Schulte et al 1996). Further analysis revealed that raf-1 concentrations declined concomitantly in different compartments of the cell, confirming the hypothesis that the association of raf-1 with Hsp90 may have significance for the transport of raf-1 to the cell membrane (Schulte et al 1995). These findings were confirmed with other models, although the disruption of the Hsp90–raf-1 complex was not always seen (Stancato et al 1997). The treatment of malignant cells with GA offers the option to abrogate the constitutive activation of raf-1 caused by ras mutations.
fak
Focal adhesion kinase (fak) is a tyrosine kinase located predominantly in focal adhesions, ie, the cellular sites of attachment to the extracellular matrix (Schaller et al 1992). Fak is expressed in all human tissues. Fak mRNA and protein expression show a positive correlation with tumor invasiveness (Owens et al 1995). In metastases, more fak can be found than in premalignant lesions. They in turn contain more fak than homologous benign cells. This was described for colon, breast, and thyroid cancers and certain sarcomas (Weiner et al 1994). Fak−/− cells produced by gene knockout exhibit an increased number of focal adhesions, implicating fak in the turnover of these subcellular structures (Ilic et al 1995). Initially, it was not clear whether fak was a tumor-repressing or tumor-promoting protein, since no malignant transformation of cells was found in the usual tests. Recently, however, it was reported that activated fak is able to transform Madin Darby Canine Kidney (MDCK) cells (Frisch et al 1996). Integrin-dependent signal transduction via fak suppresses apoptosis (Frisch and Francis 1994). Taken together, fak is a promising target for pharmacological intervention.
Treatment with GA results in a time- and dose-dependent degradation of fak protein, which is mediated by the proteasome (Ochel et al 1999). Currently, GA is the only substance with the pharmacological ability to interdict fak function.
The relevance of the described molecular effects of GA to its cytotoxic property, at least in vitro, has recently been described. Twenty-eight different derivatives of GA were tested for their cytotoxicity. Loss of mutated p53, erbB2, and raf-1 occurred concomitantly and was only observed with those derivatives that displayed antiproliferative activity (An et al 1997).
RESISTANCE
MCF-7 breast cancer cells resistant to doxorubicin (MCF-7/ADRR) are cross-resistant to GA (Benchekroun et al 1994b). For the same cytotoxic effect, higher concentrations of doxorubicin or GA had to be used in resistant vs wildtype cells. Since verapamil, known to bind to and inhibit the multidrug resistance (MDR) pump, enhanced GA cytotoxicity, the conclusion was that GA is a substrate of the MDR pump and that overexpression of MDR may be a mechanism of GA resistance. In addition, GA-induced generation of free radicals was reduced in MCF-7/ADRR as opposed to the parental cells.
However, further studies with cells selected for GA resistance (MCF-7/GAR) showed that there is no difference in GA efflux between wildtype and MCF-7/GAR cells (Benchekroun et al 1994a). Verapamil had no cytotoxicity-enhancing effect, although MDR expression was twice as high in the resistant cells. In addition, free radical formation by GA was found not to contribute to this resistance.
Interestingly, Hsp90β can be found in direct physical association with the MDR protein that is only present in chemotherapy-resistant but not in wildtype cells (Bertram et al 1996).
PRECLINICAL EVALUATION
Extensive pharmacological evaluation has shown that drug concentrations that have an antiproliferative effect in vitro are attainable in mice and dogs (Supko et al 1995). Three to 4 hours after application of the maximum tolerated dose, plasma concentrations fell into the subtherapeutic range. Liver toxic effects with elevations of transaminases were predominant. Further changes included elevations in creatinine phosphokinase, lactic dehydrogenase, and blood urea nitrogen as well as leukocytosis and reticulocytosis.
FUNCTIONAL OR STRUCTURAL GA ANALOGS
Because of the toxicity of GA, a search was initiated to characterize substances more suitable for clinical development. One of those 17-(allylamino)-17-demethoxygeldanamycin (NSC330507) is structurally related to GA (Schnur et al 1995b). It shares the ability to bind to Hsp90 and to deplete erbB2, raf-1, and mutated p53 (Schulte and Neckers 1998). This compound is currently undergoing a phase 1 clinical trial at 5 institutions in the United States and Great Britain.
Radicicol is a functional analog without structural similarity to GA (Mirrington et al 1965). It also binds to the amino terminal nucleotide binding domain of Hsp90, exerting effects comparable to GA on the aforementioned regulatory proteins (Schulte et al 1998). Radicicol derivatives have shown promising in vivo activity in various murine cancer models (Soga et al 1999).
OTHER SUBSTANCES INTERACTING WITH Hsp90
Recent research revealed that the most effective drugs for the treatment of epithelial tumors, paclitaxel and cisplatin, both bind to Hsp90 (Byrd et al 1999; Itoh et al 1999). The binding site for cisplatin has been determined to be near the carboxy terminus. Hsp90-dependent inhibition of aggregation of citrate synthase was reversed by cisplatin (Byrd et al 1999). The region of Hsp90 bound by paclitaxel has not yet been determined. Some effects of paclitaxel, eg, the expression of tumor necrosis factor, are blocked by GA (Itoh et al 1999).
SUMMARY AND PROSPECTS
GA exerts antitumor activity in a multitude of preclinical models. It influences the cellular concentrations of several oncologically significant, signal-transducing proteins through the induction of their proteasome-dependent degradation. A correlation exists between GA cytotoxicity and this protein modulation. It is currently unknown to what extent GA-induced molecular alterations contribute to its anticancer activity in case such an effect is clinically demonstrable. BAs represent the first class of antitumor drugs for which binding to Hsp90 was identified as the major, novel mechanism of action. GA is thus a model compound for the development of structural or functional analogs with a more favorable toxicity profile. To prepare for the application of these compounds in multimodal therapy, studies are under way to determine their interaction with ionizing radiation. Similarly, GA is a formidable tool to elucidate the currently unknown significance of members of the Hsp90 family in the radiation response.
Acknowledgments
We thank Dr Neckers, Tumor Cell Biology Section, Medicine Branch, National Cancer Institute, National Institutes of Health, Bethesda, MD, who critically reviewed the manuscript. We also thank Mrs Lobe, Clinic for Radiation Therapy, Otto-von-Guericke-University, Magdeburg, Germany, for secretarial assistance.
REFERENCES
- Allen IW, Ritchie DA. Cloning and analysis of DNA sequences from Streptomyces hygroscopicus encoding geldanamycin biosynthesis. Mol Gen Genet. 1994;243:593–599. doi: 10.1007/BF00284208. [DOI] [PubMed] [Google Scholar]
- Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. 1988;53:549–554. doi: 10.1016/0092-8674(88)90571-5. [DOI] [PubMed] [Google Scholar]
- An WG, Schnur RC, Neckers L, Blagosklonny MV. Depletion of p185erbB2, raf-1 and mutant p53 proteins by geldanamycin derivatives correlates with antiproliferative activity. Cancer Chemother Pharmacol. 1997;40:60–64. doi: 10.1007/s002800050626. [DOI] [PubMed] [Google Scholar]
- An WG, Schulte TW, and Neckers LM 1999 p210BCR-ABL exists in a complex with Hsp90 and is destabilized after treatment with Hsp90 inhibitors. Proc Am Assoc Cancer Res. 40:119. Abstract 793. [Google Scholar]
- Bargmann CI, Hung MC, Weinberg RA. The neu oncogene encodes an epidermal growth factor receptor-related protein. Nature. 1986;319:226–230. doi: 10.1038/319226a0. [DOI] [PubMed] [Google Scholar]
- BeBoer C, Dietz A. The description and antibiotic production of Streptomyces hygroscopicus var. geldanus. J Antibiot (Tokyo) 1976;29:1182–1188. doi: 10.7164/antibiotics.29.1182. [DOI] [PubMed] [Google Scholar]
- Benchekroun MN, Schneider E, Safa AR, Townsend AJ, Sinha BK. Mechanisms of resistance to ansamycin antibiotics in human breast cancer cell lines. Mol Pharmacol. 1994a;46:677–684. [PubMed] [Google Scholar]
- Benchekroun NM, Myers CE, Sinha BK. Free radical formation by ansamycin benzoquinone in human breast tumor cells: implications for cytotoxicity and resistance. Free Radic Biol Med. 1994b;17:191–200. doi: 10.1016/0891-5849(94)90074-4. [DOI] [PubMed] [Google Scholar]
- Bertram J, Palfner K, Hiddemann W, Kneba M. Increase of P-glycoprotein-mediated drug resistance by hsp 90β. Anticancer Drugs. 1996;7:838–845. doi: 10.1097/00001813-199611000-00004. [DOI] [PubMed] [Google Scholar]
- Blagosklonny MV. Loss of function and p53 protein stabilization. Oncogene. 1997;15:1889–1893. doi: 10.1038/sj.onc.1201374. [DOI] [PubMed] [Google Scholar]
- Blagosklonny MV, Toretsky J, Bohen S, Neckers L. Mutant conformation of p53 translated in vitro or in vivo requires functional HSP90. Proc Natl Acad Sci U S A. 1996;93:8379–83. doi: 10.1073/pnas.93.16.8379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blagosklonny MV, Toretsky J, Neckers L. Geldanamycin selectively destabilizes and conformationally alters mutated p53. Oncogene. 1995;11:933–939. [PubMed] [Google Scholar]
- Bose S, Weikl T, Bügl H, Buchner J. Chaperone function of Hsp90-associated proteins. Science. 1996;274:1715–1717. doi: 10.1126/science.274.5293.1715. [DOI] [PubMed] [Google Scholar]
- Busconi L, Guan J, Denker BM. Degradation of heterotrimeric Galpha(o) subunits via the proteosome pathway is induced by the Hsp90-specific compound geldanamycin. J Biol Chem. 2000;275:1565–1569. doi: 10.1074/jbc.275.3.1565. [DOI] [PubMed] [Google Scholar]
- Byrd CA, Bornmann W, and Erdjument-Bromage H. et al. 1999 Heat shock protein 90 mediates macrophage activation by taxol and bacterial lipopolysaccharide. Proc Natl Acad Sci U S A. 96:5645–5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavany C, Mimnaugh E, and Miller P. et al. 1996 p185erbB2 binds to GRP94 in vivo: dissociation of the p185erbB2/GRP94 heterocomplex by benzoquinone ansamycins precedes depletion of p185erbB2. J Biol Chem. 271:4974–4977. [DOI] [PubMed] [Google Scholar]
- Chen C-F, Chen Y, Dai K, Chen P-L, Riley DJ, Lee W-H. A new member of the Hsp90 family of molecular chaperones interacts with the retinoblastoma protein during mitosis and after heat shock. Mol Cell Biol. 1996;16:4691–4699. doi: 10.1128/mcb.16.9.4691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HS, Singh SS, Perdew GH. The Ah receptor is a sensitive target of geldanamycin-induced protein turnover. Arch Biochem Biophys. 1997;348:190–198. doi: 10.1006/abbi.1997.0398. [DOI] [PubMed] [Google Scholar]
- Conde AG, Lau SS, Dillmann WH, Mestril R. Induction of heat shock proteins by tyrosine kinase inhibitors in rat cardiomyocytes and myogenic cells confers protection against simulated ischemia. J Mol Cell Cardiol. 1997;29:1927–1938. doi: 10.1006/jmcc.1997.0431. [DOI] [PubMed] [Google Scholar]
- Dasgupta G, Momand J. Geldanamycin prevents nuclear translocation of mutant p53. Exp Cell Res. 1997;237:29–37. doi: 10.1006/excr.1997.3766. [DOI] [PubMed] [Google Scholar]
- DeBoer C, Meulman PA, Wnuk RJ, Peterson DH. Geldanamycin, a new antibiotic. J Antibiot (Tokyo) 1970;23:442–447. doi: 10.7164/antibiotics.23.442. [DOI] [PubMed] [Google Scholar]
- Frisch SM, Francis H. Disruption of epithelial cell-matrix interactions induces apoptosis. J Cell Biol. 1994;124:619–626. doi: 10.1083/jcb.124.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch SM, Vuori K, Ruoslahti E, Chan-Hui PY. Control of adhesion-dependent cell survival by focal adhesion kinase. J Cell Biol. 1996;134:793–799. doi: 10.1083/jcb.134.3.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan Y, Au JL, Lu J, Wientjes MG. Antiproliferative and cytotoxic effects of geldanamycin, cytochalasin E, suramin and thiacetazone in human prostate xenograft tumor histocultures. Pharm Res. 1998;15:1760–1766. doi: 10.1023/a:1011921031564. [DOI] [PubMed] [Google Scholar]
- Goldman R, Levy RB, Peles E, Yarden Y. Heterodimerization of the erbB-1 and erbB-2 receptors in human breast carcinoma cells: a mechanism for receptor transregulation. Biochemistry. 1990;29:11024–11028. doi: 10.1021/bi00502a002. [DOI] [PubMed] [Google Scholar]
- Grenert JP, Sullivan WP, and Fadden P. et al. 1997 The amino-terminal domain of heat shock protein 90 (Hsp90) that binds geldanamycin is an ATP/ADP switch domain that regulates Hsp90 conformation. J Biol Chem. 272:23843–23850. [DOI] [PubMed] [Google Scholar]
- Hartmann F, Horak EM, and Cho C. et al. 1997 Effects of the tyrosine-kinase inhibitor geldanamycin on ligand-induced Her-2/neu activation, receptor expression and proliferation of Her-2-positive malignant cell lines. Int J Cancer. 70:221–229. [DOI] [PubMed] [Google Scholar]
- Hartson SD, Barrett DJ, Burn P, Matts RL. Hsp90-mediated folding of the lymphoid cell kinase p56lck. Biochemistry. 1996;35:13451–13459. doi: 10.1021/bi961332c. [DOI] [PubMed] [Google Scholar]
- Heidecker G, Huleihel M, and Cleveland JL. et al. 1990 Mutational activation of c-raf-1 and definition of the minimal transforming sequence. Mol Cell Biol. 10:2503–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey E, Brandon SE, Smale G, Lloyd D, Weber LA. Sequence and regulation of a gene encoding a human 89-kilodalton heat shock protein. Mol Cell Biol. 1989;9:2615–2626. doi: 10.1128/mcb.9.6.2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt SE, Aisner DL, and Baur J. et al. 1999 Functional requirement of p23 and Hsp90 in telomerase complexes. Genes Dev. 13:817–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilic D, Furuta Y, and Kanazawa S. et al. 1995 Reduced cell motility and enhanced focal adhesion contact formation in cells from fak-deficient mice. Nature. 377:530–544. [DOI] [PubMed] [Google Scholar]
- Itoh H, Ogura M, Komatsuda A, Wakui H, Miura AB, Tashima Y. A novel chaperone-activity-reducing mechanism of the 90-kDa molecular chaperone Hsp90. Biochem J. 1999;343:697–703. [PMC free article] [PubMed] [Google Scholar]
- Johnson JL, Toft DO. Binding of p23 and Hsp90 during assembly with the progesterone receptor. Mol Endocrinol. 1995;9:670–678. doi: 10.1210/mend.9.6.8592513. [DOI] [PubMed] [Google Scholar]
- Kim HR, Lee CH, Choi YH, Kang HS, Kim HD. Geldanamycin induces cell cycle arrest in K562 erythroleukemic cells. IUBMB Life. 1999;48:425–428. doi: 10.1080/713803539. [DOI] [PubMed] [Google Scholar]
- King CR, Kraus MH, Stuart AA. Amplification of a novel v-erbB-related gene in a human mammary carcinoma. Science. 1985;229:974–976. doi: 10.1126/science.2992089. [DOI] [PubMed] [Google Scholar]
- Kubbutat MH, Vousden KH. Proteolytic cleavage of human p53 by calpain: a potential regulator of protein stability. Mol Cell Biol. 1997;17:460–468. doi: 10.1128/mcb.17.1.460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine AJ. The p53 tumor-suppressor gene. N Engl J Med. 1992;326:1350–1351. doi: 10.1056/NEJM199205143262008. [DOI] [PubMed] [Google Scholar]
- Li LH, Clark TD, Cowie CH, Rinehart KL Jr.. Effects of geldanamycin and its derivatives on RNA-directed DNA polymerase and infectivity of Rauscher leukemia virus. Cancer Treat Rep. 1977;61:815–824. [PubMed] [Google Scholar]
- Lindquist S. The heat-shock proteins. Annu Rev Genet. 1988;22:631–677. doi: 10.1146/annurev.ge.22.120188.003215. [DOI] [PubMed] [Google Scholar]
- Little E, Ramakrishnan M, Roy B, Gazit G, La S. The glucose-regulated proteins (GRP78 and GRP94): functions, gene regulation, and applications. Crit Rev Eukaryot Gene Expr. 1994;4:1–18. doi: 10.1615/critreveukargeneexpr.v4.i1.10. [DOI] [PubMed] [Google Scholar]
- Magnuson NS, Beck T, Vahidi H, Hahn H, Smola U, Rapp UR. The raf-1 serine/threonine protein kinase. Semin Cancer Biol. 1994;5:247–253. [PubMed] [Google Scholar]
- Marais R, Light Y, Paterson HF, Marshall CJ. Ras recruits raf-1 to the plasma membrane for activation by tyrosine phosphorylation. EMBO J. 1995;14:3136–3145. doi: 10.1002/j.1460-2075.1995.tb07316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx J. How p53 suppresses cell growth. Science. 1993;262:1644–1645. doi: 10.1126/science.8259506. [DOI] [PubMed] [Google Scholar]
- McIlwrath AJ, Brunton VG, Brown R. Cell-cycle arrest and p53 accumulation induced by geldanamycin in human ovarian tumour cells. Cancer Chemother Pharmacol. 1996;37:423–428. doi: 10.1007/s002800050407. [DOI] [PubMed] [Google Scholar]
- Mileo AM, Fanuele M, Battaglia F, Scambia G, Benedetti-Panici P, Mancuso S, Ferrini U. Selective over-expression of mRNA coding for 90 kDa stress-protein in human ovarian cancer. Anticancer Res. 1990;10:903–906. [PubMed] [Google Scholar]
- Miller P, DiOrio C, Moyer M, Schnur RC, Bruskin A, Cullen W, Moyer JD. Depletion of the erbB-2 gene product p185 by benzoquinoid ansamycins. Cancer Res. 1994a;54:2724–2730. [PubMed] [Google Scholar]
- Miller P, Schnur RC, Barbacci E, Moyer MP, Moyer JD. Binding of benzoquinoid ansamycins to p100 correlates with their ability to deplete the erbB2 gene product p185. Biochem Biophys Res Commun. 1994b;201:1313–1319. doi: 10.1006/bbrc.1994.1847. [DOI] [PubMed] [Google Scholar]
- Mimnaugh EG, Chavany C, Neckers L. Polyubiquitination and proteasomal degradation of the p185c-erbB-2 receptor protein-tyrosine kinase induced by geldanamycin. J Biol Chem. 1996;271:22796–22801. doi: 10.1074/jbc.271.37.22796. [DOI] [PubMed] [Google Scholar]
- Mimnaugh EG, Worland PJ, Whitesell L, Neckers LM. Possible role for serine/threonine phosphorylation in the regulation of the heteroprotein complex between the Hsp90 stress protein and the pp60v-src tyrosine kinase. J Biol Chem. 1995;270:28654–28659. doi: 10.1074/jbc.270.48.28654. [DOI] [PubMed] [Google Scholar]
- Mirrington RN, Ritchie E, Shoppee CW, Sternhell S, Taylor WC. Some metabolites of Nectria radicicola Gerlach & Nilsson (syn. Cylindrocarpon wr.): the structure of radicicol (Monorden) Aust J Chem. 1965;19:1265–1284. [Google Scholar]
- Nagata Y, Anan T, and Yoshida T. et al. 1999 The stabilization mechanism of mutant-type p53 by impaired ubiquitination: the loss of wild-type p53 function and the Hsp90 association. Oncogene. 18:6037–6049. [DOI] [PubMed] [Google Scholar]
- Nathan DF, Vos MH, Lindquist S. In vivo functions of the Saccharomyces cerevisiae Hsp90 chaperone. Proc Natl Acad Sci U S A. 1997;94:12949–12956. doi: 10.1073/pnas.94.24.12949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigro JM, Baker SJ, and Preisinger AC. et al. 1989 Mutations in the p53 gene occur in diverse human tumour types. Nature. 342:705–708. [DOI] [PubMed] [Google Scholar]
- Obermann WM, Sondermann H, Russo AA, Pavletich NP, Hartl FU. In vivo function of Hsp90 is dependent on ATP binding and ATP hydrolysis. J Cell Biol. 1998;143:901–10. doi: 10.1083/jcb.143.4.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochel HJ, Schulte TW, Nguyen P, Trepel J, Neckers L. The benzoquinone ansamycin geldanamycin stimulates proteolytic degradation of focal adhesion kinase. Mol Genet Metab. 1999;66:24–30. doi: 10.1006/mgme.1998.2774. [DOI] [PubMed] [Google Scholar]
- Owens LV, Xu L, and Craven RJ. et al. 1995 Overexpression of the focal adhesion kinase (p125FAK) in invasive human tumors. Cancer Res. 55:2752–2755. [PubMed] [Google Scholar]
- Panaretou B, Prodromou C, Roe SM, O'Brien R, Ladbury JE, Piper PW, Pearl LH. ATP binding and hydrolysis are essential to the function of the Hsp90 molecular chaperone in vivo. EMBO J. 1998;17:4829–4836. doi: 10.1093/emboj/17.16.4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard D, Khursheed B, Garabedian MJ, Fortin MG, Lindquist S, Yamamoto KR. Reduced levels of Hsp90 compromise steroid receptor action in vivo. Nature. 1990;348:166–168. doi: 10.1038/348166a0. [DOI] [PubMed] [Google Scholar]
- Price PJ, Suk WA, Skeen PC, Spahn GJ, Chirigos MA. Geldanamycin inhibition of 3-methylcholanthrene-induced rat embryo cell transformation. Proc Soc Exp Biol Med. 1977;155:461–463. doi: 10.3181/00379727-155-39830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prodromou C, Roe SM, O'Brien R, Ladbury JE, Piper PW, Pearl LH. Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell. 1997;90:65–75. doi: 10.1016/s0092-8674(00)80314-1. [DOI] [PubMed] [Google Scholar]
- Rebbe NF, Ware J, Bertina RM, Modrich M, Stafford DW. Nucleotide sequence of a cDNA for a member of the human 90-kDa heat-shock protein family. Gene. 1987;53:235–245. doi: 10.1016/0378-1119(87)90012-6. [DOI] [PubMed] [Google Scholar]
- Rinehart KL, Shield LS 1976 Chemistry of the ansamycin antibiotics. In: Progress in the Chemistry of Organic Natural Products, ed Herz W, Grisebach H, Kirby GW. Springer, New York, NY, 232–300. [DOI] [PubMed] [Google Scholar]
- Roe SM, Prodromou C, O'Brien R, Ladbury JE, Piper PW, Pearl LH. Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J Med Chem. 1999;42:260–266. doi: 10.1021/jm980403y. [DOI] [PubMed] [Google Scholar]
- Rutherford SL, Lindquist S. Hsp90 as a capacitor for morphological evolution. Nature. 1998;396:336–342. doi: 10.1038/24550. [DOI] [PubMed] [Google Scholar]
- Sakagami M, Morrison P, Welch WJ. Benzoquinoid ansamycins (herbimycin A and geldanamycin) interfere with the maturation of growth factor receptor tyrosine kinases. Cell Stress Chaperones. 1999;4:19–28. doi: 10.1006/csac.1998.0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki K, Yasuda H, Onodera K. Growth inhibition of virus transformed cells in vitro and antitumor activity in vivo of geldanamycin and its derivatives. J Antibiot (Tokyo) 1979;32:849–851. doi: 10.7164/antibiotics.32.849. [DOI] [PubMed] [Google Scholar]
- Schaller MD, Borgman CA, Cobb BS, Vines RR, Reynolds AB, Parsons JT. pp125FAK a structurally distinctive protein-tyrosine kinase associated with focal adhesions. Proc Natl Acad Sci U S A. 1992;89:5192–5196. doi: 10.1073/pnas.89.11.5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheibel T, Siegmund HI, Jaenicke R, Ganz P, Lilie H, Buchner J. The charged region of Hsp90 modulates the function of the N-terminal domain. Proc Natl Acad Sci U S A. 1999;96:1297–1302. doi: 10.1073/pnas.96.4.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnur RC, Corman ML, and Gallaschun RJ. et al. 1995a erbB-2 oncogene inhibition by geldanamycin derivatives: synthesis, mechanism of action, and structure-activity relationships. J Med Chem. 38:3813–3820. [DOI] [PubMed] [Google Scholar]
- Schnur RC, Corman ML, and Gallaschun RJ. et al. 1995b Inhibition of the oncogene product p185erbB-2 in vitro and in vivo by geldanamycin and dihydrogeldanamycin derivatives. J Med Chem. 38:3806–3812. [DOI] [PubMed] [Google Scholar]
- Schulte TW, Akinaga S, Soga S, Sullivan W, Stensgard B, Toft D, Neckers L. Antibiotic radicicol binds to the N-terminal domain of Hsp90 and shares important biologic activities with geldanamycin. Cell Stress Chaperones. 1998;3:100–108. doi: 10.1379/1466-1268(1998)003<0100:arbttn>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte TW, An WG, Neckers LM. Geldanamycin-induced destabilization of raf-1 involves the proteasome. Biochem Biophys Res Commun. 1997;239:655–659. doi: 10.1006/bbrc.1997.7527. [DOI] [PubMed] [Google Scholar]
- Schulte TW, Blagosklonny MV, Ingui C, Neckers L. Disruption of the raf-1-Hsp90 molecular complex results in destabilization of raf-1 and loss of raf-1-ras association. J Biol Chem. 1995;270:24585–24588. doi: 10.1074/jbc.270.41.24585. [DOI] [PubMed] [Google Scholar]
- Schulte TW, Blagosklonny MV, and Romanova L. et al. 1996 Destabilization of raf-1 by geldanamycin leads to disruption of the raf-1-MEK-mitogen-activated protein kinase signaling pathway. Mol Cell Biol. 16:5839–5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte TW, Neckers LM. The benzoquinone ansamycin 17-allylamino-17-demethoxygeldanamycin binds to Hsp90 and shares important biologic activities with geldanamycin. Cancer Chemother Pharmacol. 1998;42:273–279. doi: 10.1007/s002800050817. [DOI] [PubMed] [Google Scholar]
- Schulte TW, Akinaga S, and Murakata T. et al. 1999 Interaction of radicicol with members of the heat shock protein 90 family of molecular chaperones. Mol Endocrinol. 13:1435–1448. [DOI] [PubMed] [Google Scholar]
- Schumacher RJ, Hansen WJ, Freeman BC, Alnemri E, Litwack G, Toft DO. Cooperative action of hsp70, Hsp90, and dnaJ proteins in protein renaturation. Biochemistry. 1996;35:14889–14898. doi: 10.1021/bi961825h. [DOI] [PubMed] [Google Scholar]
- Sepehrnia B, Paz IB, Dasgupta G, Momand J. Heat shock protein 84 forms a complex with mutant p53 protein predominantly within a cytoplasmic compartment of the cell. J Biol Chem. 1996;271:15084–15090. doi: 10.1074/jbc.271.25.15084. [DOI] [PubMed] [Google Scholar]
- Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–182. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- Slamon DJ, Godolphin W, and Jones LA. et al. 1989 Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science. 244:707–712. [DOI] [PubMed] [Google Scholar]
- Soga S, Neckers LM, and Schulte TW. et al. 1999 KF25706, a novel oxime derivative of radicicol, exhibits in vivo antitumor activity via selective depletion of Hsp90 binding signaling molecules. Cancer Res. 59:2931–2938. [PubMed] [Google Scholar]
- Song HY, Dunbar JD, Zhang YX, Guo D, Donner DB. Identification of a protein with homology to Hsp90 that binds the type 1 tumor necrosis factor receptor. J Biol Chem. 1995;270:3574–3581. [PubMed] [Google Scholar]
- Srivastava BI, DiCioccio RA, Rinehart KL Jr,, Li LH. Preferential inhibition of terminal deoxynucleotidyltransferase activity among deoxyribonucleic acid polymerase activities of leukemic and normal cells by geldanamycin, streptoval C, streptovarone, and dapmavarone. Mol Pharmacol. 1978;14:442–447. [PubMed] [Google Scholar]
- Stancato LF, Chow YH, Hutchinson KA, Perdew GH, Jove R, Pratt WB. Raf exists in a native heterocomplex with Hsp90 and p50 that can be reconstituted in a cell-free system. J Biol Chem. 1993;268:21711–21716. [PubMed] [Google Scholar]
- Stancato LF, Silverstein AM, Owens-Grillo JK, Chow YH, Jove R, Pratt WB. The Hsp90-binding antibiotic geldanamycin decreases raf levels and epidermal growth factor signaling without disrupting formation of signaling complexes or reducing the specific enzymatic activity of Raf kinase. J Biol Chem. 1997;272:4013–4020. doi: 10.1074/jbc.272.7.4013. [DOI] [PubMed] [Google Scholar]
- Stebbins CE, Russo AA, Schneider C, Rosen N, Hartl FU, Pavletich NP. Crystal structure of an Hsp90-geldanamycin complex: targeting of a protein chaperone by an antitumor agent. Cell. 1997;89:239–250. doi: 10.1016/s0092-8674(00)80203-2. [DOI] [PubMed] [Google Scholar]
- Sullivan WP, Toft DO. Mutational analysis of Hsp90 binding to the progesterone receptor. J Biol Chem. 1993;268:20373–20379. [PubMed] [Google Scholar]
- Supko JG, Hickman RL, Grever MR, Malspeis L. Preclinical pharmacologic evaluation of geldanamycin as an antitumor agent. Cancer Chemother Pharmacol. 1995;36:305–315. doi: 10.1007/BF00689048. [DOI] [PubMed] [Google Scholar]
- Uehara Y, Fukazawa H, Murakami Y, Mizuno S. Irreversible inhibition of v-src tyrosine kinase activity by herbimycin A and its abrogation by sulfhydryl compounds. Biochem Biophys Res Commun. 1989;163:803–809. doi: 10.1016/0006-291x(89)92293-6. [DOI] [PubMed] [Google Scholar]
- Uehara Y, Hori M, Takeuchi T, Umezawa H. Screening of agents which convert “transformed morphology” of Rous sarcoma virus-infected rat kidney cells to “normal morphology”: identification of an active agent as herbimycin and its inhibition of intracellular src kinase. Jpn J Cancer Res. 1985;76:672–675. [PubMed] [Google Scholar]
- Uehara Y, Hori M, Takeuchi T, Umezawa H. Phenotypic change from transformed to normal induced by benzoquinonoid ansamycins accompanies inactivation of p60src in rat kidney cells infected with Rous sarcoma virus. Mol Cell Biol. 1986;6:2198–2206. doi: 10.1128/mcb.6.6.2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner TM, Liu ET, Craven RJ, Cance WG. Expression of growth factor receptors, the focal adhesion kinase, and other tyrosine kinases in human soft tissue tumors. Ann Surg Oncol. 1994;1:18–27. doi: 10.1007/BF02303537. [DOI] [PubMed] [Google Scholar]
- Whitesell L, Cook P. Stable and specific binding of heat shock protein 90 by geldanamycin disrupts glucocorticoid receptor function in intact cells. Mol Endocrinol. 1996;10:705–712. doi: 10.1210/mend.10.6.8776730. [DOI] [PubMed] [Google Scholar]
- Whitesell L, Mimnaugh EG, De Costa B, Myers CE, Neckers LM. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: essential role for stress proteins in oncogenic transformation. Proc Natl Acad Sci U S A. 1994;91:8324–8328. doi: 10.1073/pnas.91.18.8324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitesell L, Shifrin SD, Schwab G, Neckers LM. Benzoquinonoid ansamycins possess selective tumoricidal activity unrelated to src kinase inhibition. Cancer Res. 1992;52:1721–1728. [PubMed] [Google Scholar]
- Whitesell L, Sutphin P, An WG, Schulte T, Blagosklonny MV, Neckers L. Geldanamycin-stimulated destabilization of mutated p53 is mediated by the proteasome in vivo. Oncogene. 1997;14:2809–2816. doi: 10.1038/sj.onc.1201120. [DOI] [PubMed] [Google Scholar]
- Whitesell L, Sutphin PD, Pulcini EJ, Martinez JD, Cook PH. The physical association of multiple molecular chaperone proteins with mutant p53 is altered by geldanamycin, an Hsp90-binding agent. Mol Cell Biol. 1998;18:1517–1524. doi: 10.1128/mcb.18.3.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaki H, Nakajima M, Seimiya H, Saya H, Sugita M, Tsuruo T. Inhibition of the association with nuclear matrix of pRB, p70 and p40 proteins along with the specific suppression of c-myc expression by geldanamycin, an inhibitor of Src tyrosine kinase. J Antibiot (Tokyo) 1995;48:1021–1026. doi: 10.7164/antibiotics.48.1021. [DOI] [PubMed] [Google Scholar]
- Yamaki H, Suzuki H, Choi EC, Tanaka N. Inhibition of DNA synthesis in murine tumor cells by geldanamycin, an antibiotic of the benzoquinoid ansamycin group. J Antibiot (Tokyo) 1982;35:886–892. doi: 10.7164/antibiotics.35.886. [DOI] [PubMed] [Google Scholar]
- Young JC, Schneider C, Hartl FU. In vitro evidence that Hsp90 contains two independent chaperone sites. FEBS Lett. 1997;418:139–143. doi: 10.1016/s0014-5793(97)01363-x. [DOI] [PubMed] [Google Scholar]