


Abstract
The driver genetic aberrations collectively regulate core cellular processes underlying cancer development. However, identifying the modules of driver genetic alterations and characterizing their functional mechanisms are still major challenges for cancer studies. Here, we developed an integrative multi-omics method CMDD to identify the driver modules and their affecting dysregulated genes through characterizing genetic alteration-induced dysregulated networks. Applied to glioblastoma (GBM), the CMDD identified a core gene module of 17 genes, including seven known GBM drivers, and their dysregulated genes. The module showed significant association with shorter survival of GBM. When classifying driver genes in the module into two gene sets according to their genetic alteration patterns, we found that one gene set directly participated in the glioma pathway, while the other indirectly regulated the glioma pathway, mostly, via their dysregulated genes. Both of the two gene sets were significant contributors to survival and helpful for classifying GBM subtypes, suggesting their critical roles in GBM pathogenesis. Also, by applying the CMDD to other six cancers, we identified some novel core modules associated with overall survival of patients. Together, these results demonstrate integrative multi-omics data can identify driver modules and uncover their dysregulated genes, which is useful for interpreting cancer genome.
INTRODUCTION
Genetic alteration was a major mechanism underlying the transformation of normal cells to cancerous cells (1,2). Driver alterations can cooperatively disrupt a cascade of crucial biological pathways and thus provide cells with significant growth advantages (3,4). However, due to the extensive complexity of genetic alterations in cancer genome, how to identify the driver modules and characterize their functional mechanism during carcinogenesis is still a major challenge.
Initially, multiple computational methods were proposed to identify driver genes based on somatic mutation rates in cancer patient populations (5–7). With the whole-genome measurements of somatic mutations and copy number variants in the mass of cancer samples, several cancer studies revealed that driver genetic alterations involved in the same biological pathways showed mutual exclusivity (8,9). Based on the observation of mutual exclusivity, some computational methods were developed to identify causal gene modules in which genomic alterations of their member genes cover the majority of cancer samples and exhibit significant mutual exclusivity (10–13). Similarly, there are also some computational methods based on co-occurrence alteration events for identifying driver genes (14,15). Recently, combination of DNA copy number/mutation profiles and gene expression profiles were used to find driver genes by quantifying the effect of genetic alterations on the transcriptional changes (16,17). For instance, Masica et al. (18) proposed a fisher-based method to identify driver genes by calculating the correlations between somatic mutations and expression of outlying genes. DriverNet was developed to find the minimum number of driver genes whose genetic alterations can explain transcriptional changes of all genes across cancer samples, to the largest extent, by integrating the matched genome and transcriptome data (19). Although many computational methods have been proposed to identify potential driver genetic alterations, it is very limited to understand how driver genes result in extensive transcriptional changes and in turn contribute to malignant phenotypes (20,21).
In general, gene expression can be regulated by multi-layer factors such as copy number, methylation, transcription factors (TFs) and miRNAs (22–24). Recurrent copy number alterations of ∼76% genes exhibited significant correlations with gene expression in glioblastoma (24). The promoter methylation of genes can repress their own gene expression, such as the methylation of O-6-methylguanine-DNA methyltransferase (MGMT) in glioblastoma (25). In addition, TFs like oncogenic v-myc avian myelocytomatosis viral oncogene homolog (MYC) were capable of shaping tumor-specific gene expression profile (26). Also, most miRNAs predominantly decrease the mRNA levels of their targets by binding to 3′ untranslated region (UTR) (27). These factors constitute a regulatory system for maintaining homeostasis of normal cells (28–31). Diverse genetic alterations may coordinately disrupt the delicate balance by perturbing different types of regulatory relationships (32), which can finally lead to the transition from normal cell to cancer cell (33,34). Based on the principle, Jornsten et al. combined mRNA transcription regulation and turnover in a steady-state condition and constructed a CNA-driven network using lasso regression to identify driver copy number alterations (CNAs) and detect their effect on transcription in glioblastoma (GBM) (35). Subsequently, an integrative regression model further combined regulatory sequence information of TFs and miRNAs to explain the expression changes in each tumor sample, aiming to inferring transcriptional and miRNA-mediated regulatory program in GBM (36). However, little is known about the effect of driver alterations on the complex regulatory mechanisms of gene expression. Therefore, characterizing dysregulated network induced by genetic alterations in cancer will not only help us discover causal genes but also provide the details for understanding the molecular mechanism of causal genes.
In this work, we proposed a systematic method, called CMDD (Core Modules Driving Dysregulation in cancer) to identify driver modules and their dysregulated genes based on multilayer factor-mediated dysregulated networks through integrating multi-dimensional genomic data including DNA mutation, copy number, methylation, gene expression and miRNA expression profiles, as well as regulatory networks and protein interaction network. We applied our method to GBM and identified a core module consisting of 17 genes and their dysregulated genes. Seven well-known GBM-associated genes were included in the module. Moreover, we found that the module was significantly involved in many cancer-related pathways and was associated with survival. We further explored the dysregulated genes associated with the module, and revealed that two genes subsets participated in GBM-related important pathways with different ways. Also, we applied CMDD to other types of cancer and identified some novel cancer-associated modules.
MATERIALS AND METHODS
Multi-dimensional genomic data
The multi-dimensional GBM-associated data sets containing DNA mutation, copy number, DNA methylation, mRNA and miRNA expression data were collected from TCGA data portal (https://tcga-data.nci.nih.gov/tcga/). We extracted a somatic mutation data (level 2) involving 291 GBM samples directly from TCGA. Through removing silent mutations, a total of 8299 mutation genes were obtained. For a CNV array data (SNP6.0) referring to 530 GBM samples, the segmentations were identified using the circular binary segmentation method-based copy numbers estimated by calculation of normalized log2-tranformed ratios (level 3) (7). The level 3 data of DNA methylation data of 296 samples detected by JHU-USC HumanMenthylation27 were downloaded and the average methylation value for a given gene was calculated. The gene expression profile of 12042 genes in 538 samples and the miRNA expression profile of 470 human miRNAs in 500 samples were obtained. We also obtained clinical information of 465 GBM samples. Finally, 121 GBM samples having all of these genomic data were determined.
MiRNA target interactions between 1909 miRNAs and 16708 genes were downloaded from the database miRecords (37), which was an integrated miRNA-target resource. We obtained experimentally validated TF-gene regulatory relationships from the TRANSFAC (38) and predicted novel regulatory relationships using conserved TF binding sites from UCSC genome browser. The protein interaction network was obtained from the Human Protein Reference Database (HPRD) (39).
Overview of the method CMDD
We proposed a systematic method CMDD to identify cancer core gene modules and their affecting dysregulated genes through multilayer factor-mediated dysregulated networks (Figure 1). The detailed description of the method is as follows.
Figure 1.
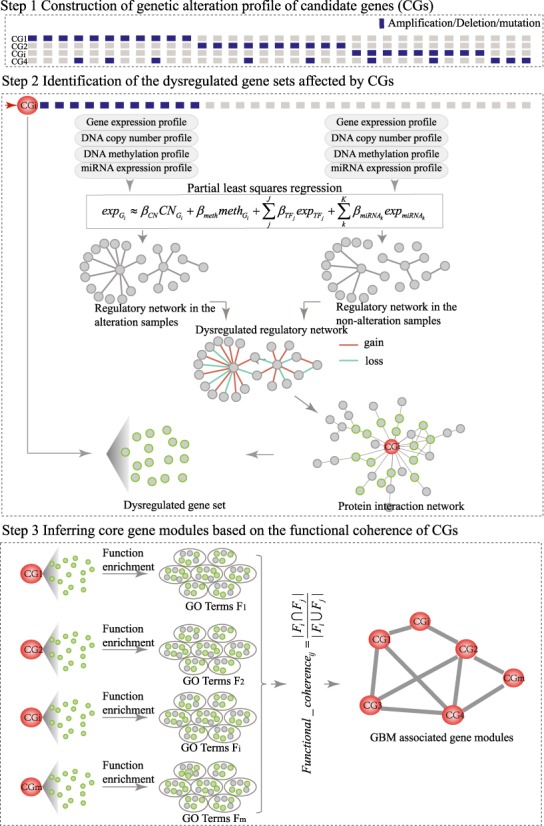
The workflow of the systematic method for identifying cancer core gene modules through integrating multi-dimensional genomic data.
Construction of genetic alteration profiles of candidate genes
We combined the DNA copy number data and DNA mutation data to construct the genetic alteration profile. The GISTIC (version 2) was applied to the DNA copy number data, and five types of discretized copy number calls (homozygous deletion, heterozygous deletion, diploid, gain and amplification) for each gene in all cancer samples were determined. Only the amplification and homozygous deletions were considered as copy number alterations. Combining with DNA mutation data, we generated a binary profile D of genetic alteration, in which dij was scored as 1 if the ith gene in the jth sample shows genetic alteration (i.e. amplification/homozygous deletion/mutation), and 0 otherwise.
Next, we used two criteria to select the candidate genes (CGs) through integrating the binary profile of genetic alteration and gene expression profile across cancer samples: (i) CGs should alter in at least 10 samples; ii) for a given CG, it should be differentially expressed between patients with and without genetic alterations of this CG using t-test at false discovery rate (FDR) ≤ 0.05. Then, the binary profile of the selected CGs was obtained.
Identification of dysregulated gene sets affected by CGs
To identify the dysregulated gene sets of CGs, we built CG-associated regulatory networks under the conditions with/without genetic alterations of CGs and constructed corresponding dysregulated networks by network comparisons.
Construction of regulatory networks under the conditions with and without genetic alterations
For every CG, we grouped the cancer samples into two groups according to its alteration status (one group with genetic alteration and the other without). Then, we constructed linear regression models for each gene Gi (i = 1,2,…,n) separately in the two groups of samples to explain gene expression changes using multi-layer regulatory factors including DNA copy number, methylation, TF and miRNA regulations as covariates.
Given a gene i (Gi) in a specific condition containing N samples, there are J TFs 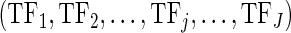 and K miRNAs (
and K miRNAs (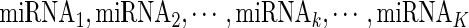 ) binding Gi. A linear regression model was trained as
) binding Gi. A linear regression model was trained as
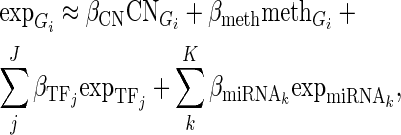 |
where the 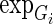 is the expression levels of Gi in N samples,
is the expression levels of Gi in N samples, 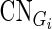 is the copy number calls of Gi,
is the copy number calls of Gi, 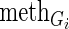 is the methylation values of Gi,
is the methylation values of Gi, 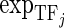 is the expression levels of the jth TF regulating Gi and
is the expression levels of the jth TF regulating Gi and 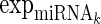 is the expression levels of the kth miRNA targeting Gi.
is the expression levels of the kth miRNA targeting Gi. 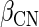 ,
,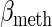 ,
, 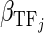 and
and 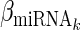 represent regression coefficients of
represent regression coefficients of 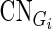 ,
, 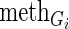 ,
, 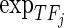 and
and 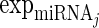 , respectively. Only the genes that were differentially expressed between cancer samples and normal samples and showed high expression variability (within the top 70%) across cancer samples (see Supplementary Materials for details) were used to train regression models. As the large number of variables and their possible high collinearity, we used the partial least squares (40) model to train the linear regression model (see Supplementary Materials). Then, using the functions (‘plsr’, ‘RMSEP’ and ‘jack.test’) in R package ‘pls’, the significance of the effect of input variables on gene expression variation was estimated using 10-fold cross-validation. Subsequently, we determined the regulatory factors (including copy number, methylation, TFs and miRNAs) with significant coefficients (FDR ≤ 0.001) for each gene and two regulatory networks corresponding to different cancer groups were formed.
, respectively. Only the genes that were differentially expressed between cancer samples and normal samples and showed high expression variability (within the top 70%) across cancer samples (see Supplementary Materials for details) were used to train regression models. As the large number of variables and their possible high collinearity, we used the partial least squares (40) model to train the linear regression model (see Supplementary Materials). Then, using the functions (‘plsr’, ‘RMSEP’ and ‘jack.test’) in R package ‘pls’, the significance of the effect of input variables on gene expression variation was estimated using 10-fold cross-validation. Subsequently, we determined the regulatory factors (including copy number, methylation, TFs and miRNAs) with significant coefficients (FDR ≤ 0.001) for each gene and two regulatory networks corresponding to different cancer groups were formed.
Construction of dysregulated networks affected by CGs
For each CG, we compared the two regulatory networks in the groups with/without genetic alterations. The relationships that were only presented in one of the two regulatory networks were identified and defined as the dysregulated regulatory relationships. To determine the significance of the dysregulated relationships, the genetic alteration profile was permuted 1000 times, preserving alteration frequency of CGs. We re-built 1000 random dysregulated networks and calculated the frequencies of the real dysregulated relationships in the 1000 random networks. Only the dysregulated relationships with less than 10% of frequency were used to form the dysregulated network.
Extraction of dysregulated genes affected by CGs
Next, in order to obtain dysregulated genes affected by CGs, the genes in the dysregulated network of a given CG were mapped onto the protein–protein interaction network. The genes within two-step distance from this CG in the protein interaction network were selected as the dysregulated genes of the CG.
Inferring core gene modules based on the functional coherence of CGs
We assumed that different cancer-associated genes disturbed the same or similar functions during tumorigenesis. Based on the hypothesis, for each CG, we determined the GO biological functions significantly enriched by its dysregulated genes using hypergeometric distribution test (P < 0.001). Then, we calculated a functional coherence score for each pair of CGs as follows:
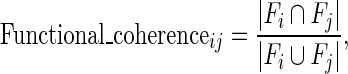 |
where 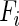 and
and 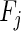 represent significantly enriched function sets associated with the ith and jth CGs, respectively,
represent significantly enriched function sets associated with the ith and jth CGs, respectively, 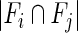 represents the number of their common functions and
represents the number of their common functions and 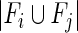 represents the total number of unique functions associated with them. Finally, the CG pairs with coherence scores larger than a threshold that was determined through analyzing the distribution of coherence scores were selected to constitute cancer core gene modules.
represents the total number of unique functions associated with them. Finally, the CG pairs with coherence scores larger than a threshold that was determined through analyzing the distribution of coherence scores were selected to constitute cancer core gene modules.
Survival analysis
We used the survival analysis to test the association of the genetic alteration of single genes or modules with cancer overall survivals. Survival analysis was performed using the R package ‘survival’.
Survival analysis for single genes
For a given single gene, we divided the cancer samples into two groups according to the genetic alteration status of the gene: the alteration group containing the samples with genetic alteration of this gene and the non-alteration group without. The significance of survival difference between the alteration and non-alteration groups was estimated using Kaplan–Meier analysis and log-rank test.
Survival analysis for core modules
For a given module, we divided the cancer samples into two groups according to the genetic alteration status of all genes in the module: the alteration group containing the samples with genetic alteration in at least one gene of the module and the non-alteration group without any alterations in the genes of the module. Likewise, the significance of survival difference between alteration and non-alteration groups was estimated using Kaplan–Meier analysis and log-rank test.
RESULTS
Identifying the core gene module in GBM
Identifying cancer-associated core module of genetic alterations was a key step for understanding fundamental mechanisms of carcinogenesis. We developed a systematic method CMDD based on dysregulated genes affected by genetic alterations. By applying CMDD to the multi-dimensional data sets of 121 GBM samples including DNA mutation, copy number, methylation, gene expression and miRNA expression profiles, we identified the dysregulated genes for each CG (see the Materials and Methods section for details) and determined the GO biological processes significantly enriched by the dysregulated genes of each CG (P = 0.001, hypergeometric distribution test). Notably, their functional coherence scores showed a bimodal distribution. We set the threshold of 0.4 that can distinguish the two peaks to find CGs with highly consistent functions (Figure 2A). Then, a core gene module composed of 17 genes including EGFR, PDGFRA, RB1, CDKN2A, PTEN, MDM4, CDK4, PIK3C2B, KIT, PHKG1, DDIT3, DCTN2, KIF5A, OS9, METTL1, AVIL and CTDSP2 was identified (Figure 2B).
Figure 2.
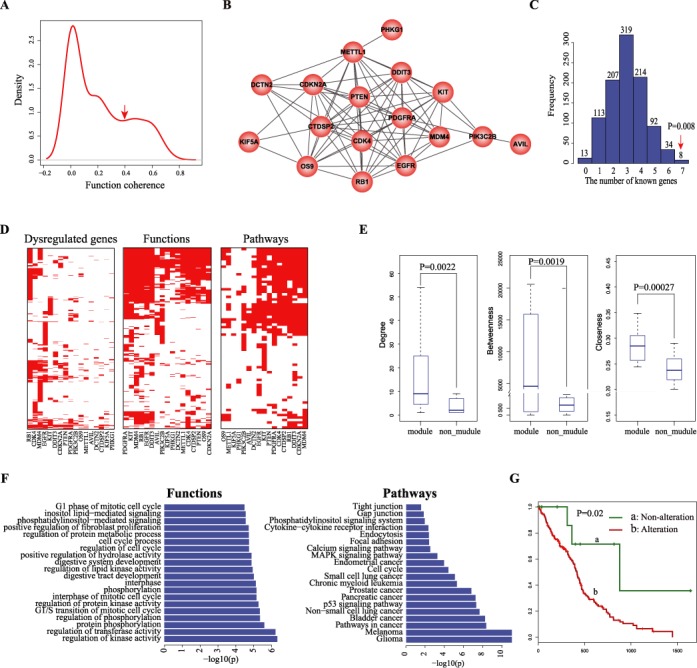
Identification of the core module in GBM. (A) The distribution of functional coherence scores of pairwise candidate genes.The arrow represents the position of the selected threshold for determining core functional modules. (B) The GBM core gene module composed of 17 highly interconnected genes. (C) The distribution of the numbers of known GBM associated genes in randomly selected gene sets. The arrow represents the real number of known GBM associated in the module. (D) The heatmaps of dysregulated genes (left), and their enriched functions (middle) and pathways (right) across the 17 genes in the module. (E) Comparisons of three topological features including degree (right), betweenness (middle) and closeness (right) of the 17 genes in module with the rest candidate genes not in the module. (F) The biological functions (left) and pathways (right) significantly enriched by the 17 genes in the module. (G) Kaplan–Meier estimates of overall survival for GBM patients in TCGA data set, with patients stratified into two groups according to the alteration status of member genes in the module.
Among the 17 genes, seven (including EGFR, PDGFRA, RB1, CDKN2A, PTEN, MDM4 and CDK4) have been reported to be associated with GBM (P = 0.0014, hypergeometric distribution test). PDGFRA and EGFR are the gene signatures for proneural and classical GBM subclass, respectively (41). The dysfunctions of EGFR and PDGFRA could lead to glioma formation in mouse model. (42,43). Mutation of RB1 disturbs its interaction with E2F, thus leading to cell cycle disorder and affecting therapeutic efficacy in GBM (44,45). Meanwhile, we randomly selected 17 genes from the original CGs 1000 times and found that the core module identified has more known GBM genes (P = 0.008; Figure 2C). In addition to known GBM-associated genes, two novel genes including METTL1 and CTDSP2 showing the highest degree in the core module were identified (Supplementary Figure S1). We found that the dysregulated genes of both METTL1 and CTDSP2 were significantly enriched in many cancer-related functions and pathways. Moreover, the survival analyses based on their genetic alteration profiles and expression profiles consistently showed that METTL1 and CTDSP2 were associated with shorter survival in GBM (Supplementary Figures S2 and S3; see Supplementary Materials for details). These findings suggested that METTL1 and CTDSP2 were the potential novel genes whose genetic alterations may contribute to the development of GBM.
In addition, we found that three topological features (including degree, betweenness and closeness) of the 17 genes in the protein interaction network were significantly higher than those of the rest CGs (Wilcoxon–Wilcox test, P = 0.002 for degree, P = 0.0019 for betweenness and P = 0.00027 for closeness; Figure 2E), highlighting their close functional associations. Then, we performed function and pathway enrichment analyses directly using the 17 genes in the module. We found that the module participated in cancer-associated functions and pathways such as ‘Glioma’, ‘G1 phase of mitotic cell cycle’ and ‘regulation of protein kinase activity’ (Figure 2F). Also, the prognostic effect of the module on GBM clinical outcomes was analyzed. We classified GBM samples into two groups: the alteration group contained patients harboring the alteration of at least one member gene in the module and the non-alteration group containing patients without any alterations of member genes. We found the alteration of the module was associated with shorter survival (P = 0.02, log-rank test; Figure 2G). Together, the GBM-associated core gene module identified by our method not only identified the known and novel GBM-associated genes but also determined their functional relationships in GBM, which could be an effective prognostic indicator of GBM.
Characterization of dysregulated genes of the core module
Next, we characterized different property of the dysregulated genes of the core module. Through the expression correlation analysis of dysregulated genes, we found that dysregulated genes induced by member genes in the module tend to show significantly higher expression correlations compared to randomly selected genes (P < 0.05, Kolmogorov–Smirnov test, Supplementary Figure S4; see Supplementary Materials for details). Enrichment analyses showed that the dysregulated genes can significantly capture the functions of their corresponding member genes (P < 0.05, hypergeometric test; Supplementary Figure S5). Moreover, after knockdown of CDK4 (GSE8866) (46) and RB1 (GSE31534) (47), both of their dysregulated genes showed significant enrichments in the differentially expressed genes (P = 0.089 for CDK4 and P = 0.02 for RB1). Notably, despite the high overlapping of functions and pathways between these 17 genes, only a few common dysregulated genes were observed (Figure 2D), suggesting that these genes in the module influenced similar biological processes through different dysregulated genes. In summary, our results showed that dysregulated genes could effectively reflect the abnormal molecular events associated with genetic alteration of CGs.
GBM-associated dysregulated network mediated by multi-factors
To further characterize how the genetic alterations of member genes in the core module affected the regulatory relationships of their dysregulated genes, we constructed a network by connecting the 17 member genes in the module to their dysregulated genes (Figure 3). In this network, we found that most dysregulated genes linked with only one or two member genes. Among the dysregulated genes, genes affected by multiple member genes were more likely to be cancer-associated genes reported in OMIM (48), CGC (49) or GAD (50) such as ESR1, AKT1 and BRCA1 (Supplementary Figure S6). Especially, ESR1 was found to be connected by the largest number of member genes (15/17 member genes; Figure 3). Previous studies have demonstrated that the encoded protein of ESR1 was involved in pathological processes of multiple cancers (51,52). And Uhlmann et al. discovered a putative association between ESR1 and gliomagenesis (53). Furthermore, we also found high expression of ESR1 was associated with favorable survival in TCGA GBM data set (P = 0.007, log-rank test; Supplementary Figure S7). These findings further support the importance of the member genes in GBM.
Figure 3.
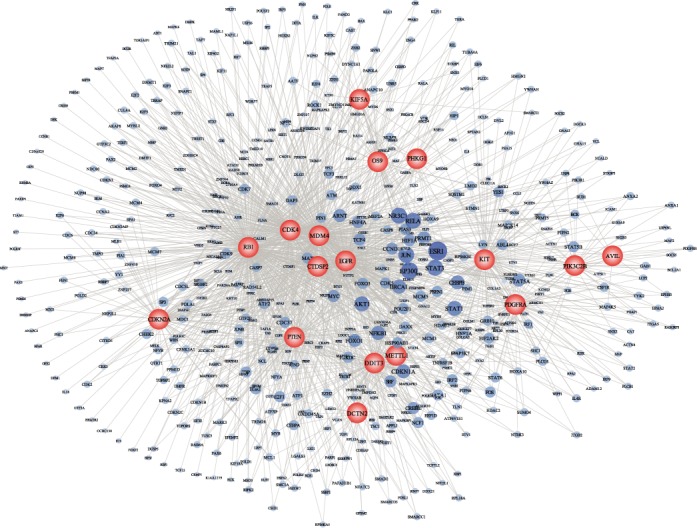
Comprehensive dysregulated network induced by the core module. Two types of nodes represent the member genes and dysregulated gens, respectively. The node size for member genes is proportion to the number of member genes connecting to the dysregulated genes.
In addition, we observed that the dysregulated genes of the module were consistently implicated in many cancer-related biological processes (such as ‘epidermal growth factor receptor signaling pathway’, ‘glial cell differentiation’, ‘apoptotic signaling pathway’ and ‘MAPK cascade’) and pathways (such as Glioma, ‘p53 signaling pathway’ and ‘Notch signaling pathway’) (Figure 4A). For each member gene, the regulatory factors of its dysregulated genes were analyzed. We found that TFs accounted for most dysregulated genes (Figure 4B), suggesting that disturbance of these cancer-related pathways is primarily dependent on dysfunction of TF-mediated regulations. We further analyzed a well-known GBM-associated pathway ‘epidermal growth factor receptor signaling pathway’. Nine of the 17 genes influenced dysregulated genes by distinct regulatory relationships and in turn regulated this pathway. For example, the TF-mediated dysregulation of AKT was consistently observed in dysregulated networks of eight member genes. The copy number-mediated dysregulation of PLCG1 was observed for DDIT3 and RB1, while additional TF-mediated dysregulation occurred in PIK3C2B and KIT. MiRNA-mediated dysregulation of ADAM12 and DNA methylation-mediated dysregulation of ADCY7 appeared in RB1 and EGFR, respectively (Figure 4C). Another three cancer-associated biological processes including ‘glial cell differentiation’, ‘apoptotic signaling pathway’ and ‘MAPK cascade’ showed similar multi-factor-mediated dysregulation (Supplementary Figure S8).
Figure 4.
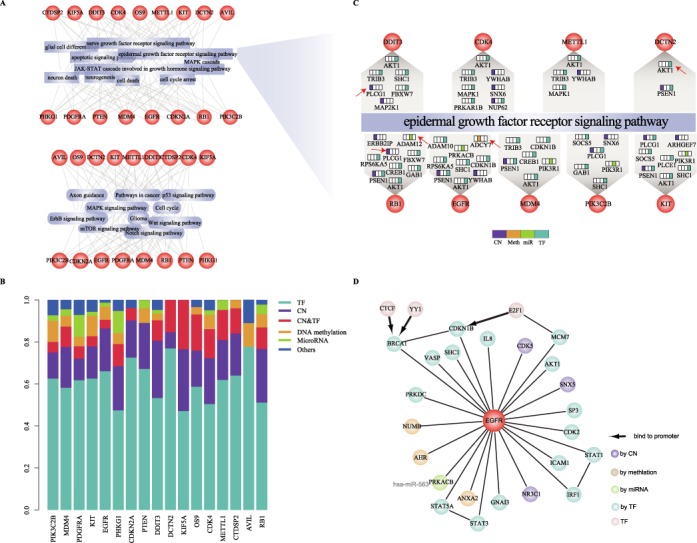
Estimation of multi-factor mediated dysregulations affected by the module. (A) The cancer associated biological processes (top) and pathways (bottom) enriched by the dysregulated genes of 17 member genes. (B) The transcription factors account for most of dysfunctions of dysregulated genes. (C) Nine member genes affect ‘epidermal growth factor receptor signaling pathway’ through distinct dysregulation of their dysregulated genes. The dysregulation of dysregulated genes is arranged in order: copy number, methylation, miRNA, TF. The genes mentioned in the text are labeled by arrows. (D) The literature-curated dysregulated relationships linked with EGFR.
Furthermore, by literature searching, we confirmed that many dysregulated relationships linked with EGFR had been demonstrated (Figure 4D). For example, genetic alteration of EGFR disturbed the expression of STAT1 and IRF1 (54). Also, the TF STAT1-mediated dysregulation of IRF1 has been confirmed in the proliferation- and apoptosis-associated signaling pathway (55). SHC1, regulated by TF FOSL1, was reported to specifically co-purified with mutated EGFR, and inhibition of SHC1 showed significant association with drug resistance in EGFR mutant cells (56). The overexpression of Numb induced by genetic alteration of EGFR may depend on methylation dysfunction (57). In addition, in the condition of EGFR mutation, miR-563-mediated dysfunction of PRKACB, a subunit of PKA, was found to promote glioma cell growth and invasion (58). CDK5 with copy number-mediated dysregulation is a downstream gene in the EGFR-family signaling (59). Taken together, the genetic alterations of member genes in the core modules could induce the multifactor-mediated regulation of dysregulated genes, especially TF-mediated regulation, and in turn disturb the cancer associated functions and pathways.
Dysregulated Glioma pathway induced by the core module contributing to survival
We investigated how the core module and their dysregulated genes participated in Glioma pathway, and contributed to survival. The patients with at least one altered member gene were divided into two subgroups using unsupervised hierarchical clustering based on the genetic alteration profiles of the core gene module (Figure 5A). We found that one subgroup (b1) showed frequent genetic alterations of one gene set (I) including nine genes (EGFR, PTEN, PDGFRA, CDKN2A, MDM4, KIT, PIK3C2B, RB1 and PHKG1), while the other (b2) had the frequent genetic alterations of another gene set (II) including eight genes (CDK4, METTL1, OS9, AVIL, CTDSP2, KIF5A, DCTN2 and DDIT3). Investigation of the dysregulated genes showed that both of these two gene sets were associated with the ‘Glioma’ pathway. Notably, among the gene set I, five genes including PDGFRA, RB1, PTEN, EGFR and CDKN2A were directly involved in the ‘Glioma’ pathway (Figure 5B). In contrast, the gene set II seemed to indirectly regulate the pathway through influencing their dysregulated genes (Figure 5C). Meanwhile, these two gene sets showed local differences in the pathway. MAPK1 dysregulation was only observed in the pathway indirectly influenced by the gene set II. MAPK1 is the convergence point in pathway cross-talk (60), which has been widely validated to be involved in cell cycle (61), angiogenesis (62), invasiveness (63) and other cancer-associated behaviors (64). The dysfunction of MAPK1 can lead to acquired drug resistance (65) and worse survival (66).
Figure 5.
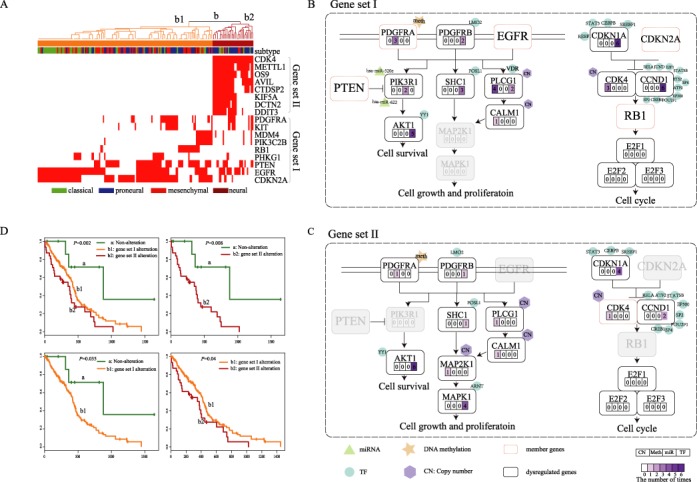
The disturbance of Glioma pathway affected by two gene sets in the core module contributing to GBM survival. (A) Two gene sets (I and II) in the core module characterized two subgroups using unsupervised hierarchical clustering of GBM patients with genomic alterations in the module. The ‘subtype’ bar represents four subtypes (including classical, proneural, mesenchymal and neural) identified using expression of genes determined in a previous study. (B) The gene set I including EGFR, PTEN, PDGFRA, CDKN2A, MDM4, KIT, PIK3C2B, RB1 and PHKG1 directly participates in Glioma pathway. (C) The gene set II including CDK4, METTL1, OS9, AVIL, CTDSP2, KIF5A, DCTN2 and DDIT3 indirectly regulates the Glioma pathway through dysregulated genes. The digits within the four rectangles represent the frequency of copy number, methylation, miRNA and TF-mediated dysregulations of dysregulated genes. The different regulatory factors are affiliated to the dysregulated genes. (D) Both of the two gene sets (I and II) contribute to the GBM survival.
In addition, through survival analysis, we found the two subgroups with distinct alteration patterns (subgroup b1 and subgroup b2 in Figure 5A) showed shorter survival compared to the group without any alterations of member genes (group a in Figure 2G). Interestingly, these two subgroups also had different survival risks (Figure 5D), and the subgroup b2 had the shortest survival. A possible explanation is the local differences in Glioma pathway between the two subgroups, such as the dysregulation of MAPK1. Furthermore, this poor-prognosis subgroup was not correlated with any GBM subtypes determined by the expression of 840 genes reported in (41), which suggested a potential new subtype of GBM. These findings indicated that these two mutual exclusive gene sets can induce similar changes of common pathways in different ways and the dysregulated genes identified can provide novel clues for explaining malignant phenotypes in GBM.
Applying CMDD to other cancer types
To reveal gene modules in other cancer types, we applied the CMDD to other six types of cancer, including Ovarian serous cystadenocarcinoma (OV), Head and Neck squamous cell carcinoma (HNSC), Lung adenocarcinoma (LUAD), Cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC), Breast invasive carcinoma (BRCA) and Prostate adenocarcinoma (PRAD) (Supplementary Table S2). Like GBM, we performed similar analyses for each type of cancer (Supplementary Figures S9–S14 and Supplementary Tables S3–S8).
For example, in OV, 31 core modules including 212 genes were identified. The function and pathway enrichment analysis showed that the genes in these core modules were significantly involved in cancer-associated functions and pathways, such as ‘mitotic cell cycle’, ‘p53 signaling pathway’ and ‘Apoptosis’ (Supplementary Figure S9B). A global dysregulated network connecting the genes in modules with their affecting dysregulated genes showed that most dysregulated genes linked with only one or two member genes (Supplementary Figure S9C) and that most genes in modules dysregulated TF-mediated regulations (Supplementary Figure S9D). Also, we found five modules showing significant associations with OV survival. For example, a core module including five completely connected genes (PUF60, SOX18, CCDC88A, MED4 and THOC7) was significantly associated with longer survival (P = 0.0059, log-rank test; Supplementary Figure S15A), suggesting that the module functioned as a protective role in the OV survival. Further, we found that the module showed better prognostic effect than any single gene in the module. By analyzing the alteration profiles of these five genes across OV samples, we found that the genetic alterations of these genes exhibited mutually exclusive patterns (Supplementary Figure S15B), indicating that different genes in the module can dysregulate the same or similar important functions and pathways in cancer. Similarly, another core module including LDHB, MRPS22 and ATP5C1 was also observed. The alterations of the three genes were mutually exclusive, and none of these genes showed significantly association with OV survival, while the prognostic effect of the whole module was significant (P = 0.007, log-rank test; Supplementary Figure S15B). We also found the survival-associated modules in other cancers, such as a clique module including NCL, DVL3, TRRAP, KDM5A, KLF5, HES1 and BARD1 (P = 0.007, log-rank test; Supplementary Figure S10E) in HNSC and a module including CSPG4, NDUFS6, SATB2 and TRIO in LUAD (P = 0.037, log-rank test; Supplementary Figure S11E).
In addition, we found that genes in some modules showed different roles in prognosis. For instance, using a clique module in HNSC including ARNT, NFE2L2, BARD1, SUB1, ATF2, PTEN and TOPBP1, the HNSC samples with the alteration of the module were divided into two subgroups. We found that these two subgroups showed significantly different prognosis (P = 0.0004, log-rank test; Supplementary Figure S16). Compared with samples without the alteration of the clique module, the subgroup with frequent alterations of ATF2, BARD1 and NFE2L2 showed significantly shorter survival time, while the other subgroup harboring frequent alterations of ARNT, SUB1, PTEN and TOPBP1 exhibited the significantly longer survival times. Similar results were also found for other clique modules, such as a module including TAF9, HOXB7, EEF1D and RXRB in OV (Supplementary Figure S17) and a module including MED21, MYC, ZHX1, KPNA2 and USF2 in LUAD (Supplementary Figure S18).
Finally, we compared the genes in the core modules among the seven types of cancer and observed that the genes in different types of cancer were obviously different. Also, the dysregulated genes affected by the core modules were mostly different, while the functions and pathways significantly enriched by the dysregulated genes exhibited high coherence (Supplementary Figure S19).
Performance evaluation of the method
We compared our method with two other methods: a frequency-based and a fisher-based method (18). Based on the frequency method, only five of the nine known GBM-associated genes were found (Figure 6A). We also applied the fisher-based method to obtain outlying genes associated with each CG based on the correlation between genetic alteration of CGs and expression of differential genes (P < 0.05). Then we ranked the CGs according to the number of outlying genes. In the top 17 genes, only two known GBM-associated genes were identified (Figure 6B). Furthermore, using differentially expressed genes between altered samples and non-altered samples (SAM, fold change ≥1.5 and FDR < 0.05) to replace dysregulated genes, we obtained three modules using the same threshold according to functional coherence of differential genes. These modules derived from distinct chromosome regions did not exhibit functional connection and contained only four known GBM-associated genes (Figure 6C), suggesting that multi-factor-mediated dysregulated networks can capture intrinsic functional associations among distinct genomic alterations.
Figure 6.
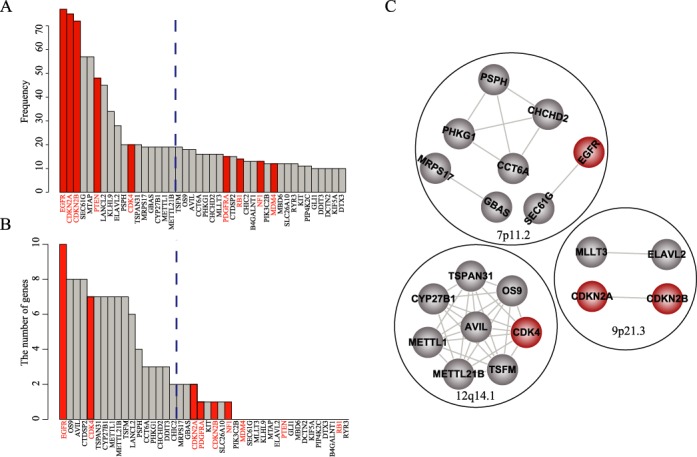
Comparison with other methods. (A) Gene list identified by the frequency-based method, which was ranked based on alteration frequency. (B) Gene list identified by the fisher-based method, which was ranked by the number of related genes. Dashed lines indicate the threshold for top 17 genes. (C) The gene modules identified using the differentially expressed genes. Genes in the same black circle are located in the same chromosomal region. EGFR, CDKN2A, CDKN2B, PTEN, CDK4, PDGFRA, RB1, NF1, MDM4 were known GBM associated genes.
DISCUSSION
In this paper, we proposed a systematic method called CMDD to identify cancer-related core gene modules and their affecting dysregulated genes through characterizing genetic alteration-induced dysregulated networks that are constructed using multi-dimensional genomic data including DNA copy number, methylation, gene expression and miRNA expression profiles, as well as protein interaction network. When applied CMDD to GBM, a core module composed of 17 genes was identified. The core module includes several well-known oncogenes and tumor suppressors associated with GBM, such as EGFR, PTEN, PDGFRA and CDKN2A, which partially provided an important validation of our method. These genes were found to influence numerous regulatory relationships, such as loss (or gain) of gene regulations by transcriptional factors, which in turn affect downstream key genes with important roles in tumorigenesis. Dysregulated genes induced by the genes in the module show significantly functional connections and are commonly involved in some important cancer-related pathways. In addition, we found novel CGs METTL1 and CTDSP2 that showed strong functional associations with known GBM-related genes. Dysregulated genes induced by METTL1 or CTDSP2 share similar functions as some known GBM-associated genes, and the amplification/high expression of METTL1 or CTDSP2 is associated with poor prognosis in GBM, suggesting that METTL1 and CTDSP2 may play an important role in the tumorigenesis and progression of GBM.
Recent studies focusing on somatic tumor genome and exome sequences revealed that most human cancers are resulted from two to eight sequential alterations (2). An interesting problem is how the small number of genetic alterations can destroy key signaling pathways associated with genome maintenance, cell fate and cell survival to cause selective growth advantage and in turn initiate cancer cell transformations. A general explanation is that genes harboring these genetic alterations are directly involved in these key signaling pathways. For example, EGFR and PDGFRA identified in the core module of GBM are the key members of RTK/Ras/PI3K/AKT signaling pathway, and their alterations can directly prompt activation of this pathway contributing to cell growth, proliferation and motility (67). Our method combined multiple layers of information (CNA, methylation, TFs and miRNAs) to infer dysregulated networks relevant to specific genetic alterations. It not only can identify the core module of genetic alterations with the direct effect on the key pathways but also can find the indirect destroy effect of these genetic alterations, which were propagated to downstream key signaling molecules via the dysregulated network. Genes in the dysregulated network of a given genetic alternation can comprehensively characterize the direct and indirect influence of this alteration. Indeed, our results showed that some key pathways, whose important member genes do not exhibit recurrent genetic alterations in certain cancer patient groups, are disturbed by indirect effects of genes harboring genetic alterations. For example, the majority of member genes in the DNA replication pathway and nod-like receptor signaling pathway were the dysregulated genes without obvious recurrent genetic alterations. Such indirect relationships between genetic alteration and downstream dysregulated genes are hard to be captured only with mutation data.
Interestingly, the core module contained two gene sets (I and II), both of which can characterize a GBM subgroup identified using unsupervised hierarchical clustering, respectively. The gene set I (including EGFR, PTEN, PDGFRA, CDKN2A, MDM4, KIT, PIK3C2B, RB1 and PHKG1) exerts important roles in the glioma signaling pathway, such as PTEN, RB1 and EGFR (9), whereas most of the gene set II (including CDK4, METTL1, OS9, AVIL, CTDSP2, KIF5A, DCTN2, DDIT3) is not the members of this pathway. Notably, our results of dysregulated genes showed that both of these two gene sets can target the glioma signaling pathway by their dysregulated genes. Furthermore, the genetic alterations of both of two gene sets were significantly associated with survival risk in GBM patients. The GBM subgroup characterized by the gene set II showed significantly shorter survival, without obvious overlap with the four expression-based subtypes of GBM previously reported. These findings suggest that the core module identified by our method is predictive for survival in GBM patients, probably characterizing new GBM subtypes.
In addition, we investigated whether integration of multi-omics data could provide more comprehensive information than using only two or three types of data. We applied our method to GBM by integrating only two or three types of data, including nine different combinations. We found that integration of multi-omics data could capture more regulatory relationships than using two or three types of data, such as regulatory relationships constructed under EGFR alteration (Supplementary Figure S20). Almost all of the regulated genes identified by different combinations were included in those identified by multi-omics data (Supplementary Figure S20C, CT: P = 1.95e-21; CMT: P = 1.4e-19; MT: P = 4.0e-18 CHT: P = 0; CH: P = 0, hypergeometric test). We further identified the core modules for different combinations. The results showed that no modules were identified in five combinations. In the rest four combinations, we identified only small modules with two to four genes, which were completely contained in the core modules identified by multi-omics data (Supplementary Figure S21A). For example, by integrating copy number and TF, we identified a module including RB1, CDK4, PTEN and DDIT3 (Supplementary Figure S21B), all of which formed a completely connected graph in the core module identified by integrating multi-omics data. It is worth noting that integration of multi-omics data could identify more known GBM-associated genes (such as PDGFRA, MDM4 and CDKN2A) and some potentially novel genes (such as METTL1 and CTDSP2) when compared with those identified by integrating two or three types of data. These results suggested that integrating multiple omics data can capture more useful information which was crucial for identifying core modules and further characterizing their downstream mechanisms.
In summary, we offered an integrative method to identify cancer-related core gene modules and their affecting dysregulated genes through inferring dysregulated regulatory networks induced by genetic alterations. Also, our approach also was applied to other cancer types for identifying causal gene modules and providing the causal gene-induced dysregulated genes, which will give a better interpretation for molecular mechanisms of cancer.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Footnotes
These authors contributed equally to the paper as first authors.
FUNDING
The National High Technology Research and Development Program of China (863 Program) [2014AA021102, in part]; the National Program on Key Basic Research Project (973 Program) [2014CB910504]; the National Natural Science Foundation of China [91129710, 61073136, 31200997, 61170154, 81070946]; the National Science Foundation of Heilongjiang Province [C201207, H0906]; Key Laboratory of Cardiovascular Medicine Research (Harbin Medical University), Ministry of Education and the Undergraduate Innovation Funds of Harbin Medical University [YJSCX2012-210HLJ]. Funding for open access charge: the National High Technology Research and Development Program of China (863 Program) [2014AA021102, in part]; the National Program on Key Basic Research Project (973 Program) [2014CB910504]; the National Natural Science Foundation of China [91129710, 61073136, 31200997, 61170154, 81070946]; the National Science Foundation of Heilongjiang Province [C201207, H0906]; Key Laboratory of Cardiovascular Medicine Research (Harbin Medical University), Ministry of Education and the Undergraduate Innovation Funds of Harbin Medical University [YJSCX2012-210HLJ].
Conflict of interest statement. None declared.
REFERENCES
- 1.Ricke R.M., van Deursen J.M. Aneuploidy in health, disease, and aging. J. Cell Biol. 2013;201:11–21. doi: 10.1083/jcb.201301061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vogelstein B., Papadopoulos N., Velculescu V.E., Zhou S., Diaz L.A., Jr, Kinzler K.W. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jonsson P.F., Bates P.A. Global topological features of cancer proteins in the human interactome. Bioinformatics. 2006;22:2291–2297. doi: 10.1093/bioinformatics/btl390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qiu Y.Q., Zhang S., Zhang X.S., Chen L. Detecting disease associated modules and prioritizing active genes based on high throughput data. BMC Bioinformatics. 2010;11:26. doi: 10.1186/1471-2105-11-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Youn A., Simon R. Identifying cancer driver genes in tumor genome sequencing studies. Bioinformatics. 2011;27:175–181. doi: 10.1093/bioinformatics/btq630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beroukhim R., Getz G., Nghiemphu L., Barretina J., Hsueh T., Linhart D., Vivanco I., Lee J.C., Huang J.H., Alexander S., et al. Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma. Proc. Natl. Acad. Sci. U.S.A. 2007;104:20007–20012. doi: 10.1073/pnas.0710052104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mermel C.H., Schumacher S.E., Hill B., Meyerson M.L., Beroukhim R., Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12:R41. doi: 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vandin F., Upfal E., Raphael B.J. De novo discovery of mutated driver pathways in cancer. Genome Res. 2012;22:375–385. doi: 10.1101/gr.120477.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leiserson M.D., Blokh D., Sharan R., Raphael B.J. Simultaneous identification of multiple driver pathways in cancer. PLoS Comput. Biol. 2013;9:e1003054. doi: 10.1371/journal.pcbi.1003054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ciriello G., Cerami E., Sander C., Schultz N. Mutual exclusivity analysis identifies oncogenic network modules. Genome Res. 2012;22:398–406. doi: 10.1101/gr.125567.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ping Y., Zhang H., Deng Y., Wang L., Zhao H., Pang L., Fan H., Xu C., Li F., Zhang Y., et al. IndividualizedPath: identifying genetic alterations contributing to the dysfunctional pathways in glioblastoma individuals. Mol. Biosyst. 2014;10:2031–2042. doi: 10.1039/c4mb00289j. [DOI] [PubMed] [Google Scholar]
- 14.Bredel M., Scholtens D.M., Harsh G.R., Bredel C., Chandler J.P., Renfrow J.J., Yadav A.K., Vogel H., Scheck A.C., Tibshirani R., et al. A network model of a cooperative genetic landscape in brain tumors. JAMA. 2009;302:261–275. doi: 10.1001/jama.2009.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klijn C., Bot J., Adams D.J., Reinders M., Wessels L., Jonkers J. Identification of networks of co-occurring, tumor-related DNA copy number changes using a genome-wide scoring approach. PLoS Comput. Biol. 2010;6:e1000631. doi: 10.1371/journal.pcbi.1000631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Akavia U.D., Litvin O., Kim J., Sanchez-Garcia F., Kotliar D., Causton H.C., Pochanard P., Mozes E., Garraway L.A., Pe'er D. An integrated approach to uncover drivers of cancer. Cell. 2010;143:1005–1017. doi: 10.1016/j.cell.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim Y.A., Wuchty S., Przytycka T.M. Identifying causal genes and dysregulated pathways in complex diseases. PLoS Comput. Biol. 2011;7:e1001095. doi: 10.1371/journal.pcbi.1001095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Masica D.L., Karchin R. Correlation of somatic mutation and expression identifies genes important in human glioblastoma progression and survival. Cancer Res. 2011;71:4550–4561. doi: 10.1158/0008-5472.CAN-11-0180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bashashati A., Haffari G., Ding J., Ha G., Lui K., Rosner J., Huntsman D.G., Caldas C., Aparicio S.A., Shah S.P. DriverNet: uncovering the impact of somatic driver mutations on transcriptional networks in cancer. Genome Biol. 2012;13:R124. doi: 10.1186/gb-2012-13-12-r124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mehnert J.M., Kluger H.M. Driver mutations in melanoma: lessons learned from bench-to-bedside studies. Curr. Oncol. Rep. 2012;14:449–457. doi: 10.1007/s11912-012-0249-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kalari S., Pfeifer G.P. Identification of driver and passenger DNA methylation in cancer by epigenomic analysis. Adv. Genet. 2010;70:277–308. doi: 10.1016/B978-0-12-380866-0.60010-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cancer Genome Atlas Research Network. Kandoth C., Schultz N., Cherniack A.D., Akbani R., Liu Y., Shen H., Robertson A.G., Pashtan I., Shen R., et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73. doi: 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weller M., Stupp R., Reifenberger G., Brandes A.A., van den Bent M.J., Wick W., Hegi M.E. MGMT promoter methylation in malignant gliomas: ready for personalized medicine. Nat. Rev. Neurol. 2010;6:39–51. doi: 10.1038/nrneurol.2009.197. [DOI] [PubMed] [Google Scholar]
- 26.Walz S., Lorenzin F., Morton J., Wiese K.E., von Eyss B., Herold S., Rycak L., Dumay-Odelot H., Karim S., Bartkuhn M., et al. Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles. Nature. 2014;511:483–487. doi: 10.1038/nature13473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo H., Ingolia N.T., Weissman J.S., Bartel D.P. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466:835–840. doi: 10.1038/nature09267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li W., Zhang S., Liu C.C., Zhou X.J. Identifying multi-layer gene regulatory modules from multi-dimensional genomic data. Bioinformatics. 2012;28:2458–2466. doi: 10.1093/bioinformatics/bts476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xiao Y., Ping Y., Fan H., Xu C., Guan J., Zhao H., Li Y., Lv Y., Jin Y., Wang L., et al. Identifying dysfunctional miRNA-mRNA regulatory modules by inverse activation, cofunction, and high interconnection of target genes: a case study of glioblastoma. Neuro-oncology. 2013;15:818–828. doi: 10.1093/neuonc/not018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xiao Y., Guan J., Ping Y., Xu C., Huang T., Zhao H., Fan H., Li Y., Lv Y., Zhao T., et al. Prioritizing cancer-related key miRNA-target interactions by integrative genomics. Nucleic Acids Res. 2012;40:7653–7665. doi: 10.1093/nar/gks538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kristensen V.N., Lingjaerde O.C., Russnes H.G., Vollan H.K., Frigessi A., Borresen-Dale A.L. Principles and methods of integrative genomic analyses in cancer. Nat. Rev. Cancer. 2014;14:299–313. doi: 10.1038/nrc3721. [DOI] [PubMed] [Google Scholar]
- 32.de la Fuente A. From ‘differential expression’ to ‘differential networking’—identification of dysfunctional regulatory networks in diseases. Trends Genet. 2010;26:326–333. doi: 10.1016/j.tig.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 33.Garnis C., Buys T.P., Lam W.L. Genetic alteration and gene expression modulation during cancer progression. Mol. Cancer. 2004;3:9. doi: 10.1186/1476-4598-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.You J.S., Jones P.A. Cancer genetics and epigenetics: two sides of the same coin. Cancer Cell. 2012;22:9–20. doi: 10.1016/j.ccr.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jornsten R., Abenius T., Kling T., Schmidt L., Johansson E., Nordling T.E., Nordlander B., Sander C., Gennemark P., Funa K., et al. Network modeling of the transcriptional effects of copy number aberrations in glioblastoma. Mol. Syst. Biol. 2011;7:486. doi: 10.1038/msb.2011.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Setty M., Helmy K., Khan A.A., Silber J., Arvey A., Neezen F., Agius P., Huse J.T., Holland E.C., Leslie C.S. Inferring transcriptional and microRNA-mediated regulatory programs in glioblastoma. Mol. Syst. Biol. 2012;8:605. doi: 10.1038/msb.2012.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xiao F., Zuo Z., Cai G., Kang S., Gao X., Li T. miRecords: an integrated resource for microRNA-target interactions. Nucleic Acids Res. 2009;37:D105–D110. doi: 10.1093/nar/gkn851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wingender E., Chen X., Hehl R., Karas H., Liebich I., Matys V., Meinhardt T., Pruss M., Reuter I., Schacherer F. TRANSFAC: an integrated system for gene expression regulation. Nucleic Acids Res. 2000;28:316–319. doi: 10.1093/nar/28.1.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Keshava Prasad T.S., Goel R., Kandasamy K., Keerthikumar S., Kumar S., Mathivanan S., Telikicherla D., Raju R., Shafreen B., Venugopal A., et al. Human Protein Reference Database–2009 update. Nucleic Acids Res. 2009;37:D767–D772. doi: 10.1093/nar/gkn892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bjornstad A., Westad F., Martens H. Analysis of genetic marker-phenotype relationships by jack-knifed partial least squares regression (PLSR) Hereditas. 2004;141:149–165. doi: 10.1111/j.1601-5223.2004.01816.x. [DOI] [PubMed] [Google Scholar]
- 41.Verhaak R.G., Hoadley K.A., Purdom E., Wang V., Qi Y., Wilkerson M.D., Miller C.R., Ding L., Golub T., Mesirov J.P., et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huse J.T., Holland E., DeAngelis L.M. Glioblastoma: molecular analysis and clinical implications. Annu. Rev. Med. 2013;64:59–70. doi: 10.1146/annurev-med-100711-143028. [DOI] [PubMed] [Google Scholar]
- 43.Ding H., Shannon P., Lau N., Wu X., Roncari L., Baldwin R.L., Takebayashi H., Nagy A., Gutmann D.H., Guha A. Oligodendrogliomas result from the expression of an activated mutant epidermal growth factor receptor in a RAS transgenic mouse astrocytoma model. Cancer Res. 2003;63:1106–1113. [PubMed] [Google Scholar]
- 44.Di Fiore R., D'Anneo A., Tesoriere G., Vento R. RB1 in cancer: different mechanisms of RB1 inactivation and alterations of pRb pathway in tumorigenesis. J. Cell. Physiol. 2013;228:1676–1687. doi: 10.1002/jcp.24329. [DOI] [PubMed] [Google Scholar]
- 45.Goldhoff P., Clarke J., Smirnov I., Berger M.S., Prados M.D., James C.D., Perry A., Phillips J.J. Clinical stratification of glioblastoma based on alterations in retinoblastoma tumor suppressor protein (RB1) and association with the proneural subtype. J. Neuropathol. Exp. Neurol. 2012;71:83–89. doi: 10.1097/NEN.0b013e31823fe8f1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Molenaar J.J., Ebus M.E., Koster J., van Sluis P., van Noesel C.J., Versteeg R., Caron H.N. Cyclin D1 and CDK4 activity contribute to the undifferentiated phenotype in neuroblastoma. Cancer Res. 2008;68:2599–2609. doi: 10.1158/0008-5472.CAN-07-5032. [DOI] [PubMed] [Google Scholar]
- 47.Wang L., Hurley D.G., Watkins W., Araki H., Tamada Y., Muthukaruppan A., Ranjard L., Derkac E., Imoto S., Miyano S., et al. Cell cycle gene networks are associated with melanoma prognosis. PLoS One. 2012;7:e34247. doi: 10.1371/journal.pone.0034247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hamosh A., Scott A.F., Amberger J., Valle D., McKusick V.A. Online Mendelian Inheritance in Man (OMIM) Hum. Mutat. 2000;15:57–61. doi: 10.1002/(SICI)1098-1004(200001)15:1<57::AID-HUMU12>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 49.Futreal P.A., Coin L., Marshall M., Down T., Hubbard T., Wooster R., Rahman N., Stratton M.R. A census of human cancer genes. Nat. Rev. Cancer. 2004;4:177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Becker K.G., Barnes K.C., Bright T.J., Wang S.A. The genetic association database. Nat. Genet. 2004;36:431–432. doi: 10.1038/ng0504-431. [DOI] [PubMed] [Google Scholar]
- 51.Thrane S., Lykkesfeldt A.E., Larsen M.S., Sorensen B.S., Yde C.W. Estrogen receptor alpha is the major driving factor for growth in tamoxifen-resistant breast cancer and supported by HER/ERK signaling. Breast Cancer Res. Treat. 2013;139:71–80. doi: 10.1007/s10549-013-2485-2. [DOI] [PubMed] [Google Scholar]
- 52.Rahman M.T., Nakayama K., Rahman M., Ishikawa M., Katagiri H., Katagiri A., Ishibashi T., Sato E., Iida K., Ishikawa N., et al. ESR1 gene amplification in endometrial carcinomas: a clinicopathological analysis. Anticancer Res. 2013;33:3775–3781. [PubMed] [Google Scholar]
- 53.Uhlmann K., Rohde K., Zeller C., Szymas J., Vogel S., Marczinek K., Thiel G., Nurnberg P., Laird P.W. Distinct methylation profiles of glioma subtypes. Int. J. Cancer. 2003;106:52–59. doi: 10.1002/ijc.11175. [DOI] [PubMed] [Google Scholar]
- 54.Han W., Carpenter R.L., Cao X., Lo H.W. STAT1 gene expression is enhanced by nuclear EGFR and HER2 via cooperation With STAT3. Mol. Carcinog. 2013;52:959–969. doi: 10.1002/mc.21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Camicia R., Bachmann S.B., Winkler H.C., Beer M., Tinguely M., Haralambieva E., Hassa P.O. BAL1/ARTD9 represses the anti-proliferative and pro-apoptotic IFNgamma-STAT1-IRF1-p53 axis in diffuse large B-cell lymphoma. J. Cell Sci. 2013;126:1969–1980. doi: 10.1242/jcs.118174. [DOI] [PubMed] [Google Scholar]
- 56.Li J., Bennett K., Stukalov A., Fang B., Zhang G., Yoshida T., Okamoto I., Kim J.Y., Song L., Bai Y., et al. Perturbation of the mutated EGFR interactome identifies vulnerabilities and resistance mechanisms. Mol. Syst. Biol. 2013;9:705. doi: 10.1038/msb.2013.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jiang X., Xing H., Kim T.M., Jung Y., Huang W., Yang H.W., Song S., Park P.J., Carroll R.S., Johnson M.D. Numb regulates glioma stem cell fate and growth by altering epidermal growth factor receptor and Skp1-Cullin-F-box ubiquitin ligase activity. Stem Cells. 2012;30:1313–1326. doi: 10.1002/stem.1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Feng H., Hu B., Vuori K., Sarkaria J.N., Furnari F.B., Cavenee W.K., Cheng S.Y. EGFRvIII stimulates glioma growth and invasion through PKA-dependent serine phosphorylation of Dock180. Oncogene. 2013;33:2504–2512. doi: 10.1038/onc.2013.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lockwood W.W., Chari R., Coe B.P., Girard L., Macaulay C., Lam S., Gazdar A.F., Minna J.D., Lam W.L. DNA amplification is a ubiquitous mechanism of oncogene activation in lung and other cancers. Oncogene. 2008;27:4615–4624. doi: 10.1038/onc.2008.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Olson J.M., Hallahan A.R. p38 MAP kinase: a convergence point in cancer therapy. Trends Mol. Med. 2004;10:125–129. doi: 10.1016/j.molmed.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 61.Carduner L., Picot C.R., Leroy-Dudal J., Blay L., Kellouche S., Carreiras F. Cell cycle arrest or survival signaling through alphav integrins, activation of PKC and ERK1/2 lead to anoikis resistance of ovarian cancer spheroids. Exp. Cell Res. 2013;320:329–342. doi: 10.1016/j.yexcr.2013.11.011. [DOI] [PubMed] [Google Scholar]
- 62.Leelahavanichkul K., Amornphimoltham P., Molinolo A.A., Basile J.R., Koontongkaew S., Gutkind J.S. A role for p38 MAPK in head and neck cancer cell growth and tumor-induced angiogenesis and lymphangiogenesis. Mol. Oncol. 2013;8:105–108. doi: 10.1016/j.molonc.2013.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Donnelly S.M., Paplomata E., Peake B.M., Sanabria E., Chen Z., Nahta R. P38 MAPK Contributes to Resistance and Invasiveness of HER2-Overexpressing Breast Cancer. Curr. Med. Chem. 2013;21:501–510. doi: 10.2174/0929867320666131119155023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.de la Cruz-Morcillo M.A., Garcia-Cano J., Arias-Gonzalez L., Garcia-Gil E., Artacho-Cordon F., Rios-Arrabal S., Valero M.L., Cimas F.J., Serrano-Oviedo L., Villas M.V., et al. Abrogation of the p38 MAPK alpha signaling pathway does not promote radioresistance but its activity is required for 5-Fluorouracil-associated radiosensitivity. Cancer Lett. 2013;335:66–74. doi: 10.1016/j.canlet.2013.01.050. [DOI] [PubMed] [Google Scholar]
- 65.Hirose Y., Katayama M., Stokoe D., Haas-Kogan D.A., Berger M.S., Pieper R.O. The p38 mitogen-activated protein kinase pathway links the DNA mismatch repair system to the G2 checkpoint and to resistance to chemotherapeutic DNA-methylating agents. Mol. Cell. Biol. 2003;23:8306–8315. doi: 10.1128/MCB.23.22.8306-8315.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ghosh S., Kumar A., Tripathi R.P., Chandna S. Connexin-43 regulates p38-mediated cell migration and invasion induced selectively in tumour cells by low doses of gamma-radiation in an ERK-1/2-independent manner. Carcinogenesis. 2013;35:383–395. doi: 10.1093/carcin/bgt303. [DOI] [PubMed] [Google Scholar]
- 67.Ohgaki H., Kleihues P. Genetic pathways to primary and secondary glioblastoma. Am. J. Pathol. 2007;170:1445–1453. doi: 10.2353/ajpath.2007.070011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.