


Abstract
The function of RNA is subtly modulated by post-transcriptional modifications. Here, we report an important crosstalk in the covalent modification of two classes of RNAs. We demonstrate that yeast Kre33 and human NAT10 are RNA cytosine acetyltransferases with, surprisingly, specificity toward both 18S rRNA and tRNAs. tRNA acetylation requires the intervention of a specific and conserved adaptor: yeast Tan1/human THUMPD1. In budding and fission yeasts, and in human cells, we found two acetylated cytosines on 18S rRNA, one in helix 34 important for translation accuracy and another in helix 45 near the decoding site. Efficient 18S rRNA acetylation in helix 45 involves, in human cells, the vertebrate-specific box C/D snoRNA U13, which, we suggest, exposes the substrate cytosine to modification through Watson–Crick base pairing with 18S rRNA precursors during small subunit biogenesis. Finally, while Kre33 and NAT10 are essential for pre-rRNA processing reactions leading to 18S rRNA synthesis, we demonstrate that rRNA acetylation is dispensable to yeast cells growth. The inactivation of NAT10 was suggested to suppress nuclear morphological defects observed in laminopathic patient cells through loss of microtubules modification and cytoskeleton reorganization. We rather propose the effects of NAT10 on laminopathic cells are due to reduced ribosome biogenesis or function.
INTRODUCTION
Post-transcriptional modifications are prevalent and ubiquitous in the RNA world, playing essential biological roles. They affect RNA structure and function and even contribute to self–non-self discrimination (1). To date, no less than 112 types of RNA modifications have been identified (2), two-thirds of which involve methylation. These modifications are distributed unevenly among the different classes of cellular RNAs and are also present on viral RNAs. Transfer RNAs (tRNAs) are by far the most heavily modified RNA class, and they show the most diverse set of modifications (3). Ribosomal RNAs (rRNAs) are also abundantly modified, but their modifications are less diverse. Among the numerous modifications found on eukaryotic rRNAs, most are 2′-O methylations (54 in yeast, 105 in human) and pseudouridylations (44 in yeast, 91 in human) catalyzed, respectively, by box C/D and box H/ACA small nucleolar ribonucleoproteins (snoRNPs) (4–6). In addition, in both yeast and human ribosomes, about 10 conserved bases, four in the small subunit and six in the large, are methylated on different atoms. Most protein enzymes involved in yeast rRNA base methylation have been identified only recently (7–14), while their human homologs still await functional characterization.
Considering the conservation of rRNA modifications across the eukaryotic kingdom, it is quite surprising that most of them are not required for growth (4). Some, however, have been implicated in the fine-tuning of translation (15–18). An individual modification can sometimes confer a competitive advantage, but often several modifications must be knocked down simultaneously before any effect is observed (15–18). In most cases, the exact function of an individual RNA modification is totally unknown. Nevertheless, all three methyltransferases (MTases) modifying the small subunit rRNA (Bud23-Trm112, Emg1/Nep1 and Dim1) are essential or important for yeast cell growth, being required for efficient production of mature 18S rRNA (7–8,10,19–21). In striking contrast, most MTases involved in large subunit modification (Rrp8, Bmt2, Rcm1, Bmt5 and Bmt6) appear non-essential (11–14), the sole exception being Nop2 (12)). rRNAs are produced by extensive processing of precursor molecules via a highly regulated pathway (22–24). The involvement of small subunit MTases in pre-rRNA processing is suggested to be part of robust quality control mechanisms ensuring that processed molecules are all properly modified (10).
In addition to 2′-O methylation, pseudouridylation and base methylation, early reports based on 2D thin-layer chromatography of rRNA digests indicated the possible presence of cytosine acetylation in yeast, Dictyostelium, chicken and rat small subunit rRNA, but not in large subunit rRNAs (25,26). N4-acetyl cytidine (ac4C) appeared as a eukaryotic-specific modification, as it was not found in Escherichia coli 16S rRNA (25). The number of rRNA acetylations, their exact positions on the ribosome and the enzymes involved in producing them have remained unknown for over 30 years (27–29).
NAT10 is a human acetyltransferase (AT) involved in histone and microtubule modification (30,31). NAT10 localizes to the nucleolus in the interphase and to the midbody during the telophase (31). Consistently, it is required for efficient nucleologenesis and cytokinesis (31). It has also been attributed a regulatory role in telomerase function and in the DNA damage response (31–34). NAT10 is a cancer biomarker whose overexpression in various soft tissue sarcomas strikingly correlates with tumor histological grading (31) and whose subcellular distribution is a potential prognostic marker in colorectal carcinoma (35). Remarkably, NAT10 inactivation was recently shown to suppress the nuclear shape defects associated with lamin mutations in laminopathic patient cells (30). This phenotypic suppression was suggested to involve cytoskeleton reorganization caused by loss of microtubule acetylation (30). Kre33, the yeast homolog of NAT10, is an essential protein detected in early nucleolar precursor ribosomes and interacting with several proteins involved in ribosome biogenesis (36,37). NAT10 also interacts with nucleolar proteins and ribosome assembly factors (30).
Altogether, these observations prompted us to investigate the roles of Kre33 and NAT10 in ribosome biogenesis and rRNA acetylation in yeast and human cells, respectively. Our work has led us to locate two acetylated cytidines in the small ribosomal subunits, to identify NAT10/Kre33 as responsible for both 18S rRNA and leucine/serine tRNA acetylation in human and yeast, to identify a specific adaptor involved in tRNA acetylation, to shed some light on the role of the vertebrate-specific box C/D snoRNA U13 in 18S rRNA acetylation and to propose a new explanation for the ability of NAT10 inactivation to correct defects of laminopathic cells.
MATERIALS AND METHODS
Culture, gene expression perturbation methods
Yeast culture
The yeast strains and human cell lines used in the present study are listed in Supplementary Table S4. Yeast strains were grown at 30°C in YPD or YPG medium (1% w/v yeast extract, 2% w/v peptone, 2% w/v glucose/galactose) or in synthetic dropout medium (0.5% w/v ammonium sulfate, 0.17% w/v yeast nitrogen base, 2–4% w/v glucose/galactose).
Yeast gene depletion
To achieve depletion of Kre33, pGAL1::3HA-kre33 cells were grown in YPGSR (yeast extract, peptone, galactose–sucrose–raffinose, 2% w/v each) to mid-log phase, washed in pre-warmed water and transferred to YPD for up to 12 h.
Yeast drop test assay
For serial dilution growth assays, yeast cells were grown overnight in YPD/YPG medium and diluted first to an OD600 of 1 and then serially 1:10. From each diluted cultures, 5 μl was spotted onto a YPD or YPG plate and incubated at 30°C or 19°C.
Yeast two-hybrid assay
To test for physical interaction between Kre33 and Tan1, we used a yeast two-hybrid assay. Yeast strain Y190 was co-transformed with different combinations of pGBT and pGAD-based plasmids carrying Kre33 and Tan1. The co-transformed colonies were initially selected on SD-Trp-Leu. For each transformation, at least three independent colonies were replica plated onto SD-Trp-Leu-His to test for activation of the PGAL1-HIS3 gene.
Human cell culture
Human cells were grown at 37°C under 5% CO2. The different cell lines used in this study and their respective growth media are listed in Supplementary Table S4.
Human cell gene depletion
HCT116 cells were reverse transfected as follows: 1.5 ml of 20-μM siRNA (Silence select, Life Technologies) or 20-μM dsiRNA (Integrated DNA Technologies) or 40-μM Aso (Integrated DNA Technologies) and 4-μl Lipofectamine RNAiMAX (Life Technologies) were mixed with 500-μl Opti-MEM (Life Technologies) in each well of a 6-well plate. After a 20-min incubation at room temperature, 1.5 × 105 cells, resuspended in 2.5-ml antibiotic-free medium, were seeded into each well. Inactivation was carried out for 72 h. All silencers and the ASO used in this study are listed in Supplementary Table S2.
Plasmid construction and site-directed mutagenesis
All plasmids used in the present study were constructed using a gap-repair strategy in budding yeast with the oligos listed in Supplementary Table S1. For introducing specific point mutations, polymerase chain reaction (PCR) site-directed mutagenesis with Single-Primer Reactions IN Parallel (SPRINP) and high-fidelity Pfu-DNA polymerase (Promega) was used (38). All plasmids carrying point mutations in Kre33 were sequenced and tested for complementation. For complementation analysis, plasmids carrying point mutations were used to transform a heterozygous Kre33 deletion mutant and tetrad analysis was performed to test complementation.
Protein biochemistry
Yeast total protein extraction
Total protein was extracted from ∼2 × 108 yeast cells resuspended in 200-μl lysis buffer (KOH pH7.5, 0.5-mM ethylenediaminetetraacetic acid (EDTA) pH8.0, 100-mM NaCl, 1-mM DTT, 20% v/v glycerol, 0.05% v/v NP40, complete protease inhibitors, Roche), mixed with an equal volume of 425–600-μm diameter glass beads (Sigma). The cells were lysed by vortexing for 5 × 30 s, with 1-min resting time on ice. The lysate was cleared by centrifugation at 20 000 g at 4°C for 15 min, and the supernatants were assayed for protein content.
Human cell total protein extraction
Total protein was extracted from ∼1 × 106 HCT116 cells. Cells were washed once with ice-cold 1x phosphate buffered saline (PBS) and lysed in modified RIPA buffer (50-mM Tris-HCl, pH 7.5, 250-mM NaCl, 2-mM EDTA, 0.5% v/v NP-40, 10% v/v glycerol) plus protease inhibitors (complete protease inhibitor, Roche) for 20 min on ice. Lysates were cleared by centrifugation at 20 000 g for 20 min at 4°C. The supernatants were assayed for protein content.
Western blotting
Protein concentrations were estimated by the Bradford protein assay (Biorad). Forty microgram total yeast or human protein extract was loaded on a Bolt 4–12% Bis-Tris plus gel (Life technologies) and transferred to a PVDF membrane (GE Healthcare). Membranes were blocked for 1 h with 5% w/v non-fat dry milk diluted in 1x TBS (Tris-borate-EDTA)/0.05% v/v Tween-20 (Sigma) and the antibodies against the following proteins were used at the indicated dilutions: NAT10 (1:1000 dilution, Santa Cruz Biotechnology), THUMPD1 (1:1000 dilution, Abcam), p53 (1:2000 dilution, Bethyl Laboratories), uL18/RPL5 (1:2000 dilution, Bethyl Laboratories), uL5/RPL11 (1:2000 dilution, Bethyl Laboratories), uS3/RPS3 (1:1000 dilution, Cell Signaling), β-actin (1:5000 dilution, Santa Cruz biotechnology), G6PDH (1:5000, Sigma), HIS (1:1000 dilution, Roche), HA (1:5000 dilution, Sigma). The membranes were then washed three times for 5 min in 1x TBS/0.05% v/v Tween-20 (Sigma) and incubated with either horseradish-peroxidase-conjugated anti-mouse IgG (1:5000 dilution, Santa Cruz Biotechnology) or horseradish-peroxidase-conjugated anti-rabbit IgG (1:5000 dilution, Santa Cruz Biotechnology). All antibodies were diluted in 1x TBS/0.05% v/v Tween-20 (Sigma) containing 1%w/v non-fat dry milk. Membranes were rinsed three times for 5 min in 1x TBS/0.05% v/v Tween-20 (Sigma) and incubated for 10 min with the Super Signal West Pico Chemiluminescent Substrate (Pierce). The luminescence signal was acquired and processed with a ChemiDoc MP (Bio-Rad).
Coimmunoprecipitation
For protein coimmunoprecipitation, ∼1 × 107 HCT116 cells were washed once in ice-cold 1x PBS, lysed in modified RIPA buffer and treated as described above for western blotting. Seven hundred fifty microgram total protein extract was mixed with Dynabeads protein G (Life technologies) covalently cross-linked with 3-μg NAT10 antibody (Santa Cruz biotechnology) by means of Bis(sulfosuccinimidyl) suberate (Thermo Scientific) according to the manufacturer's instructions and incubated for 30 min at room temperature with rotation. The beads were washed once with modified RIPA buffer and three times with 1x PBS/0.02% v/v Tween-20 (Sigma). The affinity-purified material was eluted in 1x Bolt sodium dodecyl sulphate (SDS) Sample Buffer (Life Technologies) supplemented with 50-mM Glycine pH 2.8. The eluate was then analyzed by western blotting as described above. For RNA coimmunoprecipitation, ∼1 × 107 HCT116 cells were washed once in ice-cold 1x PBS, UV irradiated (400 mJ/cm2) in a UV cross-linker CL-508 (Uvitec), and treated as described above up to the elution step. For the elution, beads were resuspended in 1x PBS supplemented with 0.01% w/v SDS and 100-μg Proteinase K (New England Biolabs) and incubated for 30 min at 55°C. RNAs were then extracted with phenol/chloroform, loaded on an RNA acrylamide gel and analyzed by northern blotting (see below for more details). As a negative control, an identical amount of mouse IgG (Santa Cruz Biotechnology) was used.
Human cells and yeast velocity gradient analysis
For ribosome subunit velocity gradient analysis, ∼2.5 × 107 HCT116 cells were washed once in ice-cold 1x PBS and lysed in buffer A (20-mM Tris-HCl pH7.4, 150-mM KCl, 3-mM MgCl2, 1-mM DTT, 0.5% v/v NP-40 and EDTA-free complete protease inhibitor, Roche) for 20 min on ice. Extracts were clarified by centrifugation at 20 000 g and 4°C for 20 min and quantified by OD260 reading. For yeast, about 2 × 109 exponentially growing cells were collected by centrifugation, rinsed in buffer K (20-mM Tris-HCl pH7.4, 50-mM KCl, 5-mM MgCl2) and collected again by centrifugation. Cell pellets were resuspended in one volume of ice-cold buffer K, supplemented with 1-mM DTT and 1× complete EDTA-free protease inhibitor cocktail (Roche). About 500 μl of ice-cold glass beads (diameter 425–600 μm, Sigma) were added to the cells. Cells were broken by 12 rounds of vigorous shaking for 30 s, with a 30-s incubation on ice after each shaking. Extracts were clarified by three successive centrifugations at 20 000 g and 4°C for 10 min and quantified by reading the OD260. About 30 A260 units were loaded onto 10–50% w/v sucrose gradients prepared in buffer A for human cells or buffer K for yeast cells, without DTT or protease inhibitors. Gradients were prepared in Beckman ultra-clear centrifugation tubes (REF344059, 14 × 89 mm). Five 2.2-ml layers of sucrose solution at decreasing concentration (50%, 40%, 30%, 20% and 10% w/v sucrose) were added one by one, with an incubation of at least 20 min at −80°C in between. Gradients were centrifuged for 165 min at 39 krpm and 4°C in an Optima L-100XP Ultracentrifuge (Beckman-Coulter) using the SW41Ti rotor. After centrifugation, fractions of 500 μl each were collected from the top of each gradient, by use of a Foxy Jr. fraction collector (Teledyne ISCO). The absorbance at 254 nm was measured during collection with a UA-6 cell (Teledyne ISCO). The first half of each fraction was mixed with 250 μl of 100% TCA, mixed vigorously and incubated for 20 min on ice. After 20 min of centrifugation at 20 000 g and 4°C. The pellets were wash once with 500-μl ice-cold acetone (Sigma) and centrifuged again, and then dried and resuspended in SDS loading buffer (50-mM Tris-HCl pH 6.8, 100-mM DTT, 2% w/v SDS, 0.1% w/v bromophenol blue and 10% v/v glycerol), incubated for 5 min at 95°C, loaded onto a precast gel (Criterion XT, Biorad) and transferred to a PVDF membrane (GE Healthcare). The second half of each fraction was used to extract RNA by adding 750 μl of Tri-reagent solution (Life Technologies). After vigorous shaking, total RNA was extracted following the manufacturer's instruction. RNA resolved on a denaturing agarose or acrylamide gel (see below for more details). The same protocol was used for polysome profiles, except that human or yeast cells were treated for 10 min with 50-μg/ml cycloheximide (Sigma) added directly to the culture medium before lysis. The same amount of cycloheximide (Sigma) was also added to all buffers.
RNA biochemistry
Yeast total RNA extraction
For extraction of total RNA from yeast cells, 50-ml cultures were grown to OD600 0.8, harvested by centrifugation, and the pellets resuspended in GTC mix (4-M guanidium isothiocyanate, 50-mM Tris-HCl pH 8.0, 10-mM EDTA pH 8.0, 2% w/v sodium lauroyl sarcosinate and 150-mM 2-mercaptoethanol). An equal volume of ice-cold glass beads (diameter 425–600 μm, Sigma) and phenol were added to the cells and the mixture was shaken vigorously for 5 min. Five volumes of GTC mix and phenol were added and the mixture was incubated at 65°C for 5 min with shaking. The mixture was cooled on ice, a mix 24:1 of chloroform and isoamyl alcohol and NaAc Mix (100-mM sodium acetate pH 5.0, 10-mM Tris-HCl pH 8.0 and 1-mM EDTA) were added and the aqueous phase was recovered by centrifugation. Following two further extractions with phenol/chloroform/isoamyl alcohol (25:24:1), RNA was recovered by ethanol precipitation.
Human cells total RNA extraction
Extraction of total RNA from HCT116 cells was carried out with Tri-reagent solution (Life Technologies) according to the manufacturer's recommendations, except that RNA pellets were first washed with absolute ethanol and then with 75% v/v ethanol. The concentration and quality of the RNA were assessed with a NanoDrop 1000 spectrophotometer (Thermo Scientific).
tRNA and 18S rRNA purification
For the purification of human tRNAs and 18S rRNAs, ∼2.5 × 107 HCT116 cells were washed once in ice-cold 1x PBS and lysed in buffer B (20-mM Tris-HCl pH7.4, 50-mM KCl, 1-mM DTT, 0.5% v/v NP-40 and EDTA-free complete protease inhibitors, Roche) for 20 min on ice. Extracts were clarified by centrifugation at 20 000 g and 4°C for 20 min. Clarified extracts were loaded on a 10–50% sucrose gradient prepared in buffer B without DTT as described above. The gradients were then analyzed as described above. Fractions of interest were pooled and the RNA extracted with Tri-reagent solution (Life Technologies) according to the manufacturer's instructions.
For isolation of yeast tRNA and 18S rRNA, ∼2 × 109 cells were harvested and total RNA was isolated as described above. Five hundred microgram total RNA was then layered onto a 5–25% sucrose gradient in TEN buffer (100-mM Tris-HCl pH 7.8, 100-mM NaCl and 1-mM EDTA in DEPC water). The gradient was made with Gradient Master 107 (Biocomp). Samples were then centrifuged at 23 000 rpm for 25 h at 4°C in an SW40 rotor in an L-70 Beckman ultracentrifuge. The gradients were fractionated in an ISCO density gradient fractionator and the absorbance profile at 254 nm was analyzed with an ISCO UA-5 absorbance monitor. tRNAs, 18S rRNA and 25S rRNA were collected and precipitated overnight with 100% ethanol.
quantitative reverse transcription (qRT)-PCR
In order to validate the efficiency of mRNA depletion by qRT-PCR 100 ng total human RNA was treated with 1-μl (1 U) RNase-free DNase I (Fermentas) in a reaction mix containing 1x reaction buffer with MgCl2 for DNase I (Fermentas). For cDNA synthesis, DNase-treated RNA was reverse transcribed in qScript cDNA SuperMix (Quanta Biosciences) following the manufacturer's instructions. cDNAs were then amplified by quantitative PCR in PerfeCta SYBR Green SuperMix, ROX (Quanta Biosciences). Amplicons for human glyceraldehyde-3-phosphate dehydrogenase were used as endogenous control for qPCR analysis. The sequences of the primers used are described in Supplementary Table S1. Data were analyzed with the StepOne software (v 2.1) (Applied Biosystem, Life Technologies) and the comparative threshold cycle (CT) method (‘Livak’ method) was used for quantification.
RNA electrophoresis
For analysis of high-molecular-weight species, 5-μg human or 10-μg yeast total RNA was dried and resuspended in agarose loading buffer (50% v/v formamide, 6% v/v formaldehyde, 20-mM HEPES, 0.5-mM EDTA, 100-μg/ml ethidium bromide, 0.025% w/v xylene cyanol, 0.025% w/v bromophenol blue, 10% v/v glycerol). RNAs were separated on agarose denaturing gels (6% v/v formaldehyle, 1.2% w/v agarose in 50-mM HEPES/1-mM EDTA. Electrophoresis was carried out for 16 h at 65V in Hepes/EDTA buffer. For analysis of the low-molecular-weight RNA species, 5-μg total RNA was dried and resuspended in acrylamide loading buffer (50% v/v formamide, 20-mM EDTA, 0.025% v/w xylene cyanol, 0.025% v/w bromophenol blue) and resolved on denaturing acrylamide gels (8% v/v acrylamide-bisacrylamide 19:1/8-M urea in 1× TBE buffer) for 5 h at 350 V.
Northern blotting
At the end of the migration, the agarose gels were washed in the following solutions: water, 75-mM NaOH, 0.5-M Tris-HCl pH 7.0/1.5-M NaCl and 10x saline sodium citrate buffer (1.5-M NaCl, 150-mM sodium citrate). After the last wash, the RNAs were transferred by capillarity overnight in 10x saline sodium citrate buffer onto nylon membranes (GE Healthcare). Transfer from acrylamide gels was done by electrotransfer in 0.5x TBE onto nylon membranes (GE Healthcare). The membranes were pre-hybridized for 1 h at 65°C in 50% v/v formamide, 5X SSPE (5-mM EDTA, 50-mM NaH2PO4, 750-mM NaCl), 5x Denhardt's solution (0.1% w/v Ficoll 400, 0.1% w/v polyvinyl pyrrolidone, 0.1% w/v bovine serum albumin (BSA) fraction V), 1% w/v SDS, 200-μg/ml fish sperm DNA solution (Roche). The 32P-labeled oligonucleotide probes were made with T4 polynucleotide kinase (New England Biolabs) according to the manufacturer's instructions, added and incubated for 1 h at 65°C and then overnight at 37°C. The probe sequences are described in Supplementary Table S1. The membranes were then washed three times for 3 min in 2x SSC and exposed to Fuji imaging plates (Fujifilm). After 48 or 72 h, signals were acquired with a Phosphorimager (FLA-7000, Fujifilm) and quantified with the native Multi Gauge Software (Fujifilm, v 3.1).
Mature rRNA quantification
The 28S/18S ratio was determined using the Agilent RNA 6000 nano kit on a Bio Analyzer 2100 (Agilent).
Primer extension
The LD2141 probe was 32P-labeled with T4 polynucleotide kinase (New England Biolabs) according to the manufacturer's instructions. LD2141 was extended into cDNAs with avian myeloblastosis virus reverse transcriptase (Promega) according to the manufacturer's instructions, 10-μg purified total RNA being used as template. Following alkaline hydrolysis, cDNAs were precipitated with ethanol, resuspended in acrylamide loading buffer and separated on a 6% v/v denaturing acrylamide gel in 0.5x TBE at 80 W for 1.5 h. After migration, the gels were dried and exposed to Fuji Imaging plates (Fujifilm). The signal was acquired with a Phosphorimager (FLA-7000, Fujifilm).
Reverse-phased high-performance liquid chromatography
Nucleoside preparation
Seventy microgram of 18S rRNA purified on a velocity gradient, whose integrity had been checked by migration on a denaturing agarose gel, was first heated for 2 min on a heating block in an Eppendorf cup at 100°C. This was followed by rapid cooling on ice. Five microliter of 10-mM ZnS04 was next added to the cup, followed by 10-μl P1 nuclease (Sigma). Nuclease digestion was carried out at 37°C for 16 h (overnight). After 16 h, 10 μl of 0.5-M Tris buffer, pH 8.3, and 10-μl bacterial alkaline phosphatase (Sigma) were added to the cup and incubated at 37°C for 2 h. The Eppendorf cup was centrifuged at 13 000 g and the nucleoside-containing supernatant was transferred to a fresh Eppendorf cup and stored at 4°C.
Nucleoside analysis
Nucleosides were analyzed by RP-HPLC on a Supelcosil LC-18-S HPLC column (25 cm x 4.6 mm, 5 μm) equipped with a pre-column (4.6 × 20 mm) at 30°C on an Agilent 1200 HPLC system, using a protocol described previously in (39). The compositions of the HPLC elution buffers were as follows: buffer A (2.5% methanol in 10-mM NH4H2P04, pH 5.3), buffer B (20% methanol in 10-mM NH4H2P04, pH 5.1), buffer C (35% acetonitrile in 10-mM NH4H2P04, pH 4.9). For high-resolution analysis, a flow rate of 1.0 ml/min was maintained throughout elution. For the first 12 min, the isocratic elution consisted of buffer A, then it was successively changed to 10% buffer B for 8 min, 25% buffer B for 5 min, 60% buffer B for 8 min, 64% buffer B for 4 min, 10% buffer B for 9 min and then back to 100% buffer A for the next 15 min. Buffer C was used to clean and recycle the column after each run. To quantify ac4C, the area of the ac4C peak was compared with and normalized with respect to canonical nucleosides such as guanosine (G) and adenosine (A).
Mung bean nuclease protection assay
The mung bean nuclease protection assay was performed exactly as described previously (11). Complementary synthetic deoxyoligonucleotides were used for hybridization and protection of specific sequences of 18S rRNA. Two thousand picomoles of synthetic deoxyoligonucleotides complementary to yeast 18S rRNA were incubated with 100 picomoles of total rRNA and digestion was carried out with mung bean nuclease (MBN Kit: M0250S, NEB) and 0.05-mg/ml RNase A (Sigma-Aldrich). Protected fragments were purified by denaturing 8-M Urea-PAGE (13%) and passive elution was carried out overnight at 4°C in 0.3-M NaAc on a shaker. Eluted fragments were then precipitated with 100% EtOH.
Cell biology
Immunofluorescence
HeLa cells stably producing a green fluorescent protein-labeled fibrillarin were used (FIB364). The cells were fixed with 2% v/v formaldehyde for 15 min and permeabilized with 2% w/v BSA/0.2% v/v Triton X-100 (Sigma) for 10 min. Non-specific binding was blocked by incubating the cells with 1x PBS/2% w/v BSA for 30 min. NAT10 was detected after a 2-h incubation in a humidified chamber at RT with a 1:500 dilution of anti-NAT10 antibody (Santa Cruz Biotechnologies). After washing with 1x PBS/2% w/v BSA, incubation with an Alexa Fluor 568 anti-mouse (Life Technologies) secondary antibody at 1:1000 dilution was carried out for 2 h at RT in a humidified chamber. The cells were washed with PBS and DNA was stained with 100-ng/ml DAPI (Sigma) for 10 min. Images were acquired with MetaMorph and a spin disc microscope (Yokogawa CSU-X1) with an HQ2 Coolsnap Roper camera and a Plan-Apochromat 100x, 1.46 numerical aperture objective lens.
Bioinformatic analysis
3D structure predictions for Kre33 and NAT10 were carried out with Phyre (40). The pdb file generated by Phyre was visualized with Chimera (41). To identify specific residues crucial for acetyl-CoA binding in the AT and adenosine triphosphate binding in helicase domain of Kre33, the 3D ligand site prediction server was used (http://www.sbg.bio.ic.ac.uk/3dligandsite/). All pairwise and multiple alignments were performed with the open-source software T-coffee (42). EsPript was used to visualize all alignments (43).
RESULTS
Yeast, plant and human 18S rRNA are acetylated
In the late 70's (25), 2D chromatography of 18S rRNA digests indicated the presence of N4-acetyl cytidine (ac4C; Figure 1A) in budding yeast, chicken and rat ribosomes. The number of acetylations, their positions in the small subunit and the enzymes involved in producing them have remained unknown for over three decades. We reinvestigated 18S rRNA acetylation in budding and fission yeasts, in plant and human cells. 18S rRNA was purified on sucrose gradients, digested with nuclease P1 and analyzed by HPLC. This revealed the unambiguous presence of ac4C across eukaryotes (Figure 1C–F and Supplementary Figure S1A–F).
Figure 1.
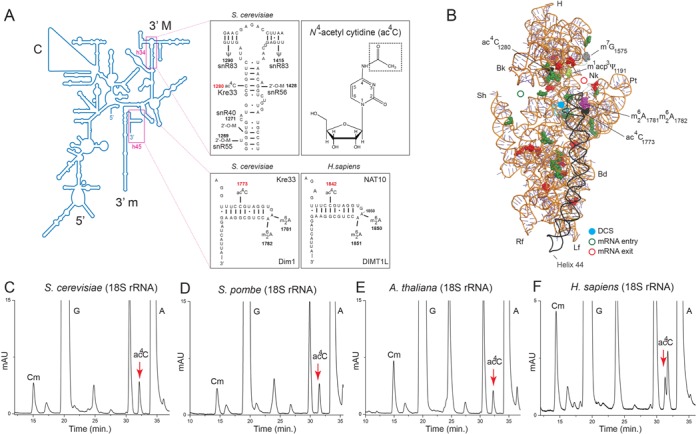
Yeast, plant and human 18S rRNA are acetylated. (A) Secondary structure of the 18S rRNA. The positions of ac4C introduced by Kre33 in yeast in helix 34 and at the base of helix 45 are indicated. The position equivalent to ac4C1773, modified by NAT10 in human, is shown (C1842). Adjacent modifications and factors involved are shown. The inset shows the chemical nature of the ac4C modification. The 5′ central (C), 3′ major (3′ M) and 3′ minor (3′ m) domains are indicated. 2′-O-M, 2′-O sugar methylation; Ψ, pseudouridine. (B) 3D representation of the small subunit based on the crystal structure of the yeast ribosome (PDB 3U5B), with post-transcriptional modifications highlighted. The decoding site (DCS, in cyan) at the base of helix 44 (in anthracite) and the mRNA entry (green circle) and exit (red circle) are indicated. Residues shown as green and red spheres are 2′-O methylated or pseudouridylated, respectively. H, head; Nk, neck; Pt; platform; Bd, body; Lf, left foot; Rf, right foot; Sh, shoulder; Bk, beak. Purified 18S rRNA from budding yeast (S. cerevisiae, (C)), fission yeast (S. pombe, (D)), plant (A. thaliana, (E)) and human cells (H. sapiens, (F)) was analyzed for the presence of ac4C by HPLC revealing the modification as a peak eluting at 32.5 min (see the Materials and Methods section and Supplementary Figure S1 for peak assignment).
Yeast Kre33 and human NAT10 are 18S rRNA ATs
Escherichia coli TmcA is a tRNA AT responsible for modification of the wobble position which impacts decoding accuracy (44). The conserved proteins yeast Kre33 and human NAT10 show limited homology to TmcA (∼25% identity) including in the AT domain and the RNA-binding motif, which in the case of TmcA binds tRNA (Figure 2A and Supplementary Figure S2). To test whether Kre33/NAT10 might act as rRNA ATs, we mutated two conserved residues (H545 and R637; Figure 2B) in the acetyl CoA binding site of yeast Kre33 and estimated 18S rRNA acetylation levels by HPLC (Figure 2C). A mutation at the position corresponding to H545 in the homologous protein TmcA totally inhibited tRNA acetylation, and the residue equivalent to R637 is predicted to be important for catalysis (45). Cells expressing, as sole source of Kre33, a construct encoding a variant with the H545A or R637A substitution were viable (Figure 2D), despite total loss of 18S rRNA acetylation (Figure 2C). Expression of a control construct encoding full-length Kre33 fully restored acetylation (Figure 2C). Importantly, Kre33-H545A and Kre33-R637A proved to be metabolically stable (Figure 2F) and as efficiently recruited to pre-ribosomes as wild-type Kre33 (Figure 2G). On velocity gradients, Kre33 was detected in association with a broad spectrum of pre-ribosomes, and this was not affected by mutations in the acetyl-CoA binding pocket (Figure 2G). We conclude that Kre33 is required for 18S rRNA acetylation and that 18S rRNA acetylation is not essential to growth of yeast cells.
Figure 2.
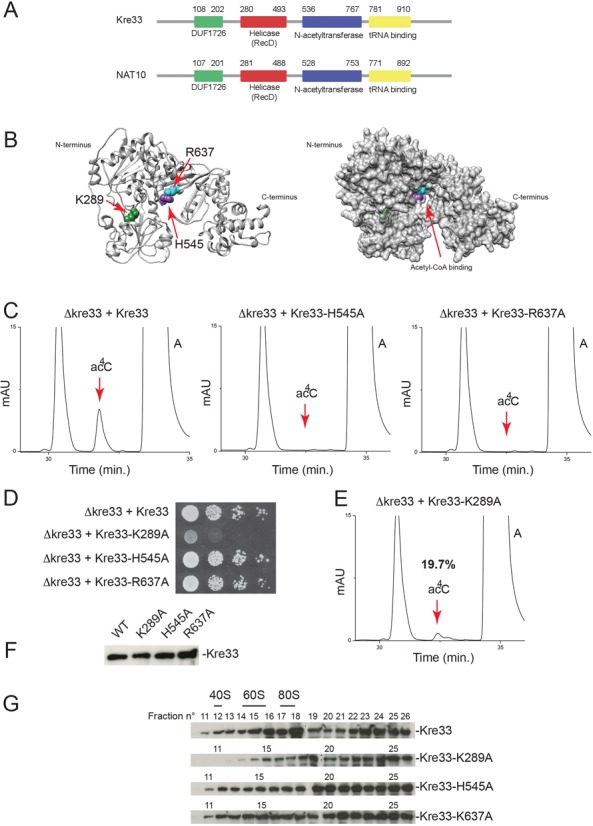
Yeast Kre33 is responsible for 18S rRNA acetylation and its helicase domain is required for efficient RNA modification. (A) Protein domain organization of yeast Kre33 and its human homolog NAT10: a conserved domain of unknown function (DUF1726) is flanked by a helicase domain (RecD), an acetyl-CoA binding domain (N-acetyltransferase) and a tRNA binding motif. (B) Left, ribbon representation of Kre33 (10–930) indicating residues mutated in this work. This model is based on PDB entry 2ZPA. Right: surface rendering highlighting the Acetyl-CoA binding pocket and residues shown to the left. (C) Mutating individual Kre33 residues lining the acetyl-CoA binding pocket leads to total loss of 18S rRNA acetylation. 18S rRNA purified from the indicated yeast strains was analyzed by HPLC for the presence of acetylation. (D) 18S rRNA acetylation is not essential to yeast cell growth in complete medium. Growth assay on plates, showing serial dilutions (1x to 103x from left to right) spotted on rich medium and incubated for 3 days at 30°C. (E) A functional Kre33 helicase domain is required for efficient 18S rRNA acetylation. 18S rRNA analyzed as in panel (C). (F) Metabolic stability of the protein constructs analyzed in this work. Identical amounts of total protein extracted from the indicated strains tested by western blotting with an anti-His antibody recognizing the tag present at the carboxy-terminal end of the constructs. (G) A functional Kre33 helicase domain is required for efficient association of the protein with pre-40S ribosomes. Total extracts of the yeast strains indicated were resolved on 10–50% sucrose gradients. Total protein was extracted from 26 fractions and tested by anti-His western blotting. Kre33 co-migrates with a large spectrum of pre-ribosomes, including early pre-90S precursors, detected in fractions heavier than 80S ribosomes (19 and above), as well as precursors of the small and large subunits (pre-40S and pre-60S), which migrate in fractions heavier than 40S (fractions 12–14) and 60S (fractions 15–17), respectively. The association of Kre33 with the pre-40S fraction is lost in the helicase-deficient mutant K289A.
To examine whether the function of Kre33 is evolutionarily conserved, we first expressed in yeast the gene encoding human NAT10 (Figure 3A and B). We found NAT10 to substitute functionally for Kre33 when produced in kre33Δ cells, complementing the growth defect of these cells (Figure 3I) and restoring 18S rRNA acetylation to ∼76% of the wild-type level (Figure 3B and data not shown). To directly prove that NAT10 acts as an 18S rRNA AT, colon carcinoma human cells were transfected with an siRNA targeting NAT10 mRNA. Depletion of NAT10 was efficient, as confirmed at both the mRNA and the protein level (see below Figure 6B, lane 8 and Figure 6C, siRNA #1), and led to a substantial (71%) loss of 18S rRNA acetylation (Figure 3C and D).
Figure 3.
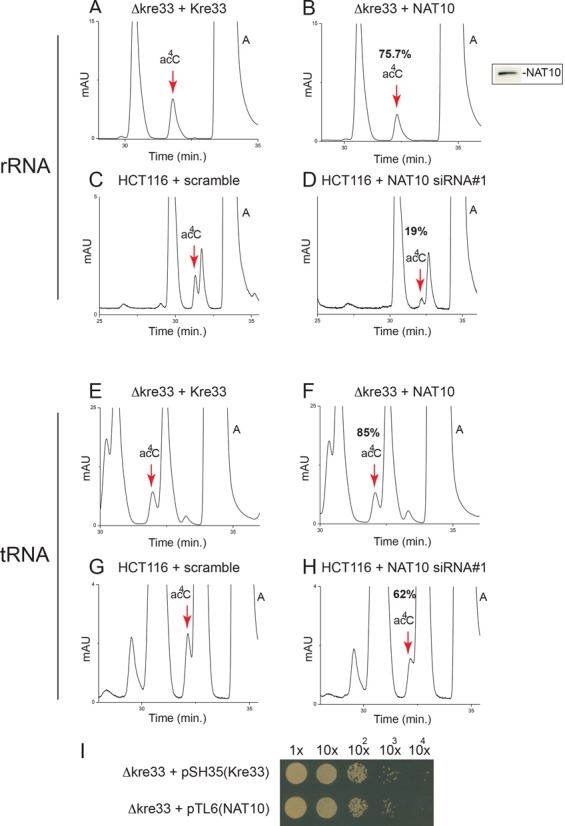
Human NAT10 is required for 18S rRNA and tRNA acetylation. (A, B and E, F) Human NAT10 substitutes for the function of yeast Kre33 in 18S rRNA and tRNA acetylation. The gene encoding NAT10 was expressed in kre33Δ cells, 18S rRNA (A, B) and tRNAs (E, F) were purified and analyzed by HPLC. Inset: western blotting with anti-NAT10 antibody established that the human construct is stably expressed in yeast cells. (C, D and G, H) NAT10 is responsible for 18S rRNA and tRNA acetylation in human cells: colon carcinoma cells (HCT116 p53+/+) transfected with an siRNA specific to NAT10 (siRNA#1) or with a non-targeting control (scramble) were incubated 72 h, 18S RNA (C, D) and tRNAs (G, H) were purified and analyzed by HPLC. (I) Human NAT10 complements growth of Δkre33 cells to the same extent as control wild-type yeast Kre33. Drop assay showing serial culture dilutions on synthetic medium incubated for 3 days at 30°C.
Figure 6.
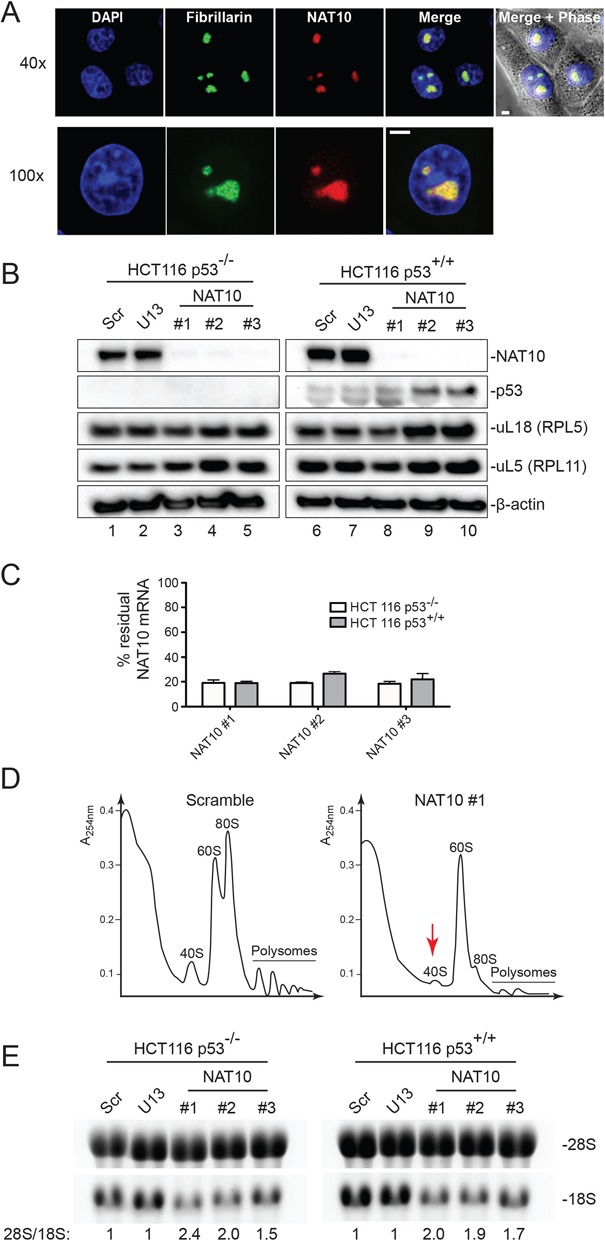
Human NAT10 is a nucleolar protein, which activates a p53-dependent nucleolar tumor surveillance pathway, and is required for 18S rRNA synthesis. (A) NAT10 is a nucleolar protein. Immunofluorescence with an antibody against NAT10 was performed in HeLa cells stably expressing a green fluorescent fibrillarin construct. DAPI, DNA stain. Images were acquired at 40× and 100× on an inverted Zeiss microscope illuminated with Cool LED and mounted with a Yokogawa spindisc head. Scale bar, 5 μm. (B) NAT10 depletion activates nucleolar surveillance, leading to p53 stabilization, and U13 depletion does not affect the metabolic stability of NAT10. Colon carcinoma cells (HCT116), expressing p53 or not, were transfected with an antisense silencer specific to U13 or NAT10 and incubated for 72 h. Three siRNAs were used for NAT10 (#1, #2 and #3); a single 2′-O modified phosphorothioate RNA/DNA hybrid antisense oligonucleotide was used for U13. As control, a non-targeting silencer (Scr) was used. Total protein was analyzed by western blotting for NAT10, p53, and the ribosomal proteins uL18(RPL5) and uL5(RPL11). β-actin was used as loading control. (C) siRNA-mediated depletion of NAT10 is efficient at the mRNA level. Total RNA extracted from HCT116 cells expressing p53 or not (p53+/+ or p53−/−), transfected with an siRNA specific to NAT10 (siRNA #1 to #3) and incubated for 72 h, was tested by qRT-PCR. (D) NAT10 is required for small ribosomal subunit accumulation. Polysomes analysis upon NAT10 depletion with siRNA #1. (E) NAT10 is required for 18S rRNA synthesis. Total RNA extracted upon U13 and NAT10 depletion was analyzed on denaturing agarose gels stained with ethidium bromide. 28S/18S ratio determined from Agilent Bioanalyzer electropherograms.
The 18S rRNA 3′ major domain helix 34 and the 3′ minor domain helix 45 are acetylated
To map the positions of ac4C modifications on the small ribosomal subunit, we used a mung bean nuclease protection assay to scan the entire yeast 18S rRNA sequence with a tiling set of forty-five 50-nt antisense oligonucleotides (Supplementary Figure S3). In this mapping strategy, modified nucleosides are protected from nuclease digestion and detectable by HPLC only if an RNA–DNA hybrid forms by annealing with a complementary oligonucleotide (11–14). In Saccharomyces cerevisiae, ac4C was detected upon protection with oligonucleotides n°32 and n°45. The former oligonucleotide is complementary to residues 1267–1317, corresponding to helix 34 in the 3′ major domain, and the latter is targeting residues 1751–1800, corresponding to helix 45 in the 3′ minor domain at the 3′-end of the 18S rRNA (Figure 1A and B and Figure 4A and B, and Supplementary Figure S3). Protection of the corresponding two rRNA regions on 18S purified from Schizosaccharomyces pombe and Homo sapiens also revealed the presence of two modifications (Supplementary Figure S4).
Figure 4.
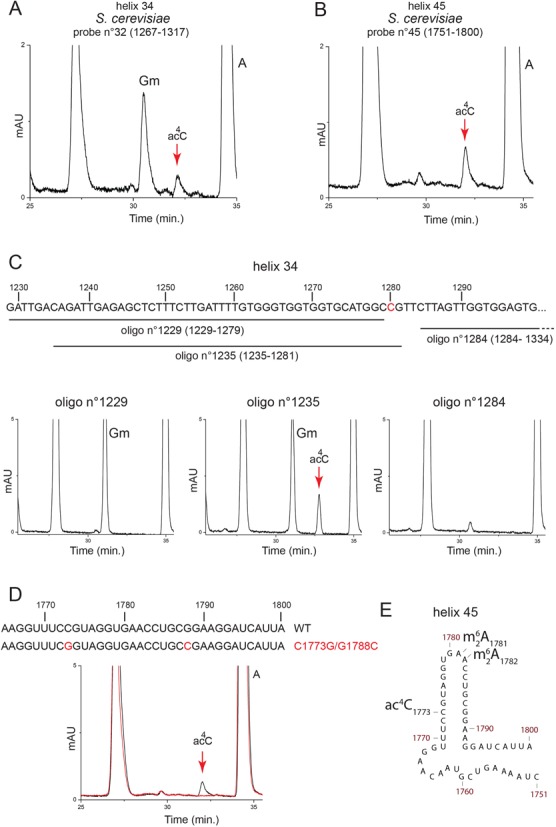
Yeast 18S rRNA is acetylated in helix 34 (3′ major domain) and in helix 45 (3′ minor domain). (A) Mung bean nuclease protection analysis. 18S rRNA was purified from wild-type budding yeast cells, hybridized with probe n°32, protecting residues 1267–1317 in helix 34 and digested with mung bean nuclease. The protected RNA fragment was gel-purified and analyzed by HPLC, revealing the presence of the acetylation. The Gm peak corresponds to Gm1271 modified by snR40 (Figure 1A). (B) Same analysis as in panel (A) but with probe n°45 complementary to a region encompassing nt 1751-to-1800 (helix 45). (C) Mapping the acetylated cytosine in helix 34 to position 1280 by differential Mung bean nuclease digestion. Purified 18S rRNA was processed as in panel (A) with probes spanning nt 1229–1279 (oligo n°1229), nt 1235–1281 (oligo n°1235) or nt 1284–1334 (oligo n°1284). The oligonucleotide n°1235 specifically protects cytosine 1280. (D) Mapping the acetylated cytosine in helix 45 to residue 1773. Yeast cells whose ribosomes contain the C1773G/G1788C mutation are not acetylated, demonstrating that C1773 is normally modified. Purified 18S rRNA was processed as in panel (A) with a probe spanning nt 1729–1779. Red HPLC trace, C1773G/G1788C mutant; black, isogenic wild-type control. (E) The secondary structure of budding yeast helix 45, at the 3′-end of 18S rRNA, with base modifications highlighted.
Several C residues, each potentially a target of acetylation, are present in helix 34 and in helix 45 (Figure 1A). To identify which one is modified, we carried out a differential mung bean nuclease digestion (for helix 34, Figure 4C), and we introduced point substitutions directly into the rDNA, using a well-established procedure (21) (for helix 45, Figure 4D).
The hybridization of helix 34 with oligonucleotide n°1235, which specifically protects cytosine 1280, but not the annealing with the flanking oligonucleotides n°1229 or n°1284, led to the HPLC detection of ac4C (Figure 4C), identifying C1280 as the acetylated residue in helix 34. To map the ac4C residue in helix 45, we used a ΔΔrDNA yeast strain lacking all genomic rDNA copies and synthesizing ribosomes exclusively from a single plasmid-borne rDNA copy (21). We observed that mutating residue C1773, at the base of helix 45, in the non-acetylable G caused failure to grow (data not shown). To explain this, we hypothesized that altering C1773 affects the highly conserved secondary structure of helix 45. Accordingly, restoration of base-pairing by simultaneously substituting C1773 into G and G1788 into C restored yeast cell growth (Figure 4D and data not shown). Total RNA extracted from yeast cells expressing ribosomes with the C1773G/G1778C mutation was analyzed in a mung bean nuclease protection assay with a 50-nt probe spanning residues 1729–1779 (probe n°1779). As we did not observe any ac4C residues in the protected fragment (Figure 4D, in red), we conclude that yeast 18S rRNA is acetylated at position C1773 (Figure 1A and B and Figure 4E).
Based on the extreme sequence conservation of helix 34 and helix 45 across eukaryotes, we extrapolate that positions 1297 and 1815 in fission yeast, and 1337 and 1842 in human cells are acetylated.
Yeast Kre33 and human NAT10 are tRNA ATs
Specific eukaryotic tRNAs, including leucine and serine in yeast, are acetylated at position 12 (46). Tan1 binds tRNA in vitro and is strictly required for yeast tRNA acetylation in vivo (47). Yet Tan1 does not carry any obvious AT motif, suggesting that another protein might be involved catalytically (47) (Figure 5A). Kre33 is a reasonable candidate, as it carries a functional AT domain. To directly test this possibility, we purified tRNAs from cells expressing the catalytically deficient Kre33-H545A or Kre33-R637A variants described above. HPLC analysis did reveal a total loss of acetylation, demonstrating that Kre33 is a ‘bona fide’ tRNA AT (Figure 5E and F). As controls, we confirmed that Tan1 is required for tRNA (Figure 5C) but not 18S rRNA (Figure 5D) acetylation (see also (47)). We found the function of Kre33 in tRNA acetylation to be evolutionarily conserved, as human NAT10 ectopically expressed in kre33Δ yeast cells proved able to acetylate tRNAs to 85% of the wild-type level (Figure 3E and F) and to suppress the growth defect observed in tan1Δ cells at high temperature (data not shown). Furthermore, NAT10 depletion in human colon carcinoma cells led to a significant 40% reduction of tRNA acetylation (Figure 3G and H), incidentally demonstrating that human tRNAs are acetylated (and see Figure 6B, lane 8 and Figure 6C for the efficiency of siRNA-mediated depletion).
Figure 5.
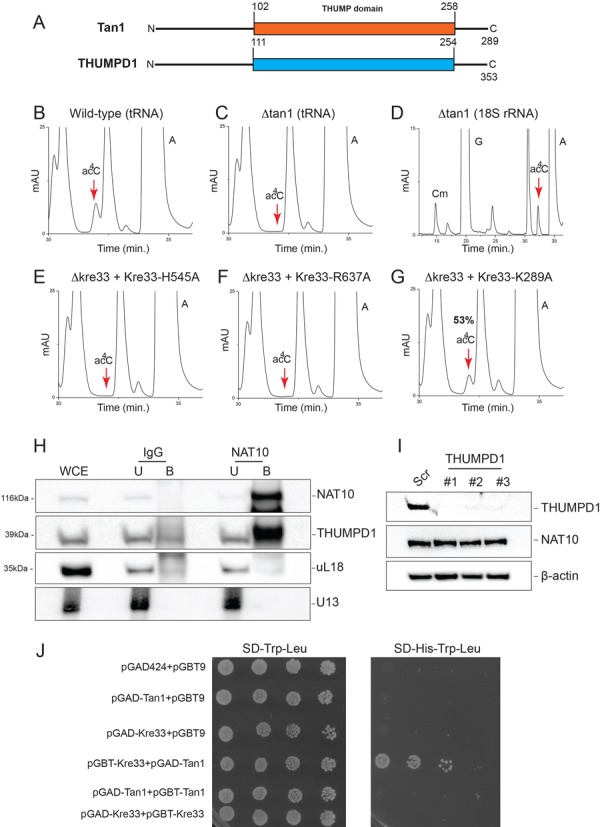
Kre33 is responsible for tRNA acetylation and Kre33 interacts in vivo with Tan1 and NAT10 with THUMPD1. (A) Yeast Tan1 and human THUMPD1 are homologous and carry a conserved tRNA binding domain (THUMP). (B–D) Tan1 is required for tRNA but not rRNA acetylation (see (47)). tRNAs (panels (B) and (C)) and 18S rRNA (panel (D)) purified from tan1Δ cells were analyzed by HPLC for the presence of ac4C. (E, F) Kre33 is required for tRNA acetylation. tRNAs purified from the indicated strains analyzed by HPLC. (G) A functional Kre33 helicase domain is required for optimal tRNA acetylation. (H) NAT10 interacts with THUMPD1 in human cells. HCT116 p53+/+ cell extracts were incubated with magnetic beads covalently linked to anti-NAT10 antibodies. The affinity-purified material was washed under stringent conditions (high salts) and analyzed by western blotting for the presence of NAT10, THUMPD1, and as a control, for ribosomal protein uL18. In parallel, RNA fractions were tested by northern blotting for the presence of U13. WCE, whole cell extract; U, unbound; B, bound. For the protein analysis, the material was loaded in a 1:0.4:10 ratio for WCE, U and B, respectively. For the RNA analysis, a 1:5:10 ratio was loaded. (I) Depletion of THUMPD1 has no impact on the metabolic stability of NAT10. Western blot analysis of total protein extract from HCT116+/+ cells transfected for 3 days with an siRNAs (#1, #2 or #3) specific to NAT10 mRNA or with a non-targeting control (Scr). The membrane was probed with antibodies against the proteins indicated on the right. (J) Kre33 and Tan1 interact in vivo in yeast cells. Kre33 and Tan1 were expressed in yeast cells, alone or in combination, from two-hybrid (2-H) assay constructs. A positive 2-H interaction between Kre33 and Tan1 reconstitutes a functional transcriptional activator (GAD and GBT domains), resulting in histidine (His) prototrophy. SD, synthetic dextrose medium. Trp, tryptophan; Leu, leucine.
Several tRNA modification enzymes are active as holoenzymes with catalytic and regulatory subunits (e.g. Trm8/Trm82 (48)). The involvement of Kre33/NAT10, carrying a functional AT domain, and Tan1/THUMPD1, harboring an RNA binding motif, in yeast and human tRNA acetylation, respectively, suggested that Kre33 and Tan1 in yeast, and NAT10 and THUMPD1 in humans, might act together in a complex. To test this possibility, we first carried out in vivo coprecipitation experiments on human cells under stringent conditions and found NAT10 to interact physically with THUMPD1, as suspected (Figure 5H). Co-activators of RNA modification enzymes are sometimes required for the metabolic stability of the catalytic subunit (e.g. Trm112 for Bud23 (7,20)). This is not the case of THUMPD1, as we found that depleting it to below western-blot detection level had no impact on the amount of NAT10 (Figure 5I). The interaction of NAT10 and THUMPD1 suggests that they work together in tRNA acetylation and that THUMPD1 acts as a specific tRNA adaptor. In agreement with this view, we found Kre33 and Tan1 to interact in a 2-hybrid assay in budding yeast cells (Figure 5J).
The helicase function of Kre33 is required for optimal 18S rRNA and tRNA acetylation
Kre33 is thus active on rRNA and tRNAs, two highly structured substrates, and in both cases the modifications are found within helices in mature molecules. This raises the possibility that the conserved RecD helicase domain of Kre33 might contribute to its efficient access to the substrate and hence to acetylation. To test this, we replaced residue K289 of Kre33 with alanine (Figure 2B). The residue equivalent to K289 in TmcA is essential to its ATPase-dependent helicase function (45). Mutating residue K289 reduced rRNA acetylation to ∼20% of its wild-type level (Figure 2E). The K289A mutation also affected tRNA acetylation, albeit to a lesser extent, leading to a residual 53% modification level (Figure 5G). Importantly, K289A was metabolically stable (Figure 2F). We conclude that a functional Kre33 helicase domain is required for optimal RNA acetylation, rRNA modification being more strongly affected than tRNA modification, in keeping with the relative complexity of these substrates. In further agreement with a role of the helicase domain of Kre33 in rRNA modification, we found Kre33-K289A to be less efficiently recruited to pre-ribosomes than wild-type Kre33 (Figure 2G). In particular, Kre33-K289A was absent from pre-40S (fractions 12–14) on velocity gradients (Figure 2G). DEAD box helicases load directly onto RNA duplexes where they can act as immobile RNA clamps, or promote locally strand separation (49). When they work as clamps, RNA helicases sometimes provide assembly platforms or nucleation centers to establish larger RNP complexes, as originally reported for eIF4AIII, in the stable clamping of the exon-junction complex onto mRNAs (50), and, more recently, for vasa, in the assembly of a germline-specific piRNA maturation complex (51). It is possible that the helicase domain of Kre33 regulates the timing and efficiency of acetylation through a similar clamping mechanism.
Kre33 and NAT10 are required for small ribosomal subunit biogenesis
All three enzymes known to catalyze small ribosomal subunit rRNA base modifications (Bud23, Emg1 and Dim1) are required for pre-rRNA processing (7–8,10,19), suggesting that this might also be the case of Kre33/NAT10.
The involvement of Kre33 in subunit biogenesis was first addressed in yeast, where the essential KRE33 gene was expressed from a repressible GAL promoter and specifically switched off upon addition of glucose to the medium (Supplementary Figure S5A). Efficient Kre33 depletion was demonstrated by western blotting (Supplementary Figure S5B). Strikingly, no 40S subunit was detected on polysome profiles of the pGAL-3HA-kre33 strain after 10 h of Kre33 depletion (Supplementary Figure S5C). Total RNA extracted at several time points during depletion, resolved on denaturing agarose gels and stained with ethidium bromide revealed that the absence of Kre33 led to strong inhibition of 18S rRNA synthesis (Supplementary Figure S5D). Specifically, the 25S/18S ratio showed an increase from 1 to >6 after 12 h of Kre33 depletion. Detailed pre-rRNA processing analysis by northern blotting with specific probes detecting the major known pre-rRNA intermediates established that Kre33 is required for early nucleolar cleavages at sites A0–A2 (Supplementary Figure S5E and F). Cleavage at the A1 and A2 sites was particularly inhibited, leading to accumulation of the aberrant 22S RNA (Supplementary Figure S5E and F). 22S RNA has previously been detected in cells depleted for Dim1, which dimethylates two adenosine residues in helix 45 (see Figure 1A) (10), or deprived of the DEAH-box RNA helicase Dhr1, involved in central pseudoknot formation and interacting directly with the MTase Bud23-Trm112 (20,52).
In human cells, we detected NAT10 in nucleoli (Figure 6A), which was compatible with a possible involvement in early pre-rRNA processing. The nucleolus has long been viewed as a cancer biomarker (53) and has recently been demonstrated to be a therapeutic target (54). As there are numerous connections between nucleolar functions and the antitumor protein p53 (55), pre-rRNA processing was analyzed upon acute NAT10 depletion in colon carcinoma cells expressing p53 or not (Figures 6 and 7). Cells were depleted of NAT10 with three different siRNAs (#1, #2 and #3). All three silencers proved effective, as judged by the residual mRNA and protein levels after a 72-h incubation (Figure 6B, lanes 3–5 and 8–10 and Figure 6C). In the absence of NAT10, significantly reduced accumulation of 40S subunits and 18S rRNA was observed (Figure 6D and E), and processing steps leading to 18S rRNA synthesis were severely inhibited (Figure 7A and B; see major loss of the 21S and 18S-E precursors). Inhibition of RNA processing was similar in the presence and absence of p53 (Figures 6E and 7A), demonstrating that NAT10-mediated inhibition of pre-rRNA processing does not depend on the presence of p53. It is also noteworthy that NAT10 localized to the nucleoli whether p53 was present or not (data not shown).
Figure 7.
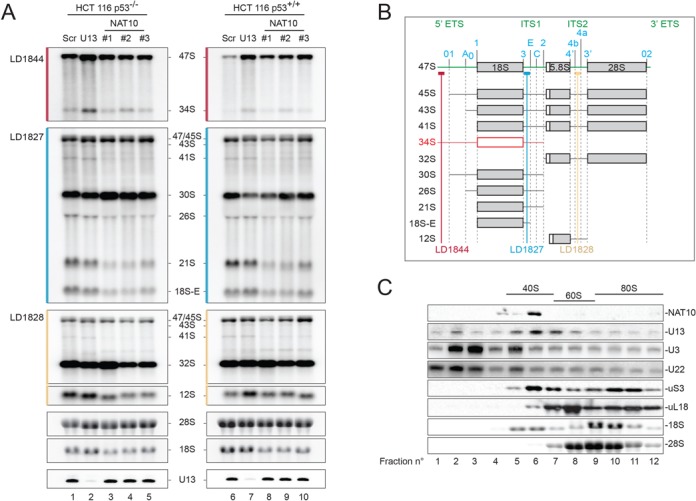
Human NAT10 is required for pre-rRNA processing, and it comigrates on velocity gradients with pre-40S ribosomes and the box C/D snoRNA U13. (A) Pre-rRNA processing analysis. Total RNA presented in Figure 6E was transferred to a nylon membrane and hybridized on northern blots with the probes indicated. The 18S and 28S were stained with ethidium bromide. U13 was detected by northern blotting with probe LD2684. (B) Major pre-rRNA processing intermediates detected by northern blot analysis, and probes used (LD1827, LD1828 and LD1844). Three out of four rRNAs are produced as a single RNA polymerase I transcript (47S). The 18S, 5.8S and 28S rRNAs are interspersed by non-coding spacer sequences (in green): the 5′ and 3′ external transcribed spacers (5′ and 3′ ETS) and internal transcribed spacers (ITS) 1 and 2. Cleavage sites (in cyan) are indicated above the 47S. (C) NAT10 and U13 strikingly comigrate with pre-40S ribosomes (fractions 6–7). Velocity gradient analysis: total HCT116+/+ cell extract was separated on a 10–50% sucrose gradient, 12 fractions were collected and analyzed by western and northern blotting for the targets indicated.
It has recently been proposed that ribosome biogenesis dysfunctions, such as that caused by the depletion of a subunit assembly factor, lead to accumulation of a small complex consisting of the 5S rRNA and the ribosomal proteins uL18(RPL5) and uL5(RPL11) (56,57). This complex interacts with and titrates Hdm2, causing p53 stabilization, cell-cycle arrest and cell death. In agreement with our finding that NAT10 is an assembly factor, its depletion led to p53 stabilization and accumulation of uL5 and uL18 (Figure 6B, best seen with siRNAs #2 and #3 in lanes 9 and 10). The activity of p53 is regulated by numerous post-translational modifications (58); interestingly, in conditions of NAT10-induced nucleolar stress, the fraction of slow-migrating p53 was increased, suggesting altered post-translational modification profiles.
U13 is required for efficient 18S rRNA acetylation but not for tRNA modification
The vertebrate-specific box C/D snoRNA U13 (59) shows two extended regions of Watson–Crick base-pair complementarity to the 3′ end of 18S rRNA (Figure 8A), suggesting that it might contribute to pre-rRNA folding during small subunit biogenesis and possibly to 18S rRNA acetylation. Strikingly, residue 1842, corresponding to the acetylated residue C1773 in budding yeast (Figure 1A), lies precisely between the two stretches of complementarity, so that base pairing during subunit biogenesis should expose it in a bulge, potentially increasing its accessibility to NAT10.
Figure 8.
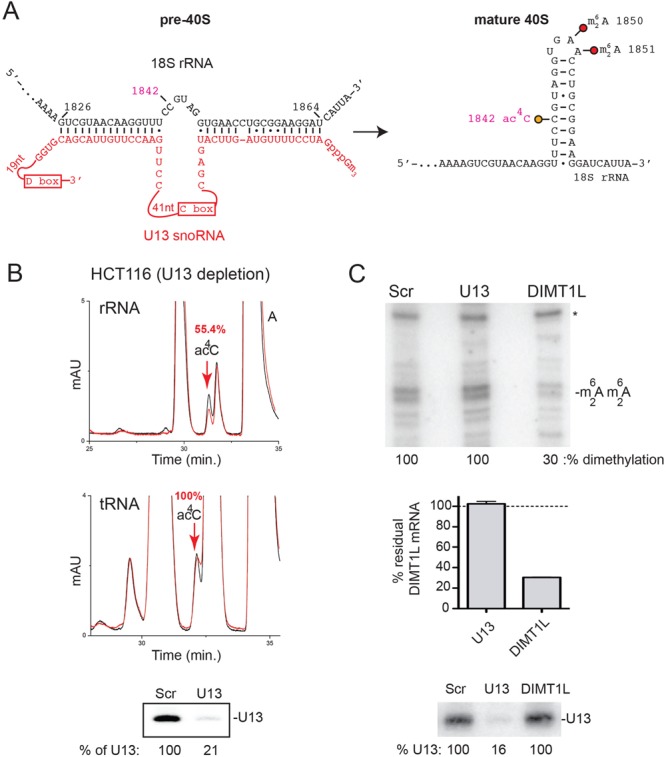
The box C/D snoRNA U13 is required for human 18S rRNA acetylation. (A) Diagrams depicting the predicted base-pairing between U13 and the 3′ end of 18S rRNA. The interaction is proposed to occur during subunit biogenesis, with substrate residue 1842 bulging out upon U13 binding. (B) U13 is required for efficient rRNA acetylation but not for tRNA acetylation. Top panel: 18S rRNA purified from HCT116 cells depleted of U13 for 72 h was analyzed by HPLC, revealing a 2-fold reduction in acetylation (in red). Control HPLC trace from unperturbed cells in black; middle panel, same analysis for purified tRNAs showing no reduction in acetylation; bottom panel, northern blot analysis of residual level of U13 (normalized with respect to the scramble control). (C) U13 is not required for DIMT1L-mediated 18S rRNA 3′ end dimethylation. Total RNA extracted from cells depleted of U13 or DIMT1L for 72 h was analyzed by primer extension to detect the 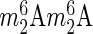 modification. The percentage of residual dimethylation was normalized with respect to a structural stop (*). The efficiencies of DIMT1L depletion (tested by qRT-PCR) and U13 depletion (tested by quantitative northern blot) are shown underneath.
modification. The percentage of residual dimethylation was normalized with respect to a structural stop (*). The efficiencies of DIMT1L depletion (tested by qRT-PCR) and U13 depletion (tested by quantitative northern blot) are shown underneath.
We found that efficient depletion of U13 (Figure 7A, lanes 2 and 7, ∼20% residual level) had no impact on 18S rRNA synthesis or nucleolar structure (Figures 6E and 7A, and data not shown), but we noted that U13 conspicuously comigrates with NAT10 on sucrose gradients, in fractions corresponding to 40S precursors (Figure 7C, fractions 6–7). This suggests that U13 might contribute to rRNA acetylation, possibly in association with NAT10, and that acetylation is a late event occurring on 40S precursor ribosomes once they have been released from larger particles. Interestingly and in contrast to U13, the U3 and U22 snoRNAs showed a distinctly different migration behavior on velocity gradients, being detected more abundantly with large pre-ribosomes (Figure 7C, fractions 9–12) and mostly as free snoRNPs, in the low-density fractions of the gradient (fractions 2–3). HPLC applied to 18S rRNA purified from cells depleted of U13 revealed a 2-fold reduction (55.4% residual level) of acetylation, while tRNA modification was unaffected (Figure 8B). Importantly, U13 depletion did not affect the metabolic stability of NAT10 (Figure 6B, lanes 2 and 7).
By analogy to the large majority of box C/D and box H/ACA snoRNAs, known to carry, respectively, an MTase or a pseudouridine synthase to RNA modification sites, we were interested in knowing whether U13 and NAT10 might interact directly in vivo under stringent conditions. While affinity purified NAT10 efficiently coprecipitated THUMPD1 (see above), U13 was not detected in association with NAT10 (Figure 5H). At this stage, we thus have no evidence that U13 and NAT10 work together directly. Rather, we favor a model in which U13 contributes to rRNA acetylation through its involvement in timely pre-rRNA folding. Since U13 is required only for efficient rRNA but not tRNA acetylation, we conclude that the effects of the snoRNA are highly specific. This was further substantiated by the finding that the neighboring dimethylation, introduced on residues A1850 and A1851 by DIMT1L (CZ, EN and DLJL, unpublished), is likewise unaffected by efficient U13 depletion (Figure 8C).
DISCUSSION
For many years, it has been assumed that yeast small ribosomal subunits contain only four modified bases: one hypermodified pseudouridine (m1acp3Ψ1191), one methylated guanosine (m7G1575) and two dimethylated adenosines (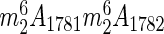 ) (Figure 1B) (4). Actually, the presence of additional acetylated cytidines on 18S rRNA was suggested in a pioneering work of the late 70's (25), but surprisingly, this observation had not been pursued for three decades, and until recently, the presence of the modification had not been confirmed with modern analytical techniques, its position on 18S rRNA had not been determined and the enzyme involved had not been identified.
) (Figure 1B) (4). Actually, the presence of additional acetylated cytidines on 18S rRNA was suggested in a pioneering work of the late 70's (25), but surprisingly, this observation had not been pursued for three decades, and until recently, the presence of the modification had not been confirmed with modern analytical techniques, its position on 18S rRNA had not been determined and the enzyme involved had not been identified.
Here we have confirmed by quantitative HPLC analysis that 18S rRNA is indeed acetylated and that the modification is widely conserved in eukaryotes, being detected in budding and fission yeasts, in plant and human cells (Figure 1C–F and Supplementary Figures S1–S4). In budding and fission yeasts and in human cells, we have shown that there are two acetylated cytosines on 40S, one in the 3′ major domain in helix 34 and another in the 3′ minor domain in helix 45, at the 3′ end of the 18S rRNA (Figure 4A and B and Supplementary Figure S4). In budding yeast, we have mapped the modification in helix 34 to C1280 and the one in helix 45 to C1773. These nucleotides correspond to C1297 and C1815 in S. pombe and to C1337 and C1842 in human, respectively. We have further shown that Kre33 and its human homolog NAT10 are responsible for 18S rRNA acetylation in budding yeast and human cells, respectively (Figures 2 and 3A–D). Recent reports, based on mass spectrometric analysis of 18S rRNA digests, corroborate our findings (27–29). We have demonstrated that Kre33 and NAT10 are required for early nucleolar processing steps leading to the synthesis of 18S rRNA (Figures 6 and 7 and Supplementary Figure S5). Using two distinct catalytically defective alleles (H545A and R637A), we have further demonstrated that small subunit biogenesis requires the modification enzyme itself rather than rRNA acetylation (Figure 2). We conclude that the physical presence of Kre33 within precursor ribosomes is required for efficient pre-rRNA processing. While Kre33 and NAT10 are reported by others to function in ribosome biogenesis (27–29,36), previous work either has not distinguished which is required, the enzyme or the modification (27,36), or has led to the erroneous conclusion that acetylation is essential to ribosome biogenesis (28,29). In human, our pre-rRNA analysis on diploid cells constitutively expressing p53 and in a paired p53-knockout cell line has enabled us to further conclude that NAT10′ depletion activates nucleolar stress and that the observed processing defects do not depend on the presence of p53 (Figures 6 and 7).
It was recently suggested that the nuclear concentration of acetyl-CoA is monitored by Kre33 as a mean to match ribosome synthesis to the cell’ energy status (28). We did not find any evidence for that as both tRNAs and 18S rRNA were properly acetylated in yeast cells inactivated for the acetyl-CoA synthetases Acs1 or Acs2 (Supplementary Figure S6).
In addition, we report that Kre33 and NAT10 are the tRNA ATs long sought in yeast and human cells, respectively (Figures 3E–H and 5). In budding yeast, Tan1 was previously known to be strictly required for tRNA acetylation, but as it lacks an AT domain, it was suggested that it might act as a tRNA adaptor or as a coactivator, rather than as a catalytic subunit (47). Our results confirm this early hypothesis, and importantly we have now identified the catalytic subunit as Kre33 or NAT10. We show that Kre33 interacts with Tan1 in yeast cells and that NAT10 coprecipitates with THUMPD1 (the human homolog of Tan1) (Figure 5H–J). Interestingly, there are several other cases of enzymes acting as heterodimeric complexes in tRNA modification, notably Gcd10/Gcd14 (60), Tad2/Tad3 (61) and Trm8/Trm82 (48). Multi-subunit enzymes are also known to modify rRNA, as recently shown for Bud23-Trm112, active in 18S rRNA N7G methylation (7,20,62). In this case, Trm112 acts as a stabilizing coactivator by masking on the catalytic subunit Bud23 a large hydrophobic core exposed to the solvent (7,20). This observation prompted us to test whether THUMPD1 might also be required for the metabolic stability of NAT10, but we found it not to be (Figure 5I).
What are the functions of 18S rRNA and tRNA acetylation?
At this stage the exact function of 18S rRNA acetylation is not known. We have clearly shown that yeast cells producing ribosomes and tRNAs that lack the modification are perfectly viable (Figures 2C and D and 5E and F), at least under optimal laboratory growth conditions. Nonetheless, the machinery involved in RNA modification is essential to early nucleolar processing steps leading to 18S rRNA synthesis and is thus required for the production of small subunits (Figures 6 and 7 and Supplementary Figure S5). Interestingly, ac4C1773 is located at the base of a highly conserved hairpin, which in addition harbors the highly conserved 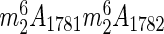 modification, making the 18S rRNA 3′ end an environment particularly rich in base modifications (Figure 1A and B). To us, this suggests a possible role in translation, especially given the conspicuous proximity of helix 45 to the decoding site, where important interactions with the mRNA and tRNAs occur (Figure 1B). The ac4C1280 modification in helix 34 is overlooking the mRNA entry channel and is within a region associated with translational fidelity defects (15,63). Also not totally understood is the exact function of C12 tRNA acetylation, introduced during tRNA biogenesis at the level of intron-containing pre-tRNAs (64). Interestingly, however, the stability of mutant forms of tRNASer appears to be affected in the absence of Tan1 (47), and tRNA acetylation is one of the numerous modifications actively monitored at the level of mature tRNAs by the Rapid tRNA Decay (RTD) surveillance pathway (65,66).
modification, making the 18S rRNA 3′ end an environment particularly rich in base modifications (Figure 1A and B). To us, this suggests a possible role in translation, especially given the conspicuous proximity of helix 45 to the decoding site, where important interactions with the mRNA and tRNAs occur (Figure 1B). The ac4C1280 modification in helix 34 is overlooking the mRNA entry channel and is within a region associated with translational fidelity defects (15,63). Also not totally understood is the exact function of C12 tRNA acetylation, introduced during tRNA biogenesis at the level of intron-containing pre-tRNAs (64). Interestingly, however, the stability of mutant forms of tRNASer appears to be affected in the absence of Tan1 (47), and tRNA acetylation is one of the numerous modifications actively monitored at the level of mature tRNAs by the Rapid tRNA Decay (RTD) surveillance pathway (65,66).
How does Kre33 or NAT10 recognize its substrates?
Kre33 and NAT10 each harbor a RecD helicase domain, which we show here to be required in yeast cells for efficient rRNA acetylation and, to a lesser extent, tRNA modification (Figures 2E and 5G). We suggest that the helicase activity of Kre33 may be required to remodel the structure of the substrate so as to provide ready access to the catalytic pocket. Alternatively, the helicase domain of Kre33 might act as a ‘molecular clamp’, or placeholder, regulating the timely access of the substrate cystosines. It is likely that the greater importance of the helicase domain in rRNA than in tRNA modification reflects the relative complexity of these two substrates. On velocity gradients, we have shown Kre33 to interact with early and late precursor ribosomes (Figure 2G), indicating that Kre33 binds nascent pre-rRNA precursors early during the assembly of the small subunit, in keeping with it being required for early nucleolar cleavages at sites A1 and A2 (Supplementary Figure S5E). Upon inactivation of its helicase domain, Kre33 appears no longer to interact with pre-40S particles. This provides a plausible explanation for the observed 5-fold reduction in rRNA acetylation and further suggests that while Kre33 binds early precursors, modification likely occurs at later stages of ribosome biogenesis. If so, Kre33 would resemble two other small subunit rRNA base modification enzymes in this respect (10,20). As both Kre33/NAT10 and Tan1/THUMPD1 carry a putative tRNA binding domain (45,47), both components of the complex might contribute to tRNA binding.
SnoRNAs are involved in pre-rRNA modification, pre-rRNA processing, and the timely folding of pre-rRNAs (24). Most snoRNAs are highly conserved and act as antisense guides to select rRNA residues for 2′-O methylation (box C/D) or uridine conversion to pseudouridines (box H/ACA) during subunit biogenesis, while others are involved in pre-rRNA processing (6,67). U13 is a vertebrate-specific snoRNA whose function has remained totally elusive (59); it has no known 2′-O methylation target site. We show here that U13 is required for efficient rRNA acetylation, but strikingly neither for tRNA acetylation nor for the dimethylation carried out by DIMT1L on two nearby adenosines (Figure 8). This suggests that U13 acts in a highly specific manner to influence rRNA acetylation. Importantly, U13 is not required for NAT10 metabolic stability (Figure 5I).
Under the stringent conditions (high salts washes) used to ascertain the specificity of the interaction between NAT10 and THUMPD1 (Figure 5H), we did not obtain any evidence of a direct interaction between NAT10 and U13. Therefore, on the basis of the potential base-pairing between U13 and the 3′ terminal helix of 18S rRNA (Figure 8A), which can occur only during ribosome assembly, we conclude that the involvement of U13 in rRNA acetylation is indirect and that U13 likely plays an important role in pre-rRNA folding during subunit biogenesis, an intervention which, we suggest, exposes the substrate cytosine on a bulge, giving NAT10 ready access to it (Figure 8A). It is unclear at this stage whether U13 and NAT10 are working together as part of the same pre-ribosomes or whether they are acting sequentially on successive particles. Considering that U13 and NAT10 clearly comigrate on sucrose gradients in a narrow peak corresponding to 40S precursors (Figure 7C), we strongly favor the view that they are part of the same pre-ribosomes.
NAT10 is reported to be a protein AT with specificity toward histones and microtubules (30,31), and we here reveal that it is active in tRNA and rRNA acetylation. To our knowledge, there are only a few other cases of enzymes involved in the modification of both proteins and RNAs ((68–70) and references therein). Remarkably, the elongator complex initially implicated in RNA polymerase II elongation (histone acetylation, (71,72)) and polarized exocytosis (microtubule modification, (73,74)) later turned out to be a ‘bona fide’ tRNA modification enzyme, and most of the in vivo phenotypes associated with elongator mutants are now known to be strictly dependent upon its primary function in the formation of modified wobble uridines in tRNAs (68,75–77) and the resulting reduced translation of specific mRNA transcripts (78,79).
Ribosomopathies are human syndromes caused by defective ribosome assembly due to mutations in ribosomal proteins or ribosome assembly factors (80). The salient features of this emerging class of human diseases are an increased pre-disposition to cancer and hematopoiesis and skeletons defects (81). NAT10 has been implicated in the DNA damage response, telomerase function and cytokinesis and reported as a cancer biomarker (31,34–35). Recently, inactivation of NAT10 has been shown to suppress morphological nuclear shape abnormalities associated with mutations in laminopathic cells, notably from patients with premature-aging progeria syndrome (30). Restoration of normal nuclear shape has been proposed to be due to loss of microtubule acetylation and consequent reorganization of the cytoskeleton (30). On the basis of our demonstration that NAT10 is a tRNA and rRNA AT required for small ribosomal subunit biogenesis, we suggest that the involvement of NAT10 in laminopathies is through ribosome biogenesis and function.
Our conclusion that RNA modification enzymes such as Kre33 and NAT10 carry specificity toward multiple substrates, in this case ribosomal and tRNAs, implies that caution must be applied when interpreting the phenotypic consequences of RNA modification enzyme inactivation. The development of transcriptome-wide RNA modification mapping strategies will undoubtedly help us in revealing the true coverage of these enzymes.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank Dr Emilien Nicolas (CMMI, ULB) for assistance with the capture of microscopic images and Ludivine Wacheul (ULB) for technical assistance. We also thank Prof. Enrico Schleiff and Benjamin Weis (U Frankfurt) for providing total RNA from plant, and Jun Yang (U Frankfurt) for scientific and technical discussions.
FUNDING
European Molecular Biology Organization [ALTF644-2014 to S.S.]; European Commission Marie Curie Action [EMBOCOFUND2012, GA-2012-600394 to S.S.]; Fonds pour la Formation à la Recherche dans l'Industrie et dans l'Agriculture [to J.-L.L.]; Deutsche Forschungsgemeinschaft [En134-9 to K.-D.E.]; State of Hesse, Germany [to K.-D.E.]; Fonds National de la Recherche (F.R.S./FNRS) [to D.L.J.L.]; the Walloon Region (DGO6) [to D.L.J.L.]; the European Research Development Fund [to D.L.J.L.]; the Université Libre de Bruxelles [to D.L.J.L.]. Funding for open access charge: Fonds National de la Recherche Scientifique (F.R.S./FNRS).
Conflict of interest statement. None declared.
REFERENCES
- 1.Motorin Y., Helm M. RNA nucleotide methylation. Wiley Interdiscip. Rev. RNA. 2011;2:611–631. doi: 10.1002/wrna.79. [DOI] [PubMed] [Google Scholar]
- 2.Cantara W.A., Crain P.F., Rozenski J., McCloskey J.A., Harris K.A., Zhang X., Vendeix F.A., Fabris D., Agris P.F. The RNA Modification Database, RNAMDB: 2011 update. Nucleic Acids Res. 2011;39:D195–D201. doi: 10.1093/nar/gkq1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grosjean H. Nucleic acids are not boring long polymers of only four types of nucleotides: a guided tour. In: Grosjean H, editor. DNA and RNA Modification Enzymes: Structure, Mechanism, Function and Evolution. Austin, TX: Landes Bioscience; 2009. pp. 1–18. [Google Scholar]
- 4.Decatur W.A., Fournier M.J. rRNA modifications and ribosome function. Trends Biochem. Sci. 2002;27:344–351. doi: 10.1016/s0968-0004(02)02109-6. [DOI] [PubMed] [Google Scholar]
- 5.Lestrade L., Weber M.J. snoRNA-LBME-db, a comprehensive database of human H/ACA and C/D box snoRNAs. Nucleic Acids Res. 2006;34:D158–D162. doi: 10.1093/nar/gkj002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Watkins N.J., Bohnsack M.T. The box C/D and H/ACA snoRNPs: key players in the modification, processing and the dynamic folding of ribosomal RNA. Wiley Interdiscip. Rev. RNA. 2012;3:397–414. doi: 10.1002/wrna.117. [DOI] [PubMed] [Google Scholar]
- 7.Figaro S., Wacheul L., Schillewaert S., Graille M., Huvelle E., Mongeard R., Zorbas C., Lafontaine D.L.J., Heurgue-Hamard V. Trm112 is required for Bud23-mediated methylation of the 18S rRNA at position G1575. Mol. Cell. Biol. 2012;32:2254–2267. doi: 10.1128/MCB.06623-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.White J., Li Z., Sardana R., Bujnicki J.M., Marcotte E.M., Johnson A.W. Bud23 methylates G1575 of 18S rRNA and is required for efficient nuclear export of pre-40S subunits. Mol. Cell. Biol. 2008;28:3151–3161. doi: 10.1128/MCB.01674-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lafontaine D.L.J., Delcour J., Glasser A.L., Desgres J., Vandenhaute J. The DIM1 gene responsible for the conserved m6(2)Am6(2)A dimethylation in the 3′-terminal loop of 18 S rRNA is essential in yeast. J. Mol. Biol. 1994;241:492–497. doi: 10.1006/jmbi.1994.1525. [DOI] [PubMed] [Google Scholar]
- 10.Lafontaine D.L.J., Vandenhaute J., Tollervey D. The 18S rRNA dimethylase Dim1p is required for pre-ribosomal RNA processing in yeast. Genes Dev. 1995;9:2470–2481. doi: 10.1101/gad.9.20.2470. [DOI] [PubMed] [Google Scholar]
- 11.Sharma S., Yang J., Duttmann S., Watzinger P., Kotter P., Entian K.D. Identification of novel methyltransferases, Bmt5 and Bmt6, responsible for the m3U methylations of 25S rRNA in Saccharomyces cerevisiae. Nucleic Acids Res. 2014;42:3246–3260. doi: 10.1093/nar/gkt1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sharma S., Yang J., Watzinger P., Kotter P., Entian K.D. Yeast Nop2 and Rcm1 methylate C2870 and C2278 of the 25S rRNA, respectively. Nucleic Acids Res. 2013;41:9062–9076. doi: 10.1093/nar/gkt679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sharma S., Watzinger P., Kotter P., Entian K.D. Identification of a novel methyltransferase, Bmt2, responsible for the N-1-methyl-adenosine base modification of 25S rRNA in Saccharomyces cerevisiae. Nucleic Acids Res. 2013;41:5428–5443. doi: 10.1093/nar/gkt195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peifer C., Sharma S., Watzinger P., Lamberth S., Kotter P., Entian K.D. Yeast Rrp8p, a novel methyltransferase responsible for m1A 645 base modification of 25S rRNA. Nucleic Acids Res. 2013;41:1151–1163. doi: 10.1093/nar/gks1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liang X.H., Liu Q., Fournier M.J. Loss of rRNA modifications in the decoding center of the ribosome impairs translation and strongly delays pre-rRNA processing. RNA. 2009;15:1716–1728. doi: 10.1261/rna.1724409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baudin-Baillieu A., Fabret C., Liang X.H., Piekna-Przybylska D., Fournier M.J., Rousset J.P. Nucleotide modifications in three functionally important regions of the Saccharomyces cerevisiae ribosome affect translation accuracy. Nucleic Acids Res. 2009;37:7665–7677. doi: 10.1093/nar/gkp816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Piekna-Przybylska D., Przybylski P., Baudin-Baillieu A., Rousset J.P., Fournier M.J. Ribosome performance is enhanced by a rich cluster of pseudouridines in the A-site finger region of the large subunit. J. Biol. Chem. 2008;283:26026–26036. doi: 10.1074/jbc.M803049200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liang X.H., Liu Q., Fournier M.J. rRNA modifications in an intersubunit bridge of the ribosome strongly affect both ribosome biogenesis and activity. Mol. Cell. 2007;28:965–977. doi: 10.1016/j.molcel.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 19.Leulliot N., Bohnsack M.T., Graille M., Tollervey D., Van Tilbeurgh H. The yeast ribosome synthesis factor Emg1 is a novel member of the superfamily of alpha/beta knot fold methyltransferases. Nucleic Acids Res. 2008;36:629–639. doi: 10.1093/nar/gkm1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Létoquart J., Huvelle E., Wacheul L., Bourgeois G., Zorbas C., Graille M., Heurgué-Hamard V., Lafontaine D.L.J. Structural and functional studies of Bud23-Trm112 reveal 18S rRNA N7-G1575 methylation occurs on late 40S precursor ribosomes. Proc. Natl. Acad. Sci. U.S.A. 2014;111:E5518–E5526. doi: 10.1073/pnas.1413089111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meyer B., Wurm J.P., Kotter P., Leisegang M.S., Schilling V., Buchhaupt M., Held M., Bahr U., Karas M., Heckel A., et al. The Bowen-Conradi syndrome protein Nep1 (Emg1) has a dual role in eukaryotic ribosome biogenesis, as an essential assembly factor and in the methylation of Psi1191 in yeast 18S rRNA. Nucleic Acids Res. 2011;39:1526–1537. doi: 10.1093/nar/gkq931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Henras A., Plisson-Chastang C., O'Donohue M.-F., Chakraborthy A., Gleizes P.-E. Overview of pre-rRNA processing in eukaryotes. WIREs RNA. 2014 doi: 10.1002/wrna.1269. doi:10.1002/wrna.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fernandez-Pevida A., Kressler D., de la Cruz J. Processing of preribosomal RNA in Saccharomyces cerevisiae. Wiley Interdiscip. Rev. RNA. 2014 doi: 10.1002/wrna.1267. doi:10.1002/wrna.1267. [DOI] [PubMed] [Google Scholar]
- 24.Lafontaine D.L.J. Noncoding RNAs in eukaryotic ribosome synthesis and function. Nat. Struct. Mol. Biol. 2015;22:11–19. doi: 10.1038/nsmb.2939. [DOI] [PubMed] [Google Scholar]
- 25.Thomas G., Gordon J., Rogg H. N4-Acetylcytidine. A previously unidentified labile component of the small subunit of eukaryotic ribosomes. J. Biol. Chem. 1978;253:1101–1105. [PubMed] [Google Scholar]
- 26.McCarroll R., Olsen G.J., Stahl Y.D., Woese C.R., Sogin M.L. Nucleotide sequence of Dictyostelium discoideum small sub-unit ribosomal ribonucleic acid inferred from gene sequence: evolutionary implications. Biochemistry. 1983;22:5858–5868. [Google Scholar]
- 27.Ito S., Horikawa S., Suzuki T., Kawauchi H., Tanaka Y., Suzuki T., Suzuki T. Human NAT10 is an ATP-dependent RNA acetyltransferase responsible for N4-acetylcytidine formation in 18S rRNA. J. Biol. Chem. 2014;289:35724–35730. doi: 10.1074/jbc.C114.602698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ito S., Akamatsu Y., Noma A., Kimura S., Miyauchi K., Ikeuchi Y., Suzuki T., Suzuki T. A single acetylation of 18 S rRNA is essential for biogenesis of the small ribosomal subunit in Saccharomyces cerevisiae. J. Biol. Chem. 2014;289:26201–26212. doi: 10.1074/jbc.M114.593996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Taoka M., Ishikawa D., Nobe Y., Ishikawa H., Yamauchi Y., Terukina G., Nakayama H., Hirota K., Takahashi N., Isobe T. RNA cytidine acetyltransferase of small-subunit ribosomal RNA: identification of acetylation sites and the responsible acetyltransferase in fission yeast, Schizosaccharomyces pombe. PloS one. 2014;9:e112156. doi: 10.1371/journal.pone.0112156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Larrieu D., Britton S., Demir M., Rodriguez R., Jackson S.P. Chemical inhibition of NAT10 corrects defects of laminopathic cells. Science. 2014;344:527–532. doi: 10.1126/science.1252651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shen Q., Zheng X., McNutt M.A., Guang L., Sun Y., Wang J., Gong Y., Hou L., Zhang B. NAT10, a nucleolar protein, localizes to the midbody and regulates cytokinesis and acetylation of microtubules. Exp. Cell Res. 2009;315:1653–1667. doi: 10.1016/j.yexcr.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 32.Fu D., Collins K. Purification of human telomerase complexes identifies factors involved in telomerase biogenesis and telomere length regulation. Mol. Cell. 2007;28:773–785. doi: 10.1016/j.molcel.2007.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lv J., Liu H., Wang Q., Tang Z., Hou L., Zhang B. Molecular cloning of a novel human gene encoding histone acetyltransferase-like protein involved in transcriptional activation of hTERT. Biochem. Biophys. Res. Commun. 2003;311:506–513. doi: 10.1016/j.bbrc.2003.09.235. [DOI] [PubMed] [Google Scholar]
- 34.Liu H., Ling Y., Gong Y., Sun Y., Hou L., Zhang B. DNA damage induces N-acetyltransferase NAT10 gene expression through transcriptional activation. Mol. Cell. Biochem. 2007;300:249–258. doi: 10.1007/s11010-006-9390-5. [DOI] [PubMed] [Google Scholar]
- 35.Zhang H., Hou W., Wang H.L., Liu H.J., Jia X.Y., Zheng X., Zou Y.X., Li X., Hou L., McNutt M.A. GSK-3beta-regulated N-acetyltransferase 10 is involved in colorectal cancer invasion. Clin. Cancer Res. 2014;20:4717–4729. doi: 10.1158/1078-0432.CCR-13-3477. [DOI] [PubMed] [Google Scholar]
- 36.Li Z., Lee I., Moradi E., Hung N.J., Johnson A.W., Marcotte E.M. Rational extension of the ribosome biogenesis pathway using network-guided genetics. PLoS Biol. 2009;7:e1000213. doi: 10.1371/journal.pbio.1000213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grandi P., Rybin V., Bassler J., Petfalski E., Strauss D., Marzioch M., Schafer T., Kuster B., Tschochner H., Tollervey D., et al. 90S pre-ribosomes include the 35S pre-rRNA, the U3 snoRNP, and 40S subunit processing factors but predominantly lack 60S synthesis factors. Mol. Cell. 2002;10:105–115. doi: 10.1016/s1097-2765(02)00579-8. [DOI] [PubMed] [Google Scholar]
- 38.Edelheit O., Hanukoglu A., Hanukoglu I. Simple and efficient site-directed mutagenesis using two single-primer reactions in parallel to generate mutants for protein structure-function studies. BMC Biotechnol. 2009;9:61. doi: 10.1186/1472-6750-9-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gehrke C.W., Kuo K.C. Ribonucleoside analysis by reversed-phase high-performance liquid chromatography. J. Chromatogr. 1989;471:3–36. doi: 10.1016/s0021-9673(00)94152-9. [DOI] [PubMed] [Google Scholar]
- 40.Kelley L.A., Sternberg M.J. Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 41.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 42.Notredame C., Higgins D.G., Heringa J. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- 43.Gouet P., Robert X., Courcelle E. ESPript/ENDscript: extracting and rendering sequence and 3D information from atomic structures of proteins. Nucleic Acids Res. 2003;31:3320–3323. doi: 10.1093/nar/gkg556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ikeuchi Y., Kitahara K., Suzuki T. The RNA acetyltransferase driven by ATP hydrolysis synthesizes N4-acetylcytidine of tRNA anticodon. EMBO J. 2008;27:2194–2203. doi: 10.1038/emboj.2008.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chimnaronk S., Suzuki T., Manita T., Ikeuchi Y., Yao M., Suzuki T., Tanaka I. RNA helicase module in an acetyltransferase that modifies a specific tRNA anticodon. EMBO J. 2009;28:1362–1373. doi: 10.1038/emboj.2009.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sprinzl M., Horn C., Brown M., Ioudovitch A., Steinberg S. Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res. 1998;26:148–153. doi: 10.1093/nar/26.1.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Johansson M.J., Bystrom A.S. The Saccharomyces cerevisiae TAN1 gene is required for N4-acetylcytidine formation in tRNA. RNA. 2004;10:712–719. doi: 10.1261/rna.5198204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alexandrov A., Martzen M.R., Phizicky E.M. Two proteins that form a complex are required for 7-methylguanosine modification of yeast tRNA. RNA. 2002;8:1253–1266. doi: 10.1017/s1355838202024019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Linder P., Jankowsky E. From unwinding to clamping—the DEAD box RNA helicase family. Nat. Rev. Mol. Cell Biol. 2011;12:505–516. doi: 10.1038/nrm3154. [DOI] [PubMed] [Google Scholar]
- 50.Ballut L., Marchadier B., Baguet A., Tomasetto C., Seraphin B., Le Hir H. The exon junction core complex is locked onto RNA by inhibition of eIF4AIII ATPase activity. Nat. Struct. Mol. Biol. 2005;12:861–869. doi: 10.1038/nsmb990. [DOI] [PubMed] [Google Scholar]
- 51.Xiol J., Spinelli P., Laussmann M.A., Homolka D., Yang Z., Cora E., Coute Y., Conn S., Kadlec J., Sachidanandam R., et al. RNA clamping by Vasa assembles a piRNA amplifier complex on transposon transcripts. Cell. 2014;157:1698–1711. doi: 10.1016/j.cell.2014.05.018. [DOI] [PubMed] [Google Scholar]
- 52.Colley A., Beggs J.D., Tollervey D., Lafontaine D.L.J. Dhr1p, a putative DEAH-box RNA helicase, is associated with the box C+D snoRNP U3. Mol. Cell. Biol. 2000;20:7238–7246. doi: 10.1128/mcb.20.19.7238-7246.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Derenzini M., Montanaro L., Trere D. What the nucleolus says to a tumour pathologist. Histopathology. 2009;54:753–762. doi: 10.1111/j.1365-2559.2008.03168.x. [DOI] [PubMed] [Google Scholar]
- 54.Bywater M.J., Poortinga G., Sanij E., Hein N., Peck A., Cullinane C., Wall M., Cluse L., Drygin D., Anderes K., et al. Inhibition of RNA polymerase I as a therapeutic strategy to promote cancer-specific activation of p53. Cancer Cell. 2012;22:51–65. doi: 10.1016/j.ccr.2012.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vlatkovic N., Boyd M.T., Rubbi C.P. Nucleolar control of p53: a cellular Achilles’ heel and a target for cancer therapy. Cell. Mol. Life Sci. 2014;71:771–791. doi: 10.1007/s00018-013-1361-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Donati G., Peddigari S., Mercer C.A., Thomas G. 5S ribosomal RNA is an essential component of a nascent ribosomal precursor complex that regulates the Hdm2-p53 checkpoint. Cell Rep. 2013;4:87–98. doi: 10.1016/j.celrep.2013.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sloan K.E., Bohnsack M.T., Watkins N.J. The 5S RNP couples p53 homeostasis to ribosome biogenesis and nucleolar stress. Cell Rep. 2013;5:237–247. doi: 10.1016/j.celrep.2013.08.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dai C., Gu W. p53 post-translational modification: deregulated in tumorigenesis. Trends Mol. Med. 2010;16:528–536. doi: 10.1016/j.molmed.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tyc K., Steitz J.A. U3, U8 and U13 comprise a new class of mammalian snRNPs localized in the cell nucleolus. EMBO J. 1989;8:3113–3119. doi: 10.1002/j.1460-2075.1989.tb08463.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Anderson J., Phan L., Hinnebusch A.G. The Gcd10p/Gcd14p complex is the essential two-subunit tRNA(1-methyladenosine) methyltransferase of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 2000;97:5173–5178. doi: 10.1073/pnas.090102597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gerber A.P., Keller W. An adenosine deaminase that generates inosine at the wobble position of tRNAs. Science. 1999;286:1146–1149. doi: 10.1126/science.286.5442.1146. [DOI] [PubMed] [Google Scholar]
- 62.Sardana R., Johnson A.W. The methyltransferase adaptor protein Trm112 is involved in biogenesis of both ribosomal subunits. Mol. Biol. Cell. 2012;23:4313–4322. doi: 10.1091/mbc.E12-05-0370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chernoff Y.O., Newnam G.P., Liebman S.W. The translational function of nucleotide C1054 in the small subunit rRNA is conserved throughout evolution: genetic evidence in yeast. Proc. Natl. Acad. Sci. U.S.A. 1996;93:2517–2522. doi: 10.1073/pnas.93.6.2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Etcheverry T., Colby D., Guthrie C. A precursor to a minor species of yeast tRNASer contains an intervening sequence. Cell. 1979;18:11–26. doi: 10.1016/0092-8674(79)90349-0. [DOI] [PubMed] [Google Scholar]
- 65.Dewe J.M., Whipple J.M., Chernyakov I., Jaramillo L.N., Phizicky E.M. The yeast rapid tRNA decay pathway competes with elongation factor 1A for substrate tRNAs and acts on tRNAs lacking one or more of several modifications. RNA. 2012;18:1886–1896. doi: 10.1261/rna.033654.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Whipple J.M., Lane E.A., Chernyakov I., D'Silva S., Phizicky E.M. The yeast rapid tRNA decay pathway primarily monitors the structural integrity of the acceptor and T-stems of mature tRNA. Genes Dev. 2011;25:1173–1184. doi: 10.1101/gad.2050711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Matera A.G., Terns R.M., Terns M.P. Non-coding RNAs: lessons from the small nuclear and small nucleolar RNAs. Nat. Rev. Mol. Cell Biol. 2007;8:209–220. doi: 10.1038/nrm2124. [DOI] [PubMed] [Google Scholar]
- 68.Esberg A., Huang B., Johansson M.J., Bystrom A.S. Elevated levels of two tRNA species bypass the requirement for elongator complex in transcription and exocytosis. Mol. Cell. 2006;24:139–148. doi: 10.1016/j.molcel.2006.07.031. [DOI] [PubMed] [Google Scholar]
- 69.Chen C., Tuck S., Bystrom A.S. Defects in tRNA modification associated with neurological and developmental dysfunctions in Caenorhabditis elegans elongator mutants. PLoS Genet. 2009;5:e1000561. doi: 10.1371/journal.pgen.1000561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tessarz P., Santos-Rosa H., Robson S.C., Sylvestersen K.B., Nelson C.J., Nielsen M.L., Kouzarides T. Glutamine methylation in histone H2A is an RNA-polymerase-I-dedicated modification. Nature. 2014;505:564–568. doi: 10.1038/nature12819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Otero G., Fellows J., Li Y., de Bizemont T., Dirac A.M., Gustafsson C.M., Erdjument-Bromage H., Tempst P., Svejstrup J.Q. Elongator, a multisubunit component of a novel RNA polymerase II holoenzyme for transcriptional elongation. Mol. Cell. 1999;3:109–118. doi: 10.1016/s1097-2765(00)80179-3. [DOI] [PubMed] [Google Scholar]
- 72.Wittschieben B.O., Otero G., de Bizemont T., Fellows J., Erdjument-Bromage H., Ohba R., Li Y., Allis C.D., Tempst P., Svejstrup J.Q. A novel histone acetyltransferase is an integral subunit of elongating RNA polymerase II holoenzyme. Mol. Cell. 1999;4:123–128. doi: 10.1016/s1097-2765(00)80194-x. [DOI] [PubMed] [Google Scholar]
- 73.Creppe C., Malinouskaya L., Volvert M.L., Gillard M., Close P., Malaise O., Laguesse S., Cornez I., Rahmouni S., Ormenese S., et al. Elongator controls the migration and differentiation of cortical neurons through acetylation of alpha-tubulin. Cell. 2009;136:551–564. doi: 10.1016/j.cell.2008.11.043. [DOI] [PubMed] [Google Scholar]
- 74.Rahl P.B., Chen C.Z., Collins R.N. Elp1p, the yeast homolog of the FD disease syndrome protein, negatively regulates exocytosis independently of transcriptional elongation. Mol. Cell. 2005;17:841–853. doi: 10.1016/j.molcel.2005.02.018. [DOI] [PubMed] [Google Scholar]
- 75.Huang B., Johansson M.J., Bystrom A.S. An early step in wobble uridine tRNA modification requires the Elongator complex. RNA. 2005;11:424–436. doi: 10.1261/rna.7247705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen C., Huang B., Eliasson M., Ryden P., Bystrom A.S. Elongator complex influences telomeric gene silencing and DNA damage response by its role in wobble uridine tRNA modification. PLoS Genet. 2011;7:e1002258. doi: 10.1371/journal.pgen.1002258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fernandez-Vazquez J., Vargas-Perez I., Sanso M., Buhne K., Carmona M., Paulo E., Hermand D., Rodriguez-Gabriel M., Ayte J., Leidel S., et al. Modification of tRNA(Lys) UUU by elongator is essential for efficient translation of stress mRNAs. PLoS Genet. 2013;9:e1003647. doi: 10.1371/journal.pgen.1003647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Johansson M.J., Esberg A., Huang B., Bjork G.R., Bystrom A.S. Eukaryotic wobble uridine modifications promote a functionally redundant decoding system. Mol. Cell. Biol. 2008;28:3301–3312. doi: 10.1128/MCB.01542-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bauer F., Matsuyama A., Candiracci J., Dieu M., Scheliga J., Wolf D.A., Yoshida M., Hermand D. Translational control of cell division by Elongator. Cell Rep. 2012;1:424–433. doi: 10.1016/j.celrep.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Narla A., Ebert B.L. Ribosomopathies: human disorders of ribosome dysfunction. Blood. 2010;115:3196–3205. doi: 10.1182/blood-2009-10-178129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Raiser D.M., Narla A., Ebert B.L. The emerging importance of ribosomal dysfunction in the pathogenesis of hematologic disorders. Leuk. Lymphoma. 2014;55:491–500. doi: 10.3109/10428194.2013.812786. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.