


Abstract
Base J (β-D-glucosyl-hydroxymethyluracil) replaces 1% of T in the Leishmania genome and is only found in telomeric repeats (99%) and in regions where transcription starts and stops. This highly restricted distribution must be co-determined by the thymidine hydroxylases (JBP1 and JBP2) that catalyze the initial step in J synthesis. To determine the DNA sequences recognized by JBP1/2, we used SMRT sequencing of DNA segments inserted into plasmids grown in Leishmania tarentolae. We show that SMRT sequencing recognizes base J in DNA. Leishmania DNA segments that normally contain J also picked up J when present in the plasmid, whereas control sequences did not. Even a segment of only 10 telomeric (GGGTTA) repeats was modified in the plasmid. We show that J modification usually occurs at pairs of Ts on opposite DNA strands, separated by 12 nucleotides. Modifications occur near G-rich sequences capable of forming G-quadruplexes and JBP2 is needed, as it does not occur in JBP2-null cells. We propose a model whereby de novo J insertion is mediated by JBP2. JBP1 then binds to J and hydroxylates another T 13 bp downstream (but not upstream) on the complementary strand, allowing JBP1 to maintain existing J following DNA replication.
INTRODUCTION
Studies on the mechanism of antigenic variation in African trypanosomes provided the first indication for the presence of an unusual nucleotide in the genome of trypanosomatids. When a telomeric expression site (ES) for variant-specific surface glycoproteins (VSGs) was switched off, some restriction enzyme recognition sites in the silenced gene became resistant to cleavage (1,2). The unusual base was eventually identified as β-D-glucosyl-hydroxymethyluracil and called base J (3). Base J has been found in all kinetoplastid flagellates analyzed (4), including major pathogens such as Trypanosoma brucei, Trypanosma cruzi and Leishmania species, and in the related unicellular alga, Euglena (5). In all these organisms, base J replaces 0.5–1% of T, mainly in telomeric repeats (4,6) and other repetitive sequences (7).
The biosynthesis of J occurs in two steps (8,9): first a specific T-residue in DNA is oxidized to form hydroxymethyluracil (HOMeU) (10); then this HOMeU is glucosylated to yield base J. While the second step is catalyzed by a single glucosyl transferase (11–13), two proteins are capable of catalyzing the first step: the J-binding protein (JBP) 1 and 2. Both contain an N-terminal dioxygenase domain (14) typical of the TET/JBP1 sub-family of Fe2+- and 2-oxoglutarate-dependent hydroxylases (15). JBP1 was initially identified as a protein that specifically binds to J-containing duplex DNA (16–19). It contains a novel DNA-binding domain (19), in which a single aspartate residue is responsible for the recognition of the glucose moiety held in a rigid edge-on configuration in the major groove of DNA by hydrogen bonding to the phosphate of the nucleotide adjacent to base J (20). Direct proof of the hydroxylase function of JBP1 was reported by Cliffe et al. (21), who demonstrated that hydroxylation of T in oligonucleotides was dependent on the presence of Fe2+, 2-oxoglutarate and O2. JBP2 was identified through the homology of its N-terminal hydroxylase domain with the corresponding domain in JBP1 (22). Despite its name JBP2 does not bind to J-DNA, but is associated with chromatin. The C-terminal half of JBP2 contains a SWI/SNF-related domain that is required for its function in J synthesis (22).
Disruption of the JBP1 gene in T. brucei resulted in loss of 95% of all J from each location in the genome where J is normally found (23) and a similar result was recently obtained with T. cruzi (24). In contrast, JBP1 is essential in Leishmania (25). The loss of JBP2 is tolerated both in trypanosomes (26) and in Leishmania (27). Initially, JBP2-null Leishmania loses little J, but during prolonged passaging the J level drops slowly to 30% of wild-type and the cells become hypersensitive to growth in bromodeoxyuridine (BrdU), a treatment also known to reduce J levels in T. brucei (8) and in Leishmania (27).
We have localized about 99% of J in Leishmania in the telomeric repeats (28), but we were initially unable to locate the remaining, chromosome-internal 1% of J. In recent papers Sabatini et al. discovered small amounts of J at the transcription initiation and termination sites of T. brucei (29), T. cruzi (24) and Leishmania major (29). We extended these results to Leishmania tarentolae and showed that this internal J (iJ) is located at convergent transcription termination sites (convergent strand switch regions, cSSRs) where the very long polycistronic transcription units of Leishmania transcribed by RNA Polymerase II terminate (30). Only some of the transcription initiation sites of Leishmania are marked by base J (30). When total J levels are reduced to 30% of wild-type in the JBP2-null, iJ levels fall even more and this is associated with massive read-through of the J-marked transcriptional stops (30). This read-through is exacerbated when iJ levels are further reduced by growth of Leishmania in BrdU resulting in cell death (30). Interestingly, the single cSSR on chromosome 28 of wild-type L. tarentolae that lacks J shows read-through in wild-type cells in the absence of BrdU (30). These results established a clear function for base J: it is essential for proper transcription termination in L. tarentolae (30). By reducing J levels approximately 32-fold with the hydroxylase inhibitor dimethyloxalylglycine, Reynolds et al. (31) also found an important transcriptional termination function for base J in L. major, although this read-through did not reduce cell viability.
A major question that remains to be addressed is the DNA sequence specificity of J insertion. How do JBP1 and 2 determine which T-residues to modify? Is this only determined by DNA sequence or does chromatin structure play a role? Why are two different enzymes required? The sequences modified are highly specific: only very few restriction enzyme sites in T. brucei DNA that contain a T are blocked (1,9), and in the telomeric repeat sequence (GGGTTA)n only the second T is modified to J (7,32). From comparisons of all sequences known to contain J, no common motif has emerged (9). A further complication is that JBP1 can maintain J in any DNA segment where it is artificially introduced in T. brucei (23). JBP1 must therefore have a J maintenance function that requires little sequence specificity. Finally, the insertion of J appears to ‘spread’ from J-containing sequences into neighboring sequences, when transcription is switched off (1,9). It is therefore likely that there are three types of recognition sequences: a primary ‘entry’ sequence required for de novo J insertion; a secondary recognition sequence required for ‘spreading’ of J into sequences adjacent to the primary recognition sequence; and a minimal sequence required for J maintenance.
Initially the Sabatini lab reported that JBP2 was essential for de novo insertion of J (26), but in later experiments JBP1 was shown to insert J in the absence of JBP2 (29,33). As J-Iess T. brucei is viable, Cliffe et al. (33) reintroduced JBP1 into the JBP1/JBP2 double null trypanosomes and found J insertion at the proper locations. This suggests that JBP1 can also insert J de novo under special circumstances.
The discovery of iJ regions in kinetoplastid DNA provided a new tool to analyze J-containing sequences. Obviously, to find out what determines the position of J in the genome, we need to know where J exactly is. To this end we have turned to Single Molecule, Real-Time (SMRT) sequencing (34). SMRT sequencing monitors the progression of single molecules of DNA polymerase in real time during base incorporation using fluorescent phospho-linked nucleotides. When the polymerase encounters an unusual base in the template it tends to pause. Pausing does not only occur at the unusual base, but also at neighboring bases, depending on the particular base modification encountered. This allows SMRT sequencing to specifically detect a variety of non-canonical bases and distinguish between closely related ones, such as MeC and hmC, based on a unique kinetic profile of the polymerase for each base (35–37).
We show here that SMRT sequencing can detect base J and that we have been able to localize J in plasmids containing Leishmania sequences grown as episomes in L. tarentolae. This has allowed us to characterize the recognition sequences for J insertion and to suggest a model for de novo insertion of J and its maintenance during replication.
MATERIALS AND METHODS
Leishmania culture and mutants
Leishmania tarentolae TarII wild-type (Cl. 1) cells were grown in SDM-79 medium (38) to a density of 108 cells/ml. The L. tarentolae JBP2-null mutant was described by Vainio et al. (27).
Cloning of L. tarentolae DNA segments into plasmids and plasmid isolation
The Leishmania expression vector pGEM 7Zf α-neo-α (39) was digested with HindIII and XbaI, gel extracted, phenol purified, and dephosphorylated with calf intestinal alkaline phosphatase (1 U/μl, Roche). The fragments to be inserted were obtained by polymerase chain reaction (PCR) amplification using L. tarentolae genomic DNA as a template, gel extracted and digested with HindIII and XbaI. After phenol extraction, the fragments to be cloned were ligated with the vector at 22°C for approximately 1.5 h. The ligated plasmids were transformed in Escherichia coli and plated on LB agar plates containing ampicillin (100 μg/ml). Plasmid DNA was isolated using commercially available plasmid isolation kits (Qiagen and Roche) following the protocol provided by the supplier with only one difference: glycogen was added at the moment of the DNA precipitation.
Transfection of plasmids into L. tarentolae and isolation of plasmids
The human T cell Nucleofector transfection kit (Lonza) was used for transfection of the recombinant plasmids obtained from E. coli into L. tarentolae cells. Electroporation was done with 1–5 μg of plasmid DNA using the Nucleofector machine of Amaxa (Lonza). After transfection, the cells were left to grow overnight prior to selection with paromomycin (100 μg/ml). The L. tarentolae cells were kept under selection to obtain stable cell lines. Isolation of the plasmids from L. tarentolae transfectants was performed using an alkaline lysis method, as for the plasmids from E. coli.
Cloning convergent strand switch regions (cSSRs) in a Leishmania expression vector
cSSR regions of the chromosomes 12, 25 and 28 were PCR amplified using primers described below and cloned blunt in the ClaI site (blunted) of the Leishmania expression vector pGEM 7Zf α-neo-α. The fragment named cSSR 25.2L covers the sequence located between position 410 965 and 412 966 of chromosome 25. A smaller fragment corresponding to the bulk of the J-peak was also cloned and named cSSR 25.2S (this fragment contains the region between positions 412 392 and 412 772 of chromosome 25). The cSSR 28.2 fragment covers the sequence located between positions 580 566 and 582 966 of chromosome 28. This corresponds to the only cSSR of Leishmania that does not have a J-peak. Primers (Invitrogen) used to amplify the cSSRs are the following:
cSSR 25.2L forward: 5′-TTTTTTCTCGAGCCTCCCTCCTCTCTTACCCCT-3′, cSSR 25.2L reverse: 5′-TTTTTTTCTAGAGCAGGCCCGTGCGTGGAGTGG-3′, cSSR 25.2S forward: 5′-TTTTTTCTCGAGCGCGCGCGCACAGCCACCGG-3′, cSSR 25.2S reverse: 5′-TTTTTTTCTAGACACGACGTCCGCCTTCTCTT-3′, cSSR 28.2 forward: 5′-TTTTTTCAGCTGCTGCTGTTCTTGCATTGG-3′, and cSSR 28.2 reverse: 5′-TTTTTTTCTAGAAACCGGCCCGATGCTTTGCC-3′, cSSR 12.1 forward: 5′-TTTTTTCAGCTGCCCGCCCCGCCTCTTTAAAACAGCC-3′, and cSSR 12.1 reverse: 5′-TTTTTTTCTAGACTTCTCCTGAGCGTGGGTG-3′.
Cloning of wild-type and mutant telomeric repeats into a Leishmania expression vector
The DNA sequences 5′-AAGCTT(GGGTTA)10TCTAGA-3′ and 5′-AAGCTT(GGGTTT)10TCTAGA-3′ were synthesized by Genscript and cloned in the DNA vector pUC57 before being subcloned in the HindIII and XbaI sites of the Leishmania expression vector pGEM 7Zf α-neo-α.
Purification of plasmid DNA for SMRT sequencing
Our standard plasmid preparations from L. tarentolae proved to be heavily contaminated with mini-circle oligomers derived from fragmented kinetoplast DNA networks. Plasmid DNA was therefore linearized with ScaI, and size-fractionated by electrophoresis through a 0.7% agarose gel in 0.5xTBE (44.5 mM Tris, 44.5 mM Boric acid, 1 mM EDTA). The plasmid band was cut out and the DNA extracted using the Qiagen or Invitrogen Purelink commercial kits.
Detection of J-DNA by immunoblotting
Total DNA or DNA fragments obtained by restriction enzyme digestion were size-fractionated through a 0.7% agarose gel in 0.5xTBE overnight and transferred onto a nitrocellulose membrane in 10xSSC buffer (1.5 M NaCl, 150 mM Na citrate) by Southern blotting according to standard protocols. The DNA was ultraviolet (UV) cross-linked and the membrane was blocked for 5 h at room temperature in 1xTBST (10 mM Tris pH 8.0, 150 mM NaCl, 0.05% Tween 20) buffer containing 5% non-fat milk powder and then incubated with 1:3000 diluted anti-J antiserum (40) overnight on a shaker at 4°C. The membrane was washed for approximately 1.5 h in 1xTBST before the secondary antibody incubation. Swine anti-rabbit horse radish peroxidase conjugate (Dako) diluted 1:5000 in 1xTBST plus 5% non-fat milk powder was used for detection of the J-containing DNA fragments. The membrane was incubated with the secondary antibody for at least 1 h on a shaker at room temperature and afterward washed again for 1.5 h in 1xTBST, followed by enhanced chemiluminescence detection and autoradiography.
Southern blots of plasmid and genomic DNA
If a blot had first been probed for J-DNA, the blot was blocked in pre-hybridization mixture containing herring sperm DNA (100 μg/ml) for at least 1 h at 42°C. Hybridization with a [α-32P] dATP labeled 25S probe followed by a neo probe was done in a formamide based buffer at 42°C overnight. The blot was washed twice with 3xSSC plus 0.1% sodium dodecyl sulfate (SDS) followed by two additional washes with 0.1xSSC plus 0.1% SDS. Following the hybridization and autoradiography, the membrane was stripped with boiling water and re-probed with the selection marker gene (neo) using the same hybridization conditions as mentioned above.
If no detection of J-DNA was required, digests of wild-type and mutant genomic DNA were size-fractionated on a 1% agarose gel in 0.5xTBE. The DNA was partially depurinated in 0.25 M HCl with gentle agitation for 15 min, followed by incubation in denaturing buffer (0.5 M NaOH, 1.5 M NaCl) and neutralization buffer (1 M Tris pH 7.4, 1.5 M NaCl) for 45 min each. The DNA was blotted by capillary transfer onto a positively charged nylon membrane in 10xSSC buffer and immobilized by UV cross-linking. Hybridization and washing conditions were as stated above. The membrane was stripped in denaturation buffer and neutralization buffer each for 30 min, followed by reprobing with another probe.
SMRT sequencing
Preparation of sequencing assays, data collection, pulse calling and read alignments were performed as described previously (35,41,42). Interpulse duration (IPD) values were tabulated for aligned template positions; to avoid outlier effects, the smallest and largest 5% of IPDs at each position were excluded. IPD ratios at each position between experimental and unmodified control templates were initially calculated using unmodified plasmid templates using whole genome amplification. Later, IPD ratios were calculated by comparison with computationally predicted IPD values for unmodified ‘in silico’ templates. To validate the use of in silico templates, IPD ratios between unmodified samples derived using amplification and in silico templates were obtained, and showed a low background (no IPD ratio >2).
To objectively score the presence or absence of base J based on IPD ratios, an algorithm was developed using data from the synthetic oligonucleotides containing J, and by comparing complete single molecule data sets from the J-less JBP2-null sample against all samples (see Supplementary Figure S1).
Consensus sequences were analyzed using the WebLogo software from the University of California, Berkeley (43).
RESULTS
Detection of base J by SMRT DNA sequencing
SMRT sequencing has thus far been able to detect many different types of unusual nucleotides in DNA, albeit with varying signal levels (37). To determine whether it could also detect base J, we subjected synthetic oligonucleotides containing base J at a known position to SMRT sequencing. Comparison of the polymerase kinetics with those obtained with a template of identical sequence but containing a T instead of J, showed that the presence of J results in a substantial kinetic signature, with characteristic pauses at 0, +1 and/or +2 and +6 nucleotides relative to the J (Figure 1). The signature is influenced by the sequence context, as seen with other unusual bases (37). We do not know yet the full range of sequence variation, since we had only access to a few J-containing oligonucleotides. In practice, however, the signature is clear and uniform enough to determine where J is located in DNA.
Figure 1.
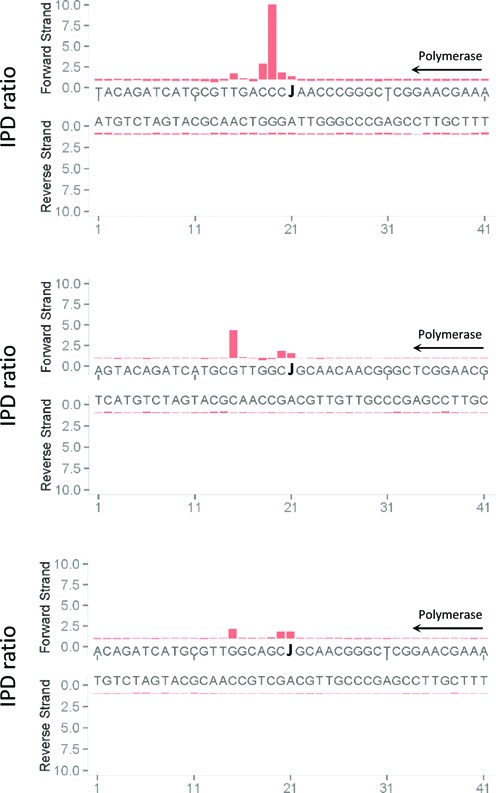
Direct detection of base J through SMRT DNA sequencing. Synthetic oligonucleotides containing J at known positions were subjected to SMRT sequencing, and the associated polymerase kinetics were compared with those obtained with a template of identical sequence but lacking the base J modification (and containing T at that position). The direction of DNA polymerase is indicated. The Y-axis shows the fold change in polymerase kinetics (interpulse duration (IPD) ratio) at each position between modified and unmodified sequences.
Insertion of J into Leishmania plasmids
We tried to SMRT sequence genomic fragments immunoprecipitated with an antiserum against J-DNA (30), but enrichment of J-containing fragments over background proved insufficient to make this approach successful. We therefore tested whether DNA sequences that normally contain J in the Leishmania genome would acquire J when inserted into a shuttle vector (Figure 2) and grown as episomes in L. tarentolae. We initially chose a cSSR on chromosome 25 (region cSSR 25.2), which contains a modest J-peak (30) and has minimal sequence similarity with other regions in the genome. Two segments of chromosome 25 were cloned, a 2 kb segment (25.2L) containing the entire cSSR, and a shortened version (25.2S) containing only the region with the J-peak. As a negative control we inserted a 2.4 kb segment containing the only cSSR in L. tarentolae that does not contain any J (chromosome 28, region 28.2). After growth in wild-type and JBP2-null L. tarentolae the plasmids were isolated by alkaline lysis, digested with restriction enzymes, and the fragments size-fractionated by agarose gel electrophoresis and probed with antibodies against base J (Figure 3). The blots show that plasmids containing the segments 25.2S and 25.2L pick up J when grown in Leishmania, whereas the plasmid with the 28.2 segment does not. Interestingly, the inserted J is not restricted to the cSSR, but ‘spreads’ into the neighboring 0.8 kb intergenic α-tubulin and the 1.7 kb intergenic α-tubulin-neo sequences. These sequences contain no J in chromosomal Leishmania DNA. The blot also shows that JBP2 is required for de novo J modification on a plasmid because in the JPB2-null mutant the plasmids do not appear to be modified. Similar experiments were performed using three additional plasmids: one containing another cSSR (cSSR 12.1); one containing 10 copies of the telomeric hexamer repeat (GGGTTA); and a modification of this sequence (GGGTTT) that lacks the T from the C-rich strand. As expected, both 12.1 and (GGGTTA)10 gained J when grown in wild-type L. tarentolae, but (GGGTTT)10 did not, despite the presence of a T (the second on the G-rich strand) that is normally modified in genomic DNA (results not shown). Note that some of the restriction sites in the digest presented in Figure 3 are partially cut. This is due to the presence of J, as we shall show below.
Figure 2.
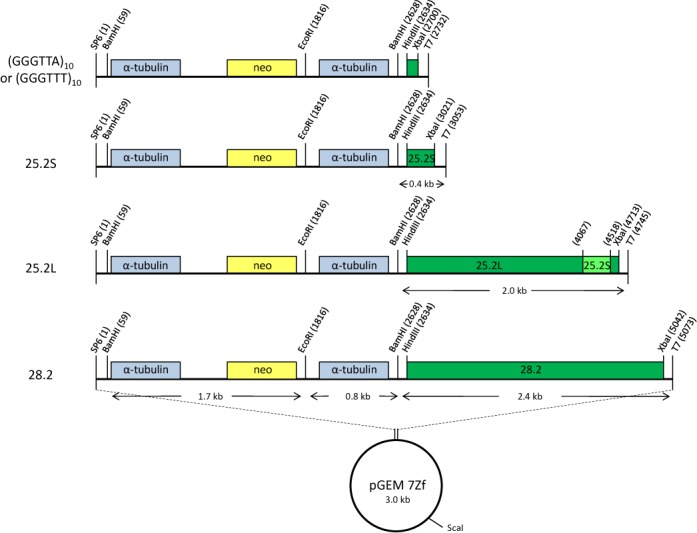
Maps of the plasmids containing convergent strand switch regions and telomeric repeats. The cSSRs and telomeric repeats were cloned into the HindIII and XbaI restriction sites of the 5.5 kb pGEM 7Zf α-neo-α backbone before transfection into wild-type L. tarentolae. The gene coding for neomycin phosphotransferase (yellow), which confers resistance to paromomycin, served as a selection marker after transfection. The intergenic α-tubulin gene fragments flanking the neomycin marker are indicated in light blue. The 25.2S plasmid has a 381 bp insert corresponding to the cSSR with a J-peak in chromosome 25 of L. tarentolae (position 412 392–412 772). The 25.2L plasmid contains a 2.0 kb insert covering the 25.2S region (positions 410 965–412 966). The plasmid 28.2 contains a 2.4 kb fragment. This fragment corresponds to the cSSR in chromosome 28 of L. tarentolae which contains no J in the wild-type (position 580 566–582 966). The telomeric sequence plasmids contain 10 copies of the wild-type telomeric repeat (GGGTTA) or a mutant version (GGGTTT). Plasmids were linearized with ScaI for gel extraction and SMRT sequencing. Plasmids were digested with BamHI, EcoRI and XbaI for DNA immunoblots.
Figure 3.
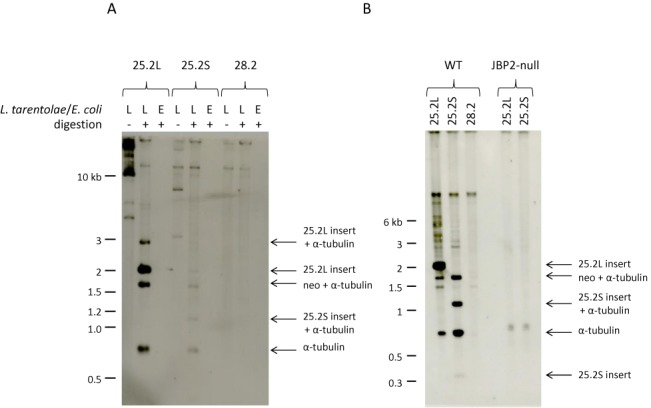
J is inserted into some plasmids transfected in L. tarentolae. The 25.2S, 25.2L and 28.2 plasmids, shown in Figure 2, were purified after amplification in L. tarentolae or E. coli. The DNA samples were digested with BamHI, EcoRI and XbaI, size-fractionated in a 0.7% agarose gel in 0.5×TBE, transferred to nitrocellulose and incubated with an anti-J antibody. (A) The plasmids 25.2L and 25.2S purified from L. tarentolae show specific bands recognized by the anti-J antibody. It is clear that J was not only formed in the inserted 2 kb 25.2L cSSR, but also in the adjacent 0.8 kb intergenic α-tubulin and the 1.7 kb α-tubulin-neo fragments. The small 25.2S insert fragment is not visible on the blot. No J-containing fragments are detected when the plasmids were isolated from E. coli or when the 28.2 plasmid was used. (B) J-containing DNA fragments are only detected after growth in the wild-type and not after growth in the JBP2-null L. tarentolae strain. This blot contains the 0.4 kb 25.2S insert band, albeit weak because these small fragments are lost during blotting. The 1.2 kb fragment in the 25.2S plasmid preparation is due to a modification of the BamHI site.
To test whether base J is inserted in the same locations in the plasmid as in the corresponding chromosomal region 25.2, we tested a battery of restriction enzymes on genomic DNA. Only cutting of the genomic region 25.2 by DdeI seemed to be affected, as shown in Supplementary Figure S2 in the Supplementary Data. A partial digest band of 1.3 kb showed up and this band disappeared when most of the iJ is absent in DNA from the JBP2–null mutant (Supplementary Figure S2B). The same 1.3 kb (1288 bp in Supplementary Figure S2A) partial digest band was also present in the 25.2L plasmid (Supplementary Data) grown in wild-type L. tarentolae, but not in the JBP2-null mutant (not shown). This supports the idea that J is inserted in the 25.2L plasmid in the same positions as in the corresponding sequence in its original genomic location.
SMRT sequencing of plasmid DNAs
To locate the J-residues in DNA, the plasmids were subjected to SMRT sequencing. Figure 4 and Table 1 present an overview of the results of the SMRT analysis of all plasmids studied. Plasmids containing (GGGTTA)10, cSSR 25.2L, cSSR 25.2S and cSSR 12.1 sequences all showed substantial peaks in the IPD ratio corresponding to the pause(s) of the DNA polymerase when encountering base J, whereas the plasmid containing cSSR 28.2 has none. As in the J-blotting experiments (Figure 3), no peaks are detectable when the cSSR 25.2S plasmid is grown in a JBP2-null mutant. ‘Spreading’ of J beyond the boundaries of the cSSR insert sequence is extensive in the 25.2S and 12.1 plasmids, but much less in the 25.2L and (GGGTTA)10 plasmids, mirroring the results from J-blotting experiments.
Figure 4.
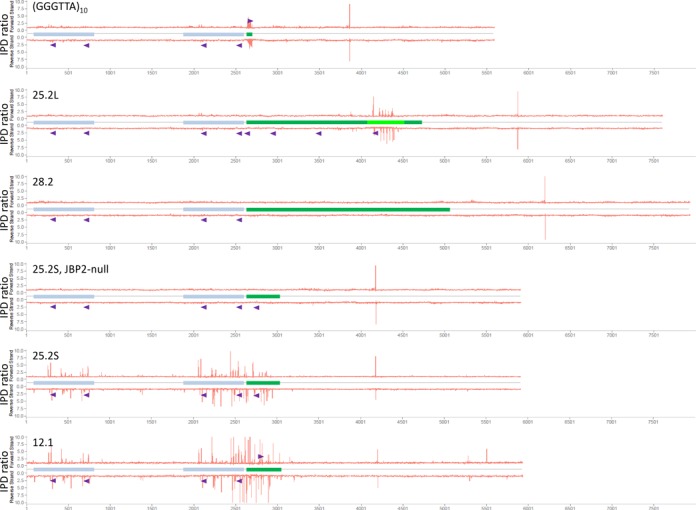
Overview of the SMRT sequencing results of the plasmids containing the cSSRs and the telomeric repeats. The plasmids depicted in Figure 2 and an additional plasmid containing the cSSR 12.1 were used for SMRT sequencing after propagation of the plasmids in L. tarentolae. The location of the intergenic α-tubulin fragments and the inserts are indicated using the same color coding as in Figure 2. The Y-axis shows the fold change in polymerase kinetics (interpulse duration (IPD) ratio) at each position between modified and unmodified sequences. The very large peak on both strands in the vector region to the right of each insert is due to digestion by ScaI at this site to linearize and purify the plasmids. The telomeric repeat insert as well as the 25.2S, 25.2L and 12.1 insert contain SMRT sequencing peaks. The 28.2 plasmid does not show any major peak, nor does the 25.2S plasmid after propagation in the JBP2-null mutant. Spreading of the SMRT sequencing signal into the α-tubulin intergenic sequences is seen in the 25.2S and 12.1 plasmids and to a minor extent in the 25.2L and telomeric repeat plasmids. The purple triangles indicate the location of a G-quadruplex in either the top or the bottom strand.
Table 1. Summary of J modifications on plasmids.
| Plasmid insert | Cell line | Total Js | Js in insert | Js in α-tub IRs | Js in vector | Fraction paired | 11 bp spacing | 12 bp spacing | 13 bp spacing |
|---|---|---|---|---|---|---|---|---|---|
| GGGTTA10 | WT Lt | 26 | 16 | 10 | 0 | 92.3% | 0% | 91.7% | 8.3% |
| cSSR 25.2L | WT Lt | 26 | 22 | 4 | 0 | 76.9% | 0% | 90% | 10% |
| cSSR 28.2 | WT Lt | 0 | 0 | 0 | 0 | – | – | – | – |
| cSSR 25.2S | JBP2-/- Lt | 0 | 0 | 0 | 0 | – | – | – | – |
| cSSR 25.2S | WT Lt | 84 | 16 | 57 | 11 | 61.9% | 7.7% | 76.9% | 15.4% |
| cSSR 12.1 | WT Lt | 97 | 23 | 60 | 14 | 61.9% | 16.7% | 70.0% | 13.3% |
| Totals | 233 | 77 | 131 | 25 | 67.0% | 9.0% | 78.2% | 12.8% | |
The SMRT sequencing results for all plasmids are shown at nucleotide-level resolution in Supplementary Figure S3. Figure 5A shows a zoom-in of the 25.2L plasmid. The most striking feature is a recurring doublet with a J at position zero and a J on the opposite strand at position +13 and occasionally position +12 or +14 (see Supplementary Figure S3 for the SMRT data of all the plasmids in this study). The main J-peaks have the overall patterns resembling the telomeric pattern in Figure 1 with a large peak due to DNA polymerase pausing two nucleotides downstream from J. The peak height varies with the fraction of molecules modified at each position, as indicated in Figure 5. This fraction can be roughly estimated because SMRT sequencing can look at individual DNA molecules. To distinguish minor peaks from noise, we chose a cut-off for the IPD ratio of 1.6, as explained in Supplementary Figure S1. Supplementary Figure S4 shows the average kinetic signature observed for Js in the 25.2 and 12.1 plasmids.
Figure 5.
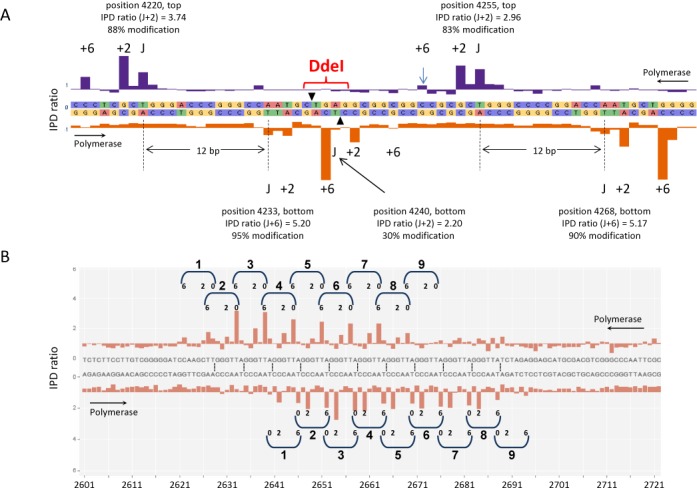
Zoom-ins of the SMRT sequencing results of the 25.2L and the telomeric repeat plasmid showing the characteristics of the J modification. (A) For the plasmid 25.2L presented in Figures 2 and 4 the region 4212 to 4270 from 25.2L is presented as in Figure 4. The typical 0, 2, 6 signature due to pausing of the DNA polymerase when passing J is seen as well the presence of couples of J modifications with one J modification on one strand and another J modification at +13 on the other strand. The estimated percentage of individual molecules contain the modification is indicated. There is a J-singlet in a DdeI site with a relative low level of modification which explains the partial digestion of this site (see also Supplementary Data). (B) SMRT sequencing of the telomeric repeat plasmid. The plasmid containing 10 copies of the telomeric GGGTTA repeat was grown in L. tarentolae, purified and sequenced using the SMRT technology. A base-resolution view of the region with the telomeric repeats based on pooling SMRT sequencing of individual molecules. The IPD ratio for each position on the top and bottom strand is plotted. The J signal has a typical 0, 2, 6 signature due to pausing of the DNA polymerase when passing J and J is present in couples with a J modification on one strand and another J modification at +13 (or +12 in case of couple 1) on the other strand.
Figure 5B shows a zoom-in of the insert in the (GGGTTA)10 plasmid, which contains a high density of modifications. The bottom strand Ts (in the CCCAAT repeats) are modified as J, while in the top strand, only the second T in GGGTTA is modified reproducing the doublet pattern highlighted in Figure 5A. As we have never seen modification of the first T in the GGGTTA strand by SMRT sequencing (Figure 5B) or in our chemical analysis (32), there is apparently a strong bias against the +14 position in this repeat sequence.
SMRT sequencing can also explain some of the partial restriction digest fragments in Figure 3. For plasmids 25.2S and 12.1 the expected insert fragment size is 387 bp. A band of this size is barely visible but an unexpected band at approximately 1.2 kb is observed in both cases. This band is explained by the SMRT sequencing data of the BamHI restriction site at position 2628: a J is found at the cut site only in these two plasmids (Supplementary Figure S3), blocking digestion at this site, in which case a 1.2 kb fragment would be expected. Partial modification of this BamHI site also explains the presence of the unexpected band of approximately 2.8 kb for 25.2L (Figure 3). SMRT sequencing also provides an explanation for some of the sites partially cut by DdeI presented in Supplementary Figure S2. The base-resolution view of a region of the cSSR of plasmid 25.2L in Figure 5A contains J doublets but also a J-singlet. This singlet has a lower modification level than the surrounding J doublets and is part of a DdeI restriction site, explaining the partial digestion observed for this site (see also Supplementary Figure S2).
SMRT sequencing confirms the ‘spreading’ of J insertion beyond the borders of the cSSR and telomeric repeat DNA segments cloned in the plasmid, but it also shows the exquisite specificity of the spreading process. This is already clear from Figure 4 and Supplementary Figure S3, but the striking conservation of the positions of the inserted Js in the two copies of the α-tubulin intergenic gene segments present in the plasmids is more clearly illustrated in Figure 6, in which both α-tubulin intergenic gene segments from the 25.2S, 12.1 and (GGGTTA)10 plasmids are aligned. Remarkably, the J positions do not seem to be affected by the size of the insert in the plasmid, or the distance from the insert, as the Js in all α-tubulin intergenic gene copies are at the same position. Note also that there is a strong discontinuity in spreading, as the neomycin resistance gene between the two α-tubulin intergenic segments is devoid of J (Figure 4). As the sensitivity of the analysis varies, there are J-peaks in the 12.1 plasmid not detected in the other ones. Moreover, the telomeric repeats in the (GGGTTA)10 plasmid appear to support less intensive spreading than the other plasmid inserts. Spreading invariably results in partial modification of sites, as also observed in the T. brucei VSG gene ES (1).
Figure 6.
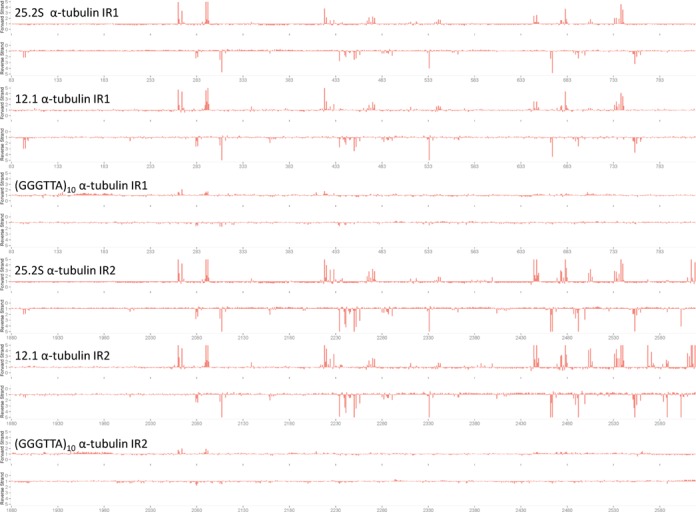
The position of base J in the intergenic α-tubulin fragment of the plasmids containing this base. The IPD ratio plot of the two different intergenic α-tubulin fragment of the pGEM 7Zf α-neo-α vector containing the cSSR 25.2S, the cSSR 12.1 and the telomeric repeat were aligned. The alignment shows the similarity of the J insertion in the two α-tubulin intergenic fragments from the different plasmids.
Sequences containing base J
In order to identify a possible sequence motif associated with J insertion sites, we aligned the sequences surrounding all J sites. No consensus emerged from the analysis (Figure 7A), other than a weak T(N)12A pattern, which is enriched by removing sequences surrounding non-paired J sites. The interpretation of this result is not straightforward, as this analysis may combine three types of recognition sequences. The simplest is the T(N)12A sequence probably required for J maintenance by JBP1 (see Discussion). The T(N)12A sequence dominates the consensus making it hard to detect the two other recognition sequences, i.e. the sequences which determine whether the plasmid picks up any J at all (the ‘entry’ sequence) and the ‘spreading’ sequence resulting in the insertion of J into plasmid vector sequences. We assume that JBP2 is responsible for both entry and spreading and that JBP2 should minimally be able to recognize one strand of the telomeric repeat. However, a strand specific consensus due to one strand, may easily get lost if there is a strong strand bias and one pools both strands. The telomeric hexamer repeats on the (GGGTTA)10 plasmid has an extreme G/C strand bias, as do many other J-containing sequences on the plasmids examined. We therefore examined the G-rich and C-rich sequence separately to find a consensus sequence hidden by the strand bias. To find the entry sequence and avoid including sequences within the inserts in the plasmids that could be due to ‘spreading’, we only included sites modified >80% according to the deconvolution analysis. This analysis resulted in a (rather weak) G-rich consensus for the G-rich strand (Figure 7B) and the reverse for the C-rich strand (data not shown). We also attempted to determine whether there were different recognition sites for ‘entry’ and ‘spreading’ by analyzing doublet sequences in the 25.2 and 12.1 inserts separately from those in the plasmid vector. The former yielded a clear G-rich consensus, with several tracts of 3–4 G-residues on either side of the J positions (Figure 7C), while the latter showed a weaker consensus.
Figure 7.
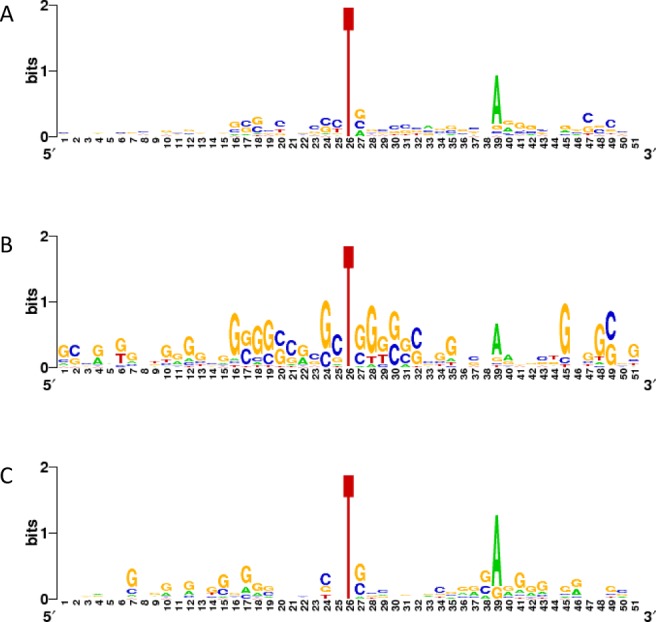
Sequences containing J. (A) An assembly of all sequences containing the characteristic J doublet. The sequences from regions containing a J modification doublet, with a J present on one strand and another J modification present at +12, +13 or +14, were aligned. Except for the T(N)12A consensus sequence, no other sequence requirements were found for J modification using this analysis. (B) G-rich sequences in ‘entry’ sequences. Sequences from the G-rich strand of the 25.2 and 12.1 plasmid insertions containing J sites modified > 80%. (C) G-rich sequences potentially involved in ‘spreading’. G-rich strands containing plasmid sequences with J in doublets.
Sequences surrounding J singlets also did not form a clear consensus sequence (data not shown), which is perhaps not surprising as they may have several possible origins. They could be part of a recently duplicated plasmid in which the +13 partner of a doublet has not yet been added; they could be introduced by JBP2 at a position where there is no suitable T at +13; or they could be part of a doublet that does not conform to the +13 rule. The sequences surrounding all J singlets can be found in Supplementary Figure S3.
DISCUSSION
The distribution of base J in the nuclear DNA of Leishmania is highly restricted: 99% is in telomeric repeats (28) and 1% at about 100 chromosome-internal positions, leaving stretches of up to 100 kb free of detectable J (30). Key to this extreme distribution are the two enzymes, JBP1 and 2, that catalyze the initial step in J synthesis, the hydroxylation of selected T-residues in DNA.
Studies on JBP1/2-nulls in trypanosomatids have suggested that JBP1 is the main player in J synthesis, as the JBP1-null loses 95% of all J in T. brucei (23,33) and T. cruzi (24) and is lethal in Leishmania (25,27), presumably because extreme loss of J is lethal (30). Early work by Cross et al. (23) suggested that JBP1, but not JBP2, is able to maintain J wherever it is ectopically introduced in the genome. When T. brucei was grown in medium containing hydroxymethyluridine, this was randomly incorporated into the DNA and converted into J. This 10-fold excess of J was only sluggishly lost in wild-type cells, but rapidly diluted out by in growing in JBP1-null cells.
A novel sequence requirement for J maintenance
This T(N)12A motif explains the unusual staggered distribution of J in telomeric repeats (Figure 5B), in which the second T in the (GGGTTA)n strand is replaced by J, but never the first T (32). Our SMRT sequencing has identified a high frequency of J doublets with a J at position zero and another J in the complementary DNA strand at position +13. It also explains why the repeat variant (GGGTTT)10 is not picking up any J in Leishmania. We propose that JBP1 is responsible for this +13 pattern and that the ability of JBP1 to maintain J in some ectopic positions in T. brucei (23) is due to the high incidental fraction of A at +13 on the same strand, about 0.25 in random DNA. The T(N12)A spacing is not very stringent, as we also find apparent doublets in which the spacing between the two Js is 11 or 13 bp.
A simple interpretation of this maintenance function of JBP1 comes from the studies on the structure–function relationships of JBP1 by the group of Perrakis (19,44). They found that when JBP1 binds to J-DNA it undergoes a rapid conformational change. Heidebrecht et al. (44) proposed that this change positions the hydroxylase domain of JBP1 on the DNA. This would allow JBP1 to hydroxylate the T at +13, and, possibly less efficiently, a T at +12 or +14. The estimated dimensions of the protein are compatible with a 11- to 13-bp spacing between the J-binding domain and the hydroxylase active center (A. Perrakis, personal communications). Modification by JBP1 appears to be directional, as we do not find J doublets with a −13 spacing or J triplets. This is not surprising as the glucose moiety of J is not free, but held in an edge-on position by hydrogen bonding to the non-bridging phosphoryl oxygen of the nucleotide at position J-1 (20). This could result in directional binding of JBP1. The site-specific interactions of JBP1 with J-DNA were extensively probed by Sabatini et al. (17,18), but unfortunately without oligonucleotides extending to the +13 position.
De novo insertion of J by JBP1?
Two observations indicate that JBP1 is usually unable to insert J de novo: we show here that none of the plasmids that pick up J in wild-type Leishmania are able to do so in the JBP2-null mutant, which contains fully active JBP1; and in earlier work with T. brucei Kieft et al. (26) showed that a newly regenerated telomere does not pick up J in the JBP2-null either. Both observations suggest that JBP2 is required for the de novo incorporation of J into DNA and that JBP1 cannot do this by itself under physiological conditions. More recently, however, the Sabatini lab has published results that contradict this simple picture. They generated a J-null mutant of T. brucei by knocking out both JBP1 and JBP2. This J-null is viable in T. brucei, in contrast to the J-nulls of Leishmania and T. cruzi. When JBP1 is reintroduced into the T. brucei J-null mutant, J is reintroduced in most positions where it normally resides in wild-type cells (29). Although this proves de novo synthesis of J by JBP1, we think that the conditions in which this occurs are unphysiological. Normally there is a large excess of J-residues in DNA over JBP1 molecules. We have estimated this ratio to be 30 in T. brucei (45). As JBP1 binds to J-DNA with high affinity, 10 000-fold higher than to T-DNA (19), there is virtually no free JBP1 in the nucleus under normal conditions. In contrast, when JBP1 is reintroduced in J-null T. brucei, there is no J-DNA to bind to and now the weak affinity for T-DNA may result in de novo insertion of J. The fact that the J ends up in locations where it normally resides, could be due to a weak preference for these sequences, to differential accessibility due to chromatin structure, but also to secondary effects, e.g. competition with transcription. Indeed, there is evidence that transcription can interfere with J maintenance: activation of a silent VSG gene ES, erases J (1,40). Ectopically introduced J is lost from DNA sites where it is not normally present (23) and this could be the result of competition with transcription since J is normally only present at locations where transcription is low (29). Indeed, the Leishmania JBP2-null loses more J at transcriptional stops (>85%) than at telomeres (70%). The loss of J at transcriptional stops is associated with massive read-through (30) and this could exacerbate J loss by interference with JBP1 action. The conclusion that the de novo synthesis of J by JBP1 in the J-null T. brucei mutant is a non-physiological reaction is in line with our results with a mutant of JBP1 in which the critical aspartate residue that is required for binding to J-DNA is replaced by an alanine. The mutant binds better to T-DNA than to J-DNA, but is unable to replace wild-type JBP1 even though the protein normally routes to the nucleus (19). This shows that JBP1 needs to bind to pre-existing J in DNA to insert J and generate doublets in an efficient fashion.
De novo insertion of J into DNA by JBP2
Our results also shed light on the sequences determining de novo insertion of J into DNA. This process requires JBP2 and it must involve a rather complex sequence, since J is only found in plasmids containing DNA sequences that have J in their normal chromosomal context. A 8-kb control plasmid did not pick up J. Nevertheless the ‘entry’ sequence for de novo J insertion cannot be very long, as an insert containing only 10 GGGTTA repeats suffices. As functional JBP1 is always present in our Leishmania cells, we do not know whether de novo synthesis results in J in both strands or only modifies one strand, followed by the addition of J to the other strand by JBP1.
As telomeric repeats are not present in the other ‘entry’ sites analyzed in plasmids, we have looked for sequences that have a similar G/C strand bias as the telomeric repeats. A potential landmark to direct J insertion are the G-rich sequences that we find near all ‘entry’ sequences. G-rich sequences are known to form G-quadruplexes in single-stranded DNA, but there is considerable evidence that such structures can also temporarily form in the G-rich strand of duplex DNA during DNA replication, during transcription of the complementary strand, and even under exceptional circumstances in duplex DNA (46) (reviewed in Patel et al. (47) and Bochman et al. (48)). Although G-quadruplexes are most efficiently formed by runs of three G-residues, the requirements are not stringent and many variants on the basic theme present in telomeric repeats have been described (47). Indeed, the G-rich flanking sequences of paired J sites (Figure 7C) have characteristic runs of 3–4 consecutive G-residues suggesting they may form G-quadruplexes. In addition (potential) G-quadruplexes are associated with J insertion sites in the 25.2L, 25.2S, 121.1 and (GGGTTA)10 inserts (as well as the α-tubulin intergenic gene segments) used in this study, but not the J-less 28.2 insert (see Figure 4). It is interesting to note that J is essential for transcription termination in Leishmania (30,31), but G-quadruplexes without J have been shown in other systems to have a (relatively weak) transcription terminating effect (47–49). However, the mere presence of G-quadruplexes in the ‘entry’ sequence does not suffice as the signal for J insertion, since there are also potential G-quadruplexes in α-tubulin intergenic gene segments in all plasmids, including the 28.2 and (GGGTTT)10 plasmids which do not pick up J. Presumably the sequence structure of the loops between the Gs in the quadruplex is important for a sequence to act as ‘entry’. More extensive mutagenesis studies are required to define these.
It is likely that local chromatin structure is involved in the de novo J synthesis. JBP2 contains a SWI/SNF domain and point mutations in this domain can abolish the ability of JBP2 to hydroxylate DNA (22). SWI/SNF proteins can move nucleosomes, but presumably might also interact with other chromatin elements, such as proteins bound to G-quadruplexes. In T. brucei the location of J coincides to a large extent with the presence of specialized nucleosomes containing the histone variants H3V and H4V (29). What determines the location of these specialized nucleosomes is not known. There is no indication, however that the histone variants co-determine J location, as the H3V-null mutants do not seem to have a grossly altered distribution of J in T. brucei (29) or L. tarentolae (P.A. Genest, S. Jan and P. Borst, unpublished). Both J insertion and H3V/H4V location could be determined, however, by long-range features of DNA that are not obvious. It remains probable that JBP2 needs help to identify these features. JBP2 does not detectably bind to DNA and its hydroxylase domain looks like a standard domain of a member of the TET/JBP family, not equipped with the ability to recognize a complex DNA structure/sequence. We have started a search for proteins that interact with JBP2 and that could provide help in sequence recognition.
Spreading of J
The ‘spreading’ of J from the primary ‘entry’ sequence to neighboring sequences was first observed by Bernards et al. (1) in T. brucei by analyzing blocked restriction enzyme recognition sites later shown to contain J (40). When a telomeric VSG ES was switched off, the silenced site accumulated J in a remarkable fashion: the modification was highly selective; in the ES most intensely studied, the 221 site, only PstI and PvuII sites became blocked. Modification at each site was partial and decreased with increasing distance from the telomeric repeats, suggesting that the modifying enzyme spread from the telomeric repeats into the adjacent DNA. Most remarkably, the degree of modification at each site increased with the length of the adjacent telomere, which is highly variable in T. brucei due to the steady growth and an occasional major contraction of the telomeric repeat region (50). This suggested that larger stretches of telomeric repeats would collect more modifying enzyme resulting in more overflow of the modifying enzyme into adjacent DNA (1).
Our present results with Leishmania confirm this speculative interpretation of Bernards et al. (1) and put it on a firm factual basis. We prove that spreading exists: when ‘entry’ sequences are introduced in a plasmid in Leishmania, J is not only inserted into the ‘entry’ sequence itself, but also in the adjacent sequences (Figure 4), that are unable to act as ‘entry’ sequence by themselves. J even appears in the vector sequences. There is a gradient in the degree of modification, the highest degree of modification being observed closest to the ‘entry’ sequence. Since we assume that JBP2 determines de novo J synthesis, we hypothesize that spreading is also primarily catalyzed by JBP2, possibly followed by insertion of a second J by JBP1 at +13. This remains to be verified, however. The degree of spreading seems to depend on the ability of the ‘entry’ sequence to accumulate sufficient modifying enzyme, as already suggested by results of Bernards et al. (1) on sub-telomeric spreading in T. brucei. Whereas we see extensive spreading from the 400-bp 25.2S segment, little spreading appears to occur from the 60-bp telomeric segment (Figures 4 and 6). Obviously, more detailed experiments are required to precisely define the relation between the number of modified sites in the entry sequence and the degree of spreading. The sequence specificity of spreading is exquisite, as illustrated in Figure 6. Remarkably, the sites modified during spreading are not dependent on the exact distance from the ‘entry’ sequence, as we find J at similar positions in the α-tubulin intergenic gene sequences adjacent to very different ‘entry’ sites.
As spreading leads to discontinuous J insertion and is completely dependent on the presence of an ‘entry’ site, it may involve association of DNA loops with the ‘entry’ sites. This association might be promoted by G-quadruplexes, as we also find the potential to form G-quadruplexes in the α-tubulin intergenic sequences that are modified, but not in the intervening neo sequence, which is not modified (Figure 4 and Supplementary Figure S3). We cannot yet exclude, however, that JBP tracks along the DNA and stops at specific sequences for T hydroxylation.
How Ts are identified by the J insertion machinery during spreading is unclear. We only find a weak consensus sequence, which appears insufficiently distinctive to explain the high specificity of the spreading sites containing J. We have also looked at the sites modified by spreading in T. brucei identified by Bernards et al. (1) and in later papers, summarized by Borst and Sabatini (9). No consensus sequence was found. It is possible that chromatin structure, e.g. nucleosome position, restricts the potential sites that can be modified. Competition between transcription and J insertion could also play a role in the distribution of J in the plasmids and this remains to be studied.
SMRT sequencing
SMRT sequencing was indispensable for identifying the exact locations of J in DNA. Immunoprecipitation of J-containing DNA fragments lacks resolution and the recently developed liquid chromatography–mass spectrometry/mass spectrometry method to detect J (6) is also unsuitable for localization. Attempts to develop (bio)chemical procedures targeting J have failed thus far. An obvious approach is to take off the glucose moiety of J, as the hydroxymethylU remaining after glucose removal, could easily be localized using the DNA glycosylase single-strand-selective monofunctional uracil-DNA glycosylase (SMUG) (10). However, none of a range of glycosidases tested could remove the glucose from J in DNA (unpublished results). This could be due to the rigid way in which the glucose is fixed on the DNA by hydrogen bonding (20).
Further improvements in J detection by SMRT sequencing are feasible. We only had access to three J-containing oligonucleotides. With many more oligonucleotides the sequence dependence of the J signal and the possible interference by closely spaced J-residues could be more precisely defined. The most recent version of SMRT sequencing is good enough, however, for roughly sequencing genomes without the need for plasmid inserts. We are therefore resequencing the entire Leishmania genome by SMRT sequencing. This will not only provide an overview of the location of J in the genome, but also give a more detailed picture of the loss of J from the genome when J synthesis is inhibited.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank Robert Sabatini and Anastassis Perrakis for critical reading of the manuscript and useful discussions.
FUNDING
National Institute of Allergy and Infectious Diseases, PHS [R01 AI103858 to P.J.M.]; Netherlands Cancer Institute [to P.B., J.K., T.C., S.T., K.L. and M.B.]. Funding for open access charge: The Netherlands Cancer Institute using its core funding supplied by the Dutch Cancer Society and the Ministry of Health.
Conflict of interest statement. J. Korlach, T. Clark, S. Turner, K. Luong and M. Boitiano are full-time employees at Pacific Biosciences, a company commercializing single-molecule sequencing technologies.
REFERENCES
- 1.Bernards A., De Lange T., Michels P.A., Lui A.Y., Huisman M.J., Borst P. Two models of activation of a single surface antigen gene of Trypanosoma brucei. Cell. 1984;36:163–170. doi: 10.1016/0092-8674(84)90085-0. [DOI] [PubMed] [Google Scholar]
- 2.Pays E., Delauw M.F., Laurent M., Steinert M. Possible DNA modification in GC dinucleotides of Trypanosoma brucei telomeric sequences; relationship with antigen gene transcription. Nucleic Acids Res. 1984;12:5235–5247. doi: 10.1093/nar/12.13.5235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gommers-Ampt J.H., Van Leeuwen F., De Beer A.L., Vliegenthart J.F., Dizdaroglu M., Kowalak J.A., Crain P.F., Borst P. β-D-glucosyl-hydroxymethyluracil: a novel modified base present in the DNA of the parasitic protozoan T. brucei. Cell. 1993;75:1129–1136. doi: 10.1016/0092-8674(93)90322-h. [DOI] [PubMed] [Google Scholar]
- 4.Van Leeuwen F., Taylor M.C., Mondragon A., Moreau H., Gobson W., Kieft R., Borst P. β-D-glucosyl-hydroxymethyluracil is a conserved DNA modification in kinetoplastid protozoans and is abundant in their telomeres. Proc. Natl. Acad. Sci. USA. 1998;95:2366–2371. doi: 10.1073/pnas.95.5.2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dooijes D., Chaves I., Kieft R., Dirks-Mulder A., Martin W., Borst P. Base J origanally found in Kinetoplastida is also a minor constituent of nuclear DNA of Euglena gracilis. Nucleic Acids Res. 2000;28:3017–3021. doi: 10.1093/nar/28.16.3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu S., Ji D., Sabatini R., Wang Y. Quantitative mass spectrometry-based analysis of β-D-glucosyl-5-hydroxymethyluracil in genomic DNA of Trypanosoma brucei. J. Am. Soc. Mass. Spectrom. 2014;25:1763–1770. doi: 10.1007/s13361-014-0960-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Leeuwen F., Kieft R., Cross M., Borst P. Tandemly repeated DNA is a target for the partial replacement of thymine by β-D-glucosyl-hydoxymethyluracil in Trypanosoma brucei. Mol. Biochem. Parasitol. 2000;109:133–145. doi: 10.1016/s0166-6851(00)00247-4. [DOI] [PubMed] [Google Scholar]
- 8.Van Leeuwen F., Kieft R., Cross M., Borst P. Biosynthesis and function of the modified DNA base β-D-glucosyl-hydroxymethyluracil in Trypanosoma brucei. Mol. Cell. Biol. 1998;18:5643–5651. doi: 10.1128/mcb.18.10.5643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Borst P., Sabatini R. Base J: discovery, biosynthesis, and possible functions. Annu. Rev. Microbiol. 2008;62:235–251. doi: 10.1146/annurev.micro.62.081307.162750. [DOI] [PubMed] [Google Scholar]
- 10.Ulbert S., Cross M., Boorstein R., Teebor G., Borst P. Expression of the human DNA glycosylase hSMUG1 in Trypanosoma brucei causes DNA damage and interferes with J biosynthesis. Nucleic Acids Res. 2002;30:3919–3926. doi: 10.1093/nar/gkf533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iyer L.M., Zhang D., Maxwell Burroughs A., Aravind L. Computational identification of novel biochemical systems involved in oxidation, glycosylation and other complex modifications of bases in DNA. Nucleic Acids Res. 2013;41:7635–7655. doi: 10.1093/nar/gkt573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bullard W., Lopes da Rosa-Spiegler J., Liu S., Wang Y., Sabatini R. Identification of the glucosyltransferase that converts hydroxymethyluracil to base J in the trypanosomatid genome. J. Biol. Chem. 2014;289:20273–20282. doi: 10.1074/jbc.M114.579821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sekar A., Merritt C., Baugh L., Stuart K., Myler P.J. Tb927.10.6900 encodes the glucosyltransferase involved in synthesis of base J in Trypanosoma brucei. Mol. Biochem. Parasitol. 2014;196:9–11. doi: 10.1016/j.molbiopara.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu Z., Genest P.A., Ter Riet B., Sweeney K., DiPaolo C., Kieft R., Christodoulou E., Perrakis A., Simmons J., Hausinger R., et al. The protein that binds to DNA base J in trypanosomatids has features of a thymidine hydroxylase. Nucleic Acids Res. 2007;35:2107–2115. doi: 10.1093/nar/gkm049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tahiliani M., Koh K., Shen Y., Pastor W., Bandukwala H., Brudno Y., Agarwal S., Iyer L., Liu D., Aravind L., et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cross M., Kieft R., Sabatini R., Wilm M., De Kort M., Van der Marel G.A., Van Boom J.H., Van Leeuwen F., Borst P. The modified base J is the target for a novel DNA-binding protein in kinetoplastid protozoans. EMBO J. 1999;18:6573–6581. doi: 10.1093/emboj/18.22.6573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sabatini R., Meeuwenoord N., van Boom J., Borst P. Recognition of base J in duplex DNA by J-binding protein. J. Biol. Chem. 2002;277:958–966. doi: 10.1074/jbc.M109000200. [DOI] [PubMed] [Google Scholar]
- 18.Sabatini R., Meeuwenoord N., van Boom J., Borst P. Site-specific interactions of JBP with base and sugar moieties in duplex J-DNA. Evidence for both major and minor groove contacts. J. Biol. Chem. 2002;277:28150–28156. doi: 10.1074/jbc.M201487200. [DOI] [PubMed] [Google Scholar]
- 19.Heidebrecht T., Christodoulou E., Chalmers M., Jan S., Ter Riet B., Grover R., Joosten R., Littler D., van Luenen H., Griffin P., et al. The structural basis for recognition of base J containing DNA by a novel DNA binding domain in JBP1. Nucleic Acids Res. 2011;39:5715–5728. doi: 10.1093/nar/gkr125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grover R., Pond S., Cui Q., Subramaniam P., Case D., Millar D., Wentworth P. O-Glycoside orientation is an essential aspect of base J recognition by the kinetoplastid DNA-binding protein JBP1. Angew. Chem. Int. Ed. 2007;46:2839–2843. doi: 10.1002/anie.200604635. [DOI] [PubMed] [Google Scholar]
- 21.Cliffe L., Hirsch G., Wang J., Ekanayake D., Bullard W., Hu M., Wang Y., Sabatini R. JBP1 and JBP2 proteins are Fe2+/2-oxoglutarate-dependent dioxygenases regulating hydroxylation of thymidine residues in trypanosome DNA. J. Biol. Chem. 2012;287:19886–19895. doi: 10.1074/jbc.M112.341974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DiPaolo C., Kieft R., Cross M., Sabatini R. Regulation of trypanosome DNA glycosylation by a SWI2/SNF2-like protein. Mol. Cell. 2005;17:441–451. doi: 10.1016/j.molcel.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 23.Cross M., Kieft R., Sabatini R., Dirks-Mulder A., Chaves I., Borst P. J-binding protein increases the level and retention of the unusual base J in trypanosome DNA. Mol. Microbiol. 2002;46:37–47. doi: 10.1046/j.1365-2958.2002.03144.x. [DOI] [PubMed] [Google Scholar]
- 24.Ekanayake D., Minning T., Weatherly B., Gunasekera K., Nilsson D., Tarleton R., Ochsenreiter T., Sabatini R. Epigenetic regulation of transcription and virulence in Trypanosoma cruzi by O-linked thymine glucosylation of DNA. Mol. Cell. Biol. 2011;31:1690–1700. doi: 10.1128/MCB.01277-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Genest P.A., Ter Riet B., Dumas C., Papadopoulou B., Van Luenen H.G.A.M., Borst P. Formation of linear inverted repeat amplicons following targeting of an essential gene in Leishmania. Nucleic Acids Res. 2005;33:1699–1709. doi: 10.1093/nar/gki304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kieft R., Brand V., Ekanayake D., Sweeney K., DiPaolo C., Reznikoff W., Sabatini R. JBP2, a SWI2/SNF2-like protein, regulates de novo telomeric DNA glycosylation in bloodstream form Trypanosoma brucei. Mol. Biochem. Parasitol. 2007;156:24–31. doi: 10.1016/j.molbiopara.2007.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vainio S., Genest P.A., Ter Riet B., Van Luenen H.G.A.M., Borst P. Evidence that J-binding protein 2 is a thymidine hydroxylase catalyzing the first step in the biosynthesis of DNA base J. Mol. Biochem. Parasitol. 2009;164:157–161. doi: 10.1016/j.molbiopara.2008.12.001. [DOI] [PubMed] [Google Scholar]
- 28.Genest P.A., Ter Riet B., Cijsouw T., van Luenen H., Borst P. Telomeric localization of the modified DNA base J in the genome of the protozoan parasite Leishmania. Nucleic Acids Res. 2007;35:2116–2124. doi: 10.1093/nar/gkm050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cliffe L., Siegel T.N., Marshall M., Cross G., Sabatini R. Two thymidine hydroxylases differentially regulate the formation of glucosylated DNA at regions flanking polymerase II polycistronic transcription units throughout the genome of Trypanosoma brucei. Nucleic Acids Res. 2010;38:3923–3935. doi: 10.1093/nar/gkq146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van Luenen H.G.A.M., Farris C., Jan S., Genest P.A., Tripathi P., Velds A., Kerkhoven R.M., Nieuwland M., Haydock A., Ramasamy G., et al. Glucosylated hydroxymethyluracil (DNA base J) prevents transcriptional read-through in Leishmania. Cell. 2012;150:909–921. doi: 10.1016/j.cell.2012.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reynolds D., Cliffe L., Förstner K.U., Hon C.-C., Siegel T.N., Sabatini R. Regulation of transcription termination by glucosylated hydroxymethyluracil, base J, in Leishmania major and Trypanosoma brucei. Nucleic Acids Res. 2014;42:9717–9729. doi: 10.1093/nar/gku714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Van Leeuwen F., Wijsman E.R., Kuyl-Yeheskiely E., Van der Marel G.A., Van Boom J.H., Borst P. The telomeric GGGTTA repeats of Trypanosoma brucei contain the hypermodified base J in both strands. Nucleic Acids Res. 1996;24:2476–2482. doi: 10.1093/nar/24.13.2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cliffe L., Kieft R., Southern T., Birkeland S., Marshall M., Sweeney K., Sabatini R. JBP1 and JBP2 are two distinct thymidine hydroxylases involved in J biosynthesis in genomic DNA of African trypanosomes. Nucleic Acids Res. 2009;37:1452–1462. doi: 10.1093/nar/gkn1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eid J., Fehr A., Gray J., Luong K., Lyle J., Otto G., Peluso P., Rank D., Baybayan P., Bettman B., et al. Real-time DNA sequencing from single polymerase molecules. Science. 2009;323:133–138. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- 35.Flusberg B., Webster D., Lee J., Travers K., Olivares E., Clark T., Korlach J., Turner S. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat. Methods. 2010;7:461–465. doi: 10.1038/nmeth.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Song C., Clark T., Lu X., Kislyuk A., Dai Q., Turner S., He C., Korlach J. Sensitive and specific single-molecule sequencing of 5-hydroxymethylcytosine. Nat. Methods. 2012;9:75–77. doi: 10.1038/nmeth.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clark T., Spittle K., Turner S., Korlach J. Direct detection and sequencing of damaged DNA bases. Genome Integrity. 2011;2:10. doi: 10.1186/2041-9414-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brun R., Schonenberger M. Cultivation and in vitro cloning or procyclic culture forms of Trypanosoma brucei in a semi-defined medium. Acta Trop. 1979;36:289–292. [PubMed] [Google Scholar]
- 39.Papadopoulou B., Roy G., Ouelette M. A novel antifolate resistance gene on the amplified H circle of Leishmania. EMBO J. 1992;11:3601–3608. doi: 10.1002/j.1460-2075.1992.tb05444.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Van Leeuwen F., Wijsman E.R., Kieft R., Van der Marel G.A., Van Boom J.H., Borst P. Localization of the modified base J in telomeric VSG gene expression sites of Trypanosoma brucei. Genes Dev. 1997;11:3232–3241. doi: 10.1101/gad.11.23.3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clarke J. Continuous base identification for single-molecule nanopore DNA sequencing. Nat. Nanotechnol. 2009;4:265–270. doi: 10.1038/nnano.2009.12. [DOI] [PubMed] [Google Scholar]
- 42.Lundquist P.M. Parallel confocal detection of single molecules in real time. Opt. Lett. 2008;33:1026–1028. doi: 10.1364/ol.33.001026. [DOI] [PubMed] [Google Scholar]
- 43.Crooks G.E., Hon G., Chandonia J.-M., Brenner S.E. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Heidebrecht T., Fish A., von Castelmur E., Johnson K.A., Zaccai G., Borst P., Perrakis A. Binding of the J-binding protein to DNA containing glucosylated hmU (base J) or 5-hmC: evidence for a rapid conformational change upon DNA binding. J. Am. Chem. Soc. 2012;134:13357–13365. doi: 10.1021/ja303423t. [DOI] [PubMed] [Google Scholar]
- 45.Toaldo C., Kieft R., Dirks-Mulder A., Sabatini R., van Luenen H., Borst P. A minor fraction of base J in kinetoplastid nuclear DNA is bound by the J-binding protein 1. Mol. Biochem. Parasitol. 2005;143:111–115. doi: 10.1016/j.molbiopara.2005.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Henderson A., Wu Y., Huang Y.C., Chavez E.A., Platt J., Johnson F.B., Brosh R.M., Sen D., Lansdorp P.M. Detection of G-quadruplex DNA in mammalian cells. Nucleic Acids Res. 2014;42:860–869. doi: 10.1093/nar/gkt957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Patel D., Phan A., Kuryavyi V. Human telomere, oncogenic promoter and 5ΓǦ-UTR G-quadruplexes: diverse higher order DNA and RNA targets for cancer therapeutics. Nucleic Acids Res. 2007;35:7429–7455. doi: 10.1093/nar/gkm711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bochman M., Paeschke K., Zakian V. DNA secondary structures: stability and function of G-quadruplex structures. Nat. Rev. Genet. 2012;13:770–780. doi: 10.1038/nrg3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Taylor J.P. Neurodegenerative diseases: G-quadruplex poses quadruple threat. Nature. 2013;507:175–177. doi: 10.1038/nature13067. [DOI] [PubMed] [Google Scholar]
- 50.Bernards A., Michels P.A., Lincke C.R., Borst P. Growth of chromosome ends in multiplying trypanosomes. Nature. 1983;303:592–597. doi: 10.1038/303592a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.