

Abstract
A series of carbocyclic analogues of naturally-occurring marine sphingolipid pachastrissamine were prepared and biologically evaluated. The analogues were efficiently synthesized via a tandem enyne/diene-ene metathesis reaction as a key step. We found that the analogue 4b exhibited comparable cytotoxicity and more potent inhibitory activity against sphingosine kinases, compared to pachastrissamine. Molecular modeling studies were conducted to provide more detailed insight into the binding mode of 4b in sphingosine kinase. In our docking model, pachastrissamine and 4b were able to effectively bind to the binding pocket of sphingosine kinase 1 as co-crystalized sphingosine. However, 4b showed a hydrophobic interaction with Phe192, which suggests that it contributes to its increased inhibitory activity against sphingosine kinase 1.
Keywords: pachastrissamine, jaspine B, carbocyclic analogue, cytotoxicity, sphingosine kinase inhibitor, molecular modeling
1. Introduction
Pachastrissamine (jaspine B, 1; Figure 1) is a natural anhydrous derivative of phytosphingosine 2, possessing a tetrahydrofuran core with three contiguous stereogenic centers. It was first isolated by Higa et al. from marine sponge Pachastrissa sp. in 2002 [1] and subsequently isolated from the Vanuatuan marine sponge Jaspis sp. by Debitus et al. in 2003 [2]. This marine natural product showed significant cytotoxicity against various cancer cell lines, such as P388, MEL28, A549, HT29 and HeLa [1,3]. It was found to have an inhibitory activity against sphingomyelin synthase, which, in turn, triggers apoptosis in tumor cells via a caspase-dependent pathway [4]. It was also reported that pachastrissamine and its stereoisomers inhibit sphingosine kinases (SphKs) and atypical protein kinase C [5]. Because of its intriguing biological activity, it has been an interesting target for synthetic chemists, and various synthetic routes to pachastrissamine have been reported [6,7,8,9,10,11,12,13,14,15,16]. However, the structure-activity relationship (SAR) of pachastrissamine remains relatively unreported. Génisson et al. described analogues with a modified aliphatic chain, which exhibited cytotoxicity comparable to or lower than pachastrissamine on two distinct cancer cell lines (B16 and A375 melanoma cell lines) [17,18]. Delgado et al. reported the stereoisomers of pachastrissamine as being 10- to 20-times less potent than the natural one [19]. Recently, Liu et al. reported pachastrissamine analogues containing a 1,2,3-triazole ring in the alkyl chain [20]. One analogue showed better increased cytotoxicity than the natural compound. These results suggest that the configuration and aliphatic chain of pachastrissamine would be essential to retain biological activity. The contribution of the tetrahydrofuran ring to its biological profile has been explored. The aza-analogues of pachastrissamine 3a were reported by Génisson et al. [21], and the sulfur and selenium analogues 3b–c were previously reported by our group [22]. They exhibited comparable potency to natural pachastrissamine, indicating that the ring oxygen atom of pachastrissamine could be replaced with bioisosteres.
Figure 1.

Chemical structures of Compounds 1–4.
In this regard, we designed carbocyclic analogues, replacing the ethereal oxygen (–O–) with a methylene group (–CH2–) as a divalent bioisostere [23]. The advantages of carbocyclic analogues of natural product have already been demonstrated by several successful examples, such as carbasugars and carbocyclic nucleosides [24,25]. As seen in these successful precedents, we expected that the carbocyclic analogue would show enhanced chemical and metabolic stability compared to the parent natural product. In addition, from this study, we would be able to decipher the SAR involved in the role of ethereal oxygen, which is valuable information in the development of new anti-cancer therapeutic agents. Herein, we report the synthesis and biological evaluation of a series of carbocyclic pachastrissamine analogues 4 (Figure 1) in which the alkyl-chain lengths have been varied.
2. Results and Discussion
2.1. Chemistry
2.1.1. Retrosynthetic Analysis
Our retrosynthetic plan for the synthesis of 4 is shown in Scheme 1. The designed analogues 4 would be accessed from diene 5 using catalytic hydrogenation and hydrolysis. We expected catalytic hydrogenation of 5 would occur from the convex face of the bicyclic system to afford the desired stereochemistry [6]. The requisite diene 5 would be derived from the ring-closing metathesis (RCM) of enyne 6 and subsequent cross-metathesis (CM) between the resulting diene and the appropriate olefin in a tandem fashion [26,27,28,29]. Substrate 6 would be derived from a known amide 7, which, in turn, can be easily prepared from commercially available (S)-allylglycine.
Scheme 1.
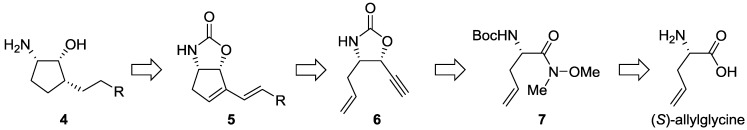
Retrosynthetic plan.
2.1.2. Synthesis
Our synthesis, as shown in Scheme 2, commenced with the preparation of a known Weinreb amide 7 from commercially available (S)-allylglycine in two steps using a previously reported procedure [30]. Treatment of amide 7 with lithium acetylide in THF yielded ynone 8 in a 71% yield. The stereoselective reduction of ynone 8 was explored with various hydride reagents. After several trials, it was found that the treatment of 8 with sodium borohydride and cerium chloride at low temperature gave an inseparable diastereomeric mixture of alcohol 9 with the best selectivity (diastereomeric ratio = 6:1) in 87% yield. The obtained alcohol 9 was treated with lithium hexamethyldisilazide in hot toluene to provide oxazolidinone 10 in a high yield (90%). At this stage, both diastereomers (10a and 10b) were separated via column chromatography, and the relative configuration of the diastereomers was assigned by a 2D-NOESY experiment (Figures S7 and S10 in the Supplementary Materials). The triisopropylsilyl group of major diastereomer 10a was easily removed with tetrabutylammonium fluoride to give the terminal acetylene 6 in a 95% yield.
Scheme 2.

Synthesis of enyne 6. TIPS: triisopropylsilyl; LiHMDS: lithium hexamethyldisilazide; TBAF: tetrabutylammonium fluoride.
With the terminal enyne 6 in hand, the tandem enyne/diene-ene metathesis reaction was carried out (Scheme 3). The reaction of 6 with the appropriate alkenes in the presence of second-generation Grubbs catalyst (3 mol%) afforded the desired diene 5a–c with exclusive (E)-geometry. The catalytic hydrogenation of diene 5a–c was accomplished to give cyclopentane 11a–c as the only stereoisomers detected by 1H NMR. Finally, hydrolysis of the oxazolidinone with KOH in refluxing EtOH afforded the desired carbocyclic analogues, 4a–c.
Scheme 3.
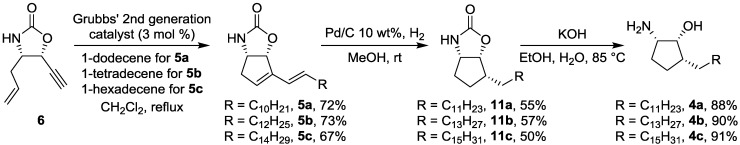
Synthesis of carbocyclic analogues, 4a–c.
2.2. Biological Evaluation
2.2.1. Cell Viability
The cytotoxic activity of analogues 4a–c was examined in various cancer cell lines (Table 1) by the sulforhodamine B (SRB) assay [31]. For a direct comparison, natural pachastrissamine (1) was employed as a positive control. It was found that the cytotoxic activities of carbocyclic analogues 4 were influenced by the length of the alkyl chain. The analogue, 4b, which has the same chain length as pachastrissamine, exhibited a comparable potency to the parent natural product, 1, against various cell lines. However, both the shorter and longer chain analogues, 4a and 4c, were less effective than pachastrissamine.
Table 1.
Cytotoxic activity of pachastrissamine (1) and its carbocyclic analogues, 4a–c.
| Compound | IC50 (μM) a | ||||
|---|---|---|---|---|---|
| HCT-116 b | SNU-638 c | MDA-MB-231 d | PC-3 e | Caki-1 f | |
| 1 | 1.0 | 1.2 | 0.7 | 0.7 | 1.7 |
| 4a | 9.7 | 12.8 | 6.1 | 7.6 | 12.1 |
| 4b | 1.0 | 1.7 | 1.8 | 1.0 | 3.2 |
| 4c | 3.2 | 4.4 | 4.1 | 3.1 | 6.6 |
a 50% inhibition concentration; b HCT-116, human colon cancer cell line; c SNU-638, human stomach cancer cell line; d MDA-MB-231, human breast cancer cell line; e PC-3, human prostate cancer cell line; f Caki-1, human renal cancer cell line.
2.2.2. Inhibitory Activity against Sphingosine Kinases
Because pachastrissamine was reported as an inhibitor of sphingosine kinases (SphKs), the inhibitory activity of analogues 4 against SphKs were examined using a sphingosine kinase inhibition assay (Table 2). There are two isoforms of mammalian SphKs (SphK1 and SphK2) catalyzing the phosphorylation of sphingosine to sphingosine 1-phosphate (S1P). S1P regulates diverse cellular processes, including cell growth, survival and differentiation [32]. In our experiments, N,N-dimethylsphingosine (DMS), as well as pachastrissamine (1) were used as a positive control [33]. It was revealed that the inhibitory activity of 4 against SphKs was influenced by the length of the attached chain. The analogue 4b showed a similar inhibitory activity to DMS, which, in turn, was more effective than 1. However, the other analogues, 4a and 4c, were found to be less effective at the inhibition of SphKs. These results suggest that the ring oxygen atom of pachastrissamine is dispensable, but the appropriate chain length is apparently required to retain its biological properties.
Table 2.
Inhibitory activity of pachastrissamine (1) and its carbocyclic analogues, 4a–c, against sphingosine kinases.
| Compound | IC50 (μM) a | |
|---|---|---|
| SphK1 | SphK2 | |
| 1 | 12.0 | 41.8 |
| 4a | 58.2 | >100 |
| 4b | 7.5 | 20.1 |
| 4c | 41.3 | >100 |
| DMS b | 6.6 | 19.9 |
a 50% inhibition concentration; b N,N-dimethylsphingosine.
2.3. Molecular Modeling
To understand the molecular interaction between 4b and the sphingosine kinase, a molecular modeling study was performed using the crystal structure of human SphK1 in complex with sphingosine (PDB Code 3VZB) [34]. As shown in Figure 2, pachastrissamine (1) and its carbocyclic analogue 4b are able to effectively bind to the binding pocket of SphK1 in virtually the same pose. They occupy nearly the same position as sphingosine. In our docking model, the alkyl chain of 4b is buried in the hydrophobic J-shaped tunnel constituted of hydrophobic residues Leu268, Ala274, His311, Phe173, Phe303, Val177 and Phe192. The hydroxyl group forms a hydrogen bond with Asp178 and a water-mediated hydrogen bond with Ser168, in the same manner as the 3-hydroxyl group of sphingosine. The amino group makes a hydrogen bond interaction with Ser168. In the hydrophilic recognition site, pachastrissamine (1) and its analogue 4b formed the same set of hydrogen bonds. However, carbocyclic analogue 4b exhibits an additional interaction with the hydrophobic residue, Phe192 (Figure 2B). This pi-alkyl interaction seems to contribute to the increased activity of 4b toward SphK1 compared to 1 [35].
Figure 2.

Docking model of the carbocyclic analogue 4b and pachastrissamine (1). (A) The overlay of the proposed binding model of carbocyclic analogue 4b (blue) and X-ray complex sphingosine (gray). A water molecule is shown as a red sphere. The direct H-bonds and water-mediated H-bonds are shown as green and blue dashed lines, respectively. The ligand binding surface of the binding pocket was colored according to hydrophobicity (brown: hydrophobic, blue: hydrophilic); (B) The overlay of the proposed binding model of 4b (blue) and 1 (orange). Hydrophobic interaction with Phe192 is shown as a pink dashed line.
3. Experimental Section
3.1. Chemistry
3.1.1. General
All chemicals were reagent grade and used as purchased. All reactions were performed under an inert atmosphere of dry nitrogen using distilled dry solvents. Reactions were monitored through thin layer chromatography (TLC) analysis using silica gel 60 F-254 thin layer plates. Compounds were visualized on the TLC plates under UV light and by spraying with either KMnO4 or anisaldehyde staining solutions. Flash column chromatography was conducted on silica gel 60 (230–400 mesh). Melting points were measured using a Buchi B-540 melting point apparatus without correction. 1H NMR (400, 500 or 600 MHz) and 13C NMR (100, 125 or 150 MHz) spectra were recorded in δ units relative to the deuterated solvent. The IR spectra were measured on a Fourier transform infrared spectrometer. High-resolution mass spectra (HRMS) were recorded using FAB.
3.1.2. tert-Butyl (S)-(5-oxo-7-(triisopropylsilyl)hept-1-en-6-yn-4-yl)carbamate (8)
To a solution of (triisopropylsilyl)acetylene (17.4 mL, 77.4 mmol) in THF (80 mL) was added n-BuLi (48.4 mL, 77.4 mmol, 1.6 M solution in hexane) at −78 °C under nitrogen. After the reaction mixture was stirred for 30 min at −78 °C, known amide 7 (10 g, 38.7 mmol) in dry THF (20 mL) was added at 0 °C. The reaction mixture was stirred for 1.5 h at 0 °C. The reaction was quenched with a saturated NH4Cl aqueous solution and extracted with EtOAc. The organic layer was washed twice with brine, dried over MgSO4, filtered and concentrated. The crude product was purified using column chromatography on silica gel (hexane/EtOAc, 20:1), to afford the desired ynone 8 (10.4 g, 71%) as a yellow oil. +8.20 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.70–5.61 (m, 1H), 5.13 (d, J = 17.2 Hz, 1H), 5.12 (d, J = 9.2 Hz, 1H), 5.10 (brs, 1H), 4.48 (d, J = 5.7 Hz, 1H), 2.70–2.59 (m, 2H), 1.42 (s, 9H), 1.17–1.01 (m, 21H); 13C NMR (100 MHz, CDCl3) δ 185.4, 155.1, 131.7, 119.5, 102.2, 100.1, 80.0, 60.8, 36.0, 28.3 (3C), 18.5 (6C), 11.0 (3C); IR (CHCl3) νmax 3358, 2947, 2869, 2150, 1719, 1682, 1495, 1368, 1171 (cm−1); HRMS (FAB) calcd. for C21H38NO3Si ([M + H]+), 380.2621; found, 380.2614.
3.1.3. tert-Butyl ((4S)-5-hydroxy-7-(triisopropylsilyl)hept-1-en-6-yn-4-yl)carbamate (9)
To a solution of ynone 8 (10 g, 26.3 mmol) in EtOH (100 mL) was added anhydrous CeCl3 (13 g, 52.6 mmol) at room temperature under nitrogen. After the reaction mixture was stirred for 15 min, NaBH4 (2.0 g, 52.6 mmol) was added at −78 °C. The reaction mixture was stirred for 3 h at −78 °C. The reaction was quenched with a saturated NH4Cl aqueous solution and extracted with EtOAc. The organic layer was washed twice with brine, dried over MgSO4, filtered and concentrated. The crude product was purified using column chromatography on silica gel (hexane/EtOAc, 5:1), to afford the desired alcohol 9 (8.7 g, 87%) as a diastereomeric mixture (6:1) as a colorless oil. −18.3 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.82–5.72 (m, 1H), 5.08 (d, J = 15.2 Hz, 1H), 5.06 (d, J = 8.9 Hz, 1H), 4.74–4.72 (m, 0.85H), 4.45–4.44 (m, 1H), 3.82 (brs, 0.85H), 3.72 (brs, 0.15H), 3.38 (brs, 0.85H), 2.56–2.46 (m, 0.15H), 2.42–2.22 (m, 1.85H), 1.40 (s, 9H), 1.04 (s, 21H); 13C NMR (100 MHz, CDCl3) δ 156.5, 134.0, 118.0, 105.2, 87.6, 79.9, 66.1, 55.1, 35.7, 28.2 (3C), 18.5 (6C), 11.1 (3C); IR (CHCl3) νmax 3428, 2945, 2868, 2170, 1696, 1504, 1170 (cm−1); HRMS (FAB) calcd. for C21H40NO3Si ([M + H]+), 382.2777; found, 382.2770.
3.1.4. (4S)-4-Allyl-5-((triisopropylsilyl)ethynyl)oxazolidin-2-one (10)
To a solution of alcohol 9 (8 g, 21 mmol) in toluene (100 mL) was added lithium hexamethyldisilazide (46.2 mL, 46.2 mmol, 1 M solution in THF) at room temperature under nitrogen. After stirring for 1.5 h at 80 °C, the reaction mixture was cooled to 0 °C. The reaction was quenched with a saturated NH4Cl aqueous solution and extracted with EtOAc. The organic layer was washed twice with brine, dried over MgSO4, filtered and concentrated. The crude product was purified using column chromatography on silica gel (hexane/EtOAc, 1.5:1), to afford the desired oxazolidinone 10a (5.0 g, 77%) and 10b (850 mg, 13%).
(4S,5R)-4-Allyl-5-((triisopropylsilyl)ethynyl)oxazolidin-2-one (10a): a white solid; m.p. 91.2–93.3 °C; −4.12 (c 0.35, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.80–5.72 (m, 1H), 5.33–5.31 (d, J = 7.8 Hz, 1H), 5.20 (d, J = 10.4 Hz, 1H), 5.19 (d, J = 16.8 Hz, 1H), 5.17 (brs, 1H), 3.88 (td, J = 4.1 Hz, 8.6 Hz, 1H), 2.59–2.53 (m, 1H), 2.44–2.36 (m, 1H), 1.07 (s, 21H); 13C NMR (100 MHz, CDCl3) δ 159.6, 132.7, 119.8, 98.5, 93.5, 70.1, 54.1, 36.8, 18.5 (6C), 11.0 (3C); IR (CHCl3) νmax 3276, 2946, 2868, 1759, 1464, 1334, 1048, 758, 676 (cm−1); HRMS (FAB) calcd. for C17H30NO2Si ([M + H]+), 308.2046; found, 308.2039.
(4S,5S)-4-Allyl-5-((triisopropylsilyl)ethynyl)oxazolidin-2-one (10b): a yellow oil; −36.2 (c 1.4, CHCl3); 1H NMR (600 MHz, CDCl3) δ 6.12 (d, J = 9.2 Hz, 1H), 5.77–5.70 (m, 1H), 5.19 (d, J = 11.5 Hz, 1H), 5.18 (d, J = 16.1 Hz, 1H), 4.80 (d, J = 6.4 Hz, 1H), 3.87 (q, J = 6.4 Hz, 1H), 2.40 (q, J = 6.7 Hz, 1H), 2.33 (q, J = 7.1 Hz, 1H), 1.05 (s, 21H); 13C NMR (150 MHz, CDCl3) δ 158.2, 131.5, 119.8, 101.7, 90.8, 70.9, 59.2, 38.7, 18.5 (6C), 11.0 (3C); IR (CHCl3) νmax 3282, 2946, 2869, 1760, 1464, 1323, 1071, 992, 883, 675 (cm−1); HRMS (FAB) calcd. for C17H30NO2Si ([M + H]+), 308.2046; found, 308.2042.
3.1.5. (4S,5R)-4-Allyl-5-ethynyloxazolidin-2-one (6)
To a solution of alcohol 10a (4.0 g, 13.0 mmol) in THF (60 mL) was added tetrabutylammonium fluoride (15.6 mL, 15.6 mmol, 1 M solution in THF) at room temperature under nitrogen. The reaction mixture was stirred for 1 h at room temperature. The reaction was quenched with a saturated NH4Cl aqueous solution and extracted with EtOAc. The organic layer was washed twice with brine, dried over MgSO4, filtered and concentrated. The crude product was purified using column chromatography on silica gel (hexane/EtOAc, 1:1), to afford the desired terminal acetylene 6 (1.87 g, 95%) as a white solid. m.p. 42.0–44.0 °C; −6.32 (c 0.54, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.81–5.70 (m, 1H), 5.28 (dd, J = 2.2 Hz, 7.8 Hz, 2H), 5.22 (d, J = 0.7 Hz, 1H), 5.19–5.18 (m, 1H), 3.91 (td, J = 4.1 Hz, 8.6 Hz, 1H) 2.72 (d, J = 2.1 Hz, 1H), 2.58–2.52 (m, 1H), 2.44–2.36 (m, 1H); 13C NMR (150 MHz, CDCl3) δ 157.2, 132.4, 120.0, 79.0, 75.8, 69.3, 53.9, 36.5; IR (CHCl3) νmax 3292, 2128, 1755, 1644, 1338, 1243, 1041 (cm−1); HRMS (FAB) calcd. for C8H10NO2 ([M + H]+), 152.0712; found, 152.0714.
3.1.6. General Procedure for the Preparation of 5
To a solution of 6 (300 mg, 2.0 mmol) with the appropriate alkene (2 mL) in CH2Cl2 (20 mL) was added Grubbs’ second-generation catalyst (50 mg, 0.059 mmol) at room temperature. The resulting mixture was refluxed for 20 h. After the mixture was cooled to room temperature, the solvent was removed under reduced pressure. The crude product was purified by column chromatography on silica gel (hexane/EtOAc, 1:3) to give diene 5.
3.1.7. (3aS,6aR)-6-((E)-Dodec-1-en-1-yl)-3,3a,4,6a-tetrahydro-2H-cyclopenta[d]oxazol-2-one (5a)
A white solid (420 mg, 72%); m.p. 83.6–84.5 °C; +23.7 (c 1.1, CHCl3); 1H NMR (500 MHz, CDCl3) δ 6.12 (d, J = 16.0 Hz, 1H), 5.94 (td, J = 7.4 Hz, 15.2 Hz, 1H), 5.68 (s, 1H), 5.63 (brs, 1H), 5.59 (d, J = 7.8 Hz, 1H), 4.44 (t, J = 7.2 Hz, 1H), 2.71 (dd, J = 6.1 Hz, 18.1 Hz, 1H), 2.47 (d, J = 18.2 Hz, 1H), 2.15–2.02 (m, 2H), 1.40–1.35 (m, 2H), 1.24 (s, 14H), 0.86 (t, J = 6.9 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 158.9, 139.8, 135.0, 128.8, 123.0, 84.9, 53.7, 40.2, 33.1, 31.9, 29.62, 29.58, 29.48, 29.33, 29.25, 29.0, 22.7, 14.1; IR (CHCl3) νmax 3270, 2922, 2854, 1739, 1716, 1385, 1238, 1018, 756 (cm−1); HRMS (FAB) calcd. for C18H30NO2 ([M + H]+), 292.2277; found, 292.2279.
3.1.8. (3aS,6aR)-6-((E)-Tetradec-1-en-1-yl)-3,3a,4,6a-tetrahydro-2H-cyclopenta[d]oxazol-2-one (5b)
A white solid (466 mg, 73%); m.p. 93.1–94.5 °C; +33.0 (c 0.43, CHCl3); 1H NMR (400 MHz, CDCl3) δ 6.12 (d, J = 15.9 Hz, 1H), 5.94 (td, J = 7.3 Hz, 15.1 Hz, 1H), 5.68 (s, 1H), 5.59 (d, J = 7.7 Hz, 1H), 5.51 (brs, 1H), 4.44 (t, J = 7.1 Hz, 1H), 2.72 (dd, J = 6.0 Hz, 18.3 Hz, 1H), 2.47 (d, J = 18.2 Hz, 1H), 2.13–2.04 (m, 2H), 1.40–1.37 (m, 2H), 1.24 (s, 18H), 0.86 (t, J = 6.7 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 158.8, 139.8, 135.1, 128.7, 123.0, 84.9, 53.7, 40.2, 33.1, 31.9, 29.7 (2C), 29.64, 29.58, 29.48, 29.35, 29.3, 29.0, 22.7, 14.1; IR (CHCl3) νmax 3244, 2917, 2852, 1735, 1471, 1386, 1248, 1016, 716 (cm−1); HRMS (FAB) calcd. for C20H34NO2 ([M + H]+), 320.2590; found, 320.2584.
3.1.9. (3aS,6aR)-6-((E)-Hexadec-1-en-1-yl)-3,3a,4,6a-tetrahydro-2H-cyclopenta[d]oxazol-2-one (5c)
A white solid (466 mg, 67%); m.p. 90.1–91.6 °C; +38.4 (c 0.95, CHCl3); 1H NMR (600 MHz, CDCl3) δ 6.12 (d, J = 16.0 Hz, 1H), 5.94 (td, J = 7.4 Hz, 15.3 Hz, 1H), 5.86 (brs, 1H), 5.68 (s, 1H), 5.59 (d, J = 8.2 Hz, 1H), 4.44 (t, J = 7.1 Hz, 1H), 2.70 (dd, J = 6.4 Hz, 17.9 Hz, 1H), 2.47 (d, J = 18.3 Hz, 1H), 2.15–2.02 (m, 2H), 1.39–1.34 (m, 2H), 1.24 (s, 22H), 0.86 (t, J = 7.1 Hz, 3H); 13C NMR (150 MHz, CDCl3) δ 159.0, 139.8, 135.0, 128.9, 123.0, 84.9, 53.8, 40.2, 33.1, 31.9, 29.67 (3C), 29.64 (2C), 29.58, 29.48, 29.35, 29.26, 29.0, 22.7, 14.1; IR (CHCl3) νmax 3284, 2922, 2853, 1737, 1716, 1385, 1237, 1015 (cm−1); HRMS (FAB) calcd. for C22H38NO2 ([M + H]+), 348.2903; found, 348.2905.
3.1.10. General Procedure for the Preparation of 11
To a solution of diene 5 (0.72 mmol) in MeOH (30 mL) was added Pd/C (40 mg, 10 wt.% Pd) at room temperature. The reaction mixture was shaken in a Parr hydrogenator for 10 h at an initial pressure of 35 psi. The catalyst was filtered off with Celite® (OCI Company Ltd., Seoul, Korea), and the solvent was removed under reduced pressure. The crude product was purified by column chromatography on silica gel (hexane/EtOAc, 1:3) to give the desired product, 11.
3.1.11. (3aS,6R,6aR)-6-Dodecylhexahydro-2H-cyclopenta[d]oxazol-2-one (11a)
A white solid (117 mg, 55%). m.p. 67.8–69.1 °C; +10.6 (c 0.50, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.71 (brs, 1H), 4.88 (t, J = 6.1 Hz, 1H), 4.22 (t, J = 6.5 Hz, 1H), 1.76–1.69 (m, 3H), 1.63–1.55 (m, 2H), 1.53–1.39 (m, 2H), 1.23 (s, 20H), 0.86 (t, J = 6.7 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 160.1, 83.2, 56.6, 46.0, 33.8, 31.9, 29.8, 29.62 (4C), 29.56, 29.3, 28.4, 28.2, 28.0, 22.7, 14.1; IR (CHCl3) νmax 3306, 2923, 2854, 1736, 1713, 1423, 1242 (cm−1); HRMS (FAB) calcd. for C18H34NO2 ([M + H]+), 296.2590; found, 296.2588.
3.1.12. (3aS,6R,6aR)-6-Tetradecylhexahydro-2H-cyclopenta[d]oxazol-2-one (11b)
A white solid (127 mg, 57%). m.p. 88.7–89.6 °C; +8.56 (c 0.52, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.59 (brs, 1H), 4.88 (t, J = 6.1 Hz, 1H), 4.23 (t, J = 6.5 Hz, 1H), 1.77–1.70 (m, 3H), 1.60–1.54 (m, 2H), 1.48–1.42 (m, 2H), 1.23 (s, 24H), 0.86 (t, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 160.0, 83.2, 56.6, 46.1, 33.9, 31.9, 29.8, 29.67 (3C), 29.64 (3C), 29.57, 29.3, 28.4, 28.3, 28.0, 22.7, 14.1; IR (CHCl3) νmax 3255, 2919, 2852, 1723, 1469, 1248 (cm−1); HRMS (FAB) calcd. for C20H38NO2 ([M + H]+), 324.2903; found, 324.2903.
3.1.13. (3aS,6R,6aR)-6-Hexadecylhexahydro-2H-cyclopenta[d]oxazol-2-one (11c)
A white solid (122 mg, 50%). m.p. 91.9–92.1 °C; +7.91 (c 0.46, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.51 (brs, 1H), 4.89 (t, J = 6.1 Hz, 1H), 4.23 (t, J = 6.5 Hz, 1H), 1.77–1.70 (m, 3H), 1.61–1.55 (m, 2H), 1.48–1.40 (m, 2H), 1.23 (s, 28H), 0.86 (t, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 159.9, 83.2, 56.6, 46.1, 33.9, 31.9, 29.8, 29.68 (5C), 29.65 (3C), 29.58, 29.3, 28.4, 28.3, 28.0, 22.7, 14.1; IR (CHCl3) νmax 3262, 2918, 2851, 1725, 1469, 1245 (cm−1); HRMS (FAB) calcd. for C22H42NO2 ([M + H]+), 352.3216; found, 352.3216.
3.1.14. General Procedure for the Preparation of 4
To a solution of 11 (0.071 mmol) in EtOH (1 mL) was added 1 M aq KOH (1 mL) at room temperature. The reaction mixture was stirred for 20 h at 85 °C. After the reaction mixture was cooled to room temperature, the solvent was removed under reduced pressure. The crude product was purified by column chromatography on silica gel (CH2Cl2/MeOH/NH4OH, 100:10:1) to give the desired product, 4.
3.1.15. (1R,2S,5R)-2-Amino-5-dodecylcyclopentan-1-ol (4a)
A white solid (17 mg, 88%). m.p. 93.2–96.2 °C; +3.96 (c 0.36, EtOH); 1H NMR (400 MHz, CDCl3) δ 3.68 (t, J = 3.5 Hz, 1H), 3.30 (td, J = 3.8 Hz, 8.4 Hz, 1H), 2.10 (brs, 2H), 1.91–1.86 (m, 1H), 1.74–1.66 (m, 2H), 1.56–1.34 (m, 1H), 1.23 (s, 23H), 0.86 (t, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 74.4, 55.4, 44.2, 31.9, 30.9, 29.95, 29.93, 29.68 (3C), 29.64, 29.3, 28.5, 28.0, 22.7, 14.1; IR (CHCl3) νmax 3054, 2922, 2852, 2727, 1469, 992 (cm−1); HRMS (FAB) calcd. for C17H36NO ([M + H]+), 270.2797; found, 270.2794.
3.1.16. (1R,2S,5R)-2-Amino-5-tetradecylcyclopentan-1-ol (4b)
A white solid (19 mg, 90%). m.p. 100.1–101.0 °C; +3.29 (c 0.25, EtOH); 1H NMR (400 MHz, CDCl3) δ 3.70 (t, J = 3.4 Hz, 1H), 3.30 (td, J = 3.9 Hz, 8.5 Hz, 1H), 2.44 (brs, 3H), 1.91–1.85 (m, 1H), 1.75–1.65 (m, 2H), 1.54–1.34 (m, 1H), 1.23 (s, 27H), 0.86 (t, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 74.4, 55.4, 44.1, 31.9, 30.8, 30.0, 29.9, 29.69 (5C), 29.65 (2C), 29.3, 28.5, 28.0, 22.7, 14.1; IR (CHCl3) νmax 3049, 2921, 2852, 2729, 1469, 994 (cm−1); HRMS (FAB) calcd. for C19H40NO ([M + H]+), 298.3110; found, 298.3117.
3.1.17. (1R,2S,5R)-2-Amino-5-hexadecylcyclopentan-1-ol (4c)
A white solid (21 mg, 91%). m.p. 102.1–103.0 °C; +1.89 (c 0.51, CHCl3); 1H NMR (400 MHz, CDCl3) δ 3.70 (t, J = 3.5 Hz, 1H), 3.30 (td, J = 3.8 Hz, 8.2 Hz, 1H), 2.41 (brs, 3H), 1.89–1.85 (m, 1H), 1.75–1.65 (m, 2H), 1.54–1.34 (m, 1H), 1.23 (s, 31H), 0.86 (t, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 74.5, 55.5, 44.2, 31.9, 30.9, 30.0, 29.9, 29.69 (7C), 29.65 (2C), 29.4, 28.5, 28.0, 22.7, 14.1; IR (CHCl3) νmax 3057, 2921, 2852, 2728, 1468, 995 (cm−1); HRMS (FAB) calcd. for C21H44NO ([M + H]+), 326.3423; found, 326.3433.
3.2. Biological Evaluation
3.2.1. Sulforhodamine B (SRB) Assay
Cells (5 × 104 cells/mL) were treated with various concentrations of test compounds in 96-well culture plates for 72 h. After incubation, cells were fixed with 10% trichloroacetic acid (TCA), dried and stained with 0.4% SRB in 1% acetic acid. The unbound dye was washed out, and the stained cells were dried and resuspended in 10 mM Tris (pH 10.0). The absorbance at 515 nm was measured, and cell proliferation was determined as follows: cell proliferation (%) = (average absorbancecompound − average absorbanceday zero)/(average absorbancecontrol − average absorbanceday zero) × 100. IC50 values were calculated by nonlinear regression analysis using TableCurve 2D v5.01 (Systat Software Inc., Richmond, CA, USA).
3.2.2. Sphingosine Kinase Inhibition Assay
In 384-well polystyrene plates, Sphk1 and Sphk2 (BPS Bioscience, San Diego, CA, USA) were incubated in kinase buffer (5 mM MOPS, pH 7.2, 2.5 mM β-glycerol-phosphate, 5 mM MgCl2, 1 mM EGTA, 0.4 mM EDTA, 0.5 mM DTT) containing 1 µM sphingosine, ATP (50 µM for SphK1; 150 µM for SphK2) and test compounds with a final concentration of 1% DMSO for 1 h at room temperature. The amount of ATP transferred was measured with the ADP-Glo™ kinase assay kit (Catalog #V9101, Promega, Madison, WI, USA) according to the manufacturer’s instructions. IC50 values were calculated using GraphPad Prism 5 software (GraphPad Software Inc., La Jolla, CA, USA).
3.3. Molecular Modeling
To understand the binding mode of the pachastrissamine and its analogue, we performed a flexible docking study using the Schrödinger Glide program with standard precision settings (Schrödinger, LLC, New York, NY, USA, http://www.schrodinger.com). The X-ray crystal structure of the human SphK1 (PDB Code 3VZB) was obtained from the Protein Data Bank (PDB, [36]). The pachastrissamine and 4b were minimized using a Merck Molecular Force Field (MMFF) with a dielectric constant of 80.0 using the MacroModel program suite. The initial structure of pachastrissamine and 4b were built based on the sphingosine structure co-crystallized with SphK1. The best docking result was visualized using Discovery Studio 4.1 (Accelrys Software, Inc., San Diego, CA, USA). The hydrogen bonding interactions were displayed as green dashed lines between the ligand and the SphK1.
4. Conclusions
We have reported the synthesis and biological evaluation of the carbocyclic analogues of pachastrissamine with varying chain lengths. The designed carbocyclic analogues were efficiently synthesized in good overall yield by the tandem ene/yne-ene metathesis as a key step. Among them, the analogue 4b exhibits comparable cytotoxic activity and more potent inhibitory activity against sphingosine kinases, compared to the parent natural product. In our docking model, 4b showed an additional interaction caused by the adjacent hydrophobic amino acid residue, which could explain its increased inhibitory activity. These results imply that bioisosteric replacement of the ethereal oxygen in the cyclic core to the methylene carbon is possible. Our findings are valuable in the development of sphingosine kinase inhibitors, which may lead to the development of promising new anti-cancer therapeutic agents. Further studies toward the biochemical and pharmacological properties of the carbocyclic analogue, based on these findings, are worthy of pursuing in the future.
Acknowledgments
This work was supported by the Mid-Career Researcher Program (No. 2013R1A2A1A01015998) of the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIP). This work was also supported by an NRF (National Research Foundation of Korea) grant funded by the Korean Government (NRF-2012-Fostering Core Leaders of the Future Basic Science Program).
Supplementary Files
Author Contributions
Yongseok Kwon, Hoon Bae and Geun-Hee Han performed the synthetic experiments. Jayoung Song and Sang Kook Lee performed the biological experiments. Joo-Youn Lee performed the molecular modeling. Yongseok Kwon, Woo-Jung Kim and Sanghee Kim wrote the manuscript. Sanghee Kim designed and developed the study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Kuroda I., Musman M., Ohtani I.I., Ichiba T., Tanaka J., Gravalos D.G., Higa T. Pachastrissamine, a cytotoxic anhydrophytosphingosine from a marine sponge, Pachastrissa sp. J. Nat. Prod. 2002;65:1505–1506. doi: 10.1021/np010659y. [DOI] [PubMed] [Google Scholar]
- 2.Ledroit V., Debitus C., Lavaud C., Massiot G. Jaspines A and B: Two new cytotoxic sphingosine derivatives from the marine sponge Jaspis sp. Tetrahedron Lett. 2003;44:225–228. doi: 10.1016/S0040-4039(02)02541-8. [DOI] [Google Scholar]
- 3.Liu J., Du Y., Dong X., Meng S., Xiao J., Cheng L. Stereoselective synthesis of jaspine B from d-xylose. Carbohydr. Res. 2006;341:2653–2657. doi: 10.1016/j.carres.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 4.Salma Y., Lafont E., Therville N., Carpentier S., Bonnafé M.-J., Levade T., Génisson Y., Andrieu-Abadie N. The natural marine anhydrophytosphingosine, jaspine B, induces apoptosis in melanoma cells by interfering with ceramide metabolism. Biochem. Pharmacol. 2009;78:477–485. doi: 10.1016/j.bcp.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 5.Yoshimitsu Y., Oishi S., Miyagaki J., Inuki S., Ohno H., Fujii N. Pachastrissamine (jaspine B) and its stereoisomers inhibit sphingosine kinases and atypical protein kinase C. Biorg. Med. Chem. 2011;19:5402–5408. doi: 10.1016/j.bmc.2011.07.061. [DOI] [PubMed] [Google Scholar]
- 6.Inuki S., Yoshimitsu Y., Oishi S., Fujii N., Ohno H. Ring-construction/stereoselective functionalization cascade: Total synthesis of pachastrissamine (jaspine B) through palladium-catalyzed bis-cyclization of propargyl chlorides and carbonates. J. Org. Chem. 2010;75:3831–3842. doi: 10.1021/jo100544v. [DOI] [PubMed] [Google Scholar]
- 7.Passiniemi M., Koskinen A.M.P. Asymmetric synthesis of pachastrissamine (jaspine B) and its diastereomers via η3-allylpalladium intermediates. Org. Biomol. Chem. 2011;9:1774–1783. doi: 10.1039/c0ob00643b. [DOI] [PubMed] [Google Scholar]
- 8.Srinivas Rao G., Venkateswara Rao B. A common strategy for the stereoselective synthesis of anhydrophytosphingosine pachastrissamine (jaspine B) and N,O,O,O-tetra-acetyl d-lyxo-phytosphingosine. Tetrahedron Lett. 2011;52:6076–6079. [Google Scholar]
- 9.Llaveria J., Díaz Y., Matheu M.I., Castillón S. Enantioselective synthesis of jaspine B (pachastrissamine) and its C-2 and/or C-3 epimers. Eur. J. Org. Chem. 2011:1514–1519. [Google Scholar]
- 10.Zhao M.-L., Zhang E., Gao J., Zhang Z., Zhao Y.-T., Qu W., Liu H.-M. An efficient and convenient formal synthesis of jaspine B from d-xylose. Carbohydr. Res. 2012;351:126–129. doi: 10.1016/j.carres.2012.01.013. [DOI] [PubMed] [Google Scholar]
- 11.Schmiedel V.M., Stefani S., Reissig H.-U. Stereodivergent synthesis of jaspine B and its isomers using a carbohydrate-derived alkoxyallene as C3-building block. Beilstein J. Org. Chem. 2013;9:2564–2569. doi: 10.3762/bjoc.9.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghosal P., Ajay S., Meena S., Sinha S., Shaw A.K. Stereoselective total synthesis of jaspine B (pachastrissamine) utilizing iodocyclization and an investigation of its cytotoxic activity. Tetrahedron Asymmetry. 2013;24:903–908. doi: 10.1016/j.tetasy.2013.07.003. [DOI] [Google Scholar]
- 13.Jana A.K., Panda G. Stereoselective synthesis of jaspine B and its C2 epimer from garner aldehyde. RSC Adv. 2013;3:16795–16801. doi: 10.1039/c3ra41778f. [DOI] [Google Scholar]
- 14.Dhand V., Chang S., Britton R. Total synthesis of the cytotoxic anhydrophytosphingosine pachastrissamine (jaspine B) J. Org. Chem. 2013;78:8208–8213. doi: 10.1021/jo4013223. [DOI] [PubMed] [Google Scholar]
- 15.Lin C.-W., Liu S.-W., Hou D.-R. Formation of tetrahydrofurans via a 5-endo-tet cyclization of aziridines—Synthesis of (−)-pachastrissamine. Org. Biomol. Chem. 2013;11:5292–5299. doi: 10.1039/c3ob40797g. [DOI] [PubMed] [Google Scholar]
- 16.Martinková M., Mezeiová E., Gonda J., Jacková D., Pomikalová K. Total synthesis of (−)-jaspine B and its 4-epi-analogue from d-xylose. Tetrahedron Asymmetry. 2014;25:750–766. doi: 10.1016/j.tetasy.2014.04.002. [DOI] [Google Scholar]
- 17.Génisson Y., Lamandé L., Salma Y., Andrieu-Abadie N., André C., Baltas M. Enantioselective access to a versatile 4-oxazolidinonecarbaldehyde and application to the synthesis of a cytotoxic jaspine B truncated analogue. Tetrahedron Asymmetry. 2007;18:857–864. doi: 10.1016/j.tetasy.2007.03.029. [DOI] [Google Scholar]
- 18.Salma Y., Ballereau S., Maaliki C., Ladeira S., Andrieu-Abadie N., Génisson Y. Flexible and enantioselective access to jaspine B and biologically active chain-modified analogues thereof. Org. Biomol. Chem. 2010;8:3227–3243. doi: 10.1039/c004218h. [DOI] [PubMed] [Google Scholar]
- 19.Canals D., Mormeneo D., Fabriàs G., Llebaria A., Casas J., Delgado A. Synthesis and biological properties of pachastrissamine (jaspine B) and diastereoisomeric jaspines. Biorg. Med. Chem. 2009;17:235–241. doi: 10.1016/j.bmc.2008.11.026. [DOI] [PubMed] [Google Scholar]
- 20.Xu J.-M., Zhang E., Shi X.-J., Wang Y.-C., Yu B., Jiao W.-W., Guo Y.-Z., Liu H.-M. Synthesis and preliminary biological evaluation of 1,2,3-triazole-jaspine B hybrids as potential cytotoxic agents. Eur. J. Med. Chem. 2014;80:593–604. doi: 10.1016/j.ejmech.2014.03.022. [DOI] [PubMed] [Google Scholar]
- 21.Rives A., Ladeira S., Levade T., Andrieu-Abadie N., Génisson Y. Synthesis of cytotoxic aza analogues of jaspine B. J. Org. Chem. 2010;75:7920–7923. doi: 10.1021/jo1014239. [DOI] [PubMed] [Google Scholar]
- 22.Jeon H., Bae H., Baek D.J., Kwak Y.-S., Kim D., Kim S. Syntheses of sulfur and selenium analogues of pachastrissamine via double displacements of cyclic sulfate. Org. Biomol. Chem. 2011;9:7237–7242. doi: 10.1039/c1ob05920c. [DOI] [PubMed] [Google Scholar]
- 23.Patani G.A., LaVoie E.J. Bioisosterism: A rational approach in drug design. Chem. Rev. 1996;96:3147–3176. doi: 10.1021/cr950066q. [DOI] [PubMed] [Google Scholar]
- 24.Arjona O., Gómez A.M., López J.C., Plumet J. Synthesis and conformational and biological aspects of carbasugars. Chem. Rev. 2007;107:1919–2036. doi: 10.1021/cr0203701. [DOI] [PubMed] [Google Scholar]
- 25.Ferrero M., Gotor V. Biocatalytic selective modifications of conventional nucleosides, carbocyclic nucleosides, and C-nucleosides. Chem. Rev. 2000;100:4319–4348. doi: 10.1021/cr000446y. [DOI] [PubMed] [Google Scholar]
- 26.Villar H., Frings M., Bolm C. Ring closing enyne metathesis: A powerful tool for the synthesis of heterocycles. Chem. Soc. Rev. 2007;36:55–66. doi: 10.1039/b508899m. [DOI] [PubMed] [Google Scholar]
- 27.Grubbs R.H. Olefin metathesis. Tetrahedron. 2004;60:7117–7140. doi: 10.1016/j.tet.2004.05.124. [DOI] [Google Scholar]
- 28.Royer F., Vilain C., Elkaïm L., Grimaud L. Selective domino ring-closing metathesis–cross-metathesis reactions between enynes and electron-deficient alkenes. Org. Lett. 2003;5:2007–2009. doi: 10.1021/ol034620r. [DOI] [PubMed] [Google Scholar]
- 29.Lee H.-Y., Kim H.Y., Tae H., Kim B.G., Lee J. One-pot three-component tandem metathesis/Diels−alder reaction. Org. Lett. 2003;5:3439–3442. doi: 10.1021/ol035194c. [DOI] [PubMed] [Google Scholar]
- 30.Borzilleri R.M., Zheng X., Schmidt R.J., Johnson J.A., Kim S.-H., DiMarco J.D., Fairchild C.R., Gougoutas J.Z., Lee F.Y.F., Long B.H., et al. A novel application of a Pd(0)-catalyzed nucleophilic substitution reaction to the regio- and stereo-selective synthesis of lactam analogues of the epothilone natural products. J. Am. Chem. Soc. 2000;122:8890–8897. doi: 10.1021/ja001899n. [DOI] [Google Scholar]
- 31.Lee S.K., Nam K.-A., Heo Y.-H. Cytotoxic activity and G2/M cell cycle arrest mediated by antofine, a phenanthroindolizidine alkaloid isolated from cynanchum paniculatum. Planta Med. 2003;69:21–25. doi: 10.1055/s-2003-37021. [DOI] [PubMed] [Google Scholar]
- 32.Pyne N.J., Pyne S. Sphingosine 1-phosphate and cancer. Nat. Rev. Cancer. 2010;10:489–503. doi: 10.1038/nrc2875. [DOI] [PubMed] [Google Scholar]
- 33.Yatomi Y., Ruan F., Megidish T., Toyokuni T., Hakomori S.-I., Igarashi Y. N,N-dimethylsphingosine inhibition of sphingosine kinase and sphingosine 1-phosphate activity in human platelets. Biochemistry. 1996;35:626–633. doi: 10.1021/bi9515533. [DOI] [PubMed] [Google Scholar]
- 34.Wang Z., Min X., Xiao S.-H., Johnstone S., Romanow W., Meininger D., Xu H., Liu J., Dai J., An S., et al. Molecular basis of sphingosine kinase 1 substrate recognition and catalysis. Structure. 2013;21:798–809. doi: 10.1016/j.str.2013.02.025. [DOI] [PubMed] [Google Scholar]
- 35.Bissantz C., Kuhn B., Stahl M. A medicinal chemist’s guide to molecular interactions. J. Med. Chem. 2010;53:5061–5084. doi: 10.1021/jm100112j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Protein Data Bank. [(accessed on 27 November 2014)]. Available online: http://www.rcsb.org.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.