

Abstract
Nuclear factor erythroid-derived 2-related factor 2 (Nrf2) was originally identified as a positive regulator of drug detoxifying enzyme gene expression during exposure to environmental electrophiles. Currently, Nrf2 is known to regulate the expression of hundreds of cytoprotective genes to counteract endogenously or exogenously generated oxidative stress. Furthermore, when activated in human tumors by somatic mutations, Nrf2 confers growth advantages and chemoresistance by regulating genes involved in various processes such as the pentose phosphate pathway and nucleotide synthesis in addition to antioxidant proteins. Interestingly, increasing evidence shows that Nrf2 is associated with mitochondrial biogenesis during environmental stresses in certain tissues such as the heart. Furthermore, SKN-1, a functional homolog of Nrf2 in C. elegans, is activated by mitochondrial reactive oxygen species and extends life span by promoting mitochondrial homeostasis (i.e., mitohormesis). Similarly, Nrf2 activation was recently observed in the heart of surfeit locus protein 1 (Surf1) -/- mice in which cellular respiration was decreased due to cytochrome c oxidase defects. In this review, we critically examine the relationship between Nrf2 and mitochondria and argue that the Nrf2 stress pathway intimately communicates with mitochondria to maintain cellular homeostasis during oxidative stress.
Keywords: mitochondria, Nrf2, heme oxygenase-1, p62, NRF-1
Introduction
Mitochondria are multifunctional organelles within the cell. Mitochondria generate energy by cellular respiration (i.e., normally 90% of cellular ATP) and also serve as an integrative platform for various intracellular signaling pathways such as apoptosis and the immune and inflammatory responses.(1) Mitochondria are also the major source of reactive oxygen species (ROS) and play an integral role in the determination of apoptosis or survival upon stress exposure.(2) Mitochondria possess their own genome, and human mitochondria contain 37 genes of which all of the 13 protein-coding genes encode oxidative phosphorylation machinery subunits.(3) However, as most mitochondrial proteins are encoded in the nuclear genome, communication between the mitochondria and nucleus is fundamental to maintain cellular homeostasis against various stresses.(3) Recently, a concept of mitohormesis that was increasingly dissected in many model organisms showed that the mild alteration of the mitochondrial function activates the mitochondrial retrograde signaling, leading to the prolonged mean life span.(4)
The Keap1-Nrf2 system was identified as a master regulator of drug detoxification and oxidative stress responses and is critical for cell survival against oxidative stress.(5) Therefore, a functional link between the Keap1-Nrf2 system and mitochondria is expected. Consistent with this idea, accumulating evidence suggests the existence of extensive protein interaction networks between the Keap1-Nrf2 system and the components of apoptotic signaling cascades. Furthermore, recent evidence has revealed various types of cross-talk between the Keap1-Nrf2 system and mitochondria.
The Keap1-Nrf2 System Regulates a Battery of Detoxification Enzymes and Antioxidant Proteins in Detoxification Organs
Nrf2 (nuclear factor erythroid-derived 2-related factor 2) belongs to the CNC (Cap’n’collar) transcription factor family, which is characterized by a highly conserved CNC domain and a basic region leucine-zipper (b-Zip) structure (Fig. 1A).(5) Nrf2 is ubiquitously expressed but is expressed at relatively higher levels in organs that interface with the environment such as the lungs and small intestine.(6) Nrf2 plays an important role in detoxifying environmental toxins by up-regulating a set of detoxification enzymes and antioxidant proteins (Fig. 1B).(5) In non-stressed cells, Nrf2 is rapidly degraded by the kelch-like ECH-associated protein 1 (Keap1)-mediated ubiquitin proteasome system (Fig. 1C and D).(5) Keap1 enhances Nrf2 degradation by acting as an adaptor protein for the Nrf2 and Cullin3-Rbx1 E3 ubiquitin ligase complex. Upon exposure to oxidative stress or electrophiles, Nrf2 translocates to the nucleus where it binds to antioxidant responsive elements (ARE) via heterodimerization with small Maf proteins and activates the expression of more than a hundred target genes, including drug detoxifying enzymes such as glutathione S-transferases or antioxidant proteins such as heme oxygenase-1 (HO-1) and thioredoxin (Fig. 1B).(5)
Fig. 1.
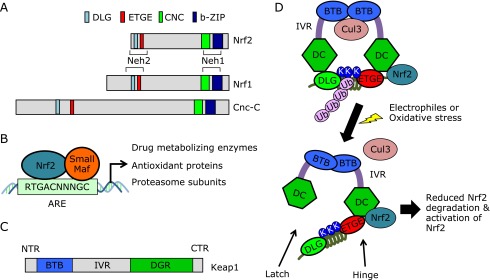
Keap1-Nrf2 stress response system. (A) Schematic domain structures of Nrf2 and Nrf2-related proteins. Nrf2 possesses six evolutionarily conserved domains called Neh (Nrf2 ECH homology) 1–6 (only Neh1 and Neh2 are shown in the figure). The Neh2 domain is a regulatory domain of Nrf2 and contains ETGE and DLG motifs. Both motifs are conserved in Nrf1, which is another member of the CNC protein family, and Cnc-C, a Drosophila Nrf2 homolog. (B) Nrf2 heterodimerizes with small Maf proteins and binds to antioxidant-responsive elements (AREs). (C) Schematic domain structures of Keap1. Keap1 shows structural similarity to the Drosophila protein Kelch and has two canonical protein interaction domains, BTB (bric-a-brac, tramtrack, broad complex) and Kelch (also called double glycine-repeat, DGR). CTR, C-terminal region; IVR, intervening region; NTR, N-terminal region. (D) Schematic representation of the Nrf2-activation mechanism. Together, the DGR and CTR of Keap1 comprise a six-bladed β-propeller structure shown as DC (DGR, CTR). Keap1 homodimerizes via its BTB domain and binds to the ETGE and DLG motifs of the Nrf2 Neh2 domain. The ETGE and DLG motifs are high- and low-affinity binding sites, respectively, for Keap1. The lysine residues (K) that are ubiquitinated localize to one side of the intervening α-helix, enhancing Keap1-mediated ubiquitination. During oxidative stress, reactive cysteines in Keap1 are oxidized, leading to conformational changes in Keap1. As a result, only binding via the low-affinity site is disrupted or the Cul3 interaction with Keap1 is disrupted leading to the inhibition of Nrf2 ubiquitination. Ub; ubiquitin.
Keap1-Nrf2-centered Protein–protein Interaction Map Suggests an Important Role for the Keap1-Nrf2 System in Mitochondrial Function
Keap1 was identified in a yeast two-hybrid screen as a factor that binds to the regulatory Neh2 domain of Nrf2 (Fig. 1A).(7) Subsequently, we have demonstrated by yeast reverse-two-hybrid screening that the ETGE motif within the Neh2 domain of Nrf2 is essential for Nrf2-Keap1 binding.(8) Another weak binding motif, called the DLG motif, was identified as a motif that was further upstream of the ETGE motifs in the Neh2 domain.(9) The Keap1 dimer binds Nrf2 via the DLG and ETGE motifs of Nrf2, which leads to the proteasomal degradation of Nrf2 in unstressed conditions (the two-site recognition mechanism).(5) Oxidative stress and electrophiles cause the oxidation of cysteine residues in Keap1, leading to a conformational change in Keap1 and the inhibition of Nrf2 ubiquitination and degradation. This mechanism explains how environmental toxins are sensed by the Keap1-Nrf2 system in detoxification organs such as the liver and intestine. Subsequent analysis demonstrated the existence of ETGE-like motifs in several other proteins, including prothymosin-α, phosphoglycerate mutase family member 5, inhibitor of kappaB kinase β and p62, mediating their interaction with Keap1 (Fig. 2).(10–14) Furthermore, Keap1 also interacts with Bcl-2 in an ETGE-independent manner.(15) Interestingly, all these proteins are involved in the mitochondrial regulation of apoptosis or in the maintenance of mitochondrial integrity and function. This extensive protein interaction between the Keap1-Nrf2 system and the mitochondria suggests extensive cross-talk between these entities.
Fig. 2.
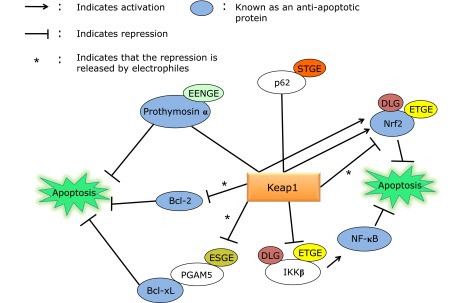
Keap1 interaction with mitochondria-related proteins. Arrows in the figure indicate activation. For example, p62 and prothymosin α activate Nrf2 by competitively binding to Keap1 via their ETGE-related motifs. See text for details.
Roles of Nrf2 in Mitochondrial Function and Quality Control
The emerging roles of Nrf2 target genes in mitochondrial function and quality control
(a) Sqstm1/p62 (hereafter called p62) was first identified as an electrophile-inducible gene in mouse macrophages, and its expression is regulated in an Nrf2-dependent manner during oxidative stress.(16,17) p62 is a multi-functional protein that functions as a selective autophagy adaptor such as xenophagy and mitophagy.(18,19) In addition to being a target gene of Nrf2, p62 was identified as an activator of Nrf2, thus forming a positive feedback loop.(20) An STGE motif in the Keap1-interacting region of p62 has lower affinity for Keap1 compared with the Nrf2 ETGE motif.(21) However, this affinity markedly increases upon the serine phosphorylation of the p62 STGE motif by mTOR, leading to the activation of Nrf2 during selective autophagy.(22) Regarding mitochondrial function, p62-knockout (KO) mice show a rapid decline in mitochondrial function with age and present an accelerated aging phenotype that is accompanied by oxidative stress.(23) In addition, p62 localizes to the mitochondria and plays a protective role by enhancing the translocation of mitochondrial transcription factor A (TFAM), a critical regulator of mitochondrial transcription and mitochondrial DNA replication, to the mitochondria.(24,25)
(b) HO-1 catalyzes the conversion of heme into biliverdin, iron and carbon monoxide (CO). HO-1 gene expression is up-regulated in response to the substrate heme as well as to various environmental stressors such as cadmium, lipopolysaccharide, nitric oxide (NO) and oxidative stress.(26) CO binds to the reduced heme of cytochrome c oxidase (COX) and inhibits cellular respiration and generates hydrogen peroxide.(27,28) Thus, CO provokes the ROS-mediated activation of nuclear respiratory factor 1 (NRF-1), which induces mitochondrial biogenesis (also discussed later).(29)
(c) NAD(P)H dehydrogenase quinone 1 (NQO1) catalyzes the two-electron reduction of quinones into hydroquinones.(30) NQO1 knockdown decreases mitochondrial membrane potential (ΔΨm) and increases oxidant levels in HCT116 colon carcinoma cells.(23) Furthermore, NQO1 rescues the decreased ΔΨm and ROS generation induced by p62 and Nrf2 knockdown.
Nrf2 regulates mitochondrial biogenesis and cellular respiration
A role for Nrf2 in mitochondrial biogenesis.
In mammals, mitochondrial biogenesis is regulated by several key factors, including peroxisome proliferator-activated receptor gamma coactivator-1 alpha (PGC-1α) and NRF-1, which cooperatively activate mitochondrial biogenesis.(31) Several recent findings indicate that Nrf2 is also involved in mitochondrial biogenesis (summarized in Table 1). Pioneering work by Piantadosi et al.(32) showed that HO-1 overexpression, which leads to CO generation, activates NRF-1 transcription in an Nrf2-dependent manner (Fig. 3). In this cascade, CO-induced H2O2 leads to the activation of the PI3K-AKT pathway, activating Nrf2, which directly binds to the AREs in the NRF-1 gene promoter.(32) Therefore, Nrf2 is indispensable for HO-1/CO-mediated mitochondrial biogenesis. AKT also phosphorylates NRF-1, which promotes NRF-1 nuclear translocation.(33) Since the work by Piantadosi et al.(32,33), Nrf2-mediated mitochondrial biogenesis has been demonstrated in several in vivo studies.(34,35,39,40) Using mitochondrial reporter mice (mt-COX8-GFP mice), Athale et al.(34) demonstrated that mitochondrial biogenesis was activated in the lungs (bronchial and alveolar type II epithelial cells) after S. aureus insufflation, which was associated with NRF-1 and TFAM induction. This response was impaired in the lungs of Nrf2 KO mice, leading to enhanced inflammation. Another study by MacGarvey et al.(35) demonstrated that inhaled CO activated mitochondrial biogenesis in the mouse liver and prevented lethality from S. aureus sepsis in an Nrf2- and AKT-dependent manner. Exposure to mild-to-moderate hypoxic conditions, including hypobaric hypoxia that mimics hypoxia at high altitudes, causes excitotoxicity and mitochondria-mediated apoptosis in hippocampal neurons, leading to cognitive impairment.(36) Acetyl-l-carnitine (ALCAR) confers neuroprotection after exposure to hypobaric hypoxia by increasing tropomyosin receptor kinase A (TrkA)-dependent Nrf2 activation in the rat hippocampus,(37) consistent with our previous finding that the NGF-TrkA pathway regulates Nrf2 activity in rat PC12 cells.(38) Hota et al.(39) demonstrated that ALCAR causes mitochondrial biogenesis by activating the Nrf2-NRF-1 cascade in an ERK1/2-dependent manner and argued that neuroprotection by ALCAR was due to intracellular calcium buffering in the mitochondria. Recently, Kim et al.(40) demonstrated that NO and resveratrol activate mitochondrial biogenesis in an Nrf2-dependent manner via a complex signaling cascade involving HO-1 induction. These researchers also demonstrated that resveratrol activates mitochondrial biogenesis in the liver in an HO-1-dependent manner during an LPS sepsis model; however, Nrf2 dependence was not analyzed.(40)
Table 1.
Nrf2-mediated mitochondrial biogenesis
| Signals | Indispensable factors other than Nrf2 | Tissue or cells | Effect of mitochondrial biogenesis | References |
|---|---|---|---|---|
| HO-1 overexpression | PI3K, Akt | HL-1 mouse cardiomyocytes | Protection against doxorubicin toxicity | 32 |
| S. aureus insufflation | NA | Mouse lung (especially AT2 cells) | Anti-inflammation, cell protection | 34 |
| Exogenous CO inhalation | Akt | Mouse liver | Anti-inflammation | 35 |
| ALCAR | MEK | Rat primary hippocampal cells | Anti-apoptosis | 39 |
| NO (SNAP) | HO-1 | HepG2, mouse liver | NA | 40 |
| Resveratrol | eNOS (NO), sGC (cGMP), HO-1 (CO), Akt | HepG2 | NA | 40 |
eNOS; endothelial nitric oxide synthase, sGC; soluble guanylate cyclase, SNAP; S-nitroso-N-acetylpenicillamine, AT2; alveolar type 2, NA; not analyzed.
Fig. 3.

Hypothetical mitochondrial retrograde signals that activate Nrf2. In specific cell types such as cardiomyocytes, HO-1-mediated CO production activates Nrf2 via the generation of H2O2 from the mitochondria. GSK3β is known to inhibit Nrf2 via β-TrCP-mediated degradation or Fyn-mediated nuclear export.(68,69) H2O2 activates the PI3K-AKT pathway and inhibits GSK3β to activate Nrf2. COX; cytochrome c oxidase, GSK3β; glycogen synthase kinase-3β.
A role for Nrf2 in oxidative phosphorylation.
Nrf2 involvement in cellular respiration was first described by Kim et al.(41) who showed that Nrf2 knockdown in the human colon tumor cell lines HT29 and HCT116 cells reduces cellular O2 consumption and cellular ATP levels, leading to the inhibition of HIF1α activation by hypoxia; however, the underlying mechanism still remains unknown. Recently, cultured neurons derived from Nrf2 KO mice have been shown to have lower ΔΨm and cytosolic ATP levels.(42) In normal healthy cells, ΔΨm is maintained by cellular respiration; however, in Nrf2 KO neurons, ΔΨm is maintained by the reverse reaction of ATP synthase using ATP, which is partially compensated by increased glycolysis, indicating a respiration defect is present in the cells. Consistently, Nrf2 KO MEFs show decreased basal O2 consumption, and Nrf2 KO neurons cannot increase respiration and hyperpolarize the mitochondria in response to malate/pyruvate, which increases the levels of the complex I substrate NADH or the complex II substrate methyl-succinate. Furthermore, mitochondria isolated from Nrf2 KO brains or livers demonstrate decreased state 4 respiration in the presence of malate or succinate, a decreased respiratory control ratio (state 3/state 4; a conventional measure of coupling of oxygen consumption to phosphorylation) and a decreased ADP/O ratio (i.e., the ratio of ADP to oxygen consumed for ATP synthesis). In addition, the oxygen consumption rate induced by malate and succinate is decreased in Nrf2 KO mitochondria as observed in the intact cells. NADH and FADH2 recovery after the treatment with mitochondrial uncoupler followed by ETC inhibitor is delayed in Nrf2 KO MEFs compared with wild-type MEFs, indicating a delay in NADH and FADH2 generation. Consistently, mitochondrial NADH pool was decreased in the Nrf2 KO MEFs compared with wild-type MEFs. The activities of respiratory chain enzymes in the brain are not different from wild-type mice; therefore, the authors argued that the availability of respiratory substrates is decreased in Nrf2 KO mitochondria. Consistent with the above observation, FADH2 generation from free fatty acids is reduced in the MEF, liver and heart mitochondria isolated form Nrf2 KO mice.(43)
Mitochondrial Retrograde Signaling Regulates Nrf2 Activity during Oxidative Stress
A permissive signal from the mitochondria for the Nrf2-HO-1 cascade
Nrf2-dependent HO-1 expression upon oxidative stress was previously demonstrated to require a permissive signal from the mitochondria.(44) In this study, the authors demonstrated that the mitochondria-targeted thiol-reactive compound, IBTP [(4-iodobutyl)triphenylphosphonium], inhibited heme- and iodoacetamide-induced HO-1 expression without affecting mitochondrial ROS production and membrane potential, thus arguing for the existence of IBTP-sensitive permissive signals from the mitochondria downstream of ROS production. Mutations in the PTEN-induced putative kinase 1 (PINK1) and Parkin genes cause autosomal-recessive forms of Parkinson’s disease (PD).(45) The PINK1-Parkin pathway plays an important role in mitochondrial quality control by sensing mitochondrial damage and subsequently inducing mitophagy of the damaged mitochondria.(46) TNF receptor-associated protein 1 (TRAP1) is a downstream substrate of PINK1 kinase that plays a role in inhibiting oxidative stress.(47) Importantly, HO-1 induction by hydrogen peroxide is severely impaired in PINK1 kinase-dead mutant-overexpressing cells or upon TRAP1 knockdown in SH-SY5Y cells.(48) Although the role of Nrf2 in HO-1 induction was not analyzed, the results argue for the possibility that mitochondrial integrity is important for HO-1 induction by oxidative stress. In control cells, oxidative stress activates the PI3K-AKT and ERK1/2 pathways; however, these pathways are inhibited by the overexpression of the kinase-dead PINK1 mutant or by the TRAP1 knockdown. In fact, PI3K-AKT and ERK1/2 signaling is indispensable for HO-1 induction by oxidative stress in SH-SY5Y cells (Fig. 4).(48)
Fig. 4.

Mitochondria-permissive signal for the Nrf2-HO-1 cascade. PINK1 directly phosphorylates Parkin and TRAP1, subsequently inducing the transfer of Parkin to the mitochondria and preventing mitochondrial oxidative damage. PI3K-AKT, ERK1/2 and DJ-1 signaling are indispensable for oxidative stress-induced Nrf2 activation. See text for details.
Loss of DJ-1 (PARK7) function has also been implicated as a familial form of PD,(49) and DJ-1 has been implicated in Nrf2 activation.(50) DJ-1 KO mice are viable and fertile, but the dopaminergic neurons of the mice were hypersensitive to mitochondrial complex 1 inhibitor 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridne (MPTP).(51) Furthermore, embryonic cortical neurons from DJ-1 KO mice are sensitive to oxidative stress. The oxidation of Cys106 in DJ-1 during oxidative stress is responsible for the localization of DJ-1 to the mitochondria as well as for its neuroprotective function.(52) DJ-1, such as Parkin and Pink1, is important for the quality control of mitochondria, and the loss of DJ-1 causes the mitochondrial depolarization and fragmentation.(53) Although DJ-1 does not improve the defective mitochondria phenotype caused by the PINK1 deficiency, DJ-1 protects rotenone-induced mitochondrial damage in the absence of PINK1, indicating that DJ-1 may act independently of PINK1/Parkin to control mitochondrial function.(53) DJ-1 activates AKT in dopaminergic neurons of the substantia nigra neurons in response to MPTP,(54) and DJ-1 is necessary for AKT to be recruited to the membrane in response to H2O2. Tanti et al.(55) recently showed that the WD-40 repeat protein SG2A mediates the interactions between Akt and DJ-1, and these 3 factors colocalize at the mitochondria and plasma membrane. Although Nrf2 is activated independently of DJ-1 in certain settings,(56) PINK1, Parkin and DJ-1 may act at the mitochondrial level to initiate a signaling cascade for Nrf2 activation during oxidative stress in a cell-specific manner (Fig. 4).
Nrf2 activation via electron transport inhibition
In contrast to the putative permissive role of mitochondria described above, recent studies in C. elegans have shown that defects in the mitochondrial ETC activate a stress response, including UPRmt, mitochondrial biogenesis and the activation of the functional Nrf2 homolog SKN-1, which lead to prolonged longevity.(57) Surfeit locus protein 1 (Surf1) is a nuclear-encoded mitochondrial inner membrane protein that aids in the assembly of the COX holoenzyme. In humans, Surf1 loss of function mutation leads to the development of Leigh’s syndrome; however, the Surf1 truncation mutation in mice prolongs the mean longevity of the mice. Interestingly, a recent study showed that a Surf1 mutation provokes mitochondrial retrograde signaling and initiates the UPRmt in skeletal muscle and Nrf2 activation in the heart.(58) Therefore, the same mutation generates different tissue-specific stress responses. Furthermore, Dassa et al.(59) showed that Nrf2 is activated in human fibroblasts isolated from the patients who harbor the mitochondria ATPase6 gene mutations. In addition, Nrf2 is activated by the treatment of oligomycin, an inhibitor of ATP synthase; however, the study examined the Nrf2 activity only via immunohistochemistry and the activity of superoxide dismutase as the downstream target of Nrf2. Further studies are needed to conclude whether the inhibition of ATP synthase does indeed activate Nrf2.
The Nrf2 system is defective in several mitochondria-related diseases
Mitochondrial dysfunction plays an important role in the pathogenesis of a wide range of diseases, including neurodegenerative diseases. Mitochondrial dysfunction has been implicated as one of the etiologic factors of PD. Indeed, recent reports have demonstrated that Nrf2 activity is decreased in olfactory neurosphere-derived cells from PD patients, indicating a defect in Nrf2 activity in PD, although the involvement of mitochondria is currently unclear.(60,61) Friedreich’s ataxia (FRDA) is caused by mutations in the FXN gene that encode the mitochondrial matrix protein frataxin, which is indispensable for iron-sulfur cluster biosynthesis in the mitochondria. Indeed, Nrf2 activation by oxidative stress is blocked in fibroblasts derived from FRDA patients or in FXN-knockdown NSC34 neurons.(62,63) The defective Nrf2 response in the fibroblasts of FRDA patients is rescued by treatment with Euk134, a catalase mimetic, indicating that chronic H2O2 generation is a causative factor of this response.(62) Furthermore, Nrf2 activity is suppressed in dorsal root ganglia (DRG) neurons in the FRDA mouse model as well as in several FXN-knockdown cell line models using DRG (ND7/23 cells) and Schwann cells (T265 cells).(64) Interestingly, Sahdeo et al.(65) also reported that Nrf2 regulates FXN expression by binding to the evolutionary conserved ARE in the upstream of FXN. Although the precise mechanisms of Nrf2 dysfunction have yet to be determined, these results indicate that mitochondrial defects lead to Nrf2 function impairment. Therefore, the impaired Nrf2 pathway in the aforementioned diseases may be a potential target for future therapeutic drugs.
Conclusions and Perspectives
We have summarized the recent advances regarding the cross-talk between the Nrf2 system and mitochondria, specifically focusing on the interaction between Nrf2-mediated antioxidant mechanisms and the regulation of mitochondrial integrity. Consistent with the data discussed above, we propose that the coordinated induction of HO-1, p62 and NQO1 by Nrf2 activation aids in the maintenance and removal of damaged mitochondria, depending on the severity of the mitochondrial damage, and provokes the biogenesis of healthy mitochondria by inducing NRF-1 expression. However, this hypothesis remains to be tested in future studies. We described that mitochondrial abnormality both activates and represses Nrf2, and detailed mechanisms should be clarified in the future studies.
In addition to energy production and ROS production, mitochondria also play an important role in heat generation in mammals.(66) Interestingly, Nrf2 is highly expressed in thermo-regulating organs such as the thyroid and brown adipose tissue in the mouse embryo and regulates the expression of uncoupling protein 3.(5,67) Therefore, we surmise that tight cooperation between Nrf2 function and the mitochondria has evolved beyond the regulation of mitochondrial quality control, which may be an interesting area to assess in future studies.
Acknowledgments
We thank Drs. Junsei Mimura, Atsushi Maruyama and Hiromi Yamazaki for their helpful advice and discussion. This work was supported by a Hirosaki University Institutional Research Grant and Grant-Aid for Scientific Research on Innovative Areas “Oxygen Biology: a new criterion for integrated understanding of life” (No. 26111010) of MEXT, Japan.
Abbreviations
- ALCAR
acetyl-l-carnitine
- ARE
antioxidant responsive element
- CO
carbon monoxide
- ΔΨm
mitochondrial membrane potential
- ETC
electron transport chain
- HO-1
heme oxygenase-1
- NQO1
NAD(P)H dehydrogenase quinone 1
- NRF-1
nuclear respiratory factor 1
- Nrf2
nuclear factor erythroid-derived 2-related factor 2
- PD
parkinson’s disease
- ROS
reactive oxygen species
Conflict of Interest
No potential conflicts of interest were disclosed.
References
- 1.Tschopp J. Mitochondria: Sovereign of inflammation? Eur J Immunol. 2011;41:1196–1202. doi: 10.1002/eji.201141436. [DOI] [PubMed] [Google Scholar]
- 2.Indo HP, Davidson M, Yen HC, et al. Evidence of ROS generation by mitochondria in cells with impaired electron transport chain and mitochondrial DNA damage. Mitochondrion. 2007;7:106–118. doi: 10.1016/j.mito.2006.11.026. [DOI] [PubMed] [Google Scholar]
- 3.Haynes CM, Fiorese CJ, Lin YF. Evaluating and responding to mitochondrial dysfunction: the mitochondrial unfolded-protein response and beyond. Trends Cell Biol. 2013;23:311–318. doi: 10.1016/j.tcb.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yun J, Finkel T. Mitohormesis. Cell Metab. 2014;19:757–766. doi: 10.1016/j.cmet.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Itoh K, Mimura J, Yamamoto M. Discovery of the negative regulator of Nrf2, Keap1: a historical overview. Antioxid Redox Signal. 2010;13:1665–1678. doi: 10.1089/ars.2010.3222. [DOI] [PubMed] [Google Scholar]
- 6.Chan K, Lu R, Chang JC, Kan YW. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc Natl Acad Sci USA. 1966;93:13943–13948. doi: 10.1073/pnas.93.24.13943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Itoh K, Wakabayashi N, Katoh Y, et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kobayashi M, Itoh K, Suzuki T, et al. Identification of the interactive interface and phylogenic conservation of the Nrf2-Keap1 system. Genes Cells. 2002;7:807–820. doi: 10.1046/j.1365-2443.2002.00561.x. [DOI] [PubMed] [Google Scholar]
- 9.Katoh Y, Iida K, Kang MI, et al. Evolutionary conserved N-terminal domain of Nrf2 is essential for the Keap1-mediated degradation of the protein by proteasome. Arch Biochem Biophys. 2005;433:342–350. doi: 10.1016/j.abb.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 10.Karapetian RN, Evstafieva AG, Abaeva IS, et al. Nuclear oncoprotein prothymosin alpha is a partner of Keap1: implications for expression of oxidative stress-protecting genes. Mol Cell Biol. 2005;25:1089–1099. doi: 10.1128/MCB.25.3.1089-1099.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Niture SK, Jaiswal AK. Inhibitor of Nrf2 (INrf2 or Keap1) protein degrades Bcl-xL via phosphoglyceratemutase 5 and controls cellular apoptosis. J Biol Chem. 2011;286:44542–44556. doi: 10.1074/jbc.M111.275073. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12.Xu Y, Fang F, Miriyala S, et al. KEAP1 is a redox sensitive target that arbitrates the opposing radiosensitive effects of parthenolide in normal and cancer cells. Cancer Res. 2013;73:4406–4417. doi: 10.1158/0008-5472.CAN-12-4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee DF, Kuo HP, Liu M, et al. KEAP1 E3 ligase-mediated downregulation of NF-κB signaling by targeting IKKβ. Mol Cell. 2009;36:131–140. doi: 10.1016/j.molcel.2009.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Komatsu M, Kurokawa H, Waguri S, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12:213–223. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- 15.Niture SK, Jaiswal AK. INrf2 (Keap1) targets Bcl-2 degradation and controls cellular apoptosis. Cell Death Differ. 2011;18:439–451. doi: 10.1038/cdd.2010.114. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 16.Ishii T, Yanagawa T, Kawane T, et al. Murine peritoneal macrophages induce a novel 60-kDa protein with structural similarity to a tyrosine kinase p56lck-associated protein in response to oxidative stress. Biochem Biophys Res Commun. 1996;226:456–460. doi: 10.1006/bbrc.1996.1377. [DOI] [PubMed] [Google Scholar]
- 17.Ishii T, Itoh K, Takahashi S, et al. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J Biol Chem. 2000;275:16023–16029. doi: 10.1074/jbc.275.21.16023. [DOI] [PubMed] [Google Scholar]
- 18.Yoshikawa Y, Ogawa M, Hain T, et al. Listeria monocytogenes ActA-mediated escape from autophagic recognition. Nat Cell Biol. 2009;11:1233–1240. doi: 10.1038/ncb1967. [DOI] [PubMed] [Google Scholar]
- 19.Geisler S, Holmström KM, Skujat D, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12:119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- 20.Kosaka K, Mimura J, Itoh K, et al. Role of Nrf2 and p62/ZIP in the neurite outgrowth by carnosic acid in PC12h cells. J Biochem. 2010;147:73–81. doi: 10.1093/jb/mvp149. [DOI] [PubMed] [Google Scholar]
- 21.Hancock R, Bertrand HC, Tsujita T, et al. Peptide inhibitors of the Keap1-Nrf2 protein-protein interaction. Free Radic Biol Med. 2012;52:444–451. doi: 10.1016/j.freeradbiomed.2011.10.486. [DOI] [PubMed] [Google Scholar]
- 22.Ichimura Y, Waguri S, Sou YS, et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell. 2013;51:618–631. doi: 10.1016/j.molcel.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 23.Kwon J, Han E, Bui CB, et al. Assurance of mitochondrial integrity and mammalian longevity by the p62-Keap1-Nrf2-Nqo1 cascade. EMBO Rep. 2012;13:150–156. doi: 10.1038/embor.2011.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seibenhener ML, Du Y, Diaz-Meco MT, Moscat J, Wooten MC, Wooten MW. A role for sequestosome 1/p62 in mitochondrial dynamics, import and genome integrity. Biochim Biophys Acta. 2013;1833:452–459. doi: 10.1016/j.bbamcr.2012.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seibenhener ML, Zhao T, Du Y, et al. Behavioral effects of SQSTM1/p62 overexpression in mice: support for a mitochondrial role in depression and anxiety. Behav Brain Res. 2013;248:94–103. doi: 10.1016/j.bbr.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alam J, Cook JL. How many transcription factors does it take to turn on the heme oxygenase-1 gene? Am J Respir Cell Mol Biol. 2007;36:166–174. doi: 10.1165/rcmb.2006-0340TR. [DOI] [PubMed] [Google Scholar]
- 27.Piantadosi CA. Biological chemistry of carbon monoxide. Antioxid Redox Signal. 2002;4:259–270. doi: 10.1089/152308602753666316. [DOI] [PubMed] [Google Scholar]
- 28.D'Amico G, Lam F, Hagen T, Moncada S. Inhibition of cellular respiration by endogenously produced carbon monoxide. J Cell Sci. 2006;119:2291–2298. doi: 10.1242/jcs.02914. [DOI] [PubMed] [Google Scholar]
- 29.Suliman HB, Carraway MS, Tatro LG, Piantadosi CA. A new activating role for CO in cardiac mitochondrial biogenesis. J Cell Sci. 2007;120:299–308. doi: 10.1242/jcs.03318. [DOI] [PubMed] [Google Scholar]
- 30.Zhu H, Li Y. NAD(P)H: quinoneoxidoreductase 1 and its potential protective role in cardiovascular diseases and related conditions. Cardiovasc Toxicol. 2012;12:39–45. doi: 10.1007/s12012-011-9136-9. [DOI] [PubMed] [Google Scholar]
- 31.Dominy JE, Puigserver P. Mitochondrial biogenesis through activation of nuclear signaling proteins. Cold Spring Harb Perspect Biol. 2013;5 doi: 10.1101/cshperspect.a015008. DOI: 10.1101/cshperspect.a015008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Piantadosi CA, Carraway MS, Babiker A, Suliman HB. Heme oxygenase-1 regulates cardiac mitochondrial biogenesis via Nrf2-mediated transcriptional control of nuclear respiratory factor-1. Circ Res. 2008;103:1232–1240. doi: 10.1161/01.RES.0000338597.71702.ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Piantadosi CA, Suliman HB. Mitochondrial transcription factor A induction by redox activation of nuclear respiratory factor 1. J Biol Chem. 2006;281:324–333. doi: 10.1074/jbc.M508805200. [DOI] [PubMed] [Google Scholar]
- 34.Athale J, Ulrich A, Macgarvey NC, et al. Nrf2 promotes alveolar mitochondrial biogenesis and resolution of lung injury in Staphylococcus aureus pneumonia in mice. Free Radic Biol Med. 2012;53:1584–1594. doi: 10.1016/j.freeradbiomed.2012.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.MacGarvey NC, Suliman HB, Bartz RR, et al. Activation of mitochondrial biogenesis by heme oxygenase-1-mediated NF-E2-related factor-2 induction rescues mice from lethal Staphylococcus aureus sepsis. Am J Respir Crit Care Med. 2012;185:851–861. doi: 10.1164/rccm.201106-1152OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hota SK, Barhwal K, Singh SB, Sairam M, Ilavazhagan G. NR1 and GluR2 expression mediates excitotoxicity in chronic hypobaric hypoxia. J Neurosci Res. 2008;86:1142–1152. doi: 10.1002/jnr.21554. [DOI] [PubMed] [Google Scholar]
- 37.Barhwal K, Hota SK, Jain V, Prasad D, Singh SB, Ilavazhagan G. Acetyl-l-carnitine (ALCAR) prevents hypobaric hypoxia-induced spatial memory impairment through extracellular related kinase-mediated nuclear factor erythroid 2-related factor 2 phosphorylation. Neuroscience. 2009;161:501–514. doi: 10.1016/j.neuroscience.2009.02.086. [DOI] [PubMed] [Google Scholar]
- 38.Kosaka K, Mimura J, Itoh K, et al. Role of Nrf2 and p62/ZIP in the neurite outgrowth by carnosic acid in PC12h cells. J Biochem. 2010;147:73–81. doi: 10.1093/jb/mvp149. [DOI] [PubMed] [Google Scholar]
- 39.Hota KB, Hota SK, Chaurasia OP, Singh SB. Acetyl-L-carnitine-mediated neuroprotection during hypoxia is attributed to ERK1/2-Nrf2-regulated mitochondrial biosynthesis. Hippocampus. 2012;22:723–736. doi: 10.1002/hipo.20934. [DOI] [PubMed] [Google Scholar]
- 40.Kim SK, Joe Y, Zheng M, et al. Resveratrol induces hepatic mitochondrial biogenesis through the sequential activation of nitric oxide and carbon monoxide production. Antioxid Redox Signal. 2014;20:2589–2605. doi: 10.1089/ars.2012.5138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim TH, Hur EG, Kang SJ, et al. NRF2 blockade suppresses colon tumor angiogenesis by inhibiting hypoxia-induced activation of HIF-1α. Cancer Res. 2011;71:2260–2275. doi: 10.1158/0008-5472.CAN-10-3007. [DOI] [PubMed] [Google Scholar]
- 42.Holmström KM, Baird L, Zhang Y, et al. Nrf2 impacts cellular bioenergetics by controlling substrate availability for mitochondrial respiration. Biol Open. 2013;2:761–770. doi: 10.1242/bio.20134853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ludtmann MH, Angelova PR, Zhang Y, Abramov AY, Dinkova-Kostova AT. Nrf2 affects the efficiency of mitochondrial fatty acid oxidation. Biochem J. 2014;457:415–424. doi: 10.1042/BJ20130863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ricart KC, Bolisetty S, Johnson MS, et al. The permissive role of mitochondria in the induction of haem oxygenase-1 in endothelial cells. Biochem J. 2009;419:427–436. doi: 10.1042/BJ20081350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scarffe LA, Stevens DA, Dawson VL, Dawson TM. Parkin and PINK1: much more than mitophagy. Trends Neurosci. 2014;37:315–324. doi: 10.1016/j.tins.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koh H, Chung J. PINK1 as a molecular checkpoint in the maintenance of mitochondrial function and integrity. Mol Cells. 2012;34:7–13. doi: 10.1007/s10059-012-0100-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pridgeon JW, Olzmann JA, Chin LS, Li L. PINK1 protects against oxidative stress by phosphorylating mitochondrial chaperone TRAP1. PLoS Biol. 2007;5:e172. doi: 10.1371/journal.pbio.0050172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chien WL, Lee TR, Hung SY, Kang KH, Lee MJ, Fu WM. Impairment of oxidative stress-induced heme oxygenase-1 expression by the defect of Parkinson-related gene of PINK1. J Neurochem. 2011;117:643–653. doi: 10.1111/j.1471-4159.2011.07229.x. [DOI] [PubMed] [Google Scholar]
- 49.Clements CM, McNally RS, Conti BJ, Mak TW, Ting JP. DJ-1, a cancer- and Parkinson’s disease-associated protein, stabilizes the antioxidant transcriptional master regulator Nrf2. Proc Natl Acad Sci USA. 2006;103:15091–15096. doi: 10.1073/pnas.0607260103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Malhotra D, Thimmulappa R, Navas-Acien A, et al. Decline in NRF2-regulated antioxidants in chronic obstructive pulmonary disease lungs due to loss of its positive regulator, DJ-1. Am J Respir Crit Care Med. 2008;178:592–604. doi: 10.1164/rccm.200803-380OC. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 51.Kim RH, Smith PD, Aleyasin H, et al. Hypersensitivity of DJ-1-deficient mice to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrindine (MPTP) and oxidative stress. Proc Natl Acad Sci USA. 2005;102:5215–5220. doi: 10.1073/pnas.0501282102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Canet-Avilés RM, Wilson MA, Miller DW, et al. The Parkinson’s disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc Natl Acad Sci U S A. 2004;101:9103–9108. doi: 10.1073/pnas.0402959101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thomas KJ, McCoy MK, Blackinton J, et al. DJ-1 acts in parallel to the PINK1/parkin pathway to control mitochondrial function and autophagy. Hum Mol Genet. 2011;20:40–50. doi: 10.1093/hmg/ddq430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Aleyasin H, Rousseaux MW, Marcogliese PC. DJ-1 protects the nigrostriatal axis from the neurotoxin MPTP by modulation of the AKT pathway. Proc Natl Acad Sci U S A. 2010;107:3186–3191. doi: 10.1073/pnas.0914876107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tanti GK, Goswami SK. SG2NA recruits DJ-1 and Akt into the mitochondria and membrane to protect cells from oxidative damage. Free Radic Biol Med. 2014;75:1–13. doi: 10.1016/j.freeradbiomed.2014.07.009. [DOI] [PubMed] [Google Scholar]
- 56.Gan L, Johnson DA, Johnson JA. Keap1-Nrf2 activation in the presence and absence of DJ-1. Eur J Neurosci. 2010;31:967–977. doi: 10.1111/j.1460-9568.2010.07138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pulliam DA, Bhattacharya A, Van Remmen H. Mitochondrial dysfunction in aging and longevity: a causal or protective role? Antioxid Redox Signal. 2013;19:1373–1387. doi: 10.1089/ars.2012.4950. [DOI] [PubMed] [Google Scholar]
- 58.Pulliam DA, Deepa SS, Liu Y, et al. Complex IV-deficient Surf1(-/-) mice initiate mitochondrial stress responses. Biochem J. 2014;462:359–371. doi: 10.1042/BJ20140291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dassa EP, Paupe V, Gonçalves S, Rustin P. The mtDNA NARP mutation activates the actin-Nrf2 signaling of antioxidant defenses. Biochem Biophys Res Commun. 2008;368:620–624. doi: 10.1016/j.bbrc.2008.01.125. [DOI] [PubMed] [Google Scholar]
- 60.Matigian N, Abrahamsen G, Sutharsan R, et al. Disease-specific, neurosphere-derived cells as models for brain disorders. Dis Model Mech. 2010;3:785–798. doi: 10.1242/dmm.005447. [DOI] [PubMed] [Google Scholar]
- 61.Abrahamsen G, Fan Y, Matigian N, et al. A patient-derived stem cell model of hereditary spastic paraplegia with SPAST mutations. Dis Model Mech. 2013;6:489–502. doi: 10.1242/dmm.010884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Paupe V, Dassa EP, Goncalves S, et al. Impaired nuclear Nrf2 translocation undermines the oxidative stress response in Friedreich ataxia. PLoS One. 2009;4:e4253. doi: 10.1371/journal.pone.0004253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.D'Oria V, Petrini S, Travaglini L, et al. Frataxin deficiency leads to reduced expression and impaired translocation of NF-E2-related factor 2 (Nrf2) in cultured motor neurons. Int J Mol Sci. 2013;14:7853–7865. doi: 10.3390/ijms14047853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shan Y, Schoenfeld RA, Hayashi G, et al. Frataxin deficiency leads to defects in expression of antioxidants and Nrf2 expression in dorsal root ganglia of the Friedreich’s ataxia YG8R mouse model. Antioxid Redox Signal. 2013;19:1481–1493. doi: 10.1089/ars.2012.4537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sahdeo S, Scott BD, McMackin MZ, et al. Dyclonine rescues frataxin deficiency in animal models and buccal cells of patients with Friedreich’s ataxia. Hum Mol Genet. 2014;23:6848–6862. doi: 10.1093/hmg/ddu408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brand MD, Couture P, Else PL, Withers KW, Hulbert AJ. Evolution of energy metabolism. Proton permeability of the inner membrane of liver mitochondria is greater in a mammal than in a reptile. Biochem J. 1991;275:81–86. doi: 10.1042/bj2750081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Anedda A, López-Bernardo E, Acosta-Iborra B, Saadeh Suleiman M, Landázuri MO, Cadenas S. The transcription factor Nrf2 promotes survival by enhancing the expression of uncouplingprotein 3 under conditions of oxidative stress. Free Radic Biol Med. 2013;61:395–407. doi: 10.1016/j.freeradbiomed.2013.04.007. [DOI] [PubMed] [Google Scholar]
- 68.Rada P, Rojo AI, Chowdhry S, McMahon M, Hayes JD, Cuadrado A. SCF/β-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol Cell Biol. 2011;31:1121–1133. doi: 10.1128/MCB.01204-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jain AK, Jaiswal AK. GSK-3beta acts upstream of Fyn kinase in regulation of nuclear export and degradation of NF-E2 related factor 2. J Biol Chem. 2007;282:16502–16510. doi: 10.1074/jbc.M611336200. [DOI] [PubMed] [Google Scholar]