

Abstract
Aims
Selisistat, a selective SirT1 inhibitor is being developed as a potentially disease-modifying therapeutic for Huntington's disease (HD). This was the first study of selisistat in HD patients and was primarily aimed at development of pharmacodynamic biomarkers.
Methods
This was a randomized, double-blind, placebo-controlled, multicentre exploratory study. Fifty-five male and female patients in early stage HD were randomized to receive 10 mg or 100 mg of selisistat or placebo once daily for 14 days. Blood sampling, clinical and safety assessments were conducted throughout the study. Candidate pharmacodynamic markers included circulating soluble huntingtin and innate immune markers.
Results
Selisistat was found to be safe and well tolerated, and systemic exposure parameters showed that the average steady-state plasma concentration achieved at the 10 mg dose level (125 nm) was comparable with the IC50 for SirT1 inhibition. No adverse effects on motor, cognitive or functional readouts were recorded. While circulating levels of soluble huntingtin were not affected by selisistat in this study, the biological samples collected have allowed development of assay technology for use in future studies. No effects on innate immune markers were seen.
Conclusions
Selisistat was found to be safe and well tolerated in early stage HD patients at plasma concentrations within the anticipated therapeutic concentration range.
Keywords: Huntington's disease, pharmacodynamics, pharmacokinetics, selisistat, SirT1 inhibition
What is Already Known about this Subject
Modulation of the acetylation status of mutant huntingtin via SirT1 inhibition has been shown to restore transcriptional dysregulation in models of Huntington's disease (HD).
Both nicotinamide and butyrate inhibit SirT1, but have insufficient potency and selectivity to test the SirT1 concept in patients with HD.
What this Study Adds
This was the first study with a selective SirT1 inhibitor in HD patients and shows that SirT1 inhibition is safe and well tolerated at plasma exposure levels providing benefit in non-clinical HD models, creating the basis for further studies of the pharmacodynamics of SirT1 modulation.
Introduction
Degradation of aggregate forming proteins is one of the main therapeutic approaches in neurodegenerative diseases to remove deposits, prevent accumulation of proteins in the brain and abrogate the fatal natural course of these diseases. Huntington's disease (HD) is a rare monogenetic, autosomal dominant disease that runs a relentless degenerative course over a period of 10 to 25 years by affecting primarily the central nervous system (CNS) leading to abnormal movements, personality changes and cognitive decline. The neuropathogenic processes are induced by the expression of mutant huntingtin (HTT). An expansion of a cytosine-adenine-guanine (CAG) triplet repeat encoding for the amino acid glutamine present in the N-terminal part of the HTT gene on chromosome 4 results in an extended polyglutamine stretch leading to misfolding and aggregate formation 1 causing changes in the brain and in peripheral tissues 2.
One mechanism being investigated as a potential approach to enhance clearance of mutant HTT is modulation of sirtuin activity. Sirtuins are NAD-dependent deacetylases which have been implicated in neuroprotection due to effects that were observed through either inhibition or overexpression of various sirtuins 3–5. It has been shown that acetylation of mutant HTT promotes its specific clearance and provides a link between protein acetylation and targeted degradation by autophagy 6.
SirT1 (silent information regulator T1) is a member of the sirtuin deacetylase family removing acetyl groups from lysine residues in histones and other proteins including mutant HTT 6–8. SirT1 is one of few deacetylases capable of deacetylating mutant HTT, and inhibition of SirT1 in transgenic Drosophila and R6/2 mice results in selective decrease in mutant HTT protein levels 8. It was therefore hypothesized that inhibition of SirT1 results in the selective clearance of mutant HTT without affecting levels of the normal protein.
Selisistat (6-chloro-2,3,4,9-tetrahydro-1H-carbazole-1-carboxamide, SEN0014196, EX-527) is a selective SirT1 inhibitor that inhibits recombinant human SirT1 with an IC50 of 98 nm. It is more than 200-fold selective over SirT2 and SirT3 and has been shown to inhibit deacetylation of several SirT1 substrates both in vitro and in vivo 9. The compound exhibits cyto- and neuroprotective activity against toxicity induced by mutant HTT in cellular and in vivo models of HD increasing survival, amelioration of psychomotor behaviour and improvement in histopathological endpoints in the most widely employed animal models of HD 10. In a previous clinical study, selisistat was shown to be well tolerated at single doses up to 600 mg and repeated doses up to 300 mg day−1 for up to 7 days to healthy volunteers 11. The availability of selective, safe SirT1 inhibitors such as selisistat therefore makes the acetylation-dependent clearance of mutant HTT a clinically testable therapeutic approach.
The primary objective of this exploratory study, representing the first administration of selisistat to HD patients, was to collect biological specimens to support the development of pharmacodynamic measures of target engagement and modulation of circulating levels of soluble HTT for use in further clinical development of the compound. This study was part of the PADDINGTON (Pharmacodynamic Approaches to Demonstration of Disease Modification in Huntington's Disease by SEN0014196) project, co-funded by the European Commission under the Seventh Framework Programme. Secondary objectives of the study included assessment of safety, tolerability and the pharmacokinetics of selisistat in HD patients as well as recording any acute phenotypical effects of repeated oral daily doses of selisistat for 14 days.
Methods
Study design
This was a randomized, double-blind, placebo-controlled, multicentre study encompassing six clinical sites in Germany (Ulm, Bochum), the United Kingdom (London, Manchester) and Poland (Cracow, Warsaw). Patients were randomly assigned to one of three parallel groups to receive either 10 or 100 mg selisistat or placebo (1:1:1). The study was conducted in accordance with the Declaration of Helsinki on the Ethical Principles for Medical Research Involving Human Subjects, adopted by the General Assembly of the World Medical Association in 1964, with subsequent amendments. The study was conducted in accordance with the protocol, the International Conference on Harmonization (ICH) guideline on Good Clinical Practices (GCP) and applicable local regulatory requirements and laws.
Study participants
All study participants provided written informed consent prior to enrolment. The protocol was reviewed and approved by the Ethics Committee of Ulm University, Ulm, Germany, the Research Ethics Committee 3 (REC3) of South West London, United Kingdom and the Bioethics Committee at the Psychiatry and Neurology Institute, Warsaw, Poland. Recruitment of trial participants was based on clinical characteristics and disease burden scores obtained in the context of REGISTRY, the large European natural history study of HD conducted by the European Huntington's Disease Network (EHDN) 12. Eligible subjects were early stage HD patients (stages I and II) aged 18 to 70 years with genetically confirmed disease (CAG repeat length ≥36). Females had to be surgically sterile or post-menopausal (2 years post-cessation of menses and/or follicular stimulating hormone >18 mIU ml−1 and serum oestradiol <110 pmol l−1). Other inclusion criteria included motor signs of HD with a motor score in the Unified Huntington's Disease Rating Scale (UHDRS'99) > 5, a total functional capacity (TFC) of ≥7 and a body weight greater than 50 kg. Consumption of alcohol was restricted to no more than 28 units (males) or 21 units (females) of alcohol per week. Patients with presence of psychosis and/or confusional states, with clinically significant laboratory or ECG abnormalities, haematological, hepatic, cardiac, or renal diseases or a history of infections with HIV or hepatitis B or C virus, or malignancies of any type were excluded. In addition, patients with prior or concomitant therapy with histone deacetylase (HDAC) inhibitors were excluded.
Participants self-administered a once daily oral dose of 10 or 100 mg selisistat or placebo at home in the morning after breakfast during a 14 day treatment period, with the exception of in-clinic visits on days 1, 2, 7 and 14, when selisistat was administered in the presence of the study staff. Time of dose was recorded in a patient diary. Study drug was supplied in size 0 hard gelatine capsules on days 1 and 7. Compliance was checked by counting capsules at each visit to the clinic. All participants returned for a post-study visit 14 days after their final dose.
Selisistat does not inhibit P-gp, CYP2B6, 2C9, 2C19, 2D6 or 3A4 and is a weak inhibitor of CYP1A2. On the basis of this profile, no restrictions regarding concomitant medication in the study were considered necessary, with the exception of HDAC inhibitors. All concomitant medication taken during the study was recorded with indication, daily dose, and start and stop dates of administration in the patient diary.
Safety and tolerability
Safety was assessed by physical examination and by monitoring of vital signs, safety laboratory measurements comprising routine haematology, serum chemistry and urinalysis pre-dose on days 1, 7, 14 and 28. Twelve-lead electrocardiograms were recorded pre-dose and 3 h after dosing on day 1 and 14 and were analyzed at a central ECG laboratory (iCardiac Technologies, Rochester, NY, USA). Adverse events were monitored throughout the study and patients were asked to record all potential adverse events (AEs) and concomitant medications in the diary, to be reviewed at all study visits.
Evaluation of clinical effects
All HD-related clinical rating assessments were performed at the visits to the clinic. All participants were evaluated with standardized clinical rating scales for HD to assess motor function, cognitive, neuropsychiatric and functional capacities. The following assessments were performed at baseline (day –1) and days 1 and 14: the UHDRS total motor score (TMS) with a range from 0 (unremarkable) to 124 points (worst score), total functional capacity (TFC) with a range from 13 (normal) to 0 points (totally dependent, bedridden), the functional assessment (FA) with up to 25 points and the independence scale (IS) in % 13,14 with higher scores indicative of higher functioning and greater independence. The neuropsychiatric battery included the EHDN Problem Behaviours Assessment – short version (PBA-s) 15, the Hospital Anxiety and Depression Scale 16 combined with the Snaith Irritability Scale (HADS-SIS) 17 and a modified version of the Frontal Systems Behavioural Scale, which was completed by the participants (FrSBe-p) 18. This modified version of the FrSBe contains 24 items to assess the frequency of certain behaviours and the level of distress caused by those behaviours, but in contrast to the original version we did not use reverse coding for several questions (items 19–24), since the reverse coding was confusing for HD patients in a previous study 18. The cognitive battery included trail making tests A and B 19, symbol digit modalities test (SDMT) 20, Stroop test 21 with evaluation of the total correct answers and the Pittsburgh sleep quality index (PSQI) 22. A mini-mental state examination (MMSE) was performed only at baseline.
Pharmacokinetics
Blood samples for determination of selisistat plasma concentrations were collected into 4 ml lithium heparin Vacutainer® tubes (Becton Dickinson UK Ltd, Oxford, UK) at pre-dose and 0.5, 1, 3, 4, 6, 8 and 24 h post-dose on days 1 and 14. After mixing, samples were placed on ice and centrifuged within 1 h of collection at 1500 g for 10 min at approximately 4°C. Plasma aliquots were stored within 2 h of collection at −70/−80°C until assayed. Plasma samples were analyzed using a fully validated LC-MS/MS procedure, with a lower limit of quantitation (LOQ) of 0.1 ng ml−1. The accuracy values, based upon the calibration standards across the range, were between 97.0% and 103% over the QC range. The pharmacokinetic analysis was conducted using WinNonlin Enterprise Version 5.2.1 (Pharsight Corporation, Mountain View, California, USA). Pharmacokinetic parameters were determined from plasma concentrations of selisistat using non-compartmental procedures. Concentration values lying outside the quantitation limits were identified, noted as < or > the LOQ value, and either excluded from computations or used as extrapolated values. Computations included the following area under the plasma concentration vs. time curves, AUC(0,τ), AUC(0,tlast), AUC(0,∞) and AUC(0,24 h), and 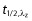 , whereas peak plasma concentration (Cmax) and time to peak concentration (tmax) were obtained directly from the plasma time−concentration curves. The plasma half-life was obtained by linear regression of the natural logarithmic concentration vs. time curve. The selection of data points for definition of the regression range was based on visual inspection of the data, and the regression range reported with the half-life. The AUC was determined by the linear trapezoidal rule. Actual blood sampling times post-dosing were used in the computation of the pharmacokinetic parameters.
, whereas peak plasma concentration (Cmax) and time to peak concentration (tmax) were obtained directly from the plasma time−concentration curves. The plasma half-life was obtained by linear regression of the natural logarithmic concentration vs. time curve. The selection of data points for definition of the regression range was based on visual inspection of the data, and the regression range reported with the half-life. The AUC was determined by the linear trapezoidal rule. Actual blood sampling times post-dosing were used in the computation of the pharmacokinetic parameters.
Pharmacodynamic assessments
Repeated serial blood samples were obtained for pharmacodynamic and transcriptomic assessments as follows: (i) for soluble HTT at baseline, 4 and 24 h post-dose on day 1 and day 14 and once during the day 28 follow-up visit and (ii) for innate immune markers at baseline, 4 h post-dose on days 1 and 14 and once during the day 28 visit. Blood samples for measurement of soluble HTT were collected in Becton Dickinson CPT™ tubes for preparation of peripheral blood mononuclear cells (PBMCs) and plasma for immune markers was collected in Becton Dickinson K2 EDTA Vacutainer® tubes. Overall, about 340 ml of blood were drawn from each subject during the study period but no more than 115 ml were drawn during any 24 h period. Biological specimen for pharmacodynamic and transcriptomic assessments was shipped to and stored at a central biorepository (BioRep SrL, Milan, Italy).
Levels of circulating soluble HTT were analyzed using a MesoScale ELISA method derived from the original ELISA assay with minor modifications 23. The six time point samples available for 54 patients enrolled in the study were assayed for soluble HTT evaluation. From one patient, insufficient amounts of plasma were available to perform HTT analysis. Quantification was performed through a 14 point standard curve ranging from 0.1 to 409 ng ml−1 by using a five parameter logistic (5PL) fitting algorithm and averaging the technical replicates. The estimated HTT levels were then normalized by total protein content.
Cytokine assays for evaluation of plasma immune markers were carried out using the MesoScale Discovery multiplex platform (Human ProInflammatory II 4-plex ultra-sensitive kit) as per the manufacturer's instructions. The operators were blinded during processing and statistical analyses were performed independently.
Statistical methods and analysis
No formal statistical assessment of sample size was conducted, as this was an exploratory clinical study. The number of subjects and the duration of the treatment were considered sufficient to achieve the objectives of the study. Statistical analyses of the clinical data in the study were performed using the statistical software SAS for Windows Version 9.1.3 (SAS Institute, Inc., Cary, NC, USA) and Stata Version 12 (StataCorp. Stata Statistical Software: College Station, TX: StataCorp LP). On account of the exploratory nature of the study, there were no specific predefined hypotheses and the statistical comparisons of clinical measures, soluble protein readouts and cytokines between treatment groups must be interpreted in an exploratory rather than a confirmatory way.
Ordinal and nominal data are described by absolute and relative frequencies and continuous data by mean, standard deviation and range. Day 14 clinical measures were compared between treatment groups using an analysis of covariance (ancova) model adjusting for baseline. Where there was suggestion of severe deviation from normal assumptions, bias corrected and accelerated bootstrap confidence intervals were calculated. Since there are two treatment groups, 97.5 bootstrap confidence intervals were used to infer whether P values were < or >0.05.
Since there were extra measurements at intermediate time points, soluble HTT and innate immune marker data were analyzed using an extension of the ancova model for randomized controlled trials with a single follow-up and baseline measure 24. Simultaneous estimation of treatment differences at all follow-up visits adjusted for baseline was achieved using a linear mixed model that allows for correlated responses at all time points, including the baseline visit. The model enforces a zero treatment effect at baseline, but imposes no restrictions from day 1 onwards. The model was fitted to log transformed data since this was found to improve normality assumptions.
Results
Patient characteristics
A total of 63 Caucasian male and female subjects were screened between March and November 2011. There were four screen failures and four consent withdrawals before randomization. Consequently, 55 patients were enrolled into the study and randomized to receive either selisistat 10 mg day−1 (n = 17), 100 mg day−1 (n = 19) or placebo (n = 19). All 55 participants completed the study and were included in the safety as well as PK analyses. One patient was excluded from the UHDRS outcome measures due to highly variable clinical presentation before, during and after the study independently of the study drug intake. Enrolled subjects were in good general health without any clinically significant medical history and without clinically significant findings from physical examination and laboratory results. The basic demographic details and clinical characteristics of motor, behavioural, functional and cognitive assessments are summarized in Table 1. There were more males than females in each of the three treatment arms, but no suggestion that the ratio of males to females was materially different between the groups. There were no statistically significant differences between the demographic and clinical baseline data, but the mean age of the 10 mg selisistat treatment group was slightly lower compared with the 100 mg and the placebo groups. Age of motor onset of HD was the lowest in the 10 mg selisistat group and highest in the 100 mg group, while the mean CAG repeat length was highest in the 10 mg group and lowest in the 100 mg group, respectively. Mean values for vital sign measurements at screening and the day before dosing (baseline) were similar for all treatment groups. None of the slight imbalances at baseline were considered sufficient to necessitate any additional adjustments in the statistical analysis.
Table 1.
Demographics and baseline characteristics of participants
| Parameter | Placebo (n = 19) | Selisistat 10 mg (n = 17) | Selisistat 100 mg (n = 19) |
|---|---|---|---|
| Age (years) | 51.9 ± 8.2 | 46.5 ± 10.2 | 54.1 ± 10.1 |
| (38–64) | (33–67) | (28–68) | |
| Gender | M = 12, F = 7 | M = 14, F = 3 | M = 13, F = 6 |
| Age at onset (years) | 46.1 ± 8.83 | 44.5 ± 10.11 | 47.6 ± 8.58 |
| (28–61) | (28–62) | (28–63) | |
| CAG repeats | 43.4 ± 2.14 | 44.1 ± 3.58 | 42.8 ± 2.48 |
| (41–50) | (40–53) | (40–49) | |
| UHDRS – TFC | 10.0 ± 2.13 | 10.6 ± 2.06 | 9.9 ± 2.23 |
| (7–13) | (7–13) | (7–13) | |
| UHDRS – TMS | 32.1 ± 11.55 | 29.5 ± 14.5 | 35.2 ± 15.37 |
| (14–53) | (8–64) | (17–81) | |
| MMSE | 26.0 ± 3.53 | 26.9 ± 2.45 | 26.9 ± 3.70 |
| (17–30) | (21–30) | (18–30) |
Means ± SD (range). MMSE, mini mental state examination; TFC, total functional capacity; TMS, total motor score; UHDRS, Unified Huntington's Disease Rating Scale.
Safety
Both dosages of selisistat were well tolerated. There were no dose- or treatment-related trends in terms of clinical laboratory evaluations, including liver function tests, haematological parameters, vital signs or cardiac function. No serious adverse events (SAEs) or adverse events (AEs) leading to discontinuation of treatment occurred.
A total of 25 patients (45.5%) reported 44 AEs in total: seven subjects (36.8%) in the placebo group, nine subjects (52.9%) in the 10 mg selisistat group and nine subjects (47.4%) of the 100 mg selisistat group. All of the AEs except four were mild.
The most frequently reported AEs were arterial hypertension (n = 7, 12.7%) and headache (n = 4, 7.3%) reported by two and one subjects in the 10 mg selisistat group, four and one subjects in the 100 mg selisistat group and by one and two subjects in the placebo group, respectively. Hypertension was the most commonly reported AE that was assessed as possibly related to the study drug by the investigator (one, three and one subjects in the 10 mg, 100 mg and placebo groups, respectively). Only a few cases of significant blood pressure changes were observed in the vital signs assessments, however, with no consistent pattern across treatment groups and the data do therefore not suggest that selisistat had an effect on systolic or diastolic blood pressure. No effects on blood pressure were observed in animal toxicity studies or in the preceding study in healthy volunteers 11.
The mean changes in the clinical chemistry, haematology and urinalysis parameters on days 7, 14 and 28 as compared with baseline were not appreciably different between groups. Two subjects from the 100 mg selisistat group developed slight increases in serum alanine transaminase levels (ALT) on days 14 and 28 (1.2- and 2.4-fold the upper limit of normal, respectively) and one subject in the placebo group showed high total bilirubin on day 7 (1.3-fold the upper limit of normal).
Based on the centrally evaluated ECGs (pre-dose and 3 h post-dose on day 1 and 14), selisistat did not have a material effect on heart rate, the PR or the QRS interval. At baseline, mean QTcF in the 100 mg selisistat group (401 ms) was 7 ms shorter than in the placebo (408 ms) and the 10 mg selisistat groups (408 ms). A mild shortening of the mean change-from-baseline QTcF interval (ΔQTcF) up to 8 ms was observed in the placebo and the 10 mg selisistat groups from day 1 to day 14, whereas no such trend was observed in the 100 mg selisistat group, in which ΔQTcF ranged between 2 and 4 ms. The resulting placebo-corrected ΔQTcF therefore increased with study duration in the selisistat 100 mg group, reaching 9.6 ms (upper bound of confidence interval 18.2 ms) on the post-dosing time point on day 14.
Clinical effects
Table 2 displays the results of all clinical, cognitive and neuropsychiatric assessments in each treatment arm, at baseline and on day 14. Also shown are estimates and 95% confidence intervals for comparisons of each active group with placebo at day 14 adjusted for baseline, and an overall test for differences between groups. There was little evidence of a treatment effect at day 14 for any of the measurements.
Table 2.
Mean (SD) of clinical outcomes at baseline and day 14 by treatment group and between group differences at day 14 adjusted for baseline
| Placebo | 10 mg | 100 mg | Comparison (mean (95% CI)) of change between treatment groups | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Baseline | Day 14 | Baseline | Day 14 | Baseline | Day14 | 10 mg vs. placebo | 100 mg vs. placebo | Joint test P value | |
| UHDRS – TMS | 32.11 (11.55) | 31.63 (12.07) | 29.53 (14.5) | 26.12 (14.68) | 35.16 (15.37) | 33.05 (10.74) | −3.45 (−7.09, −0.95)† | −1.03 (−5.97, 1.31)† | <0.05 |
| UHDRS – TFC | 10 (2.13) | 9.95 (2.2) | 10.65 (2.06) | 10.65 (2.06) | 9.89 (2.23) | 9.84 (2.24) | 0.06 (−0.07, 0.26)† | −0.0005 (−0.2917, 0.3124)† | >0.05 |
| Functional assessment | 20.37 (3.65) | 20.32 (3.68) | 21.53 (3.45) | 21.41 (3.78) | 20.26 (3.78) | 20.47 (3.47) | −0.04 (−0.5, 0.34)† | 0.26 (−0.15, 0.8)† | >0.05 |
| Independence scale | 83.68 (10.12) | 83.95 (9.8) | 86.47 (11.56) | 85.88 (11.89) | 82.37 (12.18) | 83.16 (11.57) | −0.77 (−2.06, −0.01)† | 0.49 (−0.44, 1.43)† | >0.05 |
| SDMT | 19.61 (7.77) | 21.82 (10.18) | 28.82 (15.61) | 32.29 (13.75) | 21.26 (9.18) | 22.79 (10.76) | 2.04 (−1.94, 6.02) | −0.61 (−4.29, 3.07) | 0.368 |
| Letter fluency | 17.63 (10.59) | 18.37 (10.78) | 23.71 (10.59) | 26.59 (15.66) | 16.53 (8.42) | 20.21 (8.51) | 2.27 (−2.51, 7.05) | 2.92 (−1.58, 7.43) | 0.402 |
| Category fluency | 11.58 (5.4) | 11.74 (4.56) | 13 (6.27) | 15.35 (6.92) | 11 (5.19) | 11.95 (4.64) | 2.52 (0.31, 4.73) | 0.66 (−1.48, 2.8) | 0.073 |
| Stroop colour | 38.32 (13.22) | 40 (13.79) | 50.47 (18.47) | 51.82 (19.56) | 40.11 (15.47) | 39.84 (13.91) | 1.7 (−4.59, 8) | −1.65 (−7.48, 4.18) | 0.559 |
| Stroop word | 56 (16.29) | 54.17 (20.44) | 68.06 (23.46) | 68.06 (22.55) | 56.95 (24.69) | 53.37 (18.85) | 3.98 (−2.33, 10.29) | −1.32 (−7.31, 4.68) | 0.223 |
| Stroop interference | 18.84 (10.37) | 21.79 (11.71) | 28.06 (10.79) | 30.47 (10.38) | 21.26 (9.9) | 23.74 (9.64) | 1.18 (−3.49, 5.85) | −0.02 (−4.3, 4.26) | 0.843 |
| Trail A time | 91.89 (52.67) | 96.22 (65.65) | 70.24 (34.23) | 55.18 (36.87) | 85.68 (61.34) | 84.74 (62.62) | −19.42 (−40.76, 0.15)† | −4.05 (−26.33, 16.13)† | >0.05 |
| Trail B time | 196.65 (62.05) | 191.41 (68.25) | 162.06 (60.73) | 141.41 (72.35) | 179.94 (52.64) | 173.95 (64.27) | −21.11 (−53.18, 5.9)† | −5.14 (−34.05, 24.19)† | >0.05 |
| PSQI | 5.47 (3.45) | 4.83 (3.63) | 5.19 (3.54) | 4.82 (3.52) | 6.11 (4.18) | 5.56 (4.23) | 0.16 (−1.05, 1.38) | 0.69 (−0.5, 1.87) | 0.477 |
| PBA-s mood* | 1.63 (3.11) | 2.26 (3.62) | 2.76 (3.67) | 1.82 (2.86) | 3.11 (5.45) | 2.11 (3.94) | −1.17 (−2.84, 0.39)† | −1.11 (−2.55, 0.17)† | >0.05 |
| PBA-s apathy* | 3.95 (4.14) | 4.11 (4.37) | 2.88 (3.64) | 2.59 (4.15) | 4.32 (4.11) | 3.58 (4.91) | −0.54 (−1.69, 0.91)† | −0.86 (−2.66, 1.14)† | >0.05 |
| PBA-s irritability/aggression* | 1.0 (1.63) | 1.58 (3.02) | 2.47 (3.14) | 1.94 (3.25) | 1.53 (2.86) | 1.0 (3.46) | −0.96 (−2.41, 0.73)† | −1.05 (−2.49, −0.1)† | >0.05 |
| HADS-SIS Anxiety subscore | 4.47 (3.47) | 4.11 (3.26) | 5.29 (4.78) | 4.41 (4.29) | 4.26 (3.28) | 4.63 (4.73) | −0.29 (−2.34, 1.76) | 0.68 (−1.31, 2.67) | 0.619 |
| HADS-SIS Depression subscore | 4.95 (4.58) | 4.95 (4.66) | 5.76 (4.64) | 4.82 (3.61) | 5.21 (3.69) | 5.26 (5.13) | −0.75 (−2.86, 1.36) | 0.11 (−1.93, 2.16) | 0.677 |
| HADS-SIS Irritability subscore | 4.58 (3.99) | 3.53 (3.78) | 6.65 (5.21) | 5.53 (5.34) | 3.95 (3.41) | 4.16 (3.06) | 0.62 (−1.45, 2.68) | 1.06 (−0.91, 3.02) | 0.56 |
| FrSBe-p total score | 76.61 (50.08) | 76.84 (56.81) | 97.47 (86.53) | 93.18 (74.89) | 86.58 (60.4) | 97.37 (67.57) | −0.04 (−19.95, 21.05)† | 13.9 (−3.47, 31.98)† | >0.05 |
| FRsBe executive dysfunction | 28.79 (19.55) | 28.32 (25.95) | 34.18 (33.06) | 33.59 (29.12) | 31.37 (24.24) | 39.47 (30.63) | 0.21 (−9.87, 10.5)† | 8.74 (0.83, 18.66)† | >0.05 |
| FRsBe disinihbition | 16.74 (8.81) | 16.32 (11.4) | 24.35 (22.88) | 22.41 (15.52) | 19.53 (12.73) | 20.21 (13.33) | 0.88 (−5.18, 6.07)† | 1.99 (−2.77, 6.66)† | >0.05 |
| FRsBe apathy | 32.83 (27.06) | 32.21 (28.25) | 38.94 (36.13) | 37.18 (32.57) | 35.68 (30.04) | 37.68 (33.04) | 0.09 (−10.56, 9.34)† | 3.46 (−5.91, 14.89)† | >0.05 |
For the PBA-s, only the three important components mood (depression + suicidal ideation), apathy and a composite of irritability/aggression were evaluated. †Indicates 95% bias corrected and accelerated bootstrap confidence interval instead of normal based confidence interval. Where bootstrap confidence intervals used, the joint test P value is recorded as <0.05 if either of the comparisons with placebo reached statistical significance below P = 0.025, based on assessment of 97.5% confidence intervals. In all other cases, this is the P value from the corresponding joint Wald test F value. Comparisons between 10 mg and 100 mg were not assessed as comparisons with placebo were deemed most relevant. FrSBe-p, frontal systems behavioural scale - participant self-rating; HADS, hospital anxiety and depression scale; PBA-s, problem behaviours assessment - short version; PSQI, Pittsburgh sleep quality index; SDMT, symbol digit modalities test; SIS, Snaith irritability scale; TMS, total motor score; UHDRS, Unified Huntington's Disease Rating Scale.
All groups showed an increase from baseline in the mean score for the verbal fluency test for letters, the verbal fluency test for category, the SDMT and the Stroop interference test on days 1 and 14. The mean score for the Stroop colour naming test increased on days 1 and 14 for the 10 mg and placebo groups. In almost all of these assessments improvements occurred between baseline and day 1 followed by stable values between days 1 and 14.
Pharmacokinetics
Pharmacokinetics were assessed on the first and last day of dosing and suggested essentially dose-linear behaviour, with systemic exposure parameters in line with previous studies in healthy volunteers 11. The average steady-state plasma concentration (Css,avg) at the 10 mg dose level (125 nm) was similar to the SirT1 IC50 for the compound (98 nm), while at the 100 mg dose level Css,avg (2.14 μm) was approximately 20-fold the enzymatic IC50. No significant gender difference in systemic exposure (AUC and Cmax) was observed, although a tendency for higher exposure in female patients was noted. Selisistat was found to be rapidly absorbed with a median tmax of 3 h. The between-subject variability in systemic exposure was moderately high at a mean coefficient of variation of 46%. No significant accumulation was observed at the 10 mg dose level, while the observed accumulation ratio at the 100 mg dose level was approximately 1.3, in line with data from healthy volunteers 11, where steady-state was reached within 4 days of dosing, Table 3.
Table 3.
Summary of pharmacokinetic parameters for selisistat in male and female subjects following 14 days of treatment at 10 and 100 mg once daily
| Parameter | Males | Females | ||||||
|---|---|---|---|---|---|---|---|---|
| 10 mg (n = 14) | 100 mg (n = 13) | 10 mg (n = 3) | 100 mg (n = 6) | |||||
| Day 1 | Day 14 | Day 1 | Day 14 | Day 1 | Day 14 | Day 1 | Day 14 | |
| AUC(0,τ) (μm h) | 2.87 ± 1.44 | 2.55 ± 1.14 | 33.4 ± 12.11 | 42.5 ± 21.4 | 2.94 ± 1.10 | 3.44 ± 1.53 | 44.9 ± 44.1 | 60.0 ± 49.7 |
| (50.2) | (44.7) | (36.3) | (50.4) | (37.4) | (44.5) | (98.2) | (92.8) | |
| AUC(0,∞) (μm h) | 3.02 ± 1.41 | NA | 34.3 ± 13.9 | NA | 3.03 ± 1.14 | NA | 48.1 ± 49.7 | NA |
| (46.7) | (40.4) | (37.6) | (103.3) | |||||
| Cmax (μm) | 0.65 ± 0.32 | 0.57 ± 0.22 | 5.99 ± 1.49 | 5.87 ± 1.93 | 0.77 ± 0.15 | 0.61 ± 0.07 | 7.51 ± 3.91 | 7.74 ± 4.42 |
| (49.2) | (38.6) | (24.9) | (32.9) | (19.5) | (11.5) | (52.1) | (57.1) | |
| tmax (h) | 3.00 | 2.00 | 3.00 | 3.00 | 3.00 | 3.00 | 3.00 | 3.46 |
| (1.00, 4.15) | (0.50, 4.00) | (1.00, 4.00) | (1.05, 6.00) | (3.00, 3.00) | (3.00, 3.00) | (2.97, 3.32) | (0.98, 4.00) | |
| t1/2 (h) | 2.51 ± 1.42 | 2.33 ± 0.85 | 2.86 ± 1.30 | 3.33 ± 1.56 | 2.12 ± 0.46 | 2.20 ± 0.55 | 3.20 ± 2.04 | 3.41 ± 1.64 |
| (56.6) | (36.5) | (45.5) | (46.8) | (21.7) | (25.0) | (63.8) | (48.1) | |
| RAobs | NA | 0.89 | NA | 1.27 | NA | 1.17 | NA | 1.33 |
Arithmetic means ± SD (coefficient of variation %), except for tmax, where median (range) is reported. τ, dosage interval, 24 h; AUC, area under the plasma concentration–time curve; Cmax, peak plasma concentration; NA, not applicable; RAobs, observed accumulation ratio; t1/2, half-life; tmax, time to peak concentration.
Soluble HTT
The ELISA assay effectively measured soluble total HTT in circulating blood cells. The method, described in Massai et al. 23, was capable of detecting the HTT protein extracted from PBMC in a reproducible way, and the assay showed a dynamic range of five orders of magnitude, with more than 100-fold signal-to-background ratio. However, statistical analysis of the levels of soluble HTT normalized for total protein did not show statistically significant changes between the treatment arms at any time point (Figure 1A).
Figure 1.

(A) Model predicted values and 95% confidence intervals of estimated log soluble Htt levels in peripheral blood mononuclear cells as a proportion of total protein by group at each time point. No statistically significant differences were observed between treatment groups at any time point.  , 10 mg selisistat;
, 10 mg selisistat;  , 100 mg selisistat;
, 100 mg selisistat;  , placebo. Model predicted values and 95% confidence intervals of estimated log plasma cytokine levels by group at each time point: (B) log IL-1, (C) log IL-6, (D) IL-8 and (E) TNF-α
, placebo. Model predicted values and 95% confidence intervals of estimated log plasma cytokine levels by group at each time point: (B) log IL-1, (C) log IL-6, (D) IL-8 and (E) TNF-α
Immune markers
Statistical analysis of four cytokines of interest (IL-1, IL-6, IL-8 and TNF-α) showed no statistical evidence of a treatment effect at any time point adjusted for baseline, and therefore there was no evidence to suggest that selisistat has any effect on modulation of these cytokines over the time period studied. Figure 1B–E shows model predicted estimates of the levels of each cytokine with 95% confidence interval bars, for each study arm.
Discussion
Clinical development of disease-modifying therapeutics for slowly progressive neurodegenerative conditions like HD is critically dependent on access to appropriate pharmacodynamic readouts to guide selection of doses and dosing schedules, aid in informing power calculations and define futility criteria for longitudinal efficacy studies. While symptomatic approaches typically address effects on a single clinical measurement or a subset of measurements related to a specific mechanism of action, there is no a priori possibility to identify which of the various motor, cognitive or functional domains will respond to a disease-modifying mechanism of action. Studies aimed at assessing efficacy by reducing rate of progression of disease will necessarily have a longitudinal design and may require treatment duration of 2 years or more to achieve unequivocal demonstration of clinical benefit. Running alongside other biomarker initiatives such as TRACK-HD 25,26, REGISTRY 12, PREDICT 27 and Enroll-HD 28, the PADDINGTON project is an effort to develop both mechanistic markers for SirT1-engagement and disease progression markers.
This was the first time the selective SirT1 inhibitor selisistat was administered to HD patients with the primary objective to collect biological specimens for the development of assays to assess SirT1 engagement as well as to evaluate any acute clinical effects of the compound in HD patients. Given the short treatment period of 14 days, no direct benefit was expected for patients in this trial. The treatment period was selected to ascertain that pharmacokinetic steady-state was achieved, based on projections from the previous trial in healthy volunteers 11. The dose levels were chosen with reference both to safety data from a previous trial in healthy volunteers and with the intent to achieve systemic exposure levels likely to bracket the concentration range shown to provide benefit in various non-clinical HD models 10. The average steady-state plasma concentration observed at the 10 mg dose level (125 nm) was similar to the SirT1 IC50 (98 nm) and corresponded with the lowest concentrations showing evidence of benefit in transfected cell models and the transgenic Drosophila model, while exposure at the 100 mg dose level (Css,avg of 2.14 μm) exceeded the plasma concentrations demonstrating beneficial effects on spontaneous locomotor activity and striatal degeneration in the R6/2 mouse model (370 nm) 10. At the 10 mg dose level, plasma concentrations of selisistat exceeded the IC99, IC90 and IC50 values (215, 150 and 98 nm respectively, 9) for SirT1 inhibition for 6–8 h of the dosing interval, while trough plasma concentrations at the 100 mg dose level exceeded the IC99 at all time points. While it is not known what level of SirT1 inhibition is required to achieve therapeutic benefit, data from the R6/2 mouse model, where the compound was given once daily, suggest that plasma concentrations exceeding the IC50 for 4–6 h per day are associated with beneficial effects. Accordingly, the dose range applied in the current study effectively covers the pharmacologically relevant dose as defined by the preclinical models in terms of plasma exposure and shows that the virtually complete inhibition of SirT1 achieved at the 100 mg dose level is well tolerated, at least over shorter periods of time. Data from longer term studies with selisistat will be required to determine whether a twice daily dose regimen may provide added benefit in terms of the pharmacodynamic readouts.
Selisistat was found to be safe and well tolerated when administered to early stage HD patients at 10 and 100 mg once daily over 14 days. There were no dose- or treatment-related trends in terms of clinical laboratory evaluations, vital signs or 12-lead ECG parameters and no clinically significant findings in physical examinations. In addition to the safety and tolerability data, we recorded a broad range of motor and cognitive performance measures as well as data on behaviour and sleep quality. There were no beneficial or adverse effects on any of the clinical outcome measures during the study, which is perhaps not surprising given the slowly progressive nature of HD and the proposed mechanism of action of selisistat. Indeed, the expected effect of a disease-modifying mechanism in HD may be a slowing of the progression of disease, and while pharmacodynamic or imaging markers may be able to detect differences over shorter periods of time, the motor, cognitive or functional outcome measures likely require treatment periods of 12 months or longer in order to detect significant effects 25,26.
SirT1 is widely expressed in the brain, with high levels in the cortex, hippocampus, cerebellum and hypothalamus, predominantly neurons 29. SirT1 is also expressed in peripheral tissues, including testis, skeletal muscle, liver, kidney and heart 30. Recent studies have shown that SirT1 has an important role in hypothalamic functions such as circadian control and metabolic sensoring 31, which is of special interest in the context of HD. In the lateral hypothalamus there are two groups of neurons expressing orexin and melanin concentrating hormone that have been shown to contribute in energy homeostasis, motor regulation, sleep regulation, stress response and social behaviours which are all clinically relevant in HD 32,33.
Most of the evidence for a role for SirT1 in HD derives from studies using genetic tools (e.g. genetic overexpression, RNAi approaches or knock-out systems) or using pharmacological activators whose specificity and selectivity has been questioned 34. The most direct evidence of a relevance for SirT1 modulation in HD comes from genetic overexpression and knock-out studies in mouse models of HD. In particular, Jeong et al. 6 and Jiang et al. 35 report protective effects of SirT1 overexpression in different mouse models of HD, expressing mutant HTT fragments of different length (both exon 1-based and full length). Jeong et al. also report that brain-specific knock-out (homozygous full knock-out, i.e. SirT1-/-) of SirT1 worsens the brain pathology of an exon-1 based mouse model (R6/2). Both Jiang et al. and Jeong et al. emphasize that the effects of overexpression of SirT1 in the mouse HD models need to be assessed in terms of how an increase in the expression of a protein known to be compartmentalized at the subcellular level and thought to act through multiprotein complexes can be attributed purely to increased HDAC enzymatic activity. There is increasing evidence that SirT1 may have other activities aside from HDAC activity. As an example, Pfister et al. 36 have shown that overexpression of different SirT1 mutants with catalytic-site mutations may still be neuroprotective while Seifert et al. 37 showed that SirT1 may have both HDAC-dependent and -independent roles. Finally, HDAC4, highly relevant for HD, is known to have at least two enzymatic activities (HDAC and SUMO ligase activities). Inducible or constitutive 38 overexpression of SirT1 in the brains of normal mice has been shown to have deleterious effects (impairment of metabolism and of motor function, and reference memory deficits as well as failure to deliver neuroprotection in ischaemic or MPTP-mediated neurodegeneration, respectively). Therefore, the neuroprotective effects of overexpression of SirT1 may not be solely dependent on increased HDAC activity and may engage protein substrates not normally engaged by the endogenous protein. To test the ‘activation hypothesis’, one would need a potent, specific, selective and brain penetrant pharmacological activator (to our knowledge, no such compounds have been described, 34) in order to determine if pharmacological activation of the endogenous SirT1 protein has therapeutic benefit. Conversely, knock-out approaches generally eliminate the entire gene which may not faithfully phenocopy pharmacological inhibition, particularly when both alleles are deleted. Pallos et al. 8 have performed careful genetic experiments in the same Drosophila model where selisistat has shown benefit, demonstrating protective effects of heterozygous null deletion (SirT1+/-) on the HD phenotype.
Selisistat has shown benefit in a range of both exon-1 and FL models of HD. By contrast, the data supporting benefit from SirT1 overexpression derives from genetic studies only and it must be questioned whether pharmacological inhibition of endogenous levels of SirT1 can be reasonably compared with genetic overexpression of SirT1 in the absence of any pharmacological intervention. Indeed, by extrapolating the ‘overexpression hypothesis’ to the selisistat situation, one would expect to see negative effects in terms of HD-related readouts, while selisistat instead appears to confer benefit across a wide range of HD models.
No obvious effects of short term selisistat treatment were observed in terms of the soluble HTT levels. Selisistat is thought to act by selectively increasing the clearance of mutant HTT, and while the time scale for SirT1 inhibition in tissues can be hypothesized to be tightly linked to the systemic drug levels, the dynamic response in terms of soluble HTT levels may show a more complex relationship to drug exposure. The mutant HTT exists in several forms including insoluble protein aggregates, oligomeric forms and the soluble monomers. SirT1-related perturbation of the equilibria between these compartments is likely to occur on a different time scale as compared with the immediate target modulation. In the current study, we measured soluble HTT in PBMCs, with the transition of soluble HTT from one tissue compartment to another as an additional covariate. As a consequence, studies of longer duration are likely to be required to assess modulation of the soluble HTT compartment.
SirT1 has been reported to be involved in both the innate and adaptive immune response via their regulation of NFκB and AP-1 pathways, respectively. SirT1 knock-out mice are more susceptible to inflammatory disorders including a lupus-like phenotype 39,40. Evidence also suggests simultaneous dysfunction of CNS and peripheral inflammatory pathways in HD even in incipient pathological stages of the disease 41,42 and a pattern of pro-inflammatory cytokine elevation has been observed in plasma of HD gene carriers 43. Lipopolysaccharide stimulation of HD blood monocytes reveals an inherent hyper-reactivity that is similar to that seen in microglia, suggesting a cell-autonomous effect of mutant HTT in peripheral myeloid cells as well as in the CNS 43. SirT1 has also been implicated in suppressing inflammation in macrophages, adipose and myocardial tissues, as well as in autoimmune disease 44. These observations may suggest a potential for a pro-inflammatory effect by SirT1 inhibition, which, however, was not borne out by administration of selisistat over 14 days, as demonstrated by the data in this study.
In conclusion, selisistat exhibits a safety and tolerability profile which, in combination with its pharmacokinetic properties and absence of any pro-inflammatory effects or adverse effects on the motor, cognitive and functional domains of the UHDRS, makes it a candidate for future clinical studies in HD patients.
Competing Interests
All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi%5Fdisclosure.pdf (available on request from the corresponding author) and declare: this clinical trial (ClinicalTrials.gov Identifier NCT01485952) was conducted within the PADDINGTON (Pharmacodynamic Approaches to Demonstration of Disease Modification in Huntington's Disease) project, supported by the European Community's Seventh Framework Programme (FP7/2007–2013) under grant agreement no. 261358. The drug was supplied by Siena Biotech SpA, that also co-funded the study. All authors declare that GT, CA, MDB, CL, ED, LuM, LeM, DD, EM, GP are employees of Siena Biotech with no other relationships or activities that could appear to have influenced the submitted work. GW was an employee of Siena Biotech during the period of planning and conduction of the study and subsequently a paid contractor of Siena Biotech. SS, SH, GL, RF, CF, JD, KS, DC, CS, MR, DR, MO, SB, AB, RA, ST received funding via the PADDINGTON Grant to conduct the current study. GL has provided consulting services, advisory board functions, clinical trial services and/or lectures for Allergan, Alnylam, Amarin, AOP Orphan Pharmaceuticals AG, Bayer Pharma AG, GlaxoSmithKline, Hoffmann-La-Roche, Ipsen, ISIS Pharma, Lundbeck, Neurosearch Inc, Medesis, Medivation, Medtronic, Novartis, Pfizer, Prana Biotechnology, Sangamo/Shire, Siena Biotech, Temmler Pharma and TEVA and received grant support from the CHDI Foundation, the Bundesministerium für Bildung und Forschung (BmBF) and the Deutsche Forschungsgemeinschaft (DFG) in the previous 3 years. CS has received research grants and honoraria from TEVA, Biogen, Neurosearch, Novartis, Temmler Pharma and the CHDI Foundation in the previous 3 years. ST has received consultancy fees from Siena Biotech, Simon Kucher Partnership, Roche, Takeda Pharmaceuticals, Novartis, Sanofi-Aventis, The Wellcome Trust, Isis Pharmaceuticals and TEVA in the previous 3 years. BD was a paid contractor of Siena Biotech.
This study was also supported by the National Institute for Health Research (NIHR) University College London Hospitals Biomedical Research Centre and the NIHR Manchester Biomedical Research Centre, and was endorsed by the UK Dementia and Neurodegenerative Diseases Network (DeNDRoN) and the European Huntington's Disease Network (EHDN).
We thank all patients and their families for their participation. The authors also wish to acknowledge the support from the staff at all clinical sites: Carolin Geitner, Michael Orth and Sonja Trautmann, Ulm; Gisa Ellrichmann, Barbara Kaminski, Rainer Hoffmann and Christiane Stamm, Bochum; Ralph Andre, Monika Lewis, Elin Rees and Nicola Robertson, London; Melanie Dadkhah-Taeidy, Aisling Flatley, Elizabeth Howard, Mary Jones and Dawn Rogers, Manchester; Dorota Bocwinska, Ewelina Paluch and Magdalena Wójcik, Cracow; Katarzyna Jachiñska, Rafal Rola, Halina Sienkiewicz-Jarosz and Grzegorz Witkowski, Warzaw.
References
- Rubinsztein DC, Carmichael J. Huntington's disease: molecular basis of neurodegeneration. Expert Rev Mol Med. 2003;5:1–21. doi: 10.1017/S1462399403006549. [DOI] [PubMed] [Google Scholar]
- van der Burg JM, Bjorkqvist M, Brundin P. Beyond the brain: widespread pathology in Huntington's disease. Lancet Neurol. 2009;8:765–774. doi: 10.1016/S1474-4422(09)70178-4. [DOI] [PubMed] [Google Scholar]
- Luthi-Carter R, Taylor DM, Pallos J, Lambert E, Amore A, Parker A, Moffitt H, Smith DL, Runne H, Gokce O, Kuhn A, Xiang Z, Maxwell MM, Reeves SA, Bates GP, Neri C, Thompson LM, Marsh JL, Kazantsev AG. SIRT2 inhibition achieves neuroprotection by decreasing sterol biosynthesis. Proc Natl Acad Sci USA. 2010;107:7927–7932. doi: 10.1073/pnas.1002924107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccato C, Valenza M, Cattaneo E. Molecular mechanisms and potential therapeutical targets in Huntington's disease. Physiol Rev. 2010;90:905–981. doi: 10.1152/physrev.00041.2009. [DOI] [PubMed] [Google Scholar]
- Chopra V, Quinti L, Kim J, Vollor L, Narayanan KL, Edgerly C, Cipicchio PM, Lauver MA, Choi SH, Silverman RB, Ferrante RJ, Hersch S, Kazantsev AG. The sirtuin 2 inhibitor AK-7 is neuroprotective in Huntington's disease mouse models. Cell Rep. 2012;2:1492–1497. doi: 10.1016/j.celrep.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong H, Then F, Melia TJ, Jr, Mazzulli JR, Cui L, Savas JN, Voisine C, Paganetti P, Tanese N, Hart AC, Yamamoto A, Krainc D. Acetylation targets mutant huntingtin to autophagosomes for degradation. Cell. 2009;137:60–72. doi: 10.1016/j.cell.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Wang S, Gan L, Vosler PS, Gao Y, Zigmond MJ, Chen J. Protective effects and mechanisms of sirtuins in the nervous system. Prog Neurobiol. 2011;95:373–395. doi: 10.1016/j.pneurobio.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallos J, Bodai L, Lukacsovich T, Purcell JM, Steffan JS, Thompson LM, Marsh JL. Inhibition of specific HDACs and sirtuins suppresses pathogenesis in a Drosophila model of Huntington's disease. Hum Mol Genet. 2008;17:3767–3775. doi: 10.1093/hmg/ddn273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napper AD, Hixon J, McDonagh T, Keavey K, Pons JP, Barker J, Yau WT, Amouzegh P, Flegg A, Hamelin E, Thomas RJ, Kates M, Jones S, Navia MA, Saunders JO, DiStefano PS, Curtis R. Discovery of indoles as potent and selective inhibitors of the deacetylase SirT1. J Med Chem. 2005;48:8045–8054. doi: 10.1021/jm050522v. [DOI] [PubMed] [Google Scholar]
- Smith MR, Syed A, Lukacsovich T, Purcell J, Barbaro BA, Worthge SA, Wei SR, Pollio G, Magnoni L, Scali C, Massai L, Franceschini D, Camarri M, Gianfriddo M, Diodato E, Thomas R, Gokce O, Tabrizi SJ, Caricasole A, Landwehrmeyer B, Menalled L, Ramboz S, Luthi Carter R, Westerberg G, Marsh JL. Sirtuin 1 inhibition alleviates pathology in multiple animal and cell models of Huntington's disease. Hum Mol Genet. 2014;23:2995–3007. doi: 10.1093/hmg/ddu010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerberg G, Chiesa JA, Andersen CA, Diamanti D, Magnoni L, Pollio G, Darpo B, Zhou M. Safety, pharmacokinetics, pharmacogenomics and QT concentration effect modelling of the SirT1 inhibitor selisistat in healthy volunteers. Br J Clin Pharmacol. 2014 doi: 10.1111/bcp.12513. doi: 10.1111/bcp.12513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orth M, Handley OJ, Schwenke C, Dunnett SB, Craufurd D, Ho AK, Wild E, Tabrizi SJ, Landwehrmeyer GB. Observing Huntington's disease: the European Huntington's disease network's REGISTRY. PLoS Curr. 2010;2:RRN1184. doi: 10.1371/currents.RRN1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntington Study Group. Unified Huntington's disease rating scale: reliability and consistency. Huntington Study Group. Mov Disord. 1996;11:136–142. doi: 10.1002/mds.870110204. [DOI] [PubMed] [Google Scholar]
- Marder K, Zhao H, Myers RH, Cudkowicz M, Kayson E, Kieburtz K, Orme C, Paulsen J, Penney JB, Jr, Siemers E, Shoulson I. Rate of functional decline in Huntington's disease. Huntington Study Group. Neurology. 2000;54:452–458. doi: 10.1212/wnl.54.2.452. [DOI] [PubMed] [Google Scholar]
- Craufurd D, Thompson JC, Snowden JS. Behavioral changes in Huntington disease. Neuropsychiatry Neuropsychol Behav Neurol. 2001;14:219–226. [PubMed] [Google Scholar]
- Snaith RP, Constantopoulos AA, Jardine MY, McGuffin P. A clinical scale for the self-assessment of irritability. Br J Psychiatry. 1978;132:164–171. doi: 10.1192/bjp.132.2.164. [DOI] [PubMed] [Google Scholar]
- Zigmond AS, Snaith RP. The hospital anxiety and depression scale. Acta Psychiatr Scand. 1983;67:361–370. doi: 10.1111/j.1600-0447.1983.tb09716.x. [DOI] [PubMed] [Google Scholar]
- Duff K, Paulsen JS, Beglinger LJ, Langbehn DR, Wang C, Stout JC, Ross CA, Aylward E, Carlozzi NE, Queller S. ‘Frontal’ behaviors before the diagnosis of Huntington's disease and their relationship to markers of disease progression: evidence of early lack of awareness. J Neuropsychiatry Clin Neurosci. 2010;22:196–207. doi: 10.1176/appi.neuropsych.22.2.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnett JA, Seth L. Effects of physical layout in performance on the trail making test. Psychol Assess. 1995;7:220–221. [Google Scholar]
- Smith A. The symbol-digit modalities test: a neuropsychologic test of learning and other cerebral disorders. In: Helmuth J, editor. Learning Disorders. Seattle: Special Child Publications; 1968. pp. 83–91. [Google Scholar]
- MacLeod CM. Half a century of research on the Stroop effect: an integrative review. Psychol Bull. 1991;109:163–203. doi: 10.1037/0033-2909.109.2.163. [DOI] [PubMed] [Google Scholar]
- Buysse DJ, Reynolds CF, 3rd, Monk TH, Berman SR, Kupfer DJ. The Pittsburgh Sleep Quality Index: a new instrument for psychiatric practice and research. Psychiatry Res. 1989;28:193–213. doi: 10.1016/0165-1781(89)90047-4. [DOI] [PubMed] [Google Scholar]
- Massai L, Petricca L, Magnoni L, Rovetini L, Haider S, Andre R, Tabrizi SJ, Sussmuth SD, Landwehrmeyer BG, Caricasole A, Pollio G, Bernocco S. Development of an ELISA assay for the quantification of soluble huntingtin in human blood cells. BMC Biochem. 2013;14:34. doi: 10.1186/1471-2091-14-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost C, Kenward MG, Fox NC. Optimizing the design of clinical trials where the outcome is a rate. Can estimating a baseline rate in a run-in period increase efficiency? Stat Med. 2008;27:3717–3731. doi: 10.1002/sim.3280. [DOI] [PubMed] [Google Scholar]
- Tabrizi SJ, Reilmann R, Roos RA, Durr A, Leavitt B, Owen G, Jones R, Johnson H, Craufurd D, Hicks SL, Kennard C, Landwehrmeyer B, Stout JC, Borowsky B, Scahill RI, Frost C, Langbehn DR. Potential endpoints for clinical trials in premanifest and early Huntington's disease in the TRACK-HD study: analysis of 24 month observational data. Lancet Neurol. 2012;11:42–53. doi: 10.1016/S1474-4422(11)70263-0. [DOI] [PubMed] [Google Scholar]
- Tabrizi SJ, Scahill RI, Owen G, Durr A, Leavitt BR, Roos RA, Borowsky B, Landwehrmeyer B, Frost C, Johnson H, Craufurd D, Reilmann R, Stout JC, Langbehn DR. Predictors of phenotypic progression and disease onset in premanifest and early-stage Huntington's disease in the TRACK-HD study: analysis of 36-month observational data. Lancet Neurol. 2013;12:637–649. doi: 10.1016/S1474-4422(13)70088-7. [DOI] [PubMed] [Google Scholar]
- Paulsen JS, Langbehn DR, Stout JC, Aylward E, Ross CA, Nance M, Guttman M, Johnson S, MacDonald M, Beglinger LJ, Duff K, Kayson E, Biglan K, Shoulson I, Oakes D, Hayden M. Detection of Huntington's disease decades before diagnosis: the Predict-HD study. J Neurol Neurosurg Psychiatry. 2008;79:874–880. doi: 10.1136/jnnp.2007.128728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mestre T, Giuliano J, Handley OJ, Landwehrmeyer B, Sampaio C. Enroll-HD: a global observational study enabling clinical research in Huntington's disease. J Huntington Dis. 2013;1:34–35. [Google Scholar]
- Ramadori G, Lee CE, Bookout AL, Lee S, Williams KW, Anderson J, Elmquist JK, Coppari R. Brain SirT1: anatomical distribution and regulation by energy availability. J Neurosci. 2008;28:9989–9996. doi: 10.1523/JNEUROSCI.3257-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michishita E, Park JY, Burneskis JM, Barrett JC, Horikawa I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SirT proteins. Mol Biol Cell. 2005;16:4623–4635. doi: 10.1091/mbc.E05-01-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HC, Guarente L. SIRT1 mediates central circadian control in the SCN by a mechanism that decays with aging. Cell. 2013;153:1448–1460. doi: 10.1016/j.cell.2013.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao Y, Lu M, Ge F, Marsh DJ, Qian S, Wang AH, Picciotto MR, Gao XB. Regulation of synaptic efficacy in hypocretin/orexin-containing neurons by melanin concentrating hormone in the lateral hypothalamus. J Neurosci. 2008;28:9101–9110. doi: 10.1523/JNEUROSCI.1766-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Politis M, Pavese N, Tai YF, Tabrizi SJ, Barker RA, Piccini P. Hypothalamic involvement in Huntington's disease: an in vivo PET study. Brain. 2008;131:2860–2869. doi: 10.1093/brain/awn244. [DOI] [PubMed] [Google Scholar]
- Pacholec M, Bleasdale JE, Chrunyk B, Cunningham D, Flynn D, Garofalo RS, Griffith D, Griffor M, Loulakis P, Pabst B, Qiu X, Stockman B, Thanabal V, Varghese A, Ward J, Withka J, Ahn K. SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SirT1. J Biol Chem. 2010;285:8340–8351. doi: 10.1074/jbc.M109.088682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Wang J, Fu J, Du L, Jeong H, West T, Xiang L, Peng Q, Hou Z, Cai H, Seredenina T, Arbez N, Zhu S, Sommers K, Qian J, Zhang J, Mori S, Yang XW, Tamashiro KL, Aja S, Moran TH, Luthi-Carter R, Martin B, Maudsley S, Mattson MP, Cichewicz RH, Ross CA, Holtzman DM, Krainc D, Duan W. Neuroprotective role of Sirt1 in mammalian models of Huntington's disease through activation of multiple Sirt1 targets. Nat Med. 2011;18:153–158. doi: 10.1038/nm.2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfister JA, Ma C, Morrison BE, D'Mello SR. Opposing effects of sirtuins on neuronal survival: SirT1-mediated neuroprotection is independent of its deacetylase activity. PLoS ONE. 2008;3:e4090. doi: 10.1371/journal.pone.0004090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert EL, Caron AZ, Morin K, Coulombe J, He XH, Jardine K, Dewar-Darch D, Boekelheide K, Harper ME, McBurney MW. SirT1 catalytic activity is required for male fertility and metabolic homeostasis in mice. FASEB J. 2012;26:555–566. doi: 10.1096/fj.11-193979. [DOI] [PubMed] [Google Scholar]
- Kakefuda K, Fujita Y, Oyagi A, Hyakkoku K, Kojima T, Umemura K, Tsuruma K, Shimazawa M, Ito M, Nozawa Y, Hara H. Sirtuin 1 overexpression mice show a reference memory deficit, but not neuroprotection. Biochem Biophys Res Commun. 2009;387:784–788. doi: 10.1016/j.bbrc.2009.07.119. [DOI] [PubMed] [Google Scholar]
- Sequeira J, Boily G, Bazinet S, Saliba S, He X, Jardine K, Kennedy C, Staines W, Rousseaux C, Mueller R, McBurney MW. Sirt1-null mice develop an autoimmune-like condition. Exp Cell Res. 2008;314:3069–3074. doi: 10.1016/j.yexcr.2008.07.011. [DOI] [PubMed] [Google Scholar]
- Zhang J, Lee SM, Shannon S, Gao B, Chen W, Chen A, Divekar R, McBurney MW, Braley-Mullen H, Zaghouani H, Fang D. The type III histone deacetylase Sirt1 is essential for maintenance of T cell tolerance in mice. J Clin Invest. 2009;119:3048–3058. doi: 10.1172/JCI38902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Politis M, Pavese N, Tai YF, Kiferle L, Mason SL, Brooks DJ, Tabrizi SJ, Barker RA, Piccini P. Microglial activation in regions related to cognitive function predicts disease onset in Huntington's disease: a multimodal imaging study. Hum Brain Mapp. 2011;32:258–270. doi: 10.1002/hbm.21008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai YF, Pavese N, Gerhard A, Tabrizi SJ, Barker RA, Brooks DJ, Piccini P. Microglial activation in presymptomatic Huntington's disease gene carriers. Brain. 2007;130:1759–1766. doi: 10.1093/brain/awm044. [DOI] [PubMed] [Google Scholar]
- Bjorkqvist M, Wild EJ, Thiele J, Silvestroni A, Andre R, Lahiri N, Raibon E, Lee RV, Benn CL, Soulet D, Magnusson A, Woodman B, Landles C, Pouladi MA, Hayden MR, Khalili-Shirazi A, Lowdell MW, Brundin P, Bates GP, Leavitt BR, Moller T, Tabrizi SJ. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington's disease. J Exp Med. 2008;205:1869–1877. doi: 10.1084/jem.20080178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgini F, Moller T, Kwan W, Zwilling D, Wacker JL, Hong S, Tsai LC, Cheah CS, Schwarcz R, Guidetti P, Muchowski PJ. Histone deacetylase inhibition modulates kynurenine pathway activation in yeast, microglia, and mice expressing a mutant huntingtin fragment. J Biol Chem. 2008;283:7390–7400. doi: 10.1074/jbc.M708192200. [DOI] [PubMed] [Google Scholar]