

Abstract
Activation of the calcium-sensing receptor (CaSR) by ischemia/reperfusion (I/R) favours apoptosis in cardiomyocytes, hepatocytes and neurons. Its role in renal I/R is unknown. We investigated the impact of pharmacological preactivation of the CaSR on kidney structure and function in a murine model of bilateral renal 30-min ischemia and 48-hour reperfusion, and in a 6-year cohort of kidney transplant recipients (KTR). C57BL/6J mice were administered daily with CaSR agonist, R-568, or with vehicle for 48 hours. Evaluation of serum urea and creatinine levels, renal histology and urine metabolome by nuclear magnetic resonance showed that R-568 was not nephrotoxic per se. Following I/R, serum urea and creatinine levels increased higher in R-568-treated animals than in controls. Jablonski’s score was significantly greater in R-568-treated kidneys, which showed a higher rate of cell proliferation and apoptosis in comparison to controls. Next, we retrospectively identified 36 patients (10.7% of our cohort) who were treated by CaSR agonist, cinacalcet, at the time of kidney transplantation (KTx). After matching these to 61 KTR upon type of donor, cold ischemic time, residual diuresis, and donor age, we observed that delayed graft function, i.e. need for dialysis in the first week after KTx, occurred in 42 and 23% of cinacalcet-treated and control groups, respectively (p≤0.05). These data suggest that pharmacological preactivation of the CaSR before renal I/R exacerbates kidney injury.
Keywords: Ca2+-sensing receptor, kidney, ischemia/reperfusion, cinacalcet, transplantation, delayed graft function
Introduction
The Ca2+-sensing receptor (CaSR) belongs to family C of the G-protein-coupled receptor (GPCR) superfamily, and is ubiquitously expressed [1,2]. Its principal physiological ligand is the ion Ca2+. By sensing the variations of extracellular Ca2+ ([Ca2+]e) concentration, the CaSR plays a critical role in Ca2+ homeostasis. It notably controls parathormone (PTH) secretion by the parathyroid glands and modulates Ca2+ fluxes in kidneys, intestine and bones [3-5]. The importance of the CaSR in Ca2+ homeostasis is supported by the chronic hyper- or hypocalcemia observed in patients harboring loss- or gain-of-function mutations, respectively, in the CASR gene [6]. In addition, the EC50 value for Ca2+ binding to the CaSR can be significantly modified by several physiological parameters, including ionic strength, extracellular pH, L-aromatic amino acids and polyamines, or by drugs, like the calcimimetic compounds cinacalcet and R-568 [4]. Hence, cinacalcet is used in clinical routine to treat patients with primary hyperparathyroidism or with secondary hyperparathyroidism caused by chronic kidney disease (CKD), including individuals registered on kidney transplantation (KTx) waiting list [7,8].
The CaSR has been identified in numerous tissues and cells that are not directly involved in Ca2+ homeostasis, in which its role remains unclear [9,10]. In epithelia, the CaSR particularly regulates cell proliferation, survival and Ca2+-induced polarization and differentiation [11-14]. Moreover, the CaSR has been implicated ex vivo and in vivo in the ischemia/reperfusion (I/R) cascade in cardiomyocytes [15,16] and neurons [17], as well as in hepatocytes [18]. The occurrence of I/R typically involves the reduction or interruption of organ perfusion with a subsequent reflow. Such I/R event induces significant cell metabolism modifications [19], including increased expression of the CaSR, intracellular Ca2+ overload and activation of the mitochondrial and mitogen-activated protein kinase apoptotic pathways [15-18].
Transient I/R is the primary cause of acute kidney injury (AKI), a common situation currently defined as a rapid fall of glomerular filtration rate (GFR) and/or a decline in urine output [20,21]. One of the paradigms of renal I/R is KTx. Indeed, graft procurement and storage necessarily require the temporary interruption of renal blood flow, with subsequent reperfusion after transplantation. Such unavoidable I/R events contribute to delay graft recovery post KTx [22]. The role of the CaSR in kidney I/R has not been studied thus far. In order to translationally investigate the impact of CaSR activation before renal I/R on kidney structure and function, we first took advantage of a conventional mouse model of bilateral clamping of renal pedicles. Next, we retrospectively studied a prospective cohort of kidney transplant recipients (KTR) to test whether the use of cinacalcet (Sensipar®/Mimpara®) at the time of KTx influences early graft recovery.
Materials and methods
Acute renal ischemia in mice
All animal protocols were approved by the Ethics Committee for Animal Care and Use at the University of Liege School of Medicine (protocol number #1335). Ten-week-old male C57BL/6 mice weighing ~20 g were treated with i.p. injections of CaSR agonist, R-568 (AMGEN), at 250 µg/d [23] or with a similar volume of DMSO for 2 days before surgery. Animals were then anesthetized with pentobarbital (60 mg/kg, Ceva®) by i.p. injection and, using aseptic techniques, subjected to a laparotomy with bilateral renal pedicle clamping (“ischemia”) for 30 min. Ischemia was confirmed by color change observed in kidneys following clamping. Supportive fluids were given throughout the operative period, and hypothermia was prevented by use of an isothermal heating pad and warming lights. Following surgery, animals were kept in light- and temperature-controlled conditions for 48 hours, with a twice-daily clinical evaluation of scar and general health status. At 24 h post surgery, mice were placed in metabolic cages for 24 hours with ad libitum access to food and drinking water. Urine was collected on 2%-Na+ azide solution (Sigma®) with one drop of mineral oil (Sigma®) to prevent bacterial proliferation and evaporation, respectively. Urine metabolome was analyzed using 1H-NMR (see infra). Blood was obtained by vena cava puncture at the time of sacrifice. Serum levels of Ca2+, urea and creatinine were measured on a COBAS 6000 C501 device (Roche-Hitachi®). At 48h post-surgery, the animals were once again anesthetized and subjected to a laparotomy. Kidneys were collected, fixed in 4% paraformaldehyde (Boehringer Ingelheim, Heidelberg, Germany) in 0.1 mol/L phosphate buffer, pH 7.4, prior to embedding in paraffin, and further subjected to histological analyses [24].
Immunostaining
Six-µm sections were incubated for 30 minutes with 0.3% hydrogen peroxide to block endogenous peroxidase. Antigen retrieval was performed by incubating sections in 0.01 mol/L citrate buffer, pH 6.1, for 11 minutes, in an autoclave heated at 121°C, before cooling down for 20 min, and rinsing. Following incubation with 10% normal serum for 30 minutes, sections were incubated for 45 minutes with the primary antibodies diluted in phosphate-buffered saline (PBS) containing 2% bovine serum albumin (BSA). After washing, sections were successively incubated with biotinylated secondary anti-immunoglobulin G antibodies (Dako®), avidin-biotin peroxidase (Merck®), and 3,3-diaminobenzidine (DAB, Dako®). The specificity of immunostaining was tested by incubation (i) in absence of primary antiserum, or (ii) with control IgG (Vector Lab®). The use of ApopTag kit S7101 followed the manufacturer’s recommendations (Millipore®). Following immunostaining, kidney sections were scanned using the NanoZoomer 2.0 HT (Hamamatsu®). The regions of interest were first distinguished (Supplementary Figure 1). The cortex is located between the capsula and the cortico-medullary junction (CMJ). CMJ is distinguishable from the cortex upon the absence of glomeruli, the presence of large venous spaces, and the shape of S3 straight PT. CMJ was further isolated from the medulla according to the longitudinal vs. transversal orientation of the tubules. Next, immuno-reactive cells were systematically identified in each kidney section and counted using both direct visual method and www.cytomine.be software (Supplementary Figure 1) [25]. Following a hybrid human-computer approach, tens of positive and negative cell examples were manually annotated. Then, a tree-based machine learning algorithm was used to build a positive pixel segmentation model further combined with connected-component analysis and filtering operations based on size and circularity criteria. Automatic positive cell detections were proofread by experts. The number of positive cells and area statistics were then computed for each region of interest for each image. In addition to immunostaining experiments, routine colorations, i.e. Hematoxylin-Eosine (HE) and periodic acid-Shiff, were classically performed. Sections were viewed under a Leica DM 1000 LED coupled to a Leica MC170 digital camera (Leica®). The severity of I/R-associated AKI was graded according to Jablonski’s score [26] by a pathologist specialized in renal pathology, unaware of animal groups.
1H-NMR metabolomics
Urine samples (150 μl) were supplemented with 450 μl of deuterated phosphate buffer (DPB, pH 7.4), 100 μl of a 5 mM solution of maleic acid and 10 μl of a 10 mg/ml trimethylsilyl-3-propionic acid-d4 D2O (TMSP) solution. 1H-NMR spectra were acquired using a 1D NOESY sequence with presaturation on a Bruker Avance spectrometer operating at 500.13 MHz for proton and equipped with a TCI cryoprobe at 298 K. The Noesypresat experiment used a RD-90°-T1-90°-Tm-90°-acquire sequence with a relaxation delay of 4 s, a mixing time (Tm) of 10 ms and a fixed T1 delay of 4 µs. Water suppression pulse was placed during the relaxation delay (RD). The number of transient is 128 (64K data points) and a number of 4 dummy scans is chosen. Acquisition time is fixed to 3.2769001 s. Phase and baseline corrections were performed manually over the entire range of the spectra and the δ scale was calibrated to 0 ppm using the internal standard TMSP. For statistical analysis, optimized 1H-NMR spectra were automatically baseline corrected and reduced to ASCII files using AMIX software (version 3.9; Bruker). The spectral intensities were normalized to the creatinine signal at 3.05 ppm and reduced to integrated regions of equal width (0.04 ppm) corresponding to the 0.5-10.00 ppm region. Because of the residual water signal, the region between 4.5 and 5.5 ppm was removed before further analysis. The matrices obtained were used for the statistical analysis. The output data were then submitted to a “principal component analysis” (PCA) using AMIX software.
Cohort of patients
All KTR from 2007 to 2012 at University of Liège Hospital (ULg CHU) were prospectively included in a database. Patients actively treated with cinacalcet on the day of KTx were retrospectively identified from this database and matched 1:2 with controls on (i) type of donor (living (LD), deceased after brain or circulatory death (DCD)); (ii) cold ischemic time (CIT) ± 1 hour; (iii) residual diuresis (± 500 mL); and (iv) donor age (± 5 years). Delayed graft function (DGF) was defined as the need for RRT in the first postoperative week post KTx. Baseline characteristics of both groups are summarized in Table 3.
Table 3.
Clinical features of cinacalcet-treated and control patients at the time of kidney transplantation
| Cinacalcet (n=36) | Controls (n=61) | p | |||
|---|---|---|---|---|---|
| Recipient | Age at Tx | (years) | 50.2 ± 10.3 | 49 ± 13.5 | 0.92 |
| Sex ratio | (% female) | 47 | 41 | 0.55 | |
| Dialysis vintage | (years) | 3.7 ± 2.1 | 3.3 ± 3.8 | 0.57 | |
| Resting diuresis | (ml) | 430 ± 655 | 444 ± 541 | 0.91 | |
| Multi-organ Tx | (%) | 5.6 | 1.7 | 0.28 | |
| Donor | Age | (years) | 46.8 ± 11.4 | 47 ± 11.4 | 0.93 |
| Sex ratio | (% female) | 42 | 46 | 0.67 | |
| LD | (%) | 2.8 | 1.6 | 0.70 | |
| DCD | (%) | 30.6 | 21.3 | 0.31 | |
| Transplantation | CIT | (min) | 779 ± 297 | 825 ± 255 | 0.43 |
| HLA mismatches | A | 0.8 ± 0.5 | 0.9 ± 0.5 | 0.75 | |
| B | 1.2 ± 0.7 | 1 ± 0.5 | 0.08 | ||
| DR | 0.8 ± 0.4 | 0.8 ± 0.3 | 0.99 |
Tx, transplantation; LD, living donor; DCD, donation after cardiac death; CIT, cold ischemic time; HLA, human leukocyte antigen.
Results
Administration of CaSR agonist, R-568, is not nephrotoxic in mouse
Male 10-week-old C57BL/6 mice were daily administered with CaSR agonist, R-568, or with an equivalent volume of dimethyl-sulfoxide (DMSO) by intraperitoneal (i.p.) injections for 48 hours. Urine was collected in metabolic cages for the last 24 hours, whereas blood and kidney samples were obtained after anesthesia and laparotomy. As previously described [23], R-568 administration was associated with a significant decrease of serum Ca2+ levels in comparison to DMSO treatment (Table 1). Conversely, no significant difference in serum creatinine and urea levels was observed between R-568-treated versus non-treated animals (Table 1). In order to further assess renal safety of R-568 administration in mouse, we performed 1H nuclear magnetic resonance (1H-NMR) metabolomics analysis on urine from R-568-treated and control mice. 1H-NMR profiles were undistinguishable between groups (Figure 1A). Furthermore, the relative abundance of urinary metabolites previously associated with proximal tubule (PT) toxicity [27], including glucose, acetoacetate, alanine, 3-hydroxybutyrate and valine, was similar in samples from either group (Figure 1B). Finally, comparative histological examination of kidneys from R-568-treated and DMSO-treated mice was undistinguishable in terms of glomerular or tubular injury, cell proliferation and apoptosis (Table 2). Altogether, these functional and structural data support that i.p. administration of R-568 in mice does not cause significant nephrotoxicity per se.
Table 1.
Biological parameters before and after renal ischemia/reperfusion in DMSO- and R-568-treated mice
| Creatinine (mg/dl) | BUN (g/L) | Ca2+ (mmol/L) | |
|---|---|---|---|
| Before I/R | |||
| DMSO-treated mice | 1.02 ± 0.08 | 0.63 ± 0.09 | 2.66 ± 0.21 |
| R-568-treated mice | 1.13 ± 0.28 | 0.81 ± 0.16 | 2.37 ± 0.27 |
| p (unpaired Student t-test) | ns | ns | p<0.05 |
| Following I/R | |||
| DMSO-treated mice | 0.73 ± 0.13 | 0.78 ± 0.21 | |
| R-568-treated mice | 5.74 ± 1.13 | 4.54 ± 0.82 | |
| p (unpaired Student t-test) | p<0.05 | p<0.05 |
I/R, ischemia/reperfusion; BUN, blood urea nitrogen; ns, not significant n=8 mice in each group.
Figure 1.
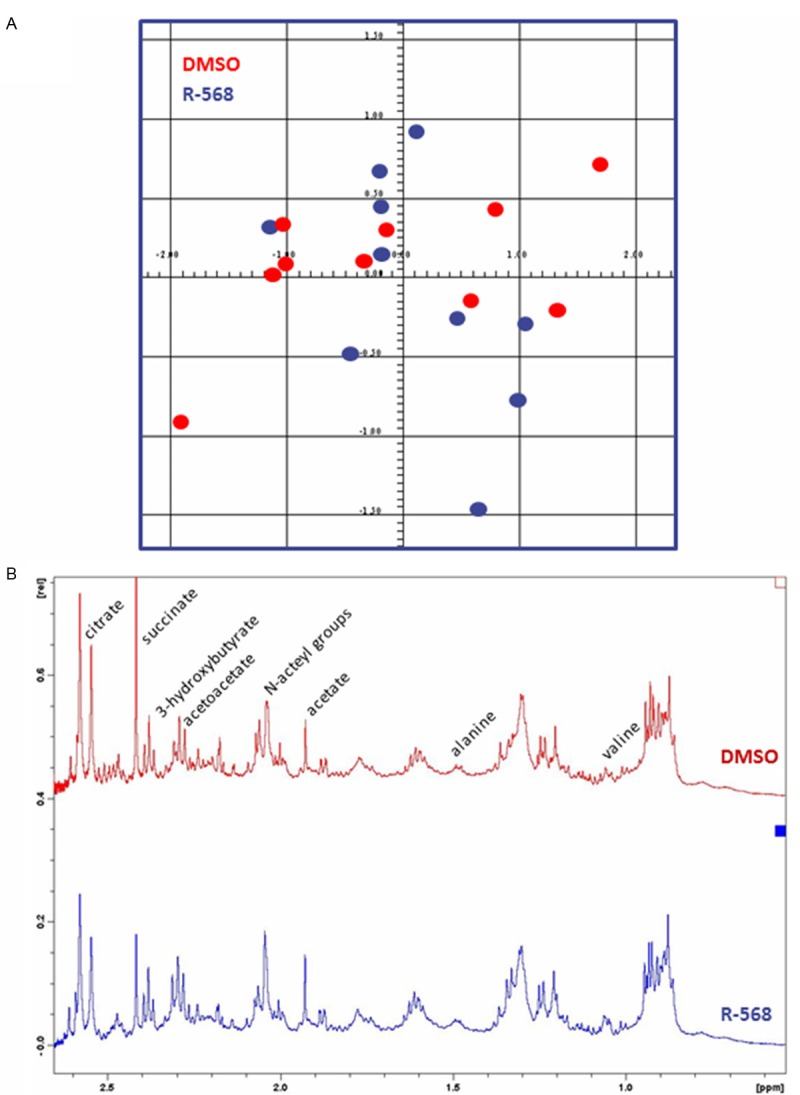
1H nuclear magnetic resonance (1H-NMR) metabolomics analysis on urine samples from DMSO-treated and R-568-treated mice. A. Representative score plot of principal component analysis (PCA, AMIX software) of urinary metabolomes of DMSO-treated (red dots) and R-568-treated (blue dots) mice. No significant difference is observed between urine metabolite profiles of both groups. B. Representative 1H-NMR spectrum of urine metabolome of a DMSO-treated (upper panel) and an R-568-treated (lower panel) mice. The relative abundance of urinary acetoacetate, alanine, 3-hydroxybutyrate and valine, is similar in samples from either group.
Table 2.
Histological parameters before and after renal ischemia/reperfusion in DMSO- and R-568-treated mice
| Jablonski score | PCNA-positive cells/mm2 | ApopTag-positive cells/mm2 | |||
|---|---|---|---|---|---|
|
|
|||||
| cortex | c-m junction | cortex | c-m junction | ||
| Before I/R | |||||
| DMSO-treated mice | 0 | 16 ± 6 | 5 ± 3 | 2 ± 1 | 2 ± 1 |
| R-568-treated mice | 0 | 13 ± 6 | 7 ± 4 | 3 ± 2 | 3 ± 2 |
| unpaired Student t-test | ns | ns | ns | ns | ns |
| Following I/R | |||||
| DMSO-treated mice | 1 ± 1 | 66 ± 12 | 23 ± 10 | 8 ± 3 | 14 ± 8 |
| R-568-treated mice | 3 ± 1 | 117 ± 31 | 78 ± 33 | 12 ± 5 | 76 ± 19 |
| unpaired Student t-test | p<0.05 | p<0.05 | p<0.05 | p<0.05 | p<0.05 |
Administration of CaSR agonist, R-568, before renal I/R exacerbates acute kidney injury in mice
Male 10-week-old C57BL/6 mice were daily administered with CaSR agonist, R-568, or with DMSO by i.p. injections for 48 hours before surgical clamping of bilateral renal pedicles for 30 minutes. After unclamping, mice were kept alive for 48 hours with ad libitum access to food and drinking water. Analysis of serum samples obtained by vena cava puncture at the time of sacrifice showed a significantly worse AKI in R-568-treated mice than in controls, as reflected by serum creatinine and urea levels (Table 1). Histological examination of kidneys from DMSO-pretreated mice revealed acute tubular necrosis (ATN), with tubulorrhexis of epithelial cells and cylinders of cellular debris in renal tubules located in the cortex and at the cortico-medullary junction (Figure 2). Jablonski’s score of ATN severity reached 1 ± 1 in DMSO-treated animals (Table 2). Furthermore, numeric quantification of cells positive for proliferative cell nuclear antigen (PCNA) and terminal deoxynucleotidyl transferase nick end labeling (TUNEL) in kidneys from DMSO-treated mice showed a higher rate of cell proliferation and apoptosis after I/R in comparison to baseline conditions (Table 2). In animals treated by CaSR agonist, R-568, before renal I/R, Jablonski’s score of ATN severity was significantly higher (3 ± 1, p<0.05) in comparison to control group (Figure 2 and Table 2). Moreover, the number of proliferating cells and apoptotic cells was significantly increased in the cortex and at the cortico-medullary junction of R-568-treated versus DMSO-treated kidneys (Figure 2 and Table 2). These observations, including functional and histological parameters, suggest that i.p. administration of CaSR agonist, R-568, before renal I/R worsens the extent of ATN in mouse.
Figure 2.
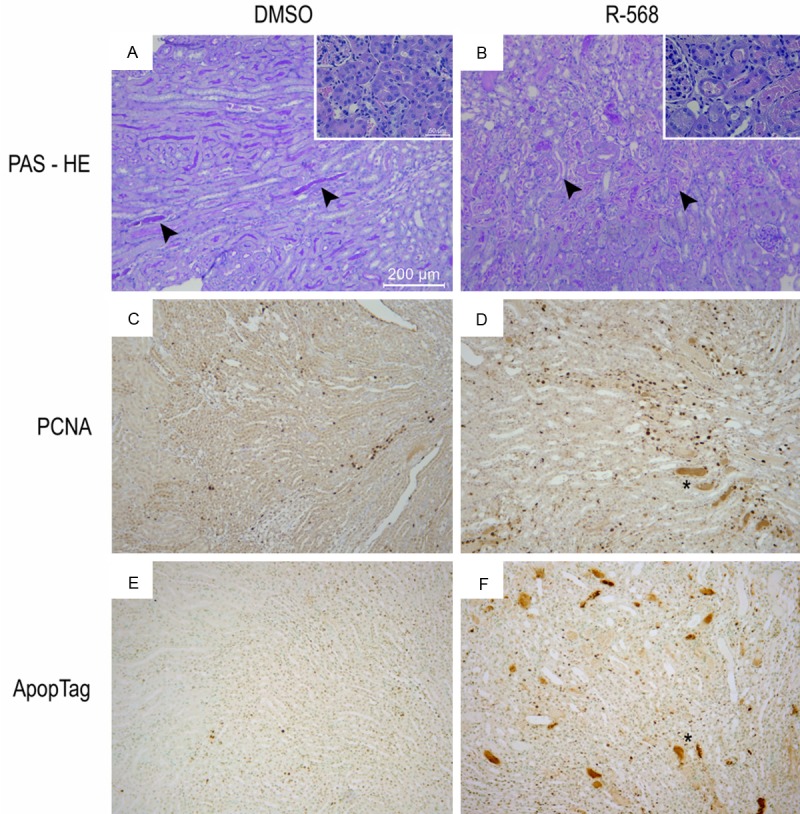
Histological examination of kidneys from DMSO-pretreated and R-568-pretreated mice following ischemia/reperfusion. Representative kidney sections following I/R injury in mice pretreated with DMSO (A, C, E) or R-568 (B, D, F). Periodic acid-Shiff coloration (A, B), immunostaining against Proliferative Cell Nuclear Antigen (PCNA) (C, D), or terminal deoxynucleotidyl transferase nick end labeling (TUNEL, ApopTag®) (E, F) were performed. Pictures are taken at the cortico-medullary junctions. Arrowheads in panels (A, B) indicate tubulorrhexis-associated intratubular debris. Scale bar represents 200 μm and 50 μm in panels (A-F) and insets, respectively.
Treatment with CaSR agonist, cinacalcet, at the time of kidney transplantation is associated with delayed graft function
At the time of KTx, I/R is an unavoidable event, which has been associated with delayed graft recovery [28]. By definition, delayed graft function (DGF) corresponds to the need for renal replacement therapy (RRT) in the first postoperative week [22]. Cinacalcet is an analog of R-568 compound, with an improved bioavailability and metabolic profile [29]. Since cinacalcet is used in CKD patients with secondary hyperparathyroidism [7], individuals registered on KTx waiting list are prone to be actively treated by cinacalcet at the time of KTx. Such a particular clinical setting provides a unique translational situation of CaSR activation before renal I/R. In our prospective 6-year database of 337 KTR, we retrospectively identified 36 (10.7%) patients who were treated by cinacalcet at the time of KTx. These patients were matched 1:2 to controls according to well-established factors of DGF, i.e. (i) type of donor, (ii) cold ischemic time (CIT), (iii) residual diuresis, and (iv) donor age. Control group included 61 patients. Characteristics of patients and donors were compared between groups using Student’s t-test or Chi-2 as appropriate, as summarized in Table 3. The occurrence of DGF was significantly superior in cinacalcet-treated patients than in controls (42% versus 23%, p=0.05). These retrospective observations in a single-centre cohort of KTR suggest that CaSR activation at the time of KTx is more often associated with a need for RRT in the first postoperative week.
Discussion
Kidney I/R is the leading cause of AKI, which accounts for up to 20% of admissions to intensive care units [30]. Furthermore, I/R cascade at the time of KTx is responsible for early graft dysfunction and enhanced graft immunogenicity, which impairs both short- and long-term graft survival [28]. Treatment of AKI remains largely supportive, including fluid maintenance, vasoactive drugs, cytoprotective therapy and extrarenal epuration [30]. Therefore, advances in deciphering the pathophysiology of renal I/R injury are urgently required to develop both preventive and curative approaches of AKI [19,31]. In our mouse model of renal I/R, we observed that administration of the CaSR agonist, R-568, before I/R was associated with a significantly worsened AKI, at both functional and architectural levels. Furthermore, the number of apoptotic cells in kidney parenchyma was significantly greater in R-568-treated animals in comparison to controls. As previously described, apoptosis particularly occurred in S3 PT segments, which are located at the cortico-medullary junction and are highly vulnerable to ischemia [32,33]. These findings concur with recent observations in various ex vivo and in vivo models of I/R [15-18]. In cardiomyocytes, the CaSR is involved in I/R-induced apoptosis through the mitochondrial pathway [16]. Exposure to the CaSR agonist, GdCl3, before I/R causes a significant increase in phosphorylated protein kinase C δ (PKCδ) translocation to the mitochondria, with enhanced release of cytochrome c (Cyt-c) and marked reduction of mitochondrial potential. Similarly, incubation of Buffalo rat liver cells with GdCl3 at the time of simulated I/R further induced CaSR expression, increased [Ca2+]i, and favored apoptosis through both mitochondrial and mitogen-activated protein kinase pathways [18]. In vivo, forebrain I/R by transient bilateral occlusion of carotid arteries in C57/BL6 mice increases CaSR expression and promotes cell death [17]. Altogether, these observations in distinct experimental models of I/R suggest a deleterious role of the CaSR in I/R-associated cellular cascades.
The CaSR has been ubiquitously identified [3,4]. In the kidney, its distribution remains debated [34]. In rats, CaSR mRNA is detected along essentially the entire nephron, including glomeruli, S1-S2 convoluted and S3 straight PT, thick ascending limbs of Henlé’s loop (TAL), distal convoluted tubules (DCT), and collecting ducts (CD) [35]. Similarly, immunohistochemistry using CaSR-specific antisera documented the localization of the CaSR protein in PT, TAL, DCT and CD, with a variable polarity upon renal cell types [36]. Still, contradictory experimentations in rats, mice and humans only found the CaSR in the TAL [37,38]. In mice, the i.p. administration of the CaSR agonist, R-568, induces hypocalcaemia [23], as we did observe in our studies. By contrast, the compound R-568 did not cause significant nephrotoxicity, as evidenced by biological and histological assessment of renal function and by 1H-NMR metabolomics analysis. 1H-NMR is currently used to assess drug-induced kidney toxicity [27]. More specifically, our findings using 1H-NMR metabolomics show that R-568 administration in mouse does not significantly modify the urinary metabolome previously linked to S3 PT toxicity. In humans, no deleterious impact on kidney function has been reported thus far after cinacalcet treatment (www.amgen.com). In KTR, Spanish multicenter observational retrospective studies reported that cinacalcet therapy initiated 20 months after KTx induced a significant and sustained decrease of both serum Ca2+ and PTH levels, but did not affect renal function during a 3-year follow-up [39]. Therefore, CaSR activation by the compound R-568 should not be regarded as nephrotoxic per se, but might make renal tubular cells more prone to apoptosis in case of I/R injury. The mechanism by which CaSR activation at the time of renal I/R enhances AKI severity is unknown. It might involve either a direct impact on tubular cells, with a rise of [Ca2+]i and stimulation of mitochondria-related apoptotic pathways, and/or an indirect effect on the blood perfusion of kidney parenchyma. Indeed, intravenous infusion of the calcimimetic, R-568, induces hypotensive effects in both normotensive and spontaneously hypertensive rats [40,41]. The cause of blood pressure drop following R-568 administration remains debated, with in vitro arguments for both CaSR-dependent and CaSR-independent production of nitric oxide (NO) [42].
KTx currently represents the best treatment for end-stage renal disease. Data from the EuroTransplant Network (www.eurotransplant.org) show graft survival rates for primary kidney transplants of 84%, 78%, and 68% after 1, 3, and 5 years, respectively. Acute vascular rejection and chronic allograft nephropathy remain the most relevant risk factors for renal graft dysfunction [43]. In addition to immunogenicity, organ preservation and I/R participate to kidney injury. DGF is a form of ATN resulting in post-transplantation oliguria, increased allograft immunogenicity and risk of acute rejection episodes, and decreased long-term survival. Factors related to the donor and prerenal, renal, or postrenal transplant factors related to the recipient can contribute to this condition [44]. The reported frequency of DGF greatly varies worldwide, from 2% to 50%, upon the ambiguity in its definition [22]. Preclinical and clinical studies have particularly demonstrated that both ischaemia and reinstitution of blood flow in ischemically damaged kidneys after hypothermic preservation play a pivotal part in the development of DGF [28]. Therefore, efforts to limit I/R injury may help minimize its deleterious impact on early graft recovery. Our retrospective analysis of a single-centre cohort of KTR suggests that cinacalcet treatment at the time of KTx is more often associated with a need for RRT in the first postoperative week. Cinacalet is an agonist of the CaSR, which is routinely prescribed to treat secondary hyperparathyroidism in CKD patients [7]. Mineral and bone disorders (MBD) are thought to play a part in extraskeletal calcification and diminished vascular compliance observed in CKD patients, thereby contributing to their increased cardiovascular risk [45]. Cinacalcet treatment may attenuate the progression of vascular and cardiac-valve calcification in hemodialysis patients, although its beneficial impact on the risk of death or major cardiovascular events remains unproven [46,47].
Our present translational study has several limitations. Our mouse model is restricted to bilateral clamping of renal vascular pedicles, without actual KTx, which does not allow us to take into account immune donor/recipient interactions. In addition, mice were only administered with the CaSR agonist, R-568, for 48 h before I/R, whereas KTR had been chronically treated with cinacalcet before KTx. Finally, the retrospective pattern of the analysis of our single-center cohort limits its interpretation. Selection bias was reduced by matching 1:2 cinacalcet-treated and control individuals according to well-established factors of DGF. As a whole, our data suggest that CaSR activation at the time of renal I/R exacerbates AKI. The regulation of CaSR activity might thus serve as a novel pharmacological target to prevent and treat such a common condition.
Acknowledgements
The authors cordially thank the surgeons (M. Meurisse, C. Coimbra Marques, A. De Roover, E. Hamoir, P. Honoré, L. Kohnen, N. Meurisse and J-P Squifflet), the physicians (S. Grosch and P. Xhignesse), the technician (J-P Cheramy-Bien), and the members of the local transplant coordination center (M-H. Delbouille, M-H. Hans, J. Mornard) for their personal and professional commitment to kidney transplantation at the University of Liège Academic Hospital in Liège, Belgium. They also thank Eric Brevers (in E. Cavalier’s lab) for the measurements of serum creatinine, urea and Ca2+ levels, as well as all members of the GIGA Bioinformatics Platform for providing access to computing servers and to the Cytomine software (http://www.cytomine.be/). This work is supported by the Fonds National de la Recherche Scientifique (FNRS, Research Credit), University of Liège (Fonds Spéciaux à la Recherche) and from Fonds Leon Frédéricq (ACiiRT grant).
Disclosure of conflict of interest
The R-568 compound was provided by AMGEN Company (Thousand Oaks, CA) under the agreements MMFA 2012578383 and RPA 2012578387.
Supporting Information
References
- 1.Brown EM, Gamba G, Riccardi D, Lombardi M, Butters R, Kifor O, Sun A, Hediger MA, Lytton J, Hebert SC. Cloning and characterization of an extracellular Ca(2+)-sensing receptor from bovine parathyroid. Nature. 1993;366:575–580. doi: 10.1038/366575a0. [DOI] [PubMed] [Google Scholar]
- 2.Brauner-Osborne H, Wellendorph P, Jensen AA. Structure, pharmacology and therapeutic prospects of family C G-protein coupled receptors. Curr Drug Targets. 2007;8:169–184. doi: 10.2174/138945007779315614. [DOI] [PubMed] [Google Scholar]
- 3.Chakravarti B, Chattopadhyay N, Brown EM. Signaling through the extracellular calcium-sensing receptor (CaSR) Adv Exp Med Biol. 2012;740:103–142. doi: 10.1007/978-94-007-2888-2_5. [DOI] [PubMed] [Google Scholar]
- 4.Geibel JP. The calcium-sensing receptor. J Nephrol. 2010;23(Suppl 16):S130–135. [PubMed] [Google Scholar]
- 5.Garg MK. The intestinal calcistat. Indian J Endocrinol Metab. 2013;17:S25–s28. doi: 10.4103/2230-8210.119497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hannan FM, Thakker RV. Calcium-sensing receptor (CaSR) mutations and disorders of calcium, electrolyte and water metabolism. Best Pract Res Clin Endocrinol Metab. 2013;27:359–371. doi: 10.1016/j.beem.2013.04.007. [DOI] [PubMed] [Google Scholar]
- 7.Torres PU. Cinacalcet HCl: a novel treatment for secondary hyperparathyroidism caused by chronic kidney disease. J Ren Nutr. 2006;16:253–258. doi: 10.1053/j.jrn.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 8.Verheyen N, Pilz S, Eller K, Kienreich K, Fahrleitner-Pammer A, Pieske B, Ritz E, Tomaschitz A. Cinacalcet hydrochloride for the treatment of hyperparathyroidism. Expert Opin Pharmacother. 2013;14:793–806. doi: 10.1517/14656566.2013.777041. [DOI] [PubMed] [Google Scholar]
- 9.Magno AL, Ward BK, Ratajczak T. The calcium-sensing receptor: a molecular perspective. Endocr Rev. 2011;32:3–30. doi: 10.1210/er.2009-0043. [DOI] [PubMed] [Google Scholar]
- 10.Riccardi D, Kemp PJ. The calcium-sensing receptor beyond extracellular calcium homeostasis: conception, development, adult physiology, and disease. Annu Rev Physiol. 2012;74:271–297. doi: 10.1146/annurev-physiol-020911-153318. [DOI] [PubMed] [Google Scholar]
- 11.Tu CL, Chang W, Xie Z, Bikle DD. Inactivation of the calcium sensing receptor inhibits E-cadherin-mediated cell-cell adhesion and calcium-induced differentiation in human epidermal keratinocytes. J Biol Chem. 2008;283:3519–3528. doi: 10.1074/jbc.M708318200. [DOI] [PubMed] [Google Scholar]
- 12.Gong Y, Renigunta V, Himmerkus N, Zhang J, Renigunta A, Bleich M, Hou J. Claudin-14 regulates renal Ca(+)(+) transport in response to CaSR signalling via a novel microRNA pathway. EMBO J. 2012;31:1999–2012. doi: 10.1038/emboj.2012.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jouret F, Wu J, Hull M, Rajendran V, Mayr B, Schofl C, Geibel J, Caplan MJ. Activation of the Ca2+-sensing receptor induces deposition of tight junction components to the epithelial cell plasma membrane. J Cell Sci. 2013;126:5132–5142. doi: 10.1242/jcs.127555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gama L, Baxendale-Cox LM, Breitwieser GE. Ca2+-sensing receptors in intestinal epithelium. Am J Physiol. 1997;273:C1168–1175. doi: 10.1152/ajpcell.1997.273.4.C1168. [DOI] [PubMed] [Google Scholar]
- 15.Jiang CM, Han LP, Li HZ, Qu YB, Zhang ZR, Wang R, Xu CQ, Li WM. Calcium-sensing receptors induce apoptosis in cultured neonatal rat ventricular cardiomyocytes during simulated ischemia/reperfusion. Cell Biol Int. 2008;32:792–800. doi: 10.1016/j.cellbi.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 16.Zheng H, Liu J, Liu C, Lu F, Zhao Y, Jin Z, Ren H, Leng X, Jia J, Hu G, Dong S, Zhong X, Li H, Yang B, Xu C, Zhang W. Calcium-sensing receptor activating phosphorylation of PKCdelta translocation on mitochondria to induce cardiomyocyte apoptosis during ischemia/reperfusion. Mol Cell Biochem. 2011;358:335–343. doi: 10.1007/s11010-011-0984-1. [DOI] [PubMed] [Google Scholar]
- 17.Kim JY, Kim N, Yenari MA, Chang W. Mild Hypothermia Suppresses Calcium-Sensing Receptor (CaSR) Induction Following Forebrain Ischemia While Increasing GABA-B Receptor 1 (GABA-B-R1) Expression. Transl Stroke Res. 2011;2:195–201. doi: 10.1007/s12975-011-0082-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xing WJ, Kong FJ, Li GW, Qiao K, Zhang WH, Zhang L, Bai SZ, Xi YH, Li HX, Tian Y, Ren H, Wu LY, Wang R, Xu CQ. Calcium-sensing receptors induce apoptosis during simulated ischaemia-reperfusion in Buffalo rat liver cells. Clin Exp Pharmacol Physiol. 2011;38:605–612. doi: 10.1111/j.1440-1681.2011.05559.x. [DOI] [PubMed] [Google Scholar]
- 19.Erpicum P, Detry O, Weekers L, Bonvoisin C, Lechanteur C, Briquet A, Beguin Y, Krzesinski JM, Jouret F. Mesenchymal stromal cell therapy in conditions of renal ischaemia/reperfusion. Nephrol Dial Transplant. 2014;29:1487–93. doi: 10.1093/ndt/gft538. [DOI] [PubMed] [Google Scholar]
- 20.Khwaja A. KDIGO Clinical Practice Guidelines for Acute Kidney Injury. Nephron Clin Pract. 2012;120:c179–184. doi: 10.1159/000339789. [DOI] [PubMed] [Google Scholar]
- 21.Schrier RW, Wang W, Poole B, Mitra A. Acute renal failure: definitions, diagnosis, pathogenesis, and therapy. J Clin Invest. 2004;114:5–14. doi: 10.1172/JCI22353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mallon DH, Summers DM, Bradley JA, Pettigrew GJ. Defining delayed graft function after renal transplantation: simplest is best. Transplantation. 2013;96:885–889. doi: 10.1097/TP.0b013e3182a19348. [DOI] [PubMed] [Google Scholar]
- 23.Lavi-Moshayoff V, Silver J, Naveh-Many T. Human PTH gene regulation in vivo using transgenic mice. Am J Physiol Renal Physiol. 2009;297:F713–719. doi: 10.1152/ajprenal.00161.2009. [DOI] [PubMed] [Google Scholar]
- 24.Gailly P, Jouret F, Martin D, Debaix H, Parreira KS, Nishita T, Blanchard A, Antignac C, Willnow TE, Courtoy PJ, Scheinman SJ, Christensen EI, Devuyst O. A novel renal carbonic anhydrase type III plays a role in proximal tubule dysfunction. Kidney Int. 2008;74:52–61. doi: 10.1038/sj.ki.5002794. [DOI] [PubMed] [Google Scholar]
- 25.Maree R, Stevens B, Rollus L, Rocks N, Lopez XM, Salmon I, Cataldo D, Wehenkel L. A rich internet application for remote visualization and collaborative annotation of digital slides in histology and cytology. Diagn Pathol. 2013;8:S26. [Google Scholar]
- 26.Jablonski P, Howden BO, Rae DA, Birrell CS, Marshall VC, Tange J. An experimental model for assessment of renal recovery from warm ischemia. Transplantation. 1983;35:198–204. doi: 10.1097/00007890-198303000-00002. [DOI] [PubMed] [Google Scholar]
- 27.Boudonck KJ, Rose DJ, Karoly ED, Lee DP, Lawton KA, Lapinskas PJ. Metabolomics for early detection of drug-induced kidney injury: review of the current status. Bioanalysis. 2009;1:1645–1663. doi: 10.4155/bio.09.142. [DOI] [PubMed] [Google Scholar]
- 28.Cooper JE, Wiseman AC. Acute kidney injury in kidney transplantation. Curr Opin Nephrol Hypertens. 2013;22:698–703. doi: 10.1097/MNH.0b013e328365b388. [DOI] [PubMed] [Google Scholar]
- 29.Nemeth EF, Heaton WH, Miller M, Fox J, Balandrin MF, Van Wagenen BC, Colloton M, Karbon W, Scherrer J, Shatzen E, Rishton G, Scully S, Qi M, Harris R, Lacey D, Martin D. Pharmacodynamics of the type II calcimimetic compound cinacalcet HCl. J Pharmacol Exp Ther. 2004;308:627–635. doi: 10.1124/jpet.103.057273. [DOI] [PubMed] [Google Scholar]
- 30.Thakar CV. Perioperative acute kidney injury. Adv Chronic Kidney Dis. 2013;20:67–75. doi: 10.1053/j.ackd.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 31.Wever KE, Menting TP, Rovers M, van der Vliet JA, Rongen GA, Masereeuw R, Ritskes-Hoitinga M, Hooijmans CR, Warle M. Ischemic preconditioning in the animal kidney, a systematic review and meta-analysis. PLoS One. 2012;7:e32296. doi: 10.1371/journal.pone.0032296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chien CT, Lee PH, Chen CF, Ma MC, Lai MK, Hsu SM. De novo demonstration and co-localization of free-radical production and apoptosis formation in rat kidney subjected to ischemia/reperfusion. J Am Soc Nephrol. 2001;12:973–982. doi: 10.1681/ASN.V125973. [DOI] [PubMed] [Google Scholar]
- 33.Bagnasco S, Good D, Balaban R, Burg M. Lactate production in isolated segments of the rat nephron. Am J Physiol. 1985;248:F522–526. doi: 10.1152/ajprenal.1985.248.4.F522. [DOI] [PubMed] [Google Scholar]
- 34.Brown EM, MacLeod RJ. Extracellular calcium sensing and extracellular calcium signaling. Physiol Rev. 2001;81:239–297. doi: 10.1152/physrev.2001.81.1.239. [DOI] [PubMed] [Google Scholar]
- 35.Riccardi D, Lee WS, Lee K, Segre GV, Brown EM, Hebert SC. Localization of the extracellular Ca(2+)-sensing receptor and PTH/PTHrP receptor in rat kidney. Am J Physiol. 1996;271:F951–956. doi: 10.1152/ajprenal.1996.271.4.F951. [DOI] [PubMed] [Google Scholar]
- 36.Riccardi D, Hall AE, Chattopadhyay N, Xu JZ, Brown EM, Hebert SC. Localization of the extracellular Ca2+/polyvalent cation-sensing protein in rat kidney. Am J Physiol. 1998;274:F611–622. doi: 10.1152/ajprenal.1998.274.3.F611. [DOI] [PubMed] [Google Scholar]
- 37.Yang T, Hassan S, Huang YG, Smart AM, Briggs JP, Schnermann JB. Expression of PTHrP, PTH/PTHrP receptor, and Ca(2+)-sensing receptor mRNAs along the rat nephron. Am J Physiol. 1997;272:F751–758. doi: 10.1152/ajprenal.1997.272.6.F751. [DOI] [PubMed] [Google Scholar]
- 38.Loupy A, Ramakrishnan SK, Wootla B, Chambrey R, de la Faille R, Bourgeois S, Bruneval P, Mandet C, Christensen EI, Faure H, Cheval L, Laghmani K, Collet C, Eladari D, Dodd RH, Ruat M, Houillier P. PTH-independent regulation of blood calcium concentration by the calcium-sensing receptor. J Clin Invest. 2012;122:3355–3367. doi: 10.1172/JCI57407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Torregrosa JV, Morales E, Diaz JM, Crespo J, Bravo J, Gomez G, Gentil MA, Rodriguez Benot A, Garcia MR, Jimenez VL, Gutierrez Dalmau A, Jimeno L, Saez MJ, Romero R, Gomez Alamillo C. Cinacalcet for hypercalcaemic secondary hyperparathyroidism after renal transplantation: a multicentre, retrospective, 3-year study. Nephrology (Carlton) 2014;19:84–93. doi: 10.1111/nep.12186. [DOI] [PubMed] [Google Scholar]
- 40.Rybczynska A, Boblewski K, Lehmann A, Orlewska C, Foks H. Pharmacological activity of calcimimetic NPS R-568 administered intravenously in rats: dose dependency. Pharmacol Rep. 2006;58:533–539. [PubMed] [Google Scholar]
- 41.Rybczynska A, Lehmann A, Jurska-Jasko A, Boblewski K, Orlewska C, Foks H, Drewnowska K. Hypertensive effect of calcilytic NPS 2143 administration in rats. J Endocrinol. 2006;191:189–195. doi: 10.1677/joe.1.06924. [DOI] [PubMed] [Google Scholar]
- 42.Bonomini M, Giardinelli A, Morabito C, Di Silvestre S, Di Cesare M, Di Pietro N, Sirolli V, Formoso G, Amoroso L, Mariggio MA, Pandolfi A. Calcimimetic R-568 and its enantiomer S-568 increase nitric oxide release in human endothelial cells. PLoS One. 2012;7:e30682. doi: 10.1371/journal.pone.0030682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bon D, Chatauret N, Giraud S, Thuillier R, Favreau F, Hauet T. New strategies to optimize kidney recovery and preservation in transplantation. Nat Rev Nephrol. 2012;8:339–347. doi: 10.1038/nrneph.2012.83. [DOI] [PubMed] [Google Scholar]
- 44.Perico N, Cattaneo D, Sayegh MH, Remuzzi G. Delayed graft function in kidney transplantation. Lancet. 2004;364:1814–1827. doi: 10.1016/S0140-6736(04)17406-0. [DOI] [PubMed] [Google Scholar]
- 45.London GM, Marchais SJ, Guerin AP, Metivier F. Arteriosclerosis, vascular calcifications and cardiovascular disease in uremia. Curr Opin Nephrol Hypertens. 2005;14:525–531. doi: 10.1097/01.mnh.0000168336.67499.c0. [DOI] [PubMed] [Google Scholar]
- 46.Raggi P, Chertow GM, Torres PU, Csiky B, Naso A, Nossuli K, Moustafa M, Goodman WG, Lopez N, Downey G, Dehmel B, Floege J. The ADVANCE study: a randomized study to evaluate the effects of cinacalcet plus low-dose vitamin D on vascular calcification in patients on hemodialysis. Nephrol Dial Transplant. 2011;26:1327–1339. doi: 10.1093/ndt/gfq725. [DOI] [PubMed] [Google Scholar]
- 47.Chertow GM, Block GA, Correa-Rotter R, Drueke TB, Floege J, Goodman WG, Herzog CA, Kubo Y, London GM, Mahaffey KW, Mix TC, Moe SM, Trotman ML, Wheeler DC, Parfrey PS. Effect of cinacalcet on cardiovascular disease in patients undergoing dialysis. N Engl J Med. 2012;367:2482–2494. doi: 10.1056/NEJMoa1205624. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.