

Summary
Background
The paternal allele of Ube3a is silenced by imprinting in neurons, and Angelman Syndrome (AS) is a disorder arising from a deletion or mutation of the maternal Ube3a allele, which thereby eliminates Ube3a neuronal expression. Sleep disorders such as short sleep duration and increased sleep onset latency are very common in AS.
Results
We found an unique link between neuronal imprinting of Ube3a and circadian rhythms in two mouse models of AS, including enfeebled circadian activity behavior and slowed molecular rhythms in ex vivo brain tissues. As a consequence of compromised circadian behavior, metabolic homeostasis is also disrupted in AS mice. Unsilencing the paternal Ube3a allele restores functional circadian periodicity in neurons deficient in maternal Ube3a, but does not affect periodicity in peripheral tissues that are not imprinted for uniparental Ube3a expression. The ubiquitin ligase encoded by Ube3a interacts with the central clock components BMAL1 and BMAL2. Moreover, inactivation of Ube3a expression elevates BMAL1 levels in brain regions that control circadian behavior of AS model mice, indicating an important role for Ube3a in modulating BMAL1 turnover.
Conclusions
Ube3a expression constitutes a direct mechanistic connection between symptoms of a human neurological disorder and the central circadian clock mechanism. The lengthened circadian period leads to delayed phase, which could explain the short sleep duration and increased sleep onset latency of AS subjects. Moreover, we report the pharmacological rescue of an AS phenotype, in this case, altered circadian period. These findings reveal potential treatments for sleep disorders in AS patients.
Keywords: circadian, imprinting, Angelman Syndrome, clock, Ube3a, Bmal1, E6-AP, ubiquitin ligase, sleep disorder
Introduction
Angelman syndrome (AS) is a neurodevelopmental disorder of imprinting characterized by mental disability, developmental delays, sleep disorders, epileptic seizures, motor difficulties, and speech impairment [1–3]. There is no specific therapy for Angelman syndrome and treatment for seizures usually becomes necessary. About 70% of AS patients have deletions of the maternal copy of chromosome 15 in the region of 15q11-q13. The Ube3a gene within this region was identified as the genetic locus for AS [4–5], although neighboring genes within the deleted region may contribute to the AS phenotype. Ube3a encodes a HECT-domain E3 ubiquitin ligase (E6-AP) that adds ubiquitin to substrates, thereby targeting them for destruction in the proteasome [6]. AS is an example of genomic imprinting that is caused by the deletion or inactivation of the maternal copy of Ube3a, while the paternal copy is imprinted and therefore silenced. Interestingly, the paternal imprinting involved in AS occurs only in the brain [7–9], and is neither imprinted in non-neural peripheral tissues [10–11] nor in glia [12–13]. Normally (i.e., in non-AS subjects), the maternal copy of Ube3a is active in neurons while the paternal copy is silenced in adult neurons. Therefore, inactivation or deletion of the maternal copy causes a gene dosage effect whereby there is a significant loss of total E6-AP activity in neurons, resulting in AS.
Sleep disorders such as short sleep duration and increased sleep onset latency are very common in AS patients (up to 75% of subjects suffer from sleep disturbances [14–15], and these sleep disruptions are one of the syndrome’s most stressful manifestations to families with an AS member [16]. Almost all of the information about sleep disruptions in AS patients are clinical/behavioral observations, with the exception of a study of daily profiles of the hormone melatonin that concluded there was a high prevalence of circadian rhythm sleep disorders among AS patients [17]. The timing of sleep is regulated by the circadian clock, and in a Drosophila model for AS based on a null mutation of the fly counterpart to Ube3a (dube3a), circadian rhythmicity and activity/rest cycles are abnormal [18]. Moreover, two other imprinted genes, Gnas and Magel2, have consequences for sleep architecture and circadian amplitude in mice [19,20], and the paternally imprinted Magel2 is located within the commonly deleted 15q11-q13 Angelman/PWS interval. A recent publication reported that a clock protein that is a central component of the mammalian circadian clock, BMAL1 (ARNTL in humans), is an ubiquitinylation target of E6-AP [21]. Despite these tantalizing connections between AS, sleep disruptions, and circadian rhythmicity, there have been no reports of the consequences of reduced Ube3a dosage in mammals in vivo that confirm an effect on circadian period, phase, and metabolism.
On the other hand, the general significance of ubiquitin-mediated turnover of circadian clock proteins in the mechanism of circadian rhythmicity has been appreciated since the first observations in model systems Drosophila and Neurospora [22–23]. Recent mammalian studies using mouse strains with mutated/knocked-out genes to the F-box proteins Fbxl3 and Fbxl21 that participate in SCF-mediated ubiquitinylation of the central clock protein CRYPTOCHROME (CRY) [24–28] demonstrate the principle that alterations of clock protein ubiquitinylation can cause circadian phenotypes, but those investigations of SCF/Fbxl have not been linked to any defects or syndromes in humans. Moreover, the E3 ligase encoded by Ube3a (E6-AP) is a single protein that associates with an E2 ubiquitin conjugating enzyme and the target substrate, whereas the SCF ubiquitin ligases are multimeric complexes [6]. This difference in the ubiquitinylation reaction between E6-AP and SCF complexes could have distinct mechanistic consequences.
We report here that mouse models of AS display altered circadian period and phase, and these phenotypes can be further exacerbated by manipulating the environmental light/dark conditions. Re-entrainment kinetics of the circadian system to shifted light/dark cycles are also altered in the AS-model mice. In addition to the change of circadian properties that is induced by the environmental conditions, there is a concomitant change in metabolism such that the AS mice gain excess body fat, which is consistent with the growing literature on the connection between circadian clocks and metabolism [29–34]. Moreover, we found that pharmacological reactivation of paternal Ube3a in AS brain slices restores circadian periodicity in E6-AP deficient neurons. We conclude that deficiency of neuronal E6-AP activity (as in AS) leads to defective ubiquitinylation of clock proteins that alters circadian clock-mediated behavior and metabolism. The data are consistent with a conclusion that altered circadian properties in AS subjects underlie the sleep disorders that characterize AS.
Results
We tested the circadian phenotypes of two different mouse models that are based on the most common genetic defect found in AS, namely a ~4Mb deletion in the maternal copy of chromosome 15 (region 15q11-q13, which includes the Ube3a locus) that leads to reduced E6-AP expression in paternally imprinted brain regions [4–5]. As shown in Fig. S1, there is excellent synteny between this region of human chromosome 15 and region 7p of mouse chromosome 7 [35]. One of these mouse models carries a 299 bp deletion within the Ube3a gene on the maternal chromosome 7 that confers a null mutation (herein called U-m-/p+)[8]. U-m-/p+ model mice exhibit phenotypes that are similar to those of AS patients, including motor dysfunction, locomotor problems, inducible seizures, context-dependent learning deficits, sleep disturbances, and changes in CaMKII phosphorylation patterns that cause hippocampal long-term potentiation (LTP) deficits [8,9,36,37]. The second mouse model is a 1.6-Mb deletion from Ube3a to Gabrb3 on maternal chromosome 7, which removes the syntenous Ube3a, Atp10a, and Gabrb3 loci (herein called UG-m-/p+, Fig. S1) [38]. UG-m-/p+ mice show increased spontaneous seizure activity and abnormal EEG, with concomitant significant impairment in motor function, learning/memory tasks, and anxiety-related measures; the researchers who generated the UG-m-/p+ mice concluded that it is an even more accurate model of AS than the U-m-/p+ mice with a maternal deletion only within Ube3a [38].
Deficiency of E6-AP expression in the brain alters circadian behavior
The free-running circadian rhythm of locomotor activity behavior is significantly prolonged in both the U-m-/p+ and UG-m-/p+ mice (Fig. 1). In U-m-/p+ mice, the rhythm of wheel-running behavior in constant darkness (DD) is 0.3 h longer than that of WT mice (p<0.05), whereas deletion of the paternal copy of Ube3a (U-m+/p-) has no significant effect on this circadian rhythm (Figs. 1A, B). The impact upon the circadian rhythm was more profound in the larger deletion UG-m-/p+ mice and resulted in a 0.6 h slowing of the period of total activity (p<0.01, Figs. 1C, D) and of wheel-running activity (Fig. S2). Not only was the timing of wheel-running activity affected in the AS-model mice (Fig. 1A, B), total activity was also reduced. Another critical circadian behavior that is affected in the AS-model mice is the kinetics of re-entrainment after a phase-shift of the light/dark (LD) cycle (Fig. 1E–H). WT mice require 4 or more days to re-entrain to a 6-h advance in the phase of the LD cycle, but the U-m-/p+ mice re-entrain to the shifted LD cycle significantly faster, within 2 d or less (p<0.01, Fig. 1G). On the other hand, both WT and U-m-/p+ mice re-entrain rapidly to delayed LD cycles (Fig. 1H). These data on period and re-entrainment kinetics denote a weakening of the central circadian oscillator strength in AS-model mice.
Figure 1. Maternal deletion of Ube3a or the Ube3a-Gabrb3 region alters circadian behavior and entrainment in mice.

A: Representative wheel-running activity records (actograms) of mice are shown in the double-plotted format. Mice (4 month old littermates) were: wildtype (WT, left), or maternal deletion (U-m-/p+, middle) and paternal deletion (U-m+/p-, right) of Ube3a. Mice were initially in a 12 h light/12 h dark cycle (blue indicates light, white indicates dark) and were then transferred to constant darkness (DD) on day 8.
B: The free-running period of wheel-running behavior in the mice depicted in panel A (n=5 or 6). Mean±SEM, * p<0.05 compared with WT, one-way ANOVA, post hoc Tukey test.
C: Representative examples of double plotted actograms of total activity in WT (left) vs. maternal deletion of the Ube3a-Gabrb3 region (UG-m-/p+, right) mice measured by infrared sensors (mice were 4 month old littermates).
D: The free-running period of total activity in the mice depicted in panel C (N=3 or 4). Mean ± SEM, ** p<0.01, 2-tailed t-test.
E–H: WT and U-m-/p+ mice were entrained to a 12 hr/12 hr light-dark (LD 12/12) cycle for at least ten days and then subjected to a 6 h advance of the LD cycle for at least 10 d, followed by 6 h delay of LD. As shown by representative actogram records of wheel-running locomotor activity (E) or statistical analyses of activity onset (F, G, and H), U-m-/p+ mice re-entrained to the phase-advanced LD significantly faster than did WT mice. (n=6 per genotype). F: mean ± SEM. p<0.001 for the genotype factor, namely WT vs. U-m/p+ (two-way ANOVA). G, H: 2-tailed t-test, ** p<0.01 in panel G. See also Figures S1 and S2.
Environmental exposure to constant light (LL), which is known to disrupt and/or suppress circadian activity patterns, has a significantly stronger effect on U-m-/p+ mice as compared with WT mice (Fig. 2). During the exposure to LL, activity levels and rhythm amplitude are suppressed in WT mice, but they are practically non-existent in U-m-/p+ mice (Fig. 2A, B, D). Moreover, after returning to DD, WT mice recover their activity levels and rhythm amplitude, but U-m-/p+ mice do not fully recover. The impact of LL on circadian period is even more interesting. As is well known, LL lengthens the period of WT mice, but we observed that LL-induced slowing of U-m-/p+ mouse periodicity is larger (Fig. 2C; the enhanced variability is due to the suppression of activity that complicates accurate period estimation). Most fascinating is the difference in circadian period after return to DD; WT mice recover their period to the pre-LL value, but U-m-/p+ mice experience an “after”-effect on their period that persists. Before exposure to LL, the difference between WT and U-m-/p+ periods is significant at the p<0.05 level (Figs. 1B, 2C), but after LL exposure, the difference is now significant at the p<0.01 level (Fig. 2C). The observation that LL-suppression of circadian rhythmicity is more deleterious to amplitude and period in U-m-/p+ mice further supports the conclusion that normal levels of E6-AP expression enhance circadian oscillator strength.
Figure 2. Environmental exposure to constant light (LL) differentially suppresses the amplitude of circadian locomotor rhythms and prolongs subsequent period when Ube3a is deficient in the central nervous system.

A: Representative wheel-running actograms of WT and U-m-/p+ mice (7 months old) exposed to LL for 4 weeks and then returned to DD for 4 weeks. (Blue, light; white, dark).
B–D: Amplitude (B), period (C), and magnitude (D) of wheel-running activity before (DD Pre LL), during (LL), and after (DD Post LL) exposure to LL. The bar are depicted as mean ± SEM, n=5 or 6, *p<0.05, ** p<0.01, ***p<0.001 compared with the corresponding WT group (2-tailed t-test).
Ube3a gene dosage effects are brain specific and can be rescued by topotecan
Mammalian circadian systems are composed of oscillators in tissues throughout the organism that are coordinated by a central pacemaker in the suprachiasmatic nuclei (SCN) of the hypothalamus [39]. When oscillations are assessed with a luciferase reporter of gene/protein expression, both the SCN and the non-neural peripheral tissues exhibit robust circadian rhythms in vitro [40,41]. We introduced the PmPer2::mPER2-LUC reporter of expression of the central PER2 clock protein [41] into WT, U-m-/p+, and UG-m-/p+ mice. SCN tissues in vitro retain the period difference between WT and U-m-/p+ strains (Fig. 3A, B), and these molecular rhythms also damp more rapidly in U-m-/p+ SCN slices (Fig. 3C). In contrast, neither the U-m-/p+ or UG-m-/p+ peripheral tissue (spleen and lung) show period differences from WT (Fig. 3D, E). This result implies that the expression of E6-AP from the active paternal allele of Ube3a in peripheral tissue of AS-model mice is sufficient to promote wild-type clock function, but in the brain tissue (SCN) where the paternal allele is inactivated, E6-AP activity is reduced to the degree that clock properties are impaired.
Figure 3. Ube3a gene dosage effects are brain specific and rescued by topotecan.

A: Maternal deletion of Ube3a alters molecular rhythms in explanted suprachiasmatic nuclei (SCN), as assessed with a luminescence reporter of mPER2 expression. SCN slices were collected from PmPer2::mPER2-LUC knockin mice in the WT and U-m-/p+ backgrounds. Tissue explants were dissected on Day 0 and recorded with a LumiCycle apparatus. Blue: WT; Red: Ube3a m-/p+; Black: WT SCN plus topotecan; Green: Ube3a m-/p+ plus topotecan (300 nM topotecan added to the medium at the beginning of culture).
B, C: period (B) and damping rate (C) analyses of SCN data (representative examples shown in panel A), plotted as mean ± SEM (n=5–7 SCN slices from 5–7 animals), *p<0.05, ** p<0.01 compared with WT or as indicated, One-way ANOVA, post hoc Tukey test.
D, E: Neither maternal deletion of Ube3a nor topotecan alters PER2 molecular rhythms in explanted peripheral tissues. D, period of PmPer2::mPER2-LUC luminescence rhythms from spleen tissue in vitro from WT, U-m-/p+, and UG-m-/p+ mice. E, period of PmPer2::mPER2-LUC luminescence rhythms from lung tissue derived from WT, U-m-/p+, and UG-m-/p+ mice plus and minus topotecan. At least 7 animals were used in each group (mean ± SEM, 300 nM topotecan was added to the lung tissues when culturing began).
However, the paternal Ube3a allele can be unsilenced. An exciting new development in the potential treatment of AS is the use of the topoisomerase inhibitor topotecan to reactivate the dormant paternal Ube3a allele in neurons [42]. We observed that nanomolar concentrations of topotecan restored the long period of the PER2 expression rhythm in U-m-/p+ SCN slices to a WT value (Fig. 3A, B), as well as preventing the accelerated damping (Fig. 3C). Topotecan does not have significant effects on the period of peripheral lung tissue where the Ube3a paternal allele is still active (Fig. 3E). There is an intriguing trend towards topotecan shortening the period expressed by WT SCN, but this difference is not statistically significant (Fig. 3B). However, since there is one active Ube3a allele in WT SCN (maternal) and two active Ube3a alleles in WT lung, the trend towards topotecan-induced shortening the period of WT SCN (Fig. 3B) without affecting the period of WT lung (Fig. 3E) implies that topotecan effects on period are not due to a non-specific side-effect (if this were true, the period in the lung should also be affected), but are attributable to the reactivation of the paternal Ube3a allele in WT SCN, thereby increasing E6-AP activity and speeding the clock.
Reciprocal regulatory network between Ube3a and the central clock gene Bmal1
Key components of the autoregulatory transcriptional feedback mechanism of the mammalian circadian clock are the heterodimeric activators BMAL1/CLOCK and BMAL1/NPAS2 that act as positive elements to drive transcription on E-box motifs in many target genes including Per1, Per2, and Avp [43–45]. The upstream region of Ube3a comprises several E-boxes and is therefore predicted to be regulated by BMAL1 in association with CLOCK and/or NPAS2. E-box-containing genes are often–but not always–regulated rhythmically [46]. The latter case appears to apply to Ube3a. In liver, Ube3a transcript levels are not rhythmic in DD in the same samples in which Per2 mRNA rhythmicity was confirmed (Fig. 4A). Immunoblot analyses of E6-AP levels at two circadian phases showed no difference in protein levels in WT hypothalamus, while confirming the tremendous reduction of E6-AP levels in U-m-/p+ brain tissue (Fig. 4B). Consistent with the presence of several potential E-boxes in the 2.1 kb upstream region of Ube3a, a PUbe3a::Luc reporter was galvanized by both BMAL1/CLOCK and BMAL1/NPAS2 co-activators (Fig. 4D). However, this PUbe3a::Luc reporter is not regulated rhythmically under the same conditions that PPer2 and PBmal1 activity is rhythmic (Fig. S3A, B), which is consistent with the lack of a rhythm in transcript or protein abundance (Fig. 4A, B). In addition, E6-AP does not directly activate the expression of other E-box-containing promoters, such as PPer1 or PAvp (Fig. S3C, D). Despite not being rhythmically regulated, however, Ube3a expression is at the behest of BMAL1. In liver of Bmal1−/− knockout mice, the level of Ube3a mRNA expression is one-third of its level in WT liver (Fig. 4C).
Figure 4. Ube3a expression is dependent upon the clock factors BMAL1, CLOCK, NPAS2, but is not rhythmically expressed.

A: Transcript levels for Ube3a (left) and Per2 (right) in liver. Ube3a and Per2 mRNA levels were quantified by real-time PCR and normalized with reference to Hprt mRNA levels. The data were normalized by setting the value to 1.0 for the mRNA levels at hour 44 in DD (= CT8). n=4–7 mice.
B, Upper panel: E6-AP and β-ACTIN protein levels in mouse hypothalamus (WT and U-m-/p+ mice) from two different circadian phases in DD (hour 26 which is CT14 in the early subjective night, and hour 38 which is CT2 in the early subjective day). Data are immunoblots of hypothalamus extracts from four separate mice (each lane is the extract from a separate mouse). The difference between the upper and middle panels of immunoblots (E6-AP) is the exposure duration. Lower panel: Densitometric analyses of the data in the upper panel. The E6-AP expression of U-m-/p+ at DD 38 h is set as 1. Statistics: p<0.0001 for genotype factor, (WT vs. U-m-/p+) by two-way ANOVA.
C: Levels of Ube3a transcript expression in WT (left) versus Bmal1−/− (right) liver in subjective night (at DD 50–56 hrs; for WT, this time equals CT 14–20). The mRNA was quantified by real-time PCR and normalized to Hprt mRNA controls. The value of WT mRNA levels was set as 1.0. At least 6 mice were used in each group. ** p<0.01, 2-tailed T test.
D: BMAL1/CLOCK (B1/C) and BMAL1/NPAS2 (B1/N) transactivation of the mUbe3a upstream region as assessed by a transient transfection assay in HEK 293 cells. Data are mean ± SEM (n =4) using a PUbe3a::Luc firefly luciferase reporter driven by 2.2 kb of the mUbe3a upstream region including 220 bp of the 5′ UTR, and normalized by a Renilla luciferase control (PCMV::Rluc). *** p<0.001 compared with empty vectors control (Con), which is set as 1.0, One-way ANOVA, post hoc Tukey test. See also Figure S3.
While Ube3a expression is controlled by BMAL1, E6-AP reciprocally regulates BMAL1 activity by modulating BMAL1 stability. BMAL1 has been recently identified as an in vitro target of E6-AP-catalyzed ubiquitinylation [21]. Consistent with those observations, we confirm in vivo that the BMAL1 protein abundance is significantly higher in the hypothalamus of U-m-/p+ mice at two opposite circadian phases, as expected if E6-AP ubiquitinylates and destabilizes BMAL1 (Fig. 5A, Two-way ANOVA for WT vs. U-m-/p+, p<0.0013). E6-AP physically interacts with BMAL1 and its paralog BMAL2 [47], as indicated by co-immunoprecipitation of E6-AP with BMAL1/BMAL2 from extracts of these proteins expressed in HEK293 cells (Fig. 5B). This interaction was not observed between E6-AP and PER1/2 or CRY1/2 (Fig. S4). Although E6-AP expression does not affect E-box-mediated transcription directly (Fig. S3C, D), it does reduce BMAL1/CLOCK transactivation of E-box activity (Fig. 5C). This E6-AP-mediated reduction in BMAL1/CLOCK activity is likely to be due to ubiquitinylation/destabilization of BMAL1, because Ube3a expression does not directly affect the activity of the Bmal1 promoter (unlike the transcriptional factor Rev-Erb-α, which is known to repress PBmal1 [48], Fig. 5D).
Figure 5. The stability of BMAL1 is regulated by Ube3a expression in vivo.
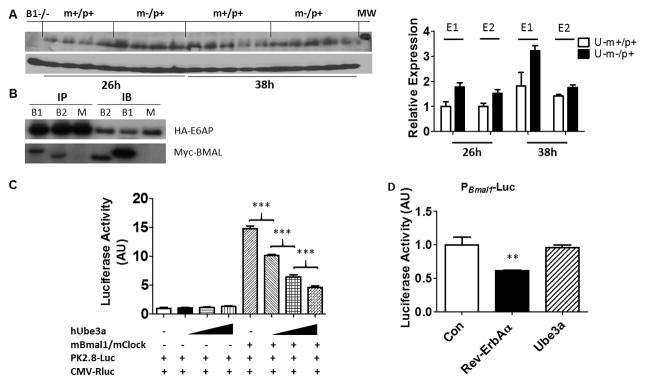
A, Left panel: Immunoblots of BMAL1 in hypothalamus of U-m-/p+ and WT mice placed in DD for 26 and 38 h (CT14 & CT2, respectively). β-Actin serves as the loading control (each lane comes from a separate mouse, one representative immunoblot is shown). Right panel: Densitometric analyses of the BMAL1 expression for two independent experiments (E1 and E2; an immunoblot for E2 is shown in the left panel). BMAL1 expression of WT at DD 26 h is set as 1 (“B1−/−” = Bmal1−/−; “MW” = a protein band of molecular weight 75 kD). Two-way ANOVA statistical analyses show significant differences for both genotype (WT vs. U-m-/p+, p = 0.0013) and for time (DD 26h vs. DD 38h, p = 0.0029).
B: Interaction between BMAL1 (B1) or BMAL2 (B2) and E6-AP in HEK293 cells transfected with Myc-mBmal1/2 and HA-hUbe3a. Immunoprecipitates were prepared using an anti-HA antibody and the immunoprecipitates (IP) and original cell lysates (IB) were separated by electrophoresis and immunoblotted with either anti-HA (for E6-AP) or anti-Myc (for BMAL1 or BMAL2)(M = mock control, pCDNA3.1 vector).
C: Ube3a expression reduces BMAL1/CLOCK activation of E-box reporter PPK2.8::Luc in HEK 293 cells. Data (mean ± SEM, n=4) were normalized to the PCMV::Rluc control and expressed relative to empty vector controls. ***p<0.01, One-way ANOVA, post hoc Tukey test.
D: Ube3a expression does not affect the promoter activity of Bmal1. Data are mean ± SEM (n =4) of firefly luciferase activity (PBmal1-Fluc) normalized to the PCMV::Rluc control. ** p<0.01 compared with empty vectors control (Con), which was set to a value of 1.0, one-way ANOVA, post hoc Tukey test. See also Figures S4 and S6.
Body weight and activity patterns are altered in AS-model mice
A blossoming literature underscores the intimate connection between circadian clocks and metabolism, including the observation that disruptions of the circadian system lead to metabolic dysfunctions [29–34]. In addition to sleep disorders, human syndromes that result from maternal and paternal deletions on chromosome 15 (AS and Prader–Willi syndrome {PWS}) have associated metabolic phenotypes including altered activity levels and weight gain/loss problems (AS: [49], PWS: [50,51]). We found similar phenotypes in the AS-model mice that have altered circadian properties. With the maternal deletion in Ube3a alone (U-m-/p+), body weight is significantly higher than in WT mice, and this difference is mostly attributable to an increase of fat mass (Fig. 6A–C). These animals were fed regular chow, so feeding a high fat diet is not necessary to promote these differences in weight gain. Moreover, the heavier weight of U-m-/p+ mice is not due to an increased food intake (Fig. 6E). On the other hand, the wheel-running activity of U-m-/p+ mice is less than that of WT mice (Figs. 2D,6D) and therefore the energy balance might therefore tip towards greater weight gain in the U-m-/p+ mice.
Figure 6. Maternal deletion of Ube3a impairs metabolic homeostasis in mice. WT and U-m-/p+ mice were fed regular chow, and their body composition, locomotor activity, and food consumption were measured.
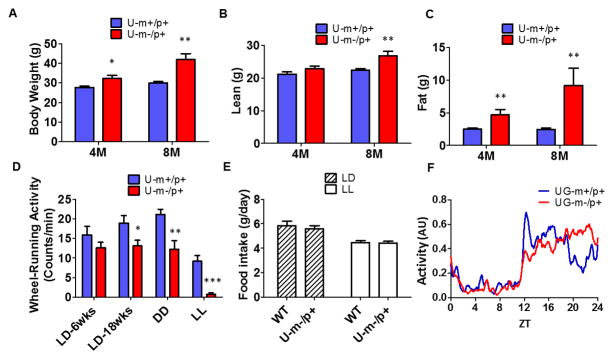
A: Body weight (n=5–6/genotype).
B, C: Body composition was measured by NMR as lean mass (B) and fat mass (C) at age 4 months (in LD) and 8 months (sequentially in LD, DD, and LL over the 8 months)(n=5–6/genotype).
D: Wheel-running locomotor activity recorded at different ages and under various light/dark conditions (n = 5–6/genotype).
E: Average daily food intake over 24 hours in LD vs. LL (n = 5–6/genotype). Each bar represents mean ± SEM; *p<0.05, ** p<0.01, ***p<0.001 compared with the corresponding WT group, 2-tailed T test.
F: Relative daily activity patterns in LD of WT (red) vs. UG-m-/p+ (blue) mice. Total locomotor activity was recorded with infrared sensors from mice in LD. ZT0 denotes lights-on, and ZT12 denotes lights-off. The lines connect the mean values over 7 d in LD of the 1 h moving averages. The data are normalized as the highest unnormalized activity level (see Fig. S5C) of each individual mouse during 24 hrs adjusted to 1 (n ≥ 3). See also Figure S5.
Similarly, the other AS-model mouse strain in which the entire Ube3a-Gabrb3 region is deleted from the maternal chromosome (UG-m-/p+) exhibits increased weight gain and altered daily activity patterns. UG-m-/p+ mice are significantly heavier than WT at an age of 10 months, even when fed regular chow (Fig. S5A). This difference may be related to a lower overall activity level, because UG-m-/p+ mice have less total activity than WT mice in LD (Fig. S5B). An interesting observation of the activity pattern of UG-m-/p+ mice in LD is that the major activity bout is distributed towards the end of the dark interval, whereas in WT mice the activity predominates in the early portion of the dark interval of LD (Figs. 6F, S5C, S5D). This patterning can be seen visually in the raw data of Fig. 1C. The phenomenon is consistent with the interpretation that the longer circadian period in DD of the AS-model mice manifests as a later phase of activity under LD as predicted by entrainment theory [52,53].
Modulating Ube3a expression in cell cultures alters circadian properties
Stifling Ube3a expression lengthens the circadian period, whether this suppression is by maternal deletion in neurons (intact animal behavior, Figs. 1,2; SCN brain slices, Fig. 3) or by siRNA knockdown in fibroblast cell cultures [21]. The converse manipulation–increasing the level of Ube3a expression has–the predictably opposite result. When transfected with a plasmid that over-expresses Ube3a, the rhythm of both mouse embryonic fibroblasts and Rat-1 fibroblasts exhibits a shorter period at low concentrations (Fig. S6A, B, D), and then suppression and disruption of the rhythm at higher concentrations (Fig. S6C, D).
Discussion
Neuronal imprinting of paternal Ube3a modulates circadian properties
Our results show that tissue-specific differences in the expression levels of Ube3a have a large effect on circadian properties because the encoded ligase E6-AP modulates the stability of the core clock protein BMAL1. Gene dosage effects of Ube3a are associated with Angelman Syndrome and autism [4, 5,7–13,54]; therefore, some key phenotypes of these disorders may be attributable to E6-AP-dependent effects upon the circadian system. The current model for the molecular mechanism of the mammalian circadian clockwork proposes autoregulatory transcriptional & translational feedback loops (TTFLs) of central clock gene expression [55–57]. There is an ensemble of different molecular components that are believed to participate in a feedback loop of clock proteins that are rhythmically abundant and interact with each other to modulate their activities. Rhythmic transcriptional “drive” is provided by the positive bHLH-PAS transcription factor, BMAL1/ARNTL, which dimerizes with CLOCK or NPAS2 [44, 45]. These BMAL1/CLOCK and BMAL1/NPAS2 heterodimers activate transcription primarily at E-box enhancers [43]. Negative feedback in the system is primarily accomplished by Per and Cry gene products.
The TTFL model posits that rhythmic abundance of at least one central clock protein is necessary to accomplish the rhythmic negative feedback, which implies that turnover is a crucial parameter of the clockwork. Indeed, the abundance of the PER and CRY negative-feedback proteins are rhythmic [58, 59]. Recent investigations have analyzed the role of SCF-mediated ubiquitinylation in regulating CRY protein turnover by the F-box proteins Fbxl3 and Fbxl21, which in turn modulates circadian period [24–28]. However, cycling of the positive factor BMAL1 has been thought to be unnecessary for basic clock function [60]. Nevertheless, the period lengthening observed in U-m-/p+ and UG-m-/p+ mice (Figs. 1–3) is reminiscent of the results obtained with mutations in F-box proteins that participate in rhythmic turnover of CRY [24–28]. Moreover, in vitro studies confirm that manipulating BMAL1 stability by changing E6-AP expression/activity has significant impact upon circadian period and amplitude (Figs. 3, S6 [21]). Apparently, when E6-AP activity is low (as when Ube3a gene dosage is reduced in the AS-model mice), BMAL1 is stabilized (Fig. 5A and ref.[21]) and the circadian oscillator is less robust. This weakening is manifested as a longer period in vivo and in vitro, enhanced damping in vitro, reduced in vivo amplitude in LL, prolonged LL-induced period after-effects, and more rapid re-entrainment to a phase-advanced LD cycle. Our results are also consistent with other studies in which enhanced expression of BMAL1 in cell cultures causes a lengthening of circadian period [61].
It is counter-intuitive that enhancing the abundance/stability of the positive transcriptional factor BMAL1 will lead to a less robust circadian pacemaker. However, two recent reports have shown that metabolic inputs to the clock, including entrainment by restricted feeding paradigms, are mediated by adjusting the stability of BMAL1 [62,63]. Taken together, these results underscore that much remains to be learned about the mechanism of the mammalian circadian clockwork, including that inputs to the clock may be mediated through the positive element BMAL1, and not only through the negative feedback elements PER and CRY. Finally, a fascinating aspect about circadian organization within the AS-model mice is that the paternal Ube3a allele is inactivated only in neurons. Therefore, U-m-/p+ and UG-m-/p+ mice become a genetic mosaic as regards Ube3a expression, and by extension, a circadian mosaic. Our data support this interpretation in the AS-model mice; as shown in Fig. 3, the circadian period and amplitude effects are observed in SCN slices (neural tissue where the paternal Ube3a allele is silenced), but not in lung or spleen (non-neural peripheral tissue where the paternal Ube3a allele is active).
Ube3a-dependent circadian modulation generates phenotypes associated with AS and PWS
Common forms of the human syndromes Angelman Syndrome (AS) and Prader-Willi Syndrome (PWS) are associated with imprinting/inactivation of Ube3a on one chromosome and deletion on the other chromosome. Sleep disorders that can lead to behavioral issues are associated with both AS and PWS, especially in childhood [16]. In the case of AS, the sleep problems include increased sleep onset latency and reduced total sleep duration [3,15–17]. Sleep disruption can also occur in AS subjects because of seizures, but even in AS subjects with no or minimal seizures, the sleep latency/duration symptoms persist. The longer periods of the U-m-/p+ and UG-m-/p+ mice can provide an explanation for these phenotypes in humans. In particular, from entrainment theory, longer circadian periods can manifest as a later phase relationship of rhythm to the light/dark cycle [52]. This happens because the longer period requires a larger advance phase shift in order to entrain to a 24 h LD cycle, and this is accomplished by illuminating a different portion of the phase-shifting landscape known as the Phase Response Curve. A later phase relationship between the activity/sleep cycle and the LD cycle will allow a longer endogenous circadian period to entrain to the 24 h environmental cycle [52], and even a small change in period can bring about a large change in the phase relationship [53]. Figs. 1C/D and 6F indicate that the nocturnal activity of AS-model mice is distributed into a later phase relationship in LD cycles, and this altered phase relationship could disturb sleep in the AS model mice [37]. In humans, a later phase relationship leads to Delayed Sleep Phase Syndrome (DSPS) in which subjects go to sleep later (i.e., increased sleep onset latency) and wake up later in the morning. Several studies confirm that DSPS in humans is associated with a longer period of the molecular clock [64–67]. AS children have increased sleep onset latency [15], and if their parents rouse them at the usual wake-up time, then they will also experience a reduced total sleep duration [15]. The application of non-invasive chronotherapeutic strategies designed for persons suffering from affective disorders [68] may enhance the entrainment of AS subjects and improve the quality of their sleep.
Metabolism and circadian rhythmicity have mutual feedback upon each other [29–34]. We observed another example of these complementary relationships in the AS-model mice. Both the U-m-/p+ and the UG-m-/p+ mice gained more weight than WT littermates under the same conditions, and this effect may be related to either a slightly lower level of overall activity (Figs. 6 and S5), and/or the suppression of rhythmicity as we concluded in a previous study [34]. AS model mice are consistently overweight [36], and metabolic phenotypes are also associated in humans with altered expression of Ube3a. For example, while AS subjects with chromosomal deletions are rarely overweight, those with uniparental disomy or imprinting center mutations are commonly overweight [69]. In addition, while obesity is a hallmark feature of its sister imprinting syndrome, PWS, mouse models of PWS are rarely overweight [70]. These correlations suggest that the AS mouse models may have rhythmic similarities to AS, but metabolic similarities to PWS [70]. Therefore, neuronal inactivation of paternal Ube3a in mice whose maternal Ube3a gene is deleted recapitulates sleep and metabolic symptoms of human syndromes that are associated with imprinting of chromosomal region 15q11-q13 (AS and PWS). It will be fascinating to test whether overexpression of maternal Ube3a, such as in uniparental disomy or imprinting center mutation genotypes of PWS, reciprocally affects circadian period and metabolic status.
By virtue of its regulation of the stability of the core clock component BMAL1, E6-AP acts as a key modulator of the circadian system, thereby affecting fundamental properties such as period, amplitude, and entrainment kinetics. New pharmaceutical treatments to unsilence paternal Ube3a are currently under development [42,49] that may treat or ameliorate the disorder at its genetic root. The circadian abnormalities identified in this study may serve as a biomarker for clinical trials of these medications in their early phases. Moreover, translational applications may result from the elucidation of the connections between Ube3a expression and circadian rhythms, as this research could lead to therapies of non-invasive alterations of environmental conditions to ameliorate at least some of the symptoms of AS. Such translational applications are suggested by the exacerbation of period and metabolic phenotypes by a clock-perturbing environmental parameter, namely LL (Fig. 2). If environmental conditions that interact with the circadian system can exacerbate undesirable phenotypes (such as LL), it is likely that environmental conditions can be identified that have an opposite impact upon circadian properties such that symptoms can be improved [68].
Experimental Procedures
Complete experimental procedures are described in the Supplemental Experimental Procedures.
Supplementary Material
Highlights.
Neuronal imprinting of Ube3a gene affects circadian activity and metabolism in mice
Pharmacological reversal of silencing rescues functional circadian physiology
Angelman Syndrome model mice are a mosaic of Ube3a expression and clock properties
Sleep disorders that are common to Angelman Syndrome may be treated by chronotherapy
Acknowledgments
For essential materials, we thank Dr. Arthur Beaudet (UG-m-/p+ model mice), Dr. Joseph Takahashi (PmPer2::mPER2-LUC knockin mice), Dr. Charles Weitz (anti-BMAL1 antibody), Dr. Heping Yan (anti-Myc monoclonal antibody), Dr. David Weaver (PAVP::FLuc reporter plasmid), Dr. Qun-yong Zhou (PPK2.8::FLuc reporter plasmid), and Dr. Nicolas Cermakian (PCMV::Myc-Bmal1/2 plasmids). Dr. David McCauley provided expert statistical advice. This research was supported by grants from the National Institutes of Health (R21HL102492-01A1 from NHLBI and R01GM088595-01 from NIGMS). SQS was partially supported by NARSAD Young Investigator Award # 17623. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Author Contributions
S.Q.S., T.J.B., and C.H.J. conceived the project. S.Q.S., R.I., and C.H.J. designed the experiments, S.Q.S., T.J.B., and R.I. performed the experiments, and S.Q.S. and C.H.J. wrote the paper.
The authors declare no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Robb SA, Pohl KRE, Wilson BJ, Brett EM. The ‘happy puppet’ syndrome of Angelman: review of the clinical features. Arch Dis Child. 1989;64:83–86. doi: 10.1136/adc.64.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Williams CA, Beaudet AL, Clayton-Smith J, Knoll JH, Kyllerman M, Laan LA, Magenis RE, Moncla A, Schinzel AA, Summers JA, et al. Angelman syndrome 2005: updated consensus for diagnostic criteria. Am J Med Genet A. 2005;140:413–418. doi: 10.1002/ajmg.a.31074. [DOI] [PubMed] [Google Scholar]
- 3.Laan LA, Haeringen vA, Brouwer OF. Angelman syndrome: a review of clinical and genetic aspects. Clin. Neurol. Neurosurg. 1999;101:161–170. doi: 10.1016/s0303-8467(99)00030-x. [DOI] [PubMed] [Google Scholar]
- 4.Matsuura T, Sutcliffe JS, Fang P, Galjaard RJ, Jiang YH, Benton CS, Rommens JM, Beaudet AL. De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat. Genet. 1997;15:74–77. doi: 10.1038/ng0197-74. [DOI] [PubMed] [Google Scholar]
- 5.Sutcliffe JS, Jiang YH, Galijaard RJ, Matsuura T, Fang P, Kubota T, Christian SL, Bressler J, Cattanach B, Ledbetter DH, et al. The E6-AP ubiquitin-protein ligase (UBE3A) gene is localized within a narrowed Angelman syndrome critical region. Genome Res. 1997;7:368–377. doi: 10.1101/gr.7.4.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lehman NL. The ubiquitin proteasome system in neuropathology. Acta Neuropathol. 2009;118:329–47. doi: 10.1007/s00401-009-0560-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Albrecht U, Sutcliffe JS, Cattanach BM, Beechey CV, Armstrong D, Eichele G, Beaudet AL. Imprinted expression of the murine Angelman syndrome gene, Ube3a, in hippocampal and Purkinje neurons. Nat. Genet. 1997;17:75–78. doi: 10.1038/ng0997-75. [DOI] [PubMed] [Google Scholar]
- 8.Jiang YH, Armstrong D, Albrecht U, Atkins CM, Noebels JL, Eichele G, Sweatt JD, Beaudet AL. Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron. 1998;21:799–811. doi: 10.1016/s0896-6273(00)80596-6. [DOI] [PubMed] [Google Scholar]
- 9.Weeber EJ, Jiang YH, Elgersma Y, Varga AW, Carrasquillo Y, Brown SE, Christian JM, Mirnikjoo B, Silva A, et al. Derangements of hippocampal calcium/calmodulin-dependent protein kinase II in a mouse model for Angelman mental retardation syndrome. J Neurosci. 2003;23:2634–44. doi: 10.1523/JNEUROSCI.23-07-02634.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nakao M, Sutcliffe JS, Durtschi B, Mutirangura A, Ledbetter DH, Beaudet AL. Imprinting analysis of three genes in the Prader-Willi/Angelman region: SNRPN, E6-associated protein, and PAR-2 (D15S225E) Hum Mol Genet. 1994;3:309–315. doi: 10.1093/hmg/3.2.309. [DOI] [PubMed] [Google Scholar]
- 11.Gustin RM, Bichell TJ, Bubser M, Daily J, Filonova I, Mrelashvili D, Deutch AY, Colbran RJ, Weeber EJ, Haas KF. Tissue-specific variation of Ube3a protein expression in rodents and in a mouse model of Angelman syndrome. Neurobiol Dis. 2010;39:283–91. doi: 10.1016/j.nbd.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dindot SV, Antalffy BA, Bhattacharjee MB, Beaudet AL. The Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus, and maternal deficiency results in abnormal dendritic spine morphology. Hum. Mol. Genet. 2008;17:111–8. doi: 10.1093/hmg/ddm288. [DOI] [PubMed] [Google Scholar]
- 13.Judson MC, Sosa-Pagan JO, Del Cid WA, Han JE, Philpot BD. Allelic specificity of Ube3a expression in the mouse brain during postnatal development. J Comp Neurol. 2014;522:1874–96. doi: 10.1002/cne.23507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith A, Wiles C, Haan E, McGill J, Wallace G, Dixon J, Selby R, Colley A, Marks R, Trent RJ. Clinical features in 27 patients with Angelman syndrome resulting from DNA deletion. J Med Genet. 1996;33:107–12. doi: 10.1136/jmg.33.2.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pelc K, Cheron G, Boyd SG, Dan B. Are there distinctive sleep problems in Angelman syndrome? Sleep Med. 2008;9:434–41. doi: 10.1016/j.sleep.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 16.Goldman SE, Bichell TJ, Surdyka K, Malow BA. Sleep in children and adolescents with Angelman syndrome: association with parent sleep and stress. J Intellect Disabil Res. 2012;56:600–8. doi: 10.1111/j.1365-2788.2011.01499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takaesu Y, Komada Y, Inoue Y. Melatonin profile and its relation to circadian rhythm sleep disorders in Angelman syndrome patients. Sleep Med. 2012;13:1164–70. doi: 10.1016/j.sleep.2012.06.015. [DOI] [PubMed] [Google Scholar]
- 18.Wu Y, Bolduc FV, Bell K, Tully T, Fang Y, Sehgal A, Fischer JA. A Drosophila model for Angelman syndrome. Proc Natl Acad Sci USA. 2008;105:12399–404. doi: 10.1073/pnas.0805291105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kozlov SV, Bogenpohl JW, Howell MP, Wevrick R, Panda S, Hogenesch JB, Muglia LJ, Van Gelder RN, Herzog ED, Stewart CL. The imprinted gene Magel2 regulates normal circadian output. Nat Genet. 2007;39:1266–1272. doi: 10.1038/ng2114. [DOI] [PubMed] [Google Scholar]
- 20.Lassi G, Ball ST, Maggi S, Colonna G, Nieus T, Cero C, Bartolomucci A, Peters J, Tucci V. Loss of Gnas imprinting differentially affects REM/NREM sleep and cognition in mice. PLoS Genet. 2012;8:e1002706. doi: 10.1371/journal.pgen.1002706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gossan NC, Zhang F, Guo B, Jin D, Yoshitane H, Yao A, Glossop N, Zhang YQ, Fukada Y, Meng QJ. The E3 ubiquitin ligase UBE3A is an integral component of the molecular circadian clock through regulating the BMAL1 transcription factor. Nucleic Acids Res. 2014;42:5765–75. doi: 10.1093/nar/gku225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Naidoo N, Song W, Hunter-Ensor M, Sehgal A. A role for the proteasome in the light response of the timeless clock protein. Science. 1999;285:1737–41. doi: 10.1126/science.285.5434.1737. [DOI] [PubMed] [Google Scholar]
- 23.He Q, Liu Y. Degradation of the Neurospora circadian clock protein FREQUENCY through the ubiquitin-proteasome pathway. Biochem Soc Trans. 2005;33:953–956. doi: 10.1042/BST20050953. [DOI] [PubMed] [Google Scholar]
- 24.Busino L, Bassermann F, Maiolica A, Lee C, Nolan PM, Godinho SI, Draetta GF, Pagano M. SCFFbxl3 controls the oscillation of the circadian clock by directing the degradation of cryptochrome proteins. Science. 2007;316:900–4. doi: 10.1126/science.1141194. [DOI] [PubMed] [Google Scholar]
- 25.Godinho SI, Maywood ES, Shaw L, Tucci V, Barnard AR, Busino L, Pagano M, Kendall R, Quwailid MM, Romero MR, et al. The after-hours mutant reveals a role for Fbxl3 in determining mammalian circadian period. Science. 2007;316:897–900. doi: 10.1126/science.1141138. [DOI] [PubMed] [Google Scholar]
- 26.Siepka SM, Yoo SH, Park J, Song W, Kumar V, Hu Y, Lee C, Takahashi JS. Circadian mutant Overtime reveals F-box protein FBXL3 regulation of cryptochrome and period gene expression. Cell. 2007;129:1011–23. doi: 10.1016/j.cell.2007.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hirano A, Yumimoto K, Tsunematsu R, Matsumoto M, Oyama M, Kozuka-Hata H, Nakagawa T, Lanjakornsiripan D, Nakayama KI, Fukada Y. FBXL21 regulates oscillation of the circadian clock through ubiquitination and stabilization of cryptochromes. Cell. 2013;152:1106–18. doi: 10.1016/j.cell.2013.01.054. [DOI] [PubMed] [Google Scholar]
- 28.Yoo SH, Mohawk JA, Siepka SM, Shan Y, Huh SK, Hong HK, Kornblum I, Kumar V, Koike N, Xu M, et al. Competing E3 ubiquitin ligases govern circadian periodicity by degradation of CRY in nucleus and cytoplasm. Cell. 2013;152:1091–105. doi: 10.1016/j.cell.2013.01.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang X, Lamia KA, Evans RM. Nuclear receptors, metabolism, and the circadian clock. Cold Spring Harb Symp Quant Biol. 2007;72:387–94. doi: 10.1101/sqb.2007.72.058. [DOI] [PubMed] [Google Scholar]
- 30.Marcheva B, Ramsey KM, Buhr ED, Kobayashi Y, Su H, Ko CH, Ivanova G, Omura C, Mo S, Vitaterna MH, et al. Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nature. 2010;466:627–31. doi: 10.1038/nature09253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Asher G, Schibler U. Crosstalk between components of circadian and metabolic cycles in mammals. Cell Metab. 2011;13:125–37. doi: 10.1016/j.cmet.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 32.Coomans CP, van den Berg SA, Houben T, van Klinken JB, van den Berg R, Pronk AC, Havekes LM, Romijn JA, van Dijk KW, Biermasz NR, et al. Detrimental effects of constant light exposure and high-fat diet on circadian energy metabolism and insulin sensitivity. FASEB J. 2013;27:1721–32. doi: 10.1096/fj.12-210898. [DOI] [PubMed] [Google Scholar]
- 33.Qian J, Block GD, Colwell CS, Matveyenko AV. Consequences of exposure to light at night on the pancreatic islet circadian clock and function in rats. Diabetes. 2013;62:3469–78. doi: 10.2337/db12-1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shi S, Ansari T, McGuinness OP, Wasserman DH, Johnson CH. Circadian disruption leads to insulin resistance and obesity. Current Biology. 2013;23:372–81. doi: 10.1016/j.cub.2013.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takumi T. The neurobiology of mouse models syntenic to human chromosome 15q. J Neurodev Disord. 2011;3:270–81. doi: 10.1007/s11689-011-9088-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang HS, Burns AJ, Nonneman RJ, Baker LK, Riddick NV, Nikolova VD, Riday TT, Yashiro K, Philpot BD, Moy SS. Behavioral deficits in an Angelman syndrome model: effects of genetic background and age. Behav Brain Res. 2013;243:79–9. doi: 10.1016/j.bbr.2012.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Colas D, WagstaffJFort P, Salvert D, Sarda N. Sleep disturbances in Ube3a maternal-deficient mice modeling Angelman syndrome. Neurobiol Dis. 1989;20:471–478. doi: 10.1016/j.nbd.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 38.Jiang YH, Pan Y, Zhu L, Landa L, Yoo J, Spencer C, Lorenzo I, Brilliant M, Noebels J, Beaudet AL. Altered ultrasonic vocalization and impaired learning and memory in Angelman syndrome mouse model with a large maternal deletion from Ube3a to Gabrb3. PLoS One. 2010;5:e12278. doi: 10.1371/journal.pone.0012278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Welsh DK, Takahashi JS, Kay SA. Suprachiasmatic nucleus: cell autonomy and network properties. Annu Rev Physiol. 2010;72:551–77. doi: 10.1146/annurev-physiol-021909-135919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yamazaki S, Numano R, Abe M, Hida A, Takahashi R, Ueda M, Block GD, Sakaki Y, Menaker M, Tei H. Resetting central and peripheral circadian oscillators in transgenic rats. Science. 2000;288:682–685. doi: 10.1126/science.288.5466.682. [DOI] [PubMed] [Google Scholar]
- 41.Yoo SH, Yamazaki S, Lowrey PL, Shimomura K, Ko CH, Buhr ED, Siepka SM, Hong HK, Oh WJ, Yoo OJ, et al. PERIOD2::LUCIFERASE real-time reporting of circadian dynamics reveals persistent circadian oscillations in mouse peripheral tissues. Proc Natl Acad Sci U S A. 2004;101:5339–46. doi: 10.1073/pnas.0308709101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang HS, Allen JA, Mabb AM, King IF, Miriyala J, Taylor-Blake B, Sciaky N, Dutton JW, Jr, Lee HM, Chen X, et al. Topoisomerase inhibitors unsilence the dormant allele of Ube3a in neurons. Nature. 2011;481:185–9. doi: 10.1038/nature10726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gekakis N, Staknis D, Nguyen HB, Davis FC, Wilsbacher LD, King DP, Takahashi JS, Weitz CJ. Role of the CLOCK protein in the mammalian circadian mechanism. Science. 1998;280:1564–1569. doi: 10.1126/science.280.5369.1564. [DOI] [PubMed] [Google Scholar]
- 44.Hogenesch JB, Gu YZ, Jain S, Bradfield CA. The basic-helix-loop-helix-PAS orphan MOP3 forms transcriptionally active complexes with circadian and hypoxia factors. Proc Natl Acad Sci USA. 1998;95:5474–5479. doi: 10.1073/pnas.95.10.5474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.DeBruyne JP, Weaver DR, Reppert SM. CLOCK and NPAS2 have overlapping roles in the suprachiasmatic circadian clock. Nat Neurosci. 2007;10:543–5. doi: 10.1038/nn1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Muñoz E, Baler R. The circadian E-box: when perfect is not good enough. Chronobiol Int. 2003;20:371–88. doi: 10.1081/cbi-120022525. [DOI] [PubMed] [Google Scholar]
- 47.Shi S, Hida A, McGuinness OP, Wasserman DH, Yamazaki S, Johnson CH. Circadian Clock Gene Bmal1 Is Not Essential; Functional Replacement with its Paralog, Bmal2. Current Biology. 2010;20:316–321. doi: 10.1016/j.cub.2009.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Preitner N, Damiola F, Lopez-Molina L, Zakany J, Duboule D, Albrecht U, Schibler U. The orphan nuclear receptor REV-ERBalpha controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell. 2002;110:251–60. doi: 10.1016/s0092-8674(02)00825-5. [DOI] [PubMed] [Google Scholar]
- 49.Meng L, Person RE, Huang W, Zhu PJ, Costa-Mattioli M, Beaudet AL. Truncation of Ube3a-ATS unsilences paternal Ube3a and ameliorates behavioral defects in the Angelman syndrome mouse model. PLoS Genet. 2013;9:e1004039. doi: 10.1371/journal.pgen.1004039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cassidy SB, Driscoll DJ. Prader-Willi syndrome. Eur J Hum Genet. 2009;17:3–13. doi: 10.1038/ejhg.2008.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Haqq AM, Muehlbauer MJ, Newgard CB, Grambow S, Freemark M. The metabolic phenotype of Prader-Willi syndrome (PWS) in childhood: heightened insulin sensitivity relative to body mass index. J Clin Endocrinol Metab. 2011;96:E225–32. doi: 10.1210/jc.2010-1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Johnson CH, Elliott JA, Foster RG. Entrainment of circadian programs. Chronobiology International. 2003;20:741–774. doi: 10.1081/cbi-120024211. [DOI] [PubMed] [Google Scholar]
- 53.Granada AE, Bordyugov G, Kramer A, Herzel H. Human chronotypes from a theoretical perspective. PLoS One. 2013;8:e59464. doi: 10.1371/journal.pone.0059464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smith SE, Zhou YD, Zhang G, Jin Z, Stoppel DC, Anderson MP. Increased gene dosage of Ube3a results in autism traits and decreased glutamate synaptic transmission in mice. Sci Transl Med. 2011;3:103ra97. doi: 10.1126/scitranslmed.3002627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hardin PE, Hall JC, Rosbash M. Feedback of the Drosophila period gene product on circadian cycling of its messenger RNA levels. Nature. 1990;343:536–40. doi: 10.1038/343536a0. [DOI] [PubMed] [Google Scholar]
- 56.Dunlap JC. Molecular bases for circadian clocks. Cell. 1999;96:271–290. doi: 10.1016/s0092-8674(00)80566-8. [DOI] [PubMed] [Google Scholar]
- 57.Reppert SM, Weaver DR. Coordination of circadian timing in mammals. Nature. 2002;418:935–941. doi: 10.1038/nature00965. [DOI] [PubMed] [Google Scholar]
- 58.Hastings MH, Field MD, Maywood ES, Weaver DR, Reppert SM. Differential regulation of mPER1 and mTIM proteins in the mouse suprachiasmatic nuclei: new insights into a core clock mechanism. J Neurosci. 1999;19:RC11: 1–7. doi: 10.1523/JNEUROSCI.19-12-j0001.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kume K, Zylka M, Sriram S, Shearman LP, Weaver DR, Jin X, Maywood ES, Hastings MH, Reppert SM. mCRY1 and mCRY2 are essential components of the negative limb of the circadian feedback loop. Cell. 1999;98:193–205. doi: 10.1016/s0092-8674(00)81014-4. [DOI] [PubMed] [Google Scholar]
- 60.Liu AC, Tran HG, Zhang EE, Priest AA, Welsh DK, Kay SA. Redundant function of REV-ERBalpha and beta and non-essential role for Bmal1 cycling in transcriptional regulation of intracellular circadian rhythms. PLoS Genet. 2008;4:e1000023. doi: 10.1371/journal.pgen.1000023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wallach T, Schellenberg K, Maier B, Kalathur RK, Porras P, Wanker EE, Futschik ME, Kramer A. Dynamic circadian protein-protein interaction networks predict temporal organization of cellular functions. PLoS Genet. 2013;9:e1003398. doi: 10.1371/journal.pgen.1003398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sahar S, Zocchi L, Kinoshita C, Borrelli E, Sassone-Corsi P. Regulation of BMAL1 protein stability and circadian function by GSK3beta-mediated phosphorylation. PLoS One. 2010;5:e8561. doi: 10.1371/journal.pone.0008561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang L, Abraham D, Lin ST, Oster H, Eichele G, Fu YH, Ptáček LJ. PKCγ participates in food entrainment by regulating BMAL1. Proc Natl Acad Sci USA. 2012;109:20679–84. doi: 10.1073/pnas.1218699110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Duffy JF, Rimmer DW, Czeisler CA. Association of intrinsic circadian period with morningness-eveningness, usual wake time, and circadian phase. Behav Neurosci. 2001;115:895–9. doi: 10.1037//0735-7044.115.4.895. [DOI] [PubMed] [Google Scholar]
- 65.Brown SA, Kunz D, Dumas A, Westermark PO, Vanselow K, Tilmann-Wahnschaffe A, Herzel H, Kramer A. Molecular insights into human daily behavior. Proc Natl Acad Sci USA. 2008;105:1602–7. doi: 10.1073/pnas.0707772105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hida A, Kitamura S, Ohsawa Y, Enomoto M, Katayose Y, Motomura Y, Moriguchi Y, Nozaki K, Watanabe M, Aritake S, et al. In vitro circadian period is associated with circadian/sleep preference. Sci Rep. 2013;3:2074. doi: 10.1038/srep02074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Micic G, de Bruyn A, Lovato N, Wright H, Gradisar M, Ferguson S, Burgess HJ, Lack L. The endogenous circadian temperature period length (tau) in delayed sleep phase disorder compared to good sleepers. J Sleep Res. 2013;22:617–24. doi: 10.1111/jsr.12072. [DOI] [PubMed] [Google Scholar]
- 68.Wirz-Justice A, Benedetti F, Terman M. Chronotherapeutics for Affective Disorders: A Clinician’s Manual for Light and Wake Therapy. Basel: Karger press; 2013. [DOI] [PubMed] [Google Scholar]
- 69.Tan WH, Bacino CA, Skinner SA, Anselm I, Barbieri-Welge R, Bauer-Carlin A, BeaudetALBichell TJ, Gentile JK, Glaze DG, et al. Angelman syndrome: Mutations influence features in early childhood. Am J Med Genet A. 2011;155A:81–90. doi: 10.1002/ajmg.a.33775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Resnick JL, Nicholls RD, Wevrick R. Prader-Willi Syndrome Animal Models Working Group. Recommendations for the investigation of animal models of Prader-Willi syndrome. Mamm Genome. 2013;24:165–178. doi: 10.1007/s00335-013-9454-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.