

Abstract
Glucocorticoids are endogenous steroid hormones that regulate several biological functions including proliferation, differentiation and apoptosis in numerous cell types in response to stress. Synthetic glucocorticoids, such as dexamethasone (Dex) are used to treat a variety of diseases ranging from allergy to depression. Glucocorticoids exert their effects by passively entering into cells and binding to a specific Glucocorticoid Receptor (GR) present in the cytoplasm. Once activated by its ligand, GR may elicit cytoplasmic (mainly suppression of p53), and nuclear (regulation of transcription of GR responsive genes), responses. Human GR is highly polymorphic and may encode > 260 different isoforms. This polymorphism is emerging as the leading cause for the variability of phenotype and response to glucocorticoid therapy observed in human populations. Studies in mice and clinical observations indicate that GR controls also the response to erythroid stress. This knowledge has been exploited in-vivo by using synthetic GR agonists for treatment of the erythropoietin-refractory congenic Diamond Blackfan Anemia and in-vitro to develop culture conditions that may theoretically generate red cells in numbers sufficient for transfusion. However, the effect exerted by GR polymorphism on the variability of the phenotype of genetic and acquired erythroid disorders observed in the human population is still poorly appreciated. This review will summarize current knowledge on the biological activity of GR and of its polymorphism in non-hematopoietic diseases and discuss the implications of these observations for erythropoiesis.
Keywords: Dexamethasone (Dex), glucocorticoid receptor (GR), single nucleotide polymorphism (SNP), erythropoietin-resistant anemia, erythrocytosis
Introduction
Considerable progress has been made in understanding the cellular compartments involved in erythropoiesis and the extrinsic (growth factors, GFs) and intrinsic (transcription factors) factors that regulate the functions of these cells [1,2].
Erythropoiesis begins at the level of the hematopoietic stem cell (HSC) which is instructed by specific GFs to generate a hierarchy of progressively lineage-restricted progenitor cells. These cells, morphologically indistinguishable from HSC, are defined by specific antigen and mRNA expression profiles. After several divisions, lineage-specific progenitor cells give rise to the first morphologically recognizable erythroblast (Ery), the proerythroblast (proErys) [1,2]. Under steady-state conditions, proErys undergo limited numbers (4-5) of divisions that generate mature erythroid precursors which accumulate the proteins necessary to perform the physiological function of red cells prior to undergo enucleation [3].
The initial phases of erythropoiesis are controlled by the early acting GFs stem cell factor (SCF), interleukin-3 (IL-3) and, in humans, granulocyte-monocyte colony stimulating factor (GM-CSF) [4]. Erythropoietin (EPO), although dispensable, synergizes with early acting GFs in inducing hematopoietic progenitor cells into proliferation [5,6]. Later on, Erys become exquisitely sensitive to EPO for proliferation, maturation and survival [7,8]. EPO exerts its effects by binding to the erythropoietin receptor (EPO-R) present on the surface of erythroid progenitors developing in the marrow [9,10]. Clinical studies have demonstrated that red cell mass and concentrations of EPO in serum are correlated in several pathologies [11,12] (Figure 1A). Under steady-state conditions, however, the red mass does not correlate with EPO concentration alone and other factors, such as sex, age and other unknown genetic determinants, play an important role in determining its variability (Figure 1B). Clinical observations suggested that these factors may be represented, at least in part, by nuclear receptors, such as the glucocorticoid (GR) [13,14] and the estrogen (ESR) [15] receptors, prompting early studies that identified the ability of synthetic GR and ESR agonists to synergize with EPO in inducing generation of erythroid bursts in cultures of either adult bone marrow or blood mononuclear cells (MNC) [15-17]. When purified hematopoietic progenitor cells, serum-free media and recombinant GFs became available, Dex and ES were shown not to affect the number of colonies generated in culture but rather the number and maturity of the cells present within individual colonies [18]. Although ESR plays an important role in the induction of anemia of post-menopausal women and in aplastic anemia, knowledge on the effect of this receptor on erythropoiesis is limited [19,20]. By contrast, research on the effects exerted by GR on erythropoiesis has provided several insights on the role of glucocorticoids in a variety of physiological and pathological conditions.
Figure 1.
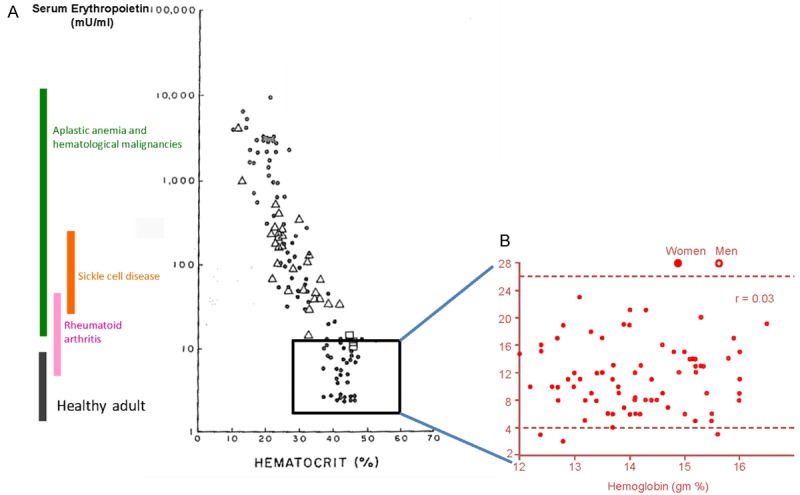
Red cell mass and EPO concentration are inversely correlated in plasma from patients with acquired and congenital anemias but not in that from non-diseased individuals. A: Correlation between red cell mass (as hematocrit) and EPO concentration in plasma (> 30 mU/mL) from anemic patients (aplastic anemia and congenital malignancies, sickle cell disease and rheumatoid arthritis) (modified from [12]). B: Lack of correlation between red cell mass (as hemoglobin, gm %) and EPO concentration in plasma (< 30 mU/mL) from non-diseased individuals. In non diseased individuals there is a statistical significant difference between the red cell mass in females (closed circles) and that in males (open circles). (Published by permission from Dr. Jerry Spivak).
Extensive studies in mice have indicated that conditions of acute or chronic blood loss (erythroid stress) activate the GR pathway which confers to Erys a self-renewal state allowing them to divide numerous times before undergoing terminal maturation [21,22]. Therefore, under conditions of stress, the final cellular output, i.e. the number of Erys produced, is determined not only by the number of hematopoietic progenitors recruited but also by the number of cell divisions allowed within the Ery compartment. This effect is mediated by activation of CXCR4 that shifts the proliferative control of erythroid cells from the SCF pathway, used in steady state conditions, to a SDF-1 (CXCL12) and BMP4 pathway used under conditions of stress [23,24]. In addition to stress erythropoiesis in adult animals, GR may also control fetal erythropoiesis by inducing a self-renewal state in the erythroid progenitor cell compartment and this effect may be mediated, at least in part, by the GR target gene ZFP36L2 [25] (also known as BRF2 and TIS11D) that controls RNA stability and/or translation [26].
The importance of GR in the regulation of human erythropoiesis has been inferred from clinical observations since 1961. Patients with Addison’s disease, a rare chronic endocrine disorder in which the adrenal gland does not produce sufficient glucocorticoids and mineralcorticoids have normocytic anemia [14]. In addition, patients who experience constitutive GR activation, either because they over-express glucocorticoids as a consequence of a pituitary corticotroph adenoma (Cushings’ disease) or because receive glucocorticoids for treatment of an underlying disease (Cushing’s syndrome), develop erythrocytosis [13]. By contrast with the murine gene, human GR is highly polymorphic and this polymorphism is emerging as a leading cause for the heterogeneity of the response to synthetic GR agonists and for the variegation of phenotypes regulated by GR observed in the human population [27]. Recent data indicate that GR polymorphism may also affect the phenotype of diseases involving the erythroid lineage [28,29]. This review will summarize current knowledge on the biological activity of GR, including effects of the polymorphism of its gene in non-erythroid systems, and discuss the implications for normal and stress erythropoiesis, EPO-resistant anemia, and erythrocytosis in humans.
The human glucocorticoid receptor (GR) gene
Human GR is encoded by GR/NR3C1 located in the 5q31.3 band of the long arms of chromosome 5. This band is deleted in patients with de novo myelodisplastic syndrome (MDS) as well as in the subgroup of MDS patients with del(5q) syndrome [30,31]. Patients with del(5q) MDS often present with EPO-resistent anemia. A retrospective FISH analyses for NR3C1 from 14 EPO-resistant del(5q) MDS and Acute Myeloid Leukemia (AML) patients identified that in 78% of the cases the breakpoint involved GR (J. Tripodi, V. Najfeld and AR Migliaccio, unpublished observations) (Table 1). Therefore, these patients should be considered GR haploinsufficient.
Table 1.
FISH with probes specific for GR (NR3C1) and EGR1 (early growth response 1) (as control) on 14 patient with MDS/AML and deletion 5q ideneified by cytogenetic analysis present in the patient archives of MSSM

|
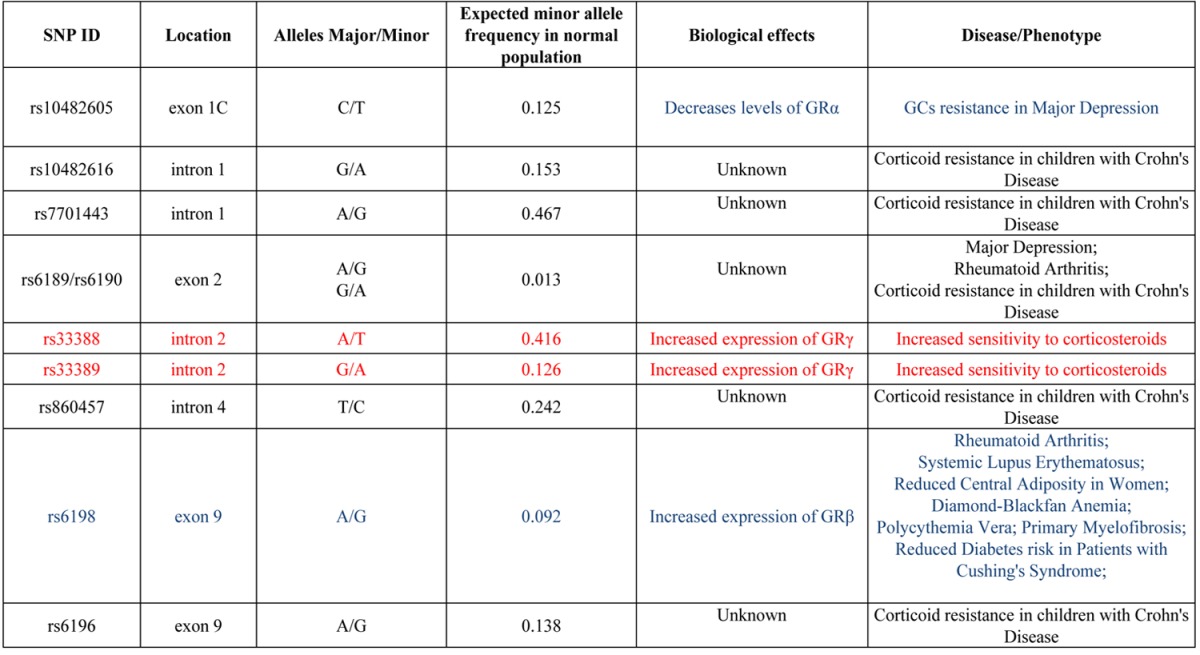
GR contains numerous single nucleotide polymorphisms (SNPs) both in the coding region and in regions associated with alternative splicing and mRNA stabilization [27], producing > 260 combinations of alternative GR isoforms which are variably expressed in the human population [27]. SNPs of human GR that have been associated with human diseases or with altered response to glucocorticoids are summarized in Table 2.
Table 2.
SNPs of human GR associated with diseases or with altered sensitivity to corticosteroids. The frequency of the minor allele in the normal population and the predicted biological consequence, when known, are also reported. SNPs that affect negatively or positively GR activity are indicated in blue and red fonts, respectively. SNPs indicated in black probably reduce the activity of GR but the mechanism is unknown. The frequencies of the minor alleles are from http://www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi. See text for further details
|
GR consists of 9 exons that are differentially spliced to produce several receptor isoforms [27,32]. The two isoforms with the most different biological activity are GRα and GRβ. GRα is the transcriptionally active isoform homologous to the murine receptor [33]. It consists of 777 amino acids (AA). GRβ is a transcriptionally inactive isoform generated by alternative splicing of exon 9 [34]. This splicing produces a mRNA encoding a protein of only 742 AA diverging from GRα in its C-terminal domain that lacks helix 12 which contains the ligand binding domain and possesses a shorter helix 11 with a unique terminal 15 AA sequence. This structure impairs ligand binding and induces nuclear retention [35]. It is debated whether GRβ is expressed in mice. Sequence analyses of murine GR indicate that murine exon 9β contains an open reading frame of 59 amino acids instead of the 15 amino acids encoded by human exon 9β [36]. However, a later study reported that an isoform similar to human GRβ may be generated in mice by alternative splicing of intron 8 [37].
GRβ is a dominant negative isoform that heterodimerizes GRα into a transcriptionally inactive complex. Confocal microscopic imaging of Cos-7 cells transfected with green fluorescent protein (GFP)-tagged GRα and GRβ indicated that nuclear translocation of GRα requires Dex stimulation but that GRβ is constitutively retained in the nucleus [35]. When complexed with GRβ, GRα is also constitutively retained in the nucleus, preventing its activation by glucocorticoids that are present in the cytoplasm.
The human population expresses numerous GRα isoforms with slightly different transcriptional activity. The most studied of them is GRγ that is generated by alternative splicing between exon 3 and 4 [38]. GRγ contains an additional Arg in the DNA-binding domain which reduces its transactivation potential by half. Reduced expression of GRγ has been associated with glucocorticoid resistance in childhood acute lymphocytic leukemia (ALL) [39].
Regulation of GR expression
GR expression is regulated by genetic and microenvironmental cues. This regulation plays an important role in fine tuning signals elicited by GR during the cell response to stress in specific tissues and is emerging as a leading cause for glucocorticoids unresponsiveness or for development of glucocorticoid resistance in patients with inflammatory and autoimmune diseases and in chronic depression [40-43].
Genetic cues that negatively regulate GR activity may be represented by SNPs that regulate levels or type of isoform expressed and/or by epigenetic modifications of GR regulatory sequences.
GRα is expressed by all cell types and its expression is negatively regulated by the SNP rs10482605 in the promoter region of the gene. The low levels of GRα transcription observed in major depression is thought to be the result of increased frequency of rs10482605 (Table 2) [44]. Methylation silencing of GR promoter regions has been instead observed in suicide victims with a history of childhood abuse [45]. Methylation silencing of GR may also be determined by underlying alterations in epigenetic programming as observed in cancer cells [46,47].
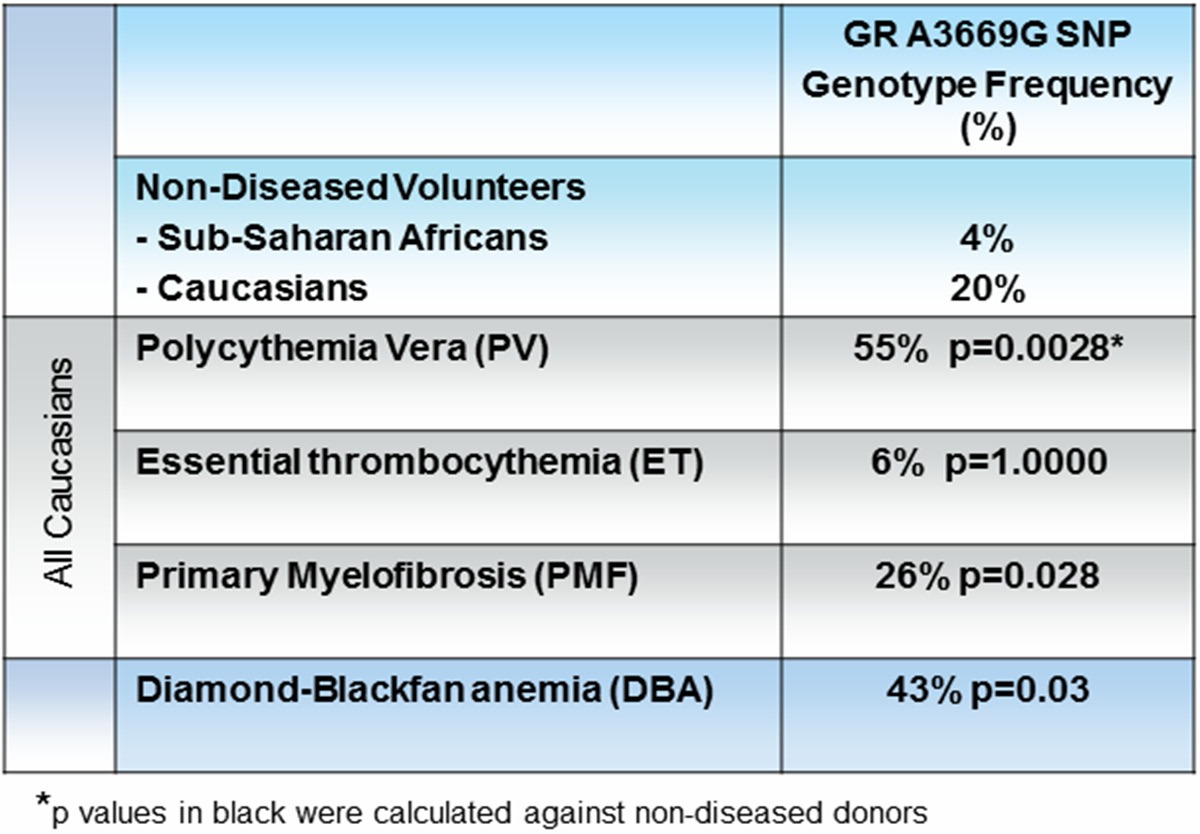
GRβ is expressed in a cell type specific fashion and its expression is positively regulated by the A3669G (rs6198) SNP in the untranslated region of exon 9 that stabilizes GRβ mRNA [35]. In the normal population, this SNP is present with an allele frequency between 4% (Sub-Saharan Africans) and 20% (Europeans) but its frequency increases in patients with autoimmune disorders (27% in systemic lupus erythematosus [48] and 42% in rheumatoid arthritis [40] and in individuals predisposed to central adiposity (30.4%) [43]. Increased GRβ expression induced by this SNP, by suppressing GRα activity, is thought to be responsible for the glucocorticoid resistance observed in these disorders. The presence of the A3669G polymorphism has also been associated with decreased risk of developing diabetes in patients with Cushing’s syndrome [49]. Among the hematopoietic disorders, the frequency of the A3669G polymorphism is increased in patients with Diamond Blackfan Anemia (DBA) or with myeloproliferative neoplasms (MPN) [28,29] (Table 3).
Table 3.
The presence of A3669G (rs6198) polymorphism in Non-Diseased Volunteers and MPN patients
|
DBA is a congenic form of erythroid aplasia often associated with mutations resulting in ribosome insufficiency [50]. The marrow of these patients is normocellular but there is no evidence of Ery maturation, suggesting that anemia in DBA is the result of defective terminal Ery maturation. DBA patients have high EPO levels and their anemia is not responsive to EPO. However, 40-50% of these patients became transfusion-independent when treated with glucocorticoids [51,52]. Erythroid cells expanded ex-vivo from DBA patients express levels of both GRα and GRβ mRNA greater than normals (Figure 2A). The relationship between levels of mRNA expression and glucocorticoid responsiveness in DBA patients is currently under investigation.
Figure 2.
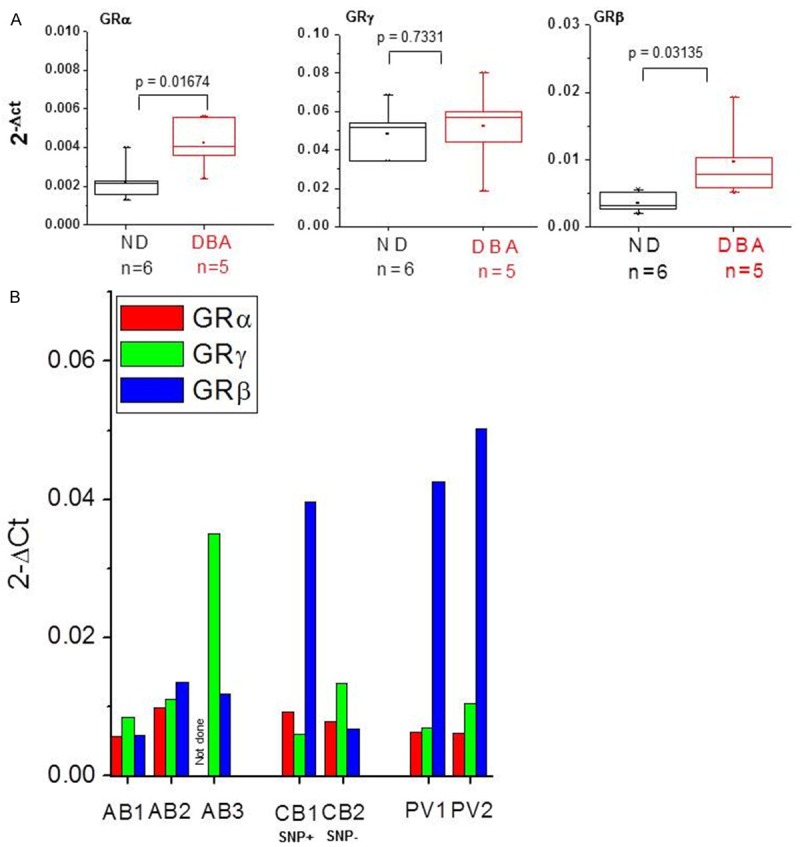
Erys expanded ex-vivo from DBA and PV patients express levels of GRβ mRNA greater than those expressed by Erys expanded from non-diseased volunteers. A: Quantitative RT-PCR analyses for the expression of GRα, GRγ and GRβ mRNA in Erys expanded from normal donors (ND, n = 6) and DBA patients (n = 5). mRNA was isolated from Erys characterized in117 and was kindly provided by Dr. Marieke von Lindern. B: Quantitative RT-PCR analyses for the expression of GRα (red bars), GRγ (green bars) and GRβ (blue bars) in Erys expanded ex-vivo from non-diseased adult blood donors (AB, n = 3), cord blood (CB, n = 2) and polycythemia vera patients (PV, n = 2). One of the CB carried the rs6198 SNP (SNP+) and the other did not (SNP-).
MPN are a class of human neoplasms characterized by the presence of the gain of function JAK2V617F mutation, which constitutively activates JAK2 [53-55], the first signaling molecule of both EPO-R and GR [10,22]. MPN are classified according to the lineage in which the myeloproliferation is manisfested into polycythemia vera (PV, erythrocytosis), essential thrombocythemia (ET, increased platelet counts) and primary myelofibrosis (PMF, ineffective megakaryocytopoiesis) [56,57]. The frequency of the rs6198 SNP is greater than normal in patients with PV (55%), suggesting that expression of GRβ may represent a host-genetic-modifier that contributes to erythroid manifestations [28], and in PMF (50%), where it may represent a susceptibility allele that confers a myeloproliferation phenotype that, when associated with JAK2V617F, may favor blast transformation determining poor survival [58]. Erythroid cells expanded in-vitro from PV patients express levels of GRβ mRNA (Figure 2B) and protein [28] greater than normal. The observation that among Erys expanded in-vitro from normal donors, those expanded from one cord blood (CB) that is rs6198 SNP-positive express levels of GRβ mRNA greater than those expanded from rs6198 SNP-negative CB supports the hypothesis that this SNP is responsible for the increased GRβ expression observed in PV (Figure 2B). While the mechanism by which GRβ expression favors blast transformation in PMF is still unknown, the manner by which it confers the erythroid phenotype to PV has been, at least in part, elucidated and is discussed later (see Biological activity of GR in erythroid cells).
Genetic evidence for an association between steroid resistance and specific GR haplotypes has been described also in Crohn’s disease [59], although the mechanism linking the SNPs associated with this disease and suppression of GR activity has not been defined (Table 2).
Genetic cues that positively regulate GR activity have also been reported. For example, the SNPs rs33389/rs33388 favor, through a mechanism still not completely understood, expression of GRγ marking a haplotype associated with increased glucocorticoid sensitivity [60] (Table 2).
In addition to GR polymorphism, response to glucocorticoids may be affected by genetic cues that alter expression of GR target gene and/or genes that encode proteins competing with GR. A genome-wide association study recognized that the glucocorticoid-induced transcript 1 gene (GLCCI1) variant rs37972/rs37973, by decreasing expression of GLCCI1, impairs the response to glucocorticoid therapy in patients with asthma [61] and data in a mouse model indicate that over-expression of the hairy and enhancer of split-1 (HES1) gene suppresses GR activity by de-repressing the expression of genes suppressed by GR [62].
Microenvironmental cues may also contribute to regulation of GR expression at the transcriptional and post-transcriptional levels [63]. In fact, the complex structure of the gene with multiple alternative starting codons, splicing and poly-adenylation sites allows a variegation of tissue-specific positive and negative transcriptional regulatory mechanisms [64].
GRα expression is negatively regulated also by its ligand [65]. Glucocorticoids may suppress both GRα expression, by inducing methylation of its promoter [45,66], and GRα activity, by inducing post-transcriptional modification of the protein. In fact, in addition to inducing phosphorylation of S211, required to observe nuclear localization and transcriptional activity [67], glucocorticoids may induce phosphorylation of S203 which results in cytoplasmic retention possibly favoring the cytoplasmic over the nuclear activity of GRα [68] (Figure 3).
Figure 3.
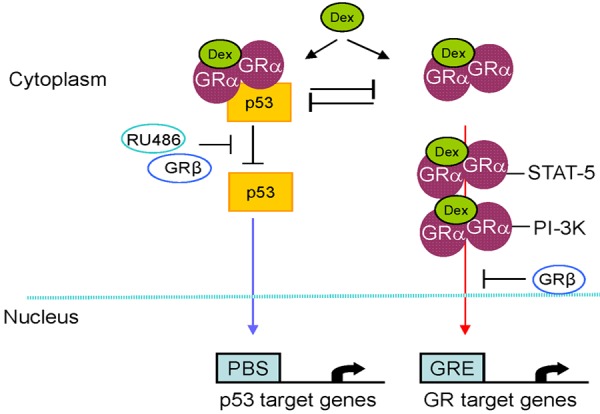
Diagram summarizing the cytoplasmic and nuclear activities of GRα observed in non-erythroid cells that may be relevant for erythropoiesis. The diagram also depicts how these GR activities may be affected by expression of GRβ or by exposure to the synthetic GR antagonists RU486. Upon binding to its ligand, exemplified in this diagram by Dex, GRα dimerizes and binds either to its transcription partners (such as STAT-5 and PI-3K) or to p53. GRα homodimers and GRα-STAT-5/PI-3K heterocomplexes migrate to the nucleus where they binds to glucocorticoid responsive elements (GRE) and to STAT-5/PI-3K consensus sequences to exert their transcriptional activity. By contrast, GRα/p53 complexes are retained in the cytoplasm inhibiting the ability of p53 to binds its consensus sequences (p53 binding sites, PBS) and to exerts its transcriptional activity. Expression of GRβ, by retaining GRα in the nucleus, inhibits the nuclear activity of GRα and stimulates that of p53. RU486, also known as mifepristone, inhibits GRα activity by preventing its dimerization.
In agreement with the observation that the promoter region of GRα contains functional binding sites for NF-kB, GRα expression has been reported to be positively regulated in HeLA cells by factors that activate NF-kB signaling [69]. In addition, human GR contains at least 15 alternative starting sites which generate mRNAs with different affinity for the ribosomal machinery. Experiments of ectopic GR expression in murine pituitary epithelial cells have indicated that basic fibroblast growth factor favors accumulation of GR protein by inducing translation of the gene from the start codon with the highest affinity for the ribosomes [70].
The alternative splicing of exon 9 leading to synthesis of GRβ mRNA is minimally active in cells from primary tissues [63] and is tightly regulated. It is mediated by Serine-Arginine-rich protein p30 in neutrophils [71] and Serine-Arginine-rich protein p40 in HeLa cells [72]. It may be induced in-vitro by components present in serum from septic patients [73] and in vivo in peripheral blood mononuclear cells exposed to IL-2 and IL-4 [74]. In mice, it is induced by treatment with tumor necrosis factor (TNF-α) and may contribute to the glucocorticoid-resistant state conferred by prolonged inflammation [69].
The alternative splicing leading to synthesis of GRγ mRNA involves tandem donor sites and intronic motifs in exon 3/4 that are highly conserved in mammals [75,76]. This splicing is constitutive and contributes to ~3-5% of all GR transcripts expressed by adult cells.
We have recently identified that in erythroid cells GRα expression is positively activated at the transcriptional and post-transcriptional level by soluble SCF [77], a form of SCF released in plasma upon cleavage of the membrane-bound form of this growth factor in response to stress [78]. The SCF signaling responsible for activation of GRα expression is the ERK pathway. This regulatory loop explains why transgenic mice carrying GR lacking its dimerization domain (GRdim mice) [21] and those carrying a SCF gene lacking the site encoding the major proteolytic domain of the protein [79] are similarly impaired in their recovery from anemia.
Biological activity of human GR, general considerations
In the absence of its ligand, GR resides mostly in the cytoplasm as a part of a hetero-oligomeric complex within the heat shock protein chaperone complex (HSPs) 90, 70, 50 [33]. HSP90 regulates ligand binding and cytoplasmic retention of GR. Glucocorticoids enter the cells by passive diffusion across the plasma membrane and binds GRα present in the cytoplasm activating the nuclear and cytoplasmic activities of this receptor (Figure 3). Binding to the ligands activates the nuclear activities of GRα by inducing its association with serine kinase p38 that phosphorylates Serine 211 [67]. pSer211GRα forms dimers that may be either translocated to the nucleus or become associated with the tyrosine kinase JAK2, leading to JAK2 activation, STAT5 phosphorylation and formation of STAT5/GRα heterocomplexes [33]. In addition to STAT5, pSer211GRα may form complexes with other signaling molecules such as PI-3K, NF-kB, and activator protein-1 (AP-1) [33]. Once in the nucleus, GRα homodimers bind to glucocorticoid-specific DNA responsive element (GRE) in the promoter regions of target genes activating and/or suppressing their expression [33]. GRα heterodimers bind instead to the consensus sequences specific for their transcription partners modulating the expression of their target genes [33]. It has been calculated that GR regulates either directly, or indirectly through its partners, expression of ~25% of the human genes.
GRβ lacks the ligand binding domain and its nuclear function was thought to be inhibition of GRα activity [33,80] (Figure 3). In fact, titration experiments in cells expressing ectopic levels of GRα and GRβ at different ratios indicated that 5-fold over-expression of GRβ is sufficient to reduce the transcriptional activity (mostly on transcriptional repression) of GRα by 50% [81,82]. The nuclear retention activity of GRβ is antagonized by Calreticulin, a Ca+2-binding protein responsible to chaperon GRα back to the cytoplasm restoring the ability of the cells to respond to glucocorticoids [83,84]. More recent observations indicate that GRβ retains AP1 and DNA binding domain and exerts a ligand-independent control on the transcription of a subset of genes not controlled by GRα [85,86]. Also the transcriptional activity of GRβ is inhibited by the GRα antagonist RU-486 [87].
The best characterized of the cytoplasmic activity of GRα is its ability to suppress p53 [88] (Figure 3). Studies in mouse models have established a central role for p53 as regulator of the transcription of genes that induce apoptosis (activation of BAX), cell cycle (activation of p21) and growth (repression of c-Myb) arrest [89,90]. In non-hematopoietic cells, once activated, GRα forms a complex with p53 through the nuclear localization signal (NLS) of p53 [91]. The formation of this complex prevents nuclear translocation of both proteins inhibiting their reciprocal nuclear activity. Therefore, treatment with the GR agonist Dex may induce cytoplasmic retention of p53, antagonizing the control of this protein on proliferation and apoptosis. Although not formally tested as yet, it may be hypothesized that GRβ, by retaining GRα in the nucleus, should indirectly activate p53.
Studies using murine embryonic fibroblasts have indicated that GRβ exerts cytoplasmic activity. As an example, GRβ mediates the proliferative effects of insulin by inhibiting the cytoplasmic activity of PTEN and activating the AKT1 growth control signaling [92].
Biological activities of human GR in erythropoiesis
The hematopoietic system responds to erythroid stress by altering the biological properties of a series of cellular elements ranging from hematopoietic stem cells (HSC) to red blood cells. The details of this process have been elucidated in vivo using animal models (mainly mouse and zebrafish) and in-vitro using surrogate assays represented by cultures of human CD34pos stem/progenitor cells stimulated with erythroid-specific growth factors and the GR agonist Dex [92-95]. This culture system was defined as Human Erythroid Massive Amplification (HEMA) culture [95] because it allows the generation of great numbers of erythroid cells (108-109 Ery/103 CD34pos cells) from discarded HSC sources [96,97].
HEMA cultures are composed of a proliferative and a differentiative phase. The proliferative phase is stimulated with Dex and estradiol in addition to SCF, interleukin-3 (IL-3), EPO. The presence of Dex allows for great expansion through the generation of waves of stress-specific cell populations with high proliferative potential primed for erythroid maturation. The differentiative phase can be initiated with Erys obtained any time from day 12-on of the proliferative phase. It is stimulated with EPO, insulin, thyroid hormone (T3) and human plasma and generates red blood cells within 7 days [98,99]. In the differentiative phase, Erys undergo distinctive morphological changes which include vesicle remodeling (degradation of cellular organelles and destruction of cytoskeleton-nuclear-membrane junctions by the autophagic machinery [100], a process controlled by p53 [101]) and activation of the HDAC2-dependent chromatin condensation necessary to generate pyknotic nuclei [102]. At the end of the differentiative culture, each Ery generates one pyrenocyte, a nucleus with a rim of cytoplasm, and one reticulocyte, an anucleated cell rich in hemoglobin. All the biochemical machinery necessary to produce a reticulocyte is negatively regulated by activation of GRα.
On the basis of growth properties and cell composition, the proliferative phase may be divided into three stages (Figure 4 [95,103]):
Figure 4.
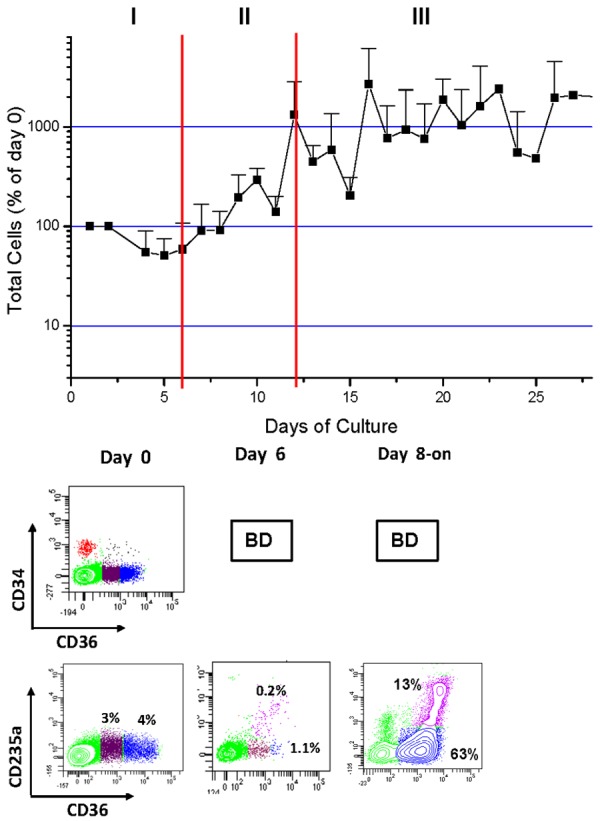
Total number (top) and phenotype (bottom) of cells generated over time in HEMA cultures of mononuclear cells from AB (See also [95,103]). Phenotype is defined by flow cytometrical analyses on the basis of CD34 (the antigen expressed by hematopoietic stem/progenitor cells [126]), CD36 (the thrombospondin receptor expressed when CD34pos cells became committed to the erythroid-megakaryocytic lineage in response to EPO [127,128]) and the erythroid marker CD235a (glycophorin A).
Stage I (day 0-6). Cell numbers increase modestly. In the first 3-days, CD34pos cells generate multilineage (CD34posCD36neg) and bipotent Ery-megakaryocytic (CD34pos/CD36pos) progenitor cells. By day 6, a population of CD34neg/CD36pos cells is detected (in red in Figure 4). This population has proEry morphology (not shown), does not express CD235a and has a phenotype similar to that of proErys generated in mice under conditions of stress [102] that includes expression of high levels of cKIT (the receptor for SCF), CD123 (the α chain of the IL-3 receptor), lack of expression of Mpl (the thrombopoietin receptor expressed instead by bipotent erythroid and megakaryocytic progenitor cells), extensive proliferation potential and ability to undergo unilineage maturation (Figure 5A) and [95,103,105].
Figure 5.
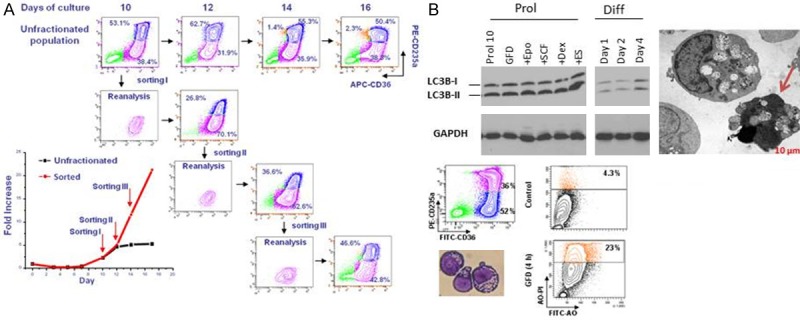
Erys generated in HEMA culture have the ability to undergo self-replication and to die by autophagy. A: Phenotype (CD235a/CD36 flow charts) and growth curve (in fold increase, FI, bottom panel on the left) of Erys generated in HEMA over time by unfractionated populations (flow charts in the top) or by proErys (CD36posCD235aneg, in pink) separated by serial sorting every two days (see also [103]). Mature Erys (CD36posCD235apos) are indicated in blue. This serial sorting/culture approach is the “culture” equivalent of serial transplantation experiments performed in mice to determine the self-replication potential of stem cells. The growth curve of unfractionated populations reaches a plateau by day 10. By contrast, the growth curve of resorted proErys remains exponential upon three sorting given the ability of sorted cells to generate new proErys, in addition to Erys (reproduced from [103] and published by permission from the editor. B: Biochemical, electron microscopy and flow cytometric evidence for activation of the autophagic machinery in Erys obtained in culture with Dex. Autophagy is a proteosome-dependent pathway developed by eukaryotic cells to survive starvation but which may lead to death [100,101] or, in the case of EBs, may promote terminal maturation [98]. One of the first steps of this pathway is formation of the autophagosome with the conversion by lipidation of the cytosolic form of the microtubule associated protein light chain 3 (LC3-I) into the vescicle-specific LC3-II form. The fusion of the autophagosome with the lysosome involves release of LC3-II from the membrane. The autophagosome machinery is mature when the ratio between LC3-1/LC3-II [100,101] is 1:2. Biochemical analyses (top panels on the left): By westen blot, Erys from the proliferative phase (Prol) express a LC3-I/LC3-II ratio of 1:2, an indication that the cells contain mature autophagosomes. This ratio is not further increased by growth factor deprivation (GFD), 15 min stimulation with EPO, SCF. Dex or estradiol (ES) or 1, 2 and 4 days exposure to EPO to induce their maturation (differentiation culture, Diff) [28]. Electron microscopy observations (Top panel on the right). Cultured Erys contain autophasomic vescicles detectable by electron microscopy. The arrow indicates an Ery presenting features of death in the process to extrude its autophagosomic vescicles. Flow cytometry observations. By flow cytometry, autophagic death is detected by acrydin orange (AO) staining. At day 10, only 4% of Erys are AOpos but the frequency of AOpos Erys increases up to 23% upon growth factor deprivation (GFD). Modified from [103] and published by permission from the editor.
Stage II (day 6-12). Cell numbers increase exponentially but CD34pos cells are no longer detected. CD36pos/CD235aneg proErys are responsible to generate new proErys, through a self renewal mechanism and to mature into CD36posCD235apos Erys (Figures 4 and 5A).
Stage III (day 12-on). cKITpos/CD36pos/CD235aneg proErys are responsible to generate new proErys and to mature into CD36posCD235apos Erys but growth reaches a plateau due to a balance between generation of new proErys and death of Erys by autophagy [103]. It may be postulated that when the autophagy machinery used by Erys to mature into reticulocytes is blocked by Dex for a prolonged period of time cell death occurs (Figure 5B).
During the first 6 days, GR activation is instrumental to induce CD34pos cells to generate the stress-specific CD36posCD235apos erythroid progenitor cells. The mechanism by which Dex stimulates CD34pos cells to generate these unilineage progenitors is still not completely elucidated and may involve, at least in part, activation of the transcription of ZFP36L2 [25], a gene encoding a protein that binds to 3’ terminus of mRNA decreasing its stability and transcription potential.
More information is available on the mechanism that in the presence of Dex retains CD36pos/CD235aneg proErys immature and confers to them a self-renewal state in the second phase of the culture. This mechanism may involve both the nuclear and cytoplasmic activity of GR.
Nuclear activity
The mechanism by which GR activation retains Erys in a proliferative state was investigated by microarray profiling of murine fetal liver cells exposed to Dex, SCF and EPO, alone or in combination [23]. This comparison identified that SCF and EPO never showed opposite effects on gene expression. By contrast, Dex alone exerted limited effects on the expression of its target genes but enhanced and/or attenuated the effects exerted by EPO and/or SCF on gene expression. Among the genes regulated by Dex observed in the library there was activation of Myb, a gene that controls Ery proliferation [106], and suppression of GATA1, that controls maturation [1].
Cytoplasmic activity
In erythroid cells, GR antagonizes the cytoplasmic activity of the receptor for EPO (EPO-R) and of p53.
Suppression of EPO-R activity
EPO-R has cytoplasmic and nuclear activities. The cytoplasmic activity of EPO-R is to fine-tune the cellular content of the transcription factor GATA1 [107]. Experiments in mouse models have established the central role of the GATA2/GATA1 switch in the control of the transition from ProErys to Erys [108]. GATA2 is expressed in early Erys and controls mostly genes involved in proliferation [109]. GATA1 is expressed in late Erys [110] and suppresses the expression of GATA2 [108], and of its downstream partners, while activating the expression of the genes required for Ery maturation [1]. EPO controls GATA1 biosynthesis by regulating both the transcription of its gene [110] and the stability [111] (by inhibiting HSP-70 activation of the caspase pathway) [107] and phosphorylation state (via the PI-3K/AKT kinase pathway) [112] of the protein. Erys exposed to Dex rapidly, within 15 min, down-regulate GATA1 expression [113]. The observation that also GRα interacts with HSP-70 suggests that Dex may down-regulate GATA1 expression also by blocking the ability of EPO to inhibit HSP-70.
The nuclear activity of EPO-R is mediated by STAT-5 [114]. Gene deletion studies in mice have indicated that STAT-5 is also the transcription partner that cooperates with GR in retaining proErys into a self-renewal state [22]. To clarify whether this interaction induces self-renewal ability also to human Erys we performed the signaling study presented in Figure 6. These studies included Erys expanded from PV patients because these cells have intrisic Dex-independent self-renewal potential [115]. In normal Erys, a previously undescribed cytoplasmic cross-talk between GR and EPO-R was identified that inhibits the transcriptional activity of both receptors. STAT-5 was phosphorylated when normal Erys were stimulated with either EPO or Dex alone but not when they were exposed to Dex and EPO in combination. In addition, Erys stimulated with EPO and Dex in combination did not contain nuclear STAT-5 DNA binding activity and expressed reduced levels of the GR-target gene GILZ. In PV Erys, as predicted by the presence of the JAK2V617F mutation, STAT-5 was constitutively activated. However, by contrast with normal Erys, PV Erys express high levels of GRβ. Threfore in these cells GRα did not form a complex with STAT-5 because it was retained by GRβ in the nucleus. As a consequence, nuclear STAT-5 DNA binding activity and expression of GILZ were barely detectable (Figure 6). In this case, formation of GRβ/GRα complexes constitutively retained in the nucleus quenched GRα/EPO-R signaling leading to ligand independent inhibition of both pathways. In addition to provide a unifying mechanism for erythrocytosis induced by exposure to excess of glucocorticoids and presence of the JAK2V617F mutation, these data indicate that individuals carrying the SNP rs6198 that favor expression of GRβ should be predisposed for a faster recovery from erythroid stress. In partial support for this hypothesis, the rs6198 SNP was found to be present at frequency greater than normal among regular Caucasian blood donors (6/12 regular donors, 50%) while it was found with a normal frequency among unselected low volume CB collected from the same geographical area (3/20 low volume CB analysed, 15%) (G. Barosi and AR Migliaccio, unpublished observations). This observation raises the possibility that the presence of rs6198 may facilitate the recovery after blood donation, increasing the likelihood for an individual to became a regular donor and may explain why African-americans, who express rs6198 at a frequency lower than that expressed by Caucasians, may be less resilient in recovering from blood donation.
Figure 6.
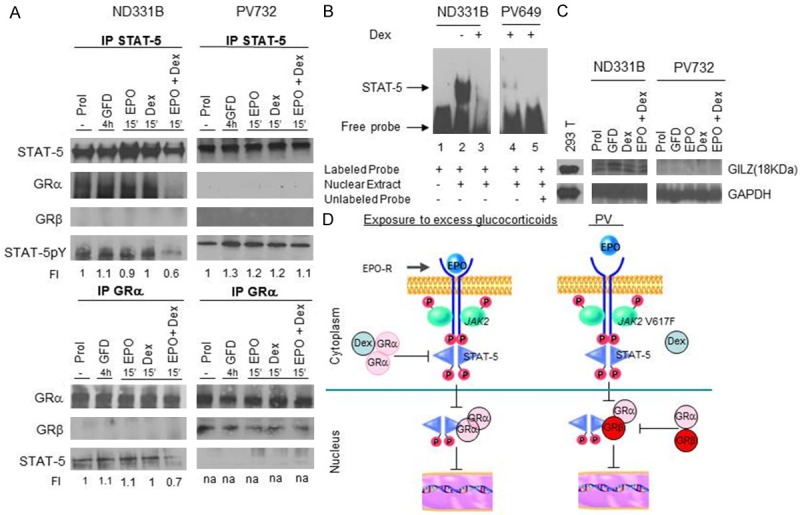
Dex and GRβ are responsible for quenching the EPO maturation signal in Erys from normal donors and from PV patients, respectively. (A) Immunoprecipitation with STAT-5 and GRα antibodies of Erys expanded from one normal donor (ND331B) and one PV (PV514) patient. The cells were analyzed at baseline and after 4 h of growth factor deprivation (GFD) and then exposure to EPO and Dex alone or in combination for 15 min. The blots were probed with antibodies against the total and phosphorylated form of STAT-5, GRα and GRβ. (B) Gel retardation assay with STAT-5-specific probes of nuclear extracts from Erys expanded from the normal donor and the PV patients and stimulated with and without Dex. (C) Western blot analyses for the expression of GILZ of Erys from the normal donor and the PV patient (the same cells as in A). (D) A model for the mechanism that quenches the maturation signal provided by EPO in Erys from normal donors and PV patients leading to erythrocytosis. Erys expanded from the normal donor exposed to Dex and EPO in combination contain low levels of STAT-5p (A), their nuclei bind poorly STAT-5 specific labeled probes (B) and express reduced levels of the GR-target GILZ gene (C). Erys expanded from PB, due to the presence of the JAK2V617F mutation, express constitutive levels of phosphorylated STAT5 however STAT5 cannot bind GRα because this protein forms a complex with GRβ (A). Therefore, also the nuclei of PV Erys bind poorly STAT-5 specific probes and do not express GILZ. Based on these data, we propose a unifying model for development of erythrocytosis through inhibition of GRα/STAT-5 interactions either by exposure to excess Dex (Cushing syndrome) or GRβ expression (PV). In both cases, the block of GRα and EPO-R signaling induces EB into self-replication. Modified from Varricchio et al Blood 2011 and published with permission from the editor.
Another important cytoplasmic interaction of GRα well studied in mice is with p53 [101], a protein required for activation of the autophagic machinery [101] that remodels the Ery cytoplasm into that of a reticulocyte [116]. Preliminary data on the interaction between GRα/p53 in human Erys are presented in Figure 7. As expected, p53 was constitutively localized in the nucleus of Erys but p21 activation was observed only when Erys were exposed to growth factor deprivation (GFD, a known activator of the p53/p21 pathway) and Dex or EPO alone but not to Dex and EPO in combination. This observation suggests that Dex antagonizes the maturation signals mediated by p53 in response to EPO. This experiment also indicated that nuclear localization of HDAC2, the histone deacethylase which mediates the chromatin condensation that precedes enucleation [102], is also observed in Erys stimulated with either Dex or EPO alone but not in those exposed to Dex and EPO in combination (Figure 7).
Figure 7.
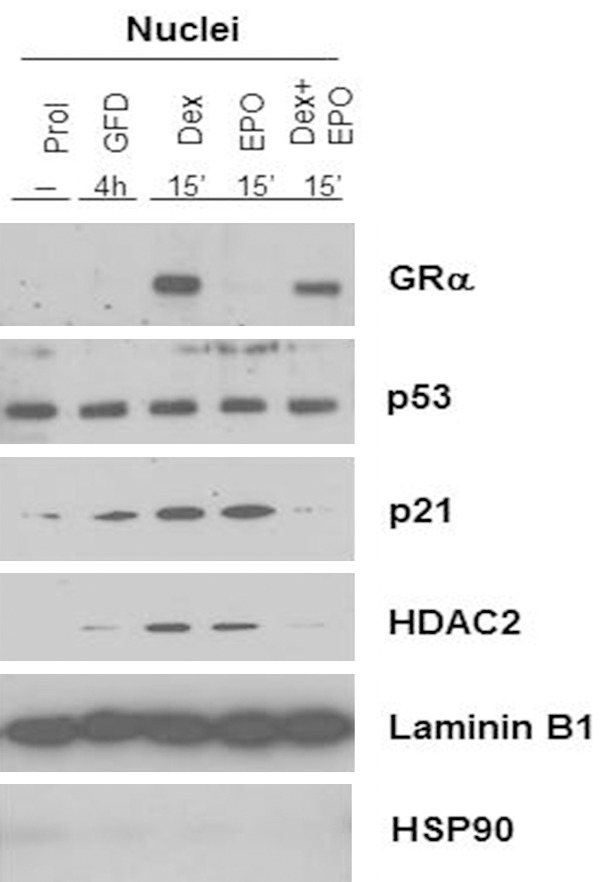
Dex antagonizes the nuclear p53 activity and HDAC2 localization induced by EPO in Erys expanded from non-diseased donors. Western blot analyses for the expression of GRα, p53, p21 (a p53 target gene) and HDAC2 in nuclear extracts from Erys expanded in-vitro (Prol), growth factor deprived (GFD) for 4 h and then exposed for 15’ to Dex and EPO alone or in combination. Expression of Laminin B1 and HSP90 is presented as loading control (nuclear-specificity) and contamination from cytoplasmic proteins, respectively. Exposure of Erys to either Dex or EPO alone, but not in combination, induces p21 expression, a marker for activation of p53 activity) and nuclear localization of HDAC2.
Summary of cell fates controlled by GR; 1) Induction of proliferation directly (c-Myb activation) and indirectly (suppression of GATA1-dependent GATA2 down-regulation and suppression of ZFP36L2); 2) reversible block of maturation (by suppressing activation of SCF/EPO target genes and translocation of HDAC2 to the nucleus and promoting GATA1 degradation); 3) promotion of autophagy by inducing dephosphorylation of AKT and down-regulation of Bcl2 but suppression of cytoplasmic remodeling by inhibiting p53.
Clinical implications of GR polymorphisms for erythroid diseases
The stimulatory effect exerted by glucocorticoids on stress erythropoiesis has been utilized for many years to treat Diamond-Blackfan Anemia (DBA) [52], an erythropoietin-resistant congenital red cell aplasia often associated with loss-of-function mutations in genes encoding proteins of either the large or small ribosome subunit [50,51]. These mutations reduce the translation efficiency of the ribosomes resulting in reduced content of key cell proteins [117]. Reductions in translation efficiency greatly impair the accumulation of hemoglobin necessary to generate functional RBCs. It has been suggested that Dex rescues the defective terminal EB maturation induced by ribosomal deficiency by retaining EBs into self-replication allowing more time for the synthesis of the erythroid proteins which are required in abundant amounts for terminal maturation [29,103]. Additional mechanisms are also possible. Since Dex inhibits p53 [91], the protein that triggers apoptosis in response to deficient ribosome biosynthesis [118], it is particularly suited to promote survival of Erys carrying loss-of-function mutations of ribosomal genes. In addition, Dex, by targeting ZFP36L2 [25], increases mRNA stability, increasing their translation efficiency. However, for reasons still unknown, approximately half of the DBA patients have a clinical response to Dex [52]. Although some DBA patients are steroid unresponsive already at diagnosis, the majority of patients acquire the non-responsive state at the end of a process during which their anemia is controlled by progressively greater doses of glucocorticoids. This transition suggests that ligand-mediated methylation silencing of GR, similar to that observed in severe depression [119], may be responsible for induction of an unresponsive state. Therefore, SNPs, such as rs6198, that favor expression of GRβ quenching the activity of GRα, may allow longer response retention. This hypothesis is testable and if proven correct suggests that rs6198 may represent a biomarker to predict glucocorticoid responsiveness and that combination therapies with demethylating agents may delay the acquisition of a non-responsive state. Glucocorticoids have significant metabolic and bone side effects [120]. It is anticipated that GR agonists lacking these effects that are under development for non-hematopoietic diseases [121] will be in the near future beneficial also for DBA. However, increasing knowledge on the biology of GR in erythropoiesis may also allow identification of additional targets (p53 and or HDAC inhibitors to mention only a few) that may be used, alone or in combination with glucocorticoids to improve treatment of DBA.
The biological activity of GR, including its polymorphism, has also implications for the therapy of MPN. Studies in transgenic JAK2 V617Fmouse models of MPN have established that the levels of JAK2V617F expressed by the transgenic lines determines whether the mice will express the PV (high levels)- or ET (low levels)-like phenotype [122]. This study also identified that deletion of Stat5, the gene encoding one of the signaling molecules immediately downstream to JAK2, normalizes blood values of JAK2V617F knock-in mice [122], indicating that STAT5 is required for development of erythroblastosis in this mouse model of PV. Since deletion of STAT5 in these mice may correspond to the neutralization of STAT5 exerted by GRβ in human erythroid cells, it is possible that in PV patients the presence of the rs6198 SNP [28], possibly in association with polymorphisms still to be identified that affect the levels of GRα activity, may represent a biomarker to predict levels of erythrocytosis. This interesting possibility is still to be demonstrated.
Last, but not least, improvement of our knowledge on the biology of GR in erythropoiesis has allowed developing the concept of blood farming, ex-vivo generation from MNC or CD34pos cells obtained from stem cell sources currently discarded (low volume CB or regular blood donations) of cultured red blood cells (cRBCs) as transfusion products [96,97]. Proof-of-principle for this concept was obtained in a mouse model of lethal anemia [123] and a first-in-man autologous transfusion (5 mL) study which demonstrated that human cRBCs have a normal life-span in vivo [124]. This has become such an active area of investigation. Since these first reports, > 525 papers may be retrieved on this subject from PubMed. These reports have defined technologies to generate from discarded stem cell sources of Caucasian origin numbers of cRBCs sufficient for 3-50 transfusions [96,97]. It is predicted that increased knowledge on the biology of GR in erythropoiesis will facilitate development of cRBCs for transfusion from donors of any ethnical background, including those with rare blood phenotypes that may represent universal donors [125].
Acknowledgements
This study was supported by grants from NHLBI (HL116329-01), the National Cancer Institute (P01-CA108671), Centro Nazionale Sangue and Associazione Italiana Ricerca sul Cancro (AIRC). The authors wish to thank Drs. Vesna Najfeld and Gianni Barosi for authorizing presentation of unpublished results and Dr. Carolyn Whitsett for critical review of the manuscript.
Disclosure of conflict of interest
None.
References
- 1.Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. 2008;132:631–644. doi: 10.1016/j.cell.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Papayannopoulou T, Migliaccio AR, Abkowitz JL, D’Andrea AD. Biology of Erythropoiesis, Erythroid Differentiation and Maturation. In: Hoffman R, Benz EJ, Shattil S, editors. Hematology: Basic Principles and Practise. ed 5. New York: Churchill Livignstone; 2009. pp. 276–294. [Google Scholar]
- 3.Hattangadi SM, Wong P, Zhang L, Flygare J, Lodish HF. From stem cell to red cell: regulation of erythropoiesis at multiple levels by multiple proteins, RNAs, and chromatin modifications. Blood. 2011;118:6258–6268. doi: 10.1182/blood-2011-07-356006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Migliaccio G, Migliaccio AR, Adamson JW. The biology of hematopoietic growth factors: studies in vitro under serum-deprived conditions. Exp Hematol. 1990;18:1049–1055. [PubMed] [Google Scholar]
- 5.Migliaccio AR, Bruno M, Migliaccio G. Evidence for direct action of human biosynthetic (recombinant) GM-CSF on erythroid progenitors in serum-free culture. Blood. 1987;70:1867–1871. [PubMed] [Google Scholar]
- 6.Grover A, Mancini E, Moore S, Mead AJ, Atkinson D, Rasmussen KD, O’Carroll D, Jacobsen SE, Nerlov C. Erythropoietin guides multipotent hematopoietic progenitor cells toward an erythroid fate. J Exp Med. 2014;211:181–188. doi: 10.1084/jem.20131189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spivak JL, Pham T, Isaacs M, Hankins WD. Erythropoietin is both a mitogen and a survival factor. Blood. 1991;77:1228–1233. [PubMed] [Google Scholar]
- 8.Koury MJ, Bondurant MC. Erythropoietin retards DNA breakdown and prevents programmed death in erythroid progenitor cells. Science. 1990;248:378–381. doi: 10.1126/science.2326648. [DOI] [PubMed] [Google Scholar]
- 9.Broudy VC, Lin N, Brice M, Nakamoto B, Papayannopoulou T. Erythropoietin receptor characteristics on primary human erythroid cells. Blood. 1991;77:2583–2590. [PubMed] [Google Scholar]
- 10.Constantinescu SN, Ghaffari S, Lodish HF. The Erythropoietin Receptor: Structure, Activation and Intracellular Signal Transduction. Trends Endocrinol Metab. 1999;10:18–23. doi: 10.1016/s1043-2760(98)00101-5. [DOI] [PubMed] [Google Scholar]
- 11.Adamson JW, Eschbach JW. Erythropoietin for end-stage renal disease. N Engl J Med. 1998;339:625–627. doi: 10.1056/NEJM199808273390910. [DOI] [PubMed] [Google Scholar]
- 12.Erslev AJ, Caro J. Physiologic and molecular biology of erythropoietin. Med Oncol Tumor Pharmacother. 1986;3:159–164. doi: 10.1007/BF02934992. [DOI] [PubMed] [Google Scholar]
- 13.Gursoy A, Dogruk Unal A, Ayturk S, Karakus S, Nur Izol A, Bascil Tutuncu N, Guvener Demirag N. Polycythemia as the first manifestation of Cushing’s disease. J Endocrinol Invest. 2006;29:742–744. doi: 10.1007/BF03344186. [DOI] [PubMed] [Google Scholar]
- 14.Ellis H. Thomas Addison: Addisonian (pernicious) anaemia, Addison’s disease of the suprarenal gland. J Perioper Pract. 2013;23:31–32. doi: 10.1177/1750458913023001-205. [DOI] [PubMed] [Google Scholar]
- 15.Dukes PP, Goldwasser E. Inhibition of erythropoiesis by estrogens. Endocrinology. 1961;69:21–29. doi: 10.1210/endo-69-1-21. [DOI] [PubMed] [Google Scholar]
- 16.Golde DW, Bersch N, Cline MJ. Potentiation of erythropoiesis in vitro by dexamethasone. J Clin Invest. 1976;57:57–62. doi: 10.1172/JCI108269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singer JW, Adamson JW. Steroids and hematopoiesis. III. The response of granulocytic and erythroid colony-forming cells to steroids of different classes. Blood. 1976;48:855–864. [PubMed] [Google Scholar]
- 18.Migliaccio AR, Whitsett C, Migliaccio G. Erythroid cells in vitro: from developmental biology to blood transfusion products. Curr Opin Hematol. 2009;16:259–268. doi: 10.1097/MOH.0b013e32832bcaa2. [DOI] [PubMed] [Google Scholar]
- 19.Young NA, Wu LC, Burd CJ, Friedman AK, Kaffenberger BH, Rajaram MV, Schlesinger LS, James H, Shupnik MA, Jarjour WN. Estrogen modulation of endosome-associated toll-like receptor 8: an IFNalpha-independent mechanism of sex-bias in systemic lupus erythematosus. Clin Immunol. 2014;151:66–77. doi: 10.1016/j.clim.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Calado RT, Yewdell WT, Wilkerson KL, Regal JA, Kajigaya S, Stratakis CA, Young NS. Sex hormones, acting on the TERT gene, increase telomerase activity in human primary hematopoietic cells. Blood. 2009;114:2236–2243. doi: 10.1182/blood-2008-09-178871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bauer A, Tronche F, Wessely O, Kellendonk C, Reichardt HM, Steinlein P, Schutz G, Beug H. The glucocorticoid receptor is required for stress erythropoiesis. Genes Dev. 1999;13:2996–3002. doi: 10.1101/gad.13.22.2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dolznig H, Grebien F, Deiner EM, Stangl K, Kolbus A, Habermann B, Kerenyi MA, Kieslinger M, Moriggl R, Beug H, Mullner EW. Erythroid progenitor renewal versus differentiation: genetic evidence for cell autonomous, essential functions of EpoR, Stat5 and the GR. Oncogene. 2006;25:2890–2900. doi: 10.1038/sj.onc.1209308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kolbus A, Blazquez-Domingo M, Carotta S, Bakker W, Luedemann S, von Lindern M, Steinlein P, Beug H. Cooperative signaling between cytokine receptors and the glucocorticoid receptor in the expansion of erythroid progenitors: molecular analysis by expression profiling. Blood. 2003;102:3136–3146. doi: 10.1182/blood-2003-03-0923. [DOI] [PubMed] [Google Scholar]
- 24.Harandi OF, Hedge S, Wu DC, McKeone D, Paulson RF. Murine erythroid short-term radioprotection requires a BMP4-dependent, self-renewing population of stress erythroid progenitors. J Clin Invest. 2010;120:4507–4519. doi: 10.1172/JCI41291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang L, Prak L, Rayon-Estrada V, Thiru P, Flygare J, Lim B, Lodish HF. ZFP36L2 is required for self-renewal of early burst-forming unit erythroid progenitors. Nature. 2013;499:92–96. doi: 10.1038/nature12215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adachi S, Homoto M, Tanaka R, Hioki Y, Murakami H, Suga H, Matsumoto M, Nakayama KI, Hatta T, Iemura S, Natsume T. ZFP36L1 and ZFP36L2 control LDLR mRNA stability via the ERK-RSK pathway. Nucleic Acids Res. 2014;42:10037–10049. doi: 10.1093/nar/gku652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou J, Cidlowski JA. The human glucocorticoid receptor: one gene, multiple proteins and diverse responses. Steroids. 2005;70:407–417. doi: 10.1016/j.steroids.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 28.Varricchio L, Masselli E, Alfani E, Battistini A, Migliaccio G, Vannucchi AM, Zhang W, Rondelli D, Godbold J, Ghinassi B, Whitsett C, Hoffman R, Migliaccio AR. The dominant negative beta isoform of the glucocorticoid receptor is uniquely expressed in erythroid cells expanded from polycythemia vera patients. Blood. 2011;118:425–436. doi: 10.1182/blood-2010-07-296921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Varricchio L, Godbold J, Scott SA, Whitsett C, Da Costa L, Pospisilova D, Garelli E, Quarello P, Ramenghi U, Migliaccio AR. Increased frequency of the glucocorticoid receptor A3669G (rs6198) polymorphism in patients with Diamond- Blackfan anemia. Blood. 2011;118:473–474. doi: 10.1182/blood-2011-03-342139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Francke U, Foellmer BE. The glucocorticoid receptor gene is in 5q31-q32 [corrected] . Genomics. 1989;4:610–612. doi: 10.1016/0888-7543(89)90287-5. [DOI] [PubMed] [Google Scholar]
- 31.Encio IJ, Detera-Wadleigh SD. The genomic structure of the human glucocorticoid receptor. J Biol Chem. 1991;266:7182–7188. [PubMed] [Google Scholar]
- 32.Turner JD, Muller CP. Structure of the glucocorticoid receptor (NR3C1) gene 5’ untranslated region: identification, and tissue distribution of multiple new human exon 1. J Mol Endocrinol. 2005;35:283–292. doi: 10.1677/jme.1.01822. [DOI] [PubMed] [Google Scholar]
- 33.Nicolaides NC, Galata Z, Kino T, Chrousos GP, Charmandari E. The human glucocorticoid receptor: molecular basis of biologic function. Steroids. 2010;75:1–12. doi: 10.1016/j.steroids.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oakley RH, Webster JC, Sar M, Parker CR Jr, Cidlowski JA. Expression and subcellular distribution of the beta-isoform of the human glucocorticoid receptor. Endocrinology. 1997;138:5028–5038. doi: 10.1210/endo.138.11.5501. [DOI] [PubMed] [Google Scholar]
- 35.Yudt MR, Jewell CM, Bienstock RJ, Cidlowski JA. Molecular origins for the dominant negative function of human glucocorticoid receptor beta. Mol Cell Biol. 2003;23:4319–4330. doi: 10.1128/MCB.23.12.4319-4330.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Otto C, Reichardt HM, Schutz G. Absence of glucocorticoid receptor-beta in mice. J Biol Chem. 1997;272:26665–26668. doi: 10.1074/jbc.272.42.26665. [DOI] [PubMed] [Google Scholar]
- 37.Hinds TD Jr, Ramakrishnan S, Cash HA, Stechschulte LA, Heinrich G, Najjar SM, Sanchez ER. Discovery of glucocorticoid receptor-beta in mice with a role in metabolism. Mol Endocrinol. 2010;24:1715–1727. doi: 10.1210/me.2009-0411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rivers C, Levy A, Hancock J, Lightman S, Norman M. Insertion of an amino acid in the DNA-binding domain of the glucocorticoid receptor as a result of alternative splicing. J Clin Endocrinol Metab. 1999;84:4283–4286. doi: 10.1210/jcem.84.11.6235. [DOI] [PubMed] [Google Scholar]
- 39.Haarman EG, Kaspers GJ, Pieters R, Rottier MM, Veerman AJ. Glucocorticoid receptor alpha, beta and gamma expression vs in vitro glucocorticod resistance in childhood leukemia. Leukemia. 2004;18:530–537. doi: 10.1038/sj.leu.2403225. [DOI] [PubMed] [Google Scholar]
- 40.Derijk RH, Schaaf MJ, Turner G, Datson NA, Vreugdenhil E, Cidlowski J, de Kloet ER, Emery P, Sternberg EM, Detera-Wadleigh SD. A human glucocorticoid receptor gene variant that increases the stability of the glucocorticoid receptor beta-isoform mRNA is associated with rheumatoid arthritis. J Rheumatol. 2001;28:2383–2388. [PubMed] [Google Scholar]
- 41.Rai T, Monoe K, Kanno Y, Saito H, Takahashi A, Irisawa A, Ohira H. Expression of human glucocorticoid receptor beta of peripheral blood mononuclear cells in patients with severe autoimmune hepatitis. Fukushima J Med Sci. 2006;52:65–70. doi: 10.5387/fms.52.65. [DOI] [PubMed] [Google Scholar]
- 42.Goleva E, Li LB, Eves PT, Strand MJ, Martin RJ, Leung DY. Increased glucocorticoid receptor beta alters steroid response in glucocorticoid-insensitive asthma. Am J Respir Crit Care Med. 2006;173:607–616. doi: 10.1164/rccm.200507-1046OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Syed AA, Irving JA, Redfern CP, Hall AG, Unwin NC, White M, Bhopal RS, Weaver JU. Association of glucocorticoid receptor polymorphism A3669G in exon 9beta with reduced central adiposity in women. Obesity (Silver Spring) 2006;14:759–764. doi: 10.1038/oby.2006.86. [DOI] [PubMed] [Google Scholar]
- 44.Kumsta R, Moser D, Streit F, Koper JW, Meyer J, Wust S. Characterization of a glucocorticoid receptor gene (GR, NR3C1) promoter polymorphism reveals functionality and extends a haplotype with putative clinical relevance. Am J Med Genet B Neuropsychiatr Genet. 2009;150B:476–482. doi: 10.1002/ajmg.b.30837. [DOI] [PubMed] [Google Scholar]
- 45.McGowan PO, Sasaki A, D’Alessio AC, Dymov S, Labonte B, Szyf M, Turecki G, Meaney MJ. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci. 2009;12:342–348. doi: 10.1038/nn.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lind GE, Kleivi K, Meling GI, Teixeira MR, Thiis-Evensen E, Rognum TO, Lothe RA. ADAMTS1, CRABP1, and NR3C1 identified as epigenetically deregulated genes in colorectal tumorigenesis. Cell Oncol. 2006;28:259–272. doi: 10.1155/2006/949506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nesset KA, Perri AM, Mueller CR. Frequent promoter hypermethylation and expression reduction of the glucocorticoid receptor gene in breast tumors. Epigenetics. 2014;9:1–9. doi: 10.4161/epi.28484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Piotrowski P, Burzynski M, Lianeri M, Mostowska M, Wudarski M, Chwalinska-Sadowska H, Jagodzinski PP. Glucocorticoid receptor beta splice variant expression in patients with high and low activity of systemic lupus erythematosus. Folia Histochem Cytobiol. 2007;45:339–342. [PubMed] [Google Scholar]
- 49.Trementino L, Appolloni G, Concettoni C, Cardinaletti M, Boscaro M, Arnaldi G. Association of glucocorticoid receptor polymorphism A3669G with decreased risk of developing diabetes in patients with Cushing’s syndrome. Eur J Endocrinol. 2012;166:35–42. doi: 10.1530/EJE-11-0722. [DOI] [PubMed] [Google Scholar]
- 50.Boria I, Garelli E, Gazda HT, Aspesi A, Quarello P, Pavesi E, Ferrante D, Meerpohl JJ, Kartal M, Da Costa L, Proust A, Leblanc T, Simansour M, Dahl N, Frojmark AS, Pospisilova D, Cmejla R, Beggs AH, Sheen MR, Landowski M, Buros CM, Clinton CM, Dobson LJ, Vlachos A, Atsidaftos E, Lipton JM, Ellis SR, Ramenghi U, Dianzani I. The ribosomal basis of Diamond-Blackfan Anemia: mutation and database update. Hum Mutat. 2010;31:1269–1279. doi: 10.1002/humu.21383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ellis SR, Lipton JM. Diamond Blackfan anemia: a disorder of red blood cell development. Curr Top Dev Biol. 2008;82:217–241. doi: 10.1016/S0070-2153(07)00008-7. [DOI] [PubMed] [Google Scholar]
- 52.Pollack MH, Endicott J, Liebowitz M, Russell J, Detke M, Spann M, Ball S, Swindle R. Examining quality of life in patients with generalized anxiety disorder: clinical relevance and response to duloxetine treatment. J Psychiatr Res. 2008;42:1042–1049. doi: 10.1016/j.jpsychires.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 53.Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, Vassiliou GS, Bench AJ, Boyd EM, Curtin N, Scott MA, Erber WN, Green AR. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054–1061. doi: 10.1016/S0140-6736(05)71142-9. [DOI] [PubMed] [Google Scholar]
- 54.James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, Lacout C, Garcon L, Raslova H, Berger R, Bennaceur-Griscelli A, Villeval JL, Constantinescu SN, Casadevall N, Vainchenker W. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144–1148. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- 55.Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, Boggon TJ, Wlodarska I, Clark JJ, Moore S, Adelsperger J, Koo S, Lee JC, Gabriel S, Mercher T, D’Andrea A, Frohling S, Dohner K, Marynen P, Vandenberghe P, Mesa RA, Tefferi A, Griffin JD, Eck MJ, Sellers WR, Meyerson M, Golub TR, Lee SJ, Gilliland DG. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7:387–397. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 56.Tefferi A, Vainchenker W. Myeloproliferative neoplasms: molecular pathophysiology, essential clinical understanding, and treatment strategies. J. Clin. Oncol. 2011;29:573–582. doi: 10.1200/JCO.2010.29.8711. [DOI] [PubMed] [Google Scholar]
- 57.Mascarenhas J, Hoffman R. Myeloproliferative neoplasms: new translational therapies. Mt Sinai J Med. 2010;77:667–683. doi: 10.1002/msj.20225. [DOI] [PubMed] [Google Scholar]
- 58.Poletto V, Rosti V, Villani L, Catarsi P, Carolei A, Campanelli R, Massa M, Martinetti M, Viarengo G, Malovini A, Migliaccio AR, Barosi G. A3669G polymorphism of glucocorticoid receptor is a susceptibility allele for primary myelofibrosis and contributes to phenotypic diversity and blast transformation. Blood. 2012;120:3112–3117. doi: 10.1182/blood-2012-05-433466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Krupoves A, Mack D, Deslandres C, Seidman E, Amre DK. Variation in the glucocorticoid receptor gene (NR3C1) may be associated with corticosteroid dependency and resistance in children with Crohn’s disease. Pharmacogenet Genomics. 2011;21:454–460. doi: 10.1097/FPC.0b013e3283476a01. [DOI] [PubMed] [Google Scholar]
- 60.Stevens A, Donn R, Ray D. Regulation of glucocorticoid receptor gamma (GRgamma) by glucocorticoid receptor haplotype and glucocorticoid. Clin Endocrinol (Oxf) 2004;61:327–331. doi: 10.1111/j.1365-2265.2004.02097.x. [DOI] [PubMed] [Google Scholar]
- 61.Tantisira KG, Lasky-Su J, Harada M, Murphy A, Litonjua AA, Himes BE, Lange C, Lazarus R, Sylvia J, Klanderman B, Duan QL, Qiu W, Hirota T, Martinez FD, Mauger D, Sorkness C, Szefler S, Lazarus SC, Lemanske RF Jr, Peters SP, Lima JJ, Nakamura Y, Tamari M, Weiss ST. Genomewide association between GLCCI1 and response to glucocorticoid therapy in asthma. N Engl J Med. 2011;365:1173–1183. doi: 10.1056/NEJMoa0911353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Revollo JR, Oakley RH, Lu NZ, Kadmiel M, Gandhavadi M, Cidlowski JA. HES1 is a master regulator of glucocorticoid receptor-dependent gene expression. Sci Signal. 2013;6:ra103. doi: 10.1126/scisignal.2004389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pujols L, Mullol J, Roca-Ferrer J, Torrego A, Xaubet A, Cidlowski JA, Picado C. Expression of glucocorticoid receptor alpha- and beta-isoforms in human cells and tissues. Am J Physiol Cell Physiol. 2002;283:C1324–1331. doi: 10.1152/ajpcell.00363.2001. [DOI] [PubMed] [Google Scholar]
- 64.Lu NZ, Cidlowski JA. Translational regulatory mechanisms generate N-terminal glucocorticoid receptor isoforms with unique transcriptional target genes. Mol Cell. 2005;18:331–342. doi: 10.1016/j.molcel.2005.03.025. [DOI] [PubMed] [Google Scholar]
- 65.Pujols L, Mullol J, Perez M, Roca-Ferrer J, Juan M, Xaubet A, Cidlowski JA, Picado C. Expression of the human glucocorticoid receptor alpha and beta isoforms in human respiratory epithelial cells and their regulation by dexamethasone. Am J Respir Cell Mol Biol. 2001;24:49–57. doi: 10.1165/ajrcmb.24.1.4024. [DOI] [PubMed] [Google Scholar]
- 66.Petropoulos S, Matthews SG, Szyf M. Adult glucocorticoid exposure leads to transcriptional and DNA methylation changes in nuclear steroid receptors in the hippocampus and kidney of mouse male offspring. Biol Reprod. 2014;90:43. doi: 10.1095/biolreprod.113.115899. [DOI] [PubMed] [Google Scholar]
- 67.Miller AL, Webb MS, Copik AJ, Wang Y, Johnson BH, Kumar R, Thompson EB. p38 Mitogen-activated protein kinase (MAPK) is a key mediator in glucocorticoid-induced apoptosis of lymphoid cells: correlation between p38 MAPK activation and site-specific phosphorylation of the human glucocorticoid receptor at serine 211. Mol Endocrinol. 2005;19:1569–1583. doi: 10.1210/me.2004-0528. [DOI] [PubMed] [Google Scholar]
- 68.Matthews L, Johnson J, Berry A, Trebble P, Cookson A, Spiller D, Rivers C, Norman M, White M, Ray D. Cell cycle phase regulates glucocorticoid receptor function. PLoS One. 2011;6:e22289. doi: 10.1371/journal.pone.0022289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Webster JC, Oakley RH, Jewell CM, Cidlowski JA. Proinflammatory cytokines regulate human glucocorticoid receptor gene expression and lead to the accumulation of the dominant negative beta isoform: a mechanism for the generation of glucocorticoid resistance. Proc Natl Acad Sci U S A. 2001;98:6865–6870. doi: 10.1073/pnas.121455098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bockmuhl Y, Murgatroyd CA, Kuczynska A, Adcock IM, Almeida OF, Spengler D. Differential regulation and function of 5’-untranslated GR-exon 1 transcripts. Mol Endocrinol. 2011;25:1100–1110. doi: 10.1210/me.2010-0436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xu Q, Leung DY, Kisich KO. Serine-arginine-rich protein p30 directs alternative splicing of glucocorticoid receptor pre-mRNA to glucocorticoid receptor beta in neutrophils. J Biol Chem. 2003;278:27112–27118. doi: 10.1074/jbc.M300824200. [DOI] [PubMed] [Google Scholar]
- 72.Yan XB, Tang CH, Huang Y, Fang H, Yu ZQ, Wu LM, Liu RY. Alternative splicing in exon 9 of glucocorticoid receptor pre-mRNA is regulated by SRp40. Mol Biol Rep. 2009;37:1427–1433. doi: 10.1007/s11033-009-9529-z. [DOI] [PubMed] [Google Scholar]
- 73.Guerrero J, Gatica HA, Rodriguez M, Estay R, Goecke IA. Septic serum induces glucocorticoid resistance and modifies the expression of glucocorticoid isoforms receptors: a prospective cohort study and in vitro experimental assay. Crit Care. 2013;17:R107. doi: 10.1186/cc12774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Leung DY, Hamid Q, Vottero A, Szefler SJ, Surs W, Minshall E, Chrousos GP, Klemm DJ. Association of glucocorticoid insensitivity with increased expression of glucocorticoid receptor beta. J Exp Med. 1997;186:1567–1574. doi: 10.1084/jem.186.9.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rivers C, Levy A, Hancock J, Lightman S, Norman M. Insertion of an amino acid in the DNA-binding domain of the glucocorticoid receptor as a result of alternative splicing. J Clin Endocrinol Metab. 1999;84:4283–4286. doi: 10.1210/jcem.84.11.6235. [DOI] [PubMed] [Google Scholar]
- 76.Rivers C, Flynn A, Qian X, Matthews L, Lightman S, Ray D, Norman M. Characterization of conserved tandem donor sites and intronic motifs required for alternative splicing in corticosteroid receptor genes. Endocrinology. 2009;150:4958–4967. doi: 10.1210/en.2009-0346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Varricchio L, Tirelli V, Masselli E, Ghinassi B, Saha N, Besmer P, Migliaccio AR. The expression of the glucocorticoid receptor in human erythroblasts is uniquely regulated by KIT ligand: implications for stress erythropoiesis. Stem Cells Dev. 2012;21:2852–2865. doi: 10.1089/scd.2011.0676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Besmer P. Kit-ligand-stem cell factor. In: Garland JM, Quesenberry PJ, Hilton DJ, editors. Colony-Stimulating Factors: Molecular and Cellular Biology. New York: Marcel Dekker; 1997. pp. 369–404. [Google Scholar]
- 79.Tajima Y, Moore MA, Soares V, Ono M, Kissel H, Besmer P. Consequences of exclusive expression in vivo of Kit-ligand lacking the major proteolytic cleavage site. Proc Natl Acad Sci U S A. 1998;95:11903–11908. doi: 10.1073/pnas.95.20.11903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Torrego A, Pujols L, Roca-Ferrer J, Mullol J, Xaubet A, Picado C. Glucocorticoid receptor isoforms alpha and beta in in vitro cytokine-induced glucocorticoid insensitivity. Am J Respir Crit Care Med. 2004;170:420–425. doi: 10.1164/rccm.200308-1143OC. [DOI] [PubMed] [Google Scholar]
- 81.Leung DY, Hamid Q, Vottero A, Szefler SJ, Surs W, Minshall E, Chrousos GP, Klemm DJ. Association of glucocorticoid insensitivity with increased expression of glucocorticoid receptor beta. J Exp Med. 1997;186:1567–1574. doi: 10.1084/jem.186.9.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bamberger CM, Bamberger AM, de Castro M, Chrousos GP. Glucocorticoid receptor beta, a potential endogenous inhibitor of glucocorticoid action in humans. J Clin Invest. 1995;95:2435–2441. doi: 10.1172/JCI117943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Michalak M, Corbett EF, Mesaeli N, Nakamura K, Opas M. Calreticulin: one protein, one gene, many functions. Biochem J. 1999;344 Pt2:281–292. [PMC free article] [PubMed] [Google Scholar]
- 84.Varricchio L, Migliaccio AR. Calreticulin in Myeloproliferative Neoplasms: The Other Side of the Alice Mirror. EMJ Hema. 2014;1:114–122. [Google Scholar]
- 85.Taniguchi Y, Iwasaki Y, Tsugita M, Nishiyama M, Taguchi T, Okazaki M, Nakayama S, Kambayashi M, Hashimoto K, Terada Y. Glucocorticoid receptor-beta and receptor-gamma exert dominant negative effect on gene repression but not on gene induction. Endocrinology. 2010;151:3204–3213. doi: 10.1210/en.2009-1254. [DOI] [PubMed] [Google Scholar]
- 86.Kino T, Manoli I, Kelkar S, Wang Y, Su YA, Chrousos GP. Glucocorticoid receptor (GR) beta has intrinsic, GRalpha-independent transcriptional activity. Biochem Biophys Res Commun. 2009;381:671–675. doi: 10.1016/j.bbrc.2009.02.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lewis-Tuffin LJ, Jewell CM, Bienstock RJ, Collins JB, Cidlowski JA. Human glucocorticoid receptor beta binds RU-486 and is transcriptionally active. Mol Cell Biol. 2007;27:2266–2282. doi: 10.1128/MCB.01439-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sengupta S, Wasylyk B. Physiological and pathological consequences of the interactions of the p53 tumor suppressor with the glucocorticoid, androgen, and estrogen receptors. Ann N Y Acad Sci. 2004;1024:54–71. doi: 10.1196/annals.1321.005. [DOI] [PubMed] [Google Scholar]
- 89.Miyashita T, Krajewski S, Krajewska M, Wang HG, Lin HK, Liebermann DA, Hoffman B, Reed JC. Tumor suppressor p53 is a regulator of bcl-2 and bax gene expression in vitro and in vivo. Oncogene. 1994;9:1799–1805. [PubMed] [Google Scholar]
- 90.Sarvaiya PJ, Schwartz JR, Geng CD, Vedeckis WV. c-Myb interacts with the glucocorticoid receptor and regulates its level in pre-B-acute lymphoblastic leukemia cells. Mol Cell Endocrinol. 2012;361:124–132. doi: 10.1016/j.mce.2012.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sengupta S, Wasylyk B. Ligand-dependent interaction of the glucocorticoid receptor with p53 enhances their degradation by Hdm2. Genes Dev. 2001;15:2367–2380. doi: 10.1101/gad.202201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Stechschulte LA, Wuescher L, Marino JS, Hill JW, Eng C, Hinds TD Jr. Glucocorticoid receptor beta stimulates Akt1 growth pathway by attenuation of PTEN. J Biol Chem. 2014;289:17885–17894. doi: 10.1074/jbc.M113.544072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.von Lindern M, Zauner W, Mellitzer G, Steinlein P, Fritsch G, Huber K, Lowenberg B, Beug H. The glucocorticoid receptor cooperates with the erythropoietin receptor and c-Kit to enhance and sustain proliferation of erythroid progenitors in vitro. Blood. 1999;94:550–559. [PubMed] [Google Scholar]
- 94.Leberbauer C, Boulme F, Unfried G, Huber J, Beug H, Mullner EW. Different steroids co-regulate long-term expansion versus terminal differentiation in primary human erythroid progenitors. Blood. 2005;105:85–94. doi: 10.1182/blood-2004-03-1002. [DOI] [PubMed] [Google Scholar]
- 95.Migliaccio G, Di Pietro R, di Giacomo V, Di Baldassarre A, Migliaccio AR, Maccioni L, Galanello R, Papayannopoulou T. In vitro mass production of human erythroid cells from the blood of normal donors and of thalassemic patients. Blood Cells Mol Dis. 2002;28:169–180. doi: 10.1006/bcmd.2002.0502. [DOI] [PubMed] [Google Scholar]
- 96.Migliaccio AR, Whitsett C, Migliaccio G. Erythroid cells in vitro: from developmental biology to blood transfusion products. Curr Opin Hematol. 2009;16:259–268. doi: 10.1097/MOH.0b013e32832bcaa2. [DOI] [PubMed] [Google Scholar]
- 97.Migliaccio AR, Whitsett C, Papayannopoulou T, Sadelain M. The potential of stem cells as an in vitro source of red blood cells for transfusion. Cell Stem Cell. 2012;10:115–119. doi: 10.1016/j.stem.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Masiello F, Tirelli V, Sanchez M, van den Akker E, Gabriella G, Marconi M, Villa MA, Rebulla P, Hashmi G, Whitsett C, Migliaccio AR. Mononuclear cells from a rare blood donor, after freezing under good manufacturing practice conditions, generate red blood cells that recapitulate the rare blood phenotype. Transfusion. 2014;54:1059–1070. doi: 10.1111/trf.12391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.van den Akker E, Satchwell TJ, Pellegrin S, Daniels G, Toye AM. The majority of the in vitro erythroid expansion potential resides in CD34(-) cells, outweighing the contribution of CD34(+) cells and significantly increasing the erythroblast yield from peripheral blood samples. Haematologica. 2010;95:1594–1598. doi: 10.3324/haematol.2009.019828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, Wang J. Essential role for Nix in autophagic maturation of erythroid cells. Nature. 2008;454:232–235. doi: 10.1038/nature07006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Maiuri MC, Galluzzi L, Morselli E, Kepp O, Malik SA, Kroemer G. Autophagy regulation by p53. Curr Opin Cell Biol. 2010;22:181–185. doi: 10.1016/j.ceb.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 102.Ji P, Yeh V, Ramirez T, Murata-Hori M, Lodish HF. Histone deacetylase 2 is required for chromatin condensation and subsequent enucleation of cultured mouse fetal erythroblasts. Haematologica. 2010;95:2013–2021. doi: 10.3324/haematol.2010.029827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Migliaccio G, Masiello F, Tirelli V, Sanchez M, Varricchio L, Whitsett C, Migliaccio AR. Under HEMA conditions, self-replication of human erythroblasts is limited by autophagic death. Blood Cells Mol Dis. 2011;47:182–197. doi: 10.1016/j.bcmd.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 104.Paulson RF, Shi L, Wu DC. Stress erythropoiesis: new signals and new stress progenitor cells. Curr Opin Hematol. 2011;18:139–145. doi: 10.1097/MOH.0b013e32834521c8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tirelli V, Ghinassi B, Migliaccio AR, Whitsett C, Masiello F, Sanchez M, Migliaccio G. Phenotypic definition of the progenitor cells with erythroid differentiation potential present in human adult blood. Stem Cells Int. 2011;2011:602483. doi: 10.4061/2011/602483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gambone JE, Dusaban SS, Loperena R, Nakata Y, Shetzline SE. The c-Myb target gene neuromedin U functions as a novel cofactor during the early stages of erythropoiesis. Blood. 2011;117:5733–5743. doi: 10.1182/blood-2009-09-242131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.De Maria R, Zeuner A, Eramo A, Domenichelli C, Bonci D, Grignani F, Srinivasula SM, Alnemri ES, Testa U, Peschle C. Negative regulation of erythropoiesis by caspase-mediated cleavage of GATA-1. Nature. 1999;401:489–493. doi: 10.1038/46809. [DOI] [PubMed] [Google Scholar]
- 108.Grass JA, Boyer ME, Pal S, Wu J, Weiss MJ, Bresnick EH. GATA-1-dependent transcriptional repression of GATA-2 via disruption of positive autoregulation and domain-wide chromatin remodeling. Proc Natl Acad Sci U S A. 2003;100:8811–8816. doi: 10.1073/pnas.1432147100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Leonard M, Brice M, Engel JD, Papayannopoulou T. Dynamics of GATA transcription factor expression during erythroid differentiation. Blood. 1993;82:1071–1079. [PubMed] [Google Scholar]
- 110.Vannucchi AM, Linari S, Lin CS, Koury MJ, Bondurant MC, Migliaccio AR. Increased expression of the distal, but not of the proximal, Gata1 transcripts during differentiation of primary erythroid cells. J Cell Physiol. 1999;180:390–401. doi: 10.1002/(SICI)1097-4652(199909)180:3<390::AID-JCP10>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 111.Migliaccio AR, Jiang Y, Migliaccio G, Nicolis S, Crotta S, Ronchi A, Ottolenghi S, Adamson JW. Transcriptional and posttranscriptional regulation of the expression of the erythropoietin receptor gene in human erythropoietin-responsive cell lines. Blood. 1993;82:3760–3769. [PubMed] [Google Scholar]
- 112.Zhao W, Kitidis C, Fleming MD, Lodish HF, Ghaffari S. Erythropoietin stimulates phosphorylation and activation of GATA-1 via the PI3-kinase/AKT signaling pathway. Blood. 2006;107:907–915. doi: 10.1182/blood-2005-06-2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Stellacci E, Di Noia A, Di Baldassarre A, Migliaccio G, Battistini A, Migliaccio AR. Interaction between the glucocorticoid and erythropoietin receptors in human erythroid cells. Exp Hematol. 2009;37:559–572. doi: 10.1016/j.exphem.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Quelle FW, Wang D, Nosaka T, Thierfelder WE, Stravopodis D, Weinstein Y, Ihle JN. Erythropoietin induces activation of Stat5 through association with specific tyrosines on the receptor that are not required for a mitogenic response. Mol Cell Biol. 1996;16:1622–1631. doi: 10.1128/mcb.16.4.1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Laubach JP, Fu P, Jiang X, Salter KH, Potti A, Arcasoy MO. Polycythemia vera erythroid precursors exhibit increased proliferation and apoptosis resistance associated with abnormal RAS and PI3K pathway activation. Exp Hematol. 2009;37:1411–1422. doi: 10.1016/j.exphem.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kang YA, Sanalkumar R, O’Geen H, Linnemann AK, Chang CJ, Bouhassira EE, Farnham PJ, Keles S, Bresnick EH. Autophagy driven by a master regulator of hematopoiesis. Mol Cell Biol. 2012;32:226–239. doi: 10.1128/MCB.06166-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Horos R, Ijspeert H, Pospisilova D, Sendtner R, Andrieu-Soler C, Taskesen E, Nieradka A, Cmejla R, Sendtner M, Touw IP, von Lindern M. Ribosomal deficiencies in Diamond-Blackfan anemia impair translation of transcripts essential for differentiation of murine and human erythroblasts. Blood. 2012;119:262–272. doi: 10.1182/blood-2011-06-358200. [DOI] [PubMed] [Google Scholar]
- 118.Dutt S, Narla A, Lin K, Mullally A, Abayasekara N, Megerdichian C, Wilson FH, Currie T, Khanna-Gupta A, Berliner N, Kutok JL, Ebert BL. Haploinsufficiency for ribosomal protein genes causes selective activation of p53 in human erythroid progenitor cells. Blood. 2011;117:2567–2576. doi: 10.1182/blood-2010-07-295238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Szczepankiewicz A, Leszczynska-Rodziewicz A, Pawlak J, Rajewska-Rager A, Dmitrzak-Weglarz M, Wilkosc M, Skibinska M, Hauser J. Glucocorticoid receptor polymorphism is associated with major depression and predominance of depression in the course of bipolar disorder. J Affect Disord. 2011;134:138–144. doi: 10.1016/j.jad.2011.06.020. [DOI] [PubMed] [Google Scholar]
- 120.Baugh JE Jr, Floyd ZE, Stephens JM. The modulation of STAT5A/GR complexes during fat cell differentiation and in mature adipocytes. Obesity (Silver Spring) 2007;15:583–590. doi: 10.1038/oby.2007.500. [DOI] [PubMed] [Google Scholar]
- 121.Trebble PJ, Woolven JM, Saunders KA, Simpson KD, Farrow SN, Matthews LC, Ray DW. A ligand-specific kinetic switch regulates glucocorticoid receptor trafficking and function. J Cell Sci. 2013;126:3159–3169. doi: 10.1242/jcs.124784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yan D, Hutchison RE, Mohi G. Critical requirement for Stat5 in a mouse model of polycythemia vera. Blood. 2012;119:3539–3549. doi: 10.1182/blood-2011-03-345215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hiroyama T, Miharada K, Sudo K, Danjo I, Aoki N, Nakamura Y. Establishment of mouse embryonic stem cell-derived erythroid progenitor cell lines able to produce functional red blood cells. PLoS One. 2008;3:e1544. doi: 10.1371/journal.pone.0001544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Giarratana MC, Rouard H, Dumont A, Kiger L, Safeukui I, Le Pennec PY, Francois S, Trugnan G, Peyrard T, Marie T, Jolly S, Hebert N, Mazurier C, Mario N, Harmand L, Lapillonne H, Devaux JY, Douay L. Proof of principle for transfusion of in vitro-generated red blood cells. Blood. 2011;118:5071–5079. doi: 10.1182/blood-2011-06-362038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Peyrard T, Bardiaux L, Krause C, Kobari L, Lapillonne H, Andreu G, Douay L. Banking of pluripotent adult stem cells as an unlimited source for red blood cell production: potential applications for alloimmunized patients and rare blood challenges. Transfus Med Rev. 2011;25:206–216. doi: 10.1016/j.tmrv.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 126.Bender JG, Unverzagt K, Walker DE, Lee W, Smith S, Williams S, Van Epps DE. Phenotypic analysis and characterization of CD34+ cells from normal human bone marrow, cord blood, peripheral blood, and mobilized peripheral blood from patients undergoing autologous stem cell transplantation. Clin Immunol Immunopathol. 1994;70:10–18. doi: 10.1006/clin.1994.1003. [DOI] [PubMed] [Google Scholar]
- 127.Chen L, Gao Z, Zhu J, Rodgers GP. Identification of CD13+CD36+ cells as a common progenitor for erythroid and myeloid lineages in human bone marrow. Exp Hematol. 2007;35:1047–55. doi: 10.1016/j.exphem.2007.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kieffer N, Bettaieb A, Legrand C, Coulombel L, Vainchenker W, Edelman L, Breton-Gorius J. Developmentally regulated expression of a 78 kDa erythroblast membrane glycoprotein immunologically related to the platelet thrombospondin receptor. Biochem J. 1989;262:835–842. doi: 10.1042/bj2620835. [DOI] [PMC free article] [PubMed] [Google Scholar]