

Abstract
Background: Cleidocranial dysplasia is a rare hereditary skeletal disorder due to heterozygous loss of function mutations in the RUNX2 gene that encodes runt-related transcription factor 2 (RUNX2). Here we report a 52 year-old woman with cleidocranial dysplasia due to a novel RUNX2 mutation. Case description: A 52 year-old Han Chinese woman presented with short stature and skeletal dysplasia that was first noted during early childhood. She was 153 cm in height and 40 kg in weight. Her skull was deformed with hypertelorism, midface hypoplasia, protrusion of chin, and dental abnormalities. Radiological examination revealed shortened clavicles and depressed skull bone and that were consistent with the clinical diagnosis of cleidocranial dysplasia. There was no family history of a similar skeletal disorder. We sequenced the RUNX2 gene and discovered a novel heterozygous mutation in exon 3 (c.476 del G, p.G159fs175X) that is predicted to cause a frameshift and premature termination that leads to the loss of the final 347 amino acid residues. This severely truncated protein is expected to be inactive. Literature review: RUNX2 gene controls osteoblast differentiation and chondrocyte maturation. Around 90 RUNX2 mutations have been discovered in patients with cleidocranial dysplasia. Clinical relevance: We identified a case of cleidocranial dysplasia due to a novel mutation of RUNX2 gene at exon 3 (c.476 del G).
Keywords: Cleidocranial dysplasia, RUNX2 gene, mutation
Introduction
Cleidocranial dysplasia (CCD) (MIM 119600), also known as cleidocranial dystosis, is a rare hereditary skeletal disorder. In most cases the disorder is inherited as an autosomal dominant trait, but in some cases the disorder appears sporadic. The main clinical features of CCD are recognized during early childhood and include proportionate short stature, delayed closure of fontanelles, prominent forehead, drooping shoulders, and abnormal dental development. The distinctive radiological features are shortened or absent clavicles, delayed ossification of the skull bones, and delayed ossification of pelvic bones [1].
Heterozygous mutations in the RUNX2 gene (OMIM 600211) that encodes runt-related transcription factor 2 (RUNX2), also termed core-binding factor alpha1 (CBFA1), at chromosome 6p21 are the principal cause of CCD [2,3]. The human RUNX2 gene encodes a 521 amino-acid length protein (GenBank: CAI19639.1) that contains a highly conserved 128-amino-acid region termed the “Runt domain” [4]. In addition, the RUNX2 protein contains an N-terminal stretch of glutamine/alanine repeats (Q/A domain) and a C-terminal proline/serine/threonine-rich (PST) domain [5,6]. The RUNX-binding site is the element binding to the DNA sequence and may regulate several bone-related genes [7].
RUNX2 is the master gene of osteoblast differentiation and also controls chondrocyte maturation [8]. The RUNX2 protein is essential for osteoblastic differentiation and skeletal morphogenesis. RUNX2 binds DNA both are as a monomer or, with more affinity, as a subunit of a heterodimeric complex, and behaves as a scaffold for nucleic acids and regulatory factors involved in skeletal gene expression. Variant transcripts that encode different protein isoforms result from the use of alternate promoters as well as alternate splicing [7]. RUNX2 binds to the core site, 5’-PYGPYGGT-3’, that is present in the promoter regions of a number genes, including osteocalcin, osteopontin, bone sialoprotein, and alpha 1 (I) collagen. RUNX2 participates in both intramembranous and endochondral ossification. Endochondral ossification is characterized by formation of cartilage model that is later replaced by bone, and accounts for most skeletal development [9]. By contrast, intramembranous bone develops directly from osteoblastic action and is limited to the cranial bones, some facial bones, and parts of the mandible and clavicle [10]. The two mechanisms of bone formation, intramembranous and endochondral ossification, are necessary to form the clavicular anlagen in the clavicle [11]. Hence, RUNX2 haploin sufficiency accounts for the distinctive bone dysplasia that is limited to the cranium and clavicle.
To date, fewer than 90 RUNX2 mutations have been described in subjects with CCD, including insertions, deletions, nonsense, and missense mutations, and CCD has been reported in people of Mongoloid ethnicity including Japanese, Korean and the Chinese living in China and Taiwan [12-33]. In most cases mutations occur in the runt domain [3,5]. Here we report a patient with a typical clinical manifestation of CCD with a novel mutation in the RUNX2 gene that leads to a truncated RUNX2 protein.
Case report
We evaluated a 52 year-old female with a history of childhood onset proportionate short stature and skeletal dysplasia. She was 153 cm in height and 40 kg in weight. Her face was unusual and showed features of hypertelorism, depressed frontal area, protrusion of chin and supernumary teeth. Radiological examination revealed marked shortening of clavicles and depressed skull bone (Figure 1A-C). There were no other family members with similar clinical characteristics (Figure 2A). The clinical evaluation was most consistent with CCD, and thus we analyzed her RUNX2 gene. The study protocol was approved by institutional review board of the hospital, and informed consent was obtained from the patient.
Figure 1.
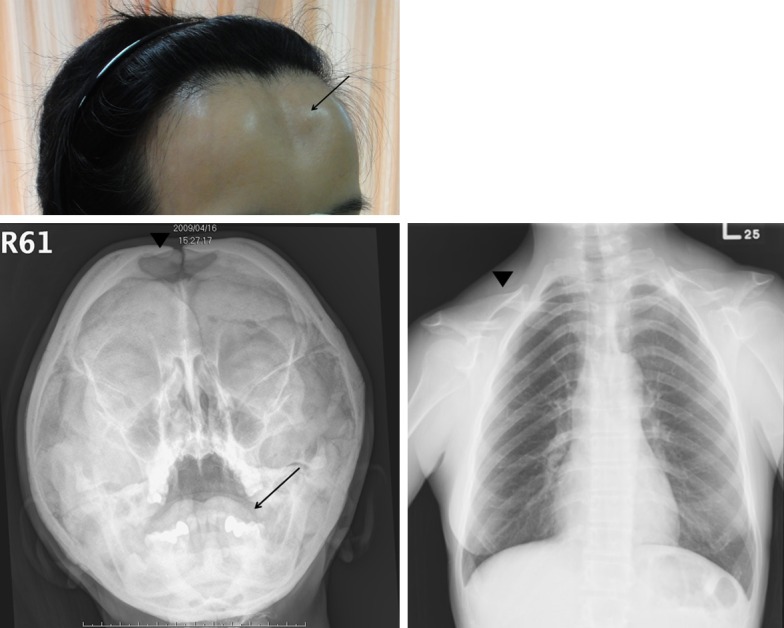
A. Depressed frontal area (arrow) was noted in our patient with cleidocranial dysplasia. B. The plain skull X ray reveals a depressed skull bone (arrowhead) and dental abnormalities (arrow). C. Hypoplasia of the clavicles was seen in the chest X ray (arrowhead).
Figure 2.

A. There was no similar skeletal disorder in the family. B. Partial sequence chromatograms of the RUNX2 gene revealed a single base deletion at nucleotide position 476 in exon 3 (c.476 del G, p.G159fs175X). C. Schematic representation of wild-type and mutant alleles, including mature mRNA sequences and predicted proteins, were shown. The mutant allele with single base deletion at nucleotide position 476 generated an abnormal 5’ splice site at codon 175, leading to early termination and protein truncation with loss of 347 amino acid residues. D. Summary of mutation spectrum of RUNX2 gene mutations among Han-Chinese with cleidocranial dysplasia was displayed.
Genomic DNA was extracted from peripheral whole blood. Genomic DNA was used to amplify exons 1 through 8 and the flanking intronic sequences of the RUNX gene using eight pairs of PCR primers that were designed as previously described [6]. Purified PCR products were sequenced in both directions at our on-site biochemistry sequencing facility using Big Dye Version 3.1 and a 3730 XL sequencer (Applied Biosystems, Foster City, CA).
The patient was found to be heterozygous for single-base deletion (c.476 del G, p.G159fs175X) in exon 3 of RUNX2, which predicts a termination site at the 159th codon and leads to a truncation in the runt domain of RUNX2 protein (Figure 2B, 2C).
Discussion
Several lines of evidence indicate that the deletion G in position 476 is related causally to CCD in this patient. 1) c.476 del G residue in the exon 3 is evolutionally conserved in human, rat and mouse RUNX2 genes; 2) the c. 476 del G mutation, results in a frame shift and premature termination at amino acid 175, which is predicted to lead to a markedly truncated protein that lacks the NLS and the PST domain. Other missense or truncated mutations in the carboxyl end of RUNX2, such as T420I, can cause CCD [6]. Hence, a truncated protein that lacks 347 amino acid residues and would certainly be expected to be inactive.
RUNX2 mutations are scattered throughout the entire gene and include deletions, insertions, or missense mutations. However, most mutations occur in the runt domain and missense mutations are the most common. Loss of the runt domain is expected to abolish ability of the protein to bind DNA [3]. The runt domain has the ability to mediate DNA binding and protein heterodimerization [26]. The C-terminal PST domain is suggested to be the transcription activation domain and is involved in functional interactions with various other transcription factor. Besides, a nuclear-localization signal (NLS) is located at the junction of the runt and PST domains and is a short basic stretch with nine amino acids [amino acids 221-229 in RUNX2]. It is necessary for nuclear localization of the protein [7,26,34].
Mundlos et al demonstrated that RUNX2 mutations segregate with the CCD phenotype and found that heterozygous loss of function is sufficient to induce the characteristic clinical findings [11]. However, there is a wide spectrum of phenotypic variability, from primary dental anomalies to complete CCD. Lou et al. suggested that there is a critical gene dosage requirement for the formation of intramembranous bone formation during embryogenesis and a decrease to 70% of wild-type Runx2 levels will result the CCD syndrome. There is a strong relationship between the phenotype of CCD and quantitative reduction in the functional activity of RUNX2 [35]. The RUNX2 mutation that we identified in this study is novel and differs from the mutations that have been previously described in Chinese patients with CCD [14,17-33] (Figure 2D), indicating that there is significant variability in RUNX2 mutations in this population.
In conclusion, the symptoms in this patient are due to c. 476 del G mutation of RUNX2 gene. To our knowledge, it is a novel mutation which was not reported before.
Acknowledgements
This study was partly supported by research grants V102B-048 and V103B-019 to L.Y.L. from Taipei Veterans General Hospital, Taipei, Taiwan and by research grants MOST 1032314-B-075-005-MY2 to L.Y.L from Taiwan’s Ministry of Science and Technology. The study protocol was approved by institutional review board of Taipei-Veterans General Hospital, Taipei, Taiwan 112 (IRB No. 2010-08-0130OB).
Disclosure of conflict of interest
None.
References
- 1.Horton WA, Hecht JT. Disorders Involving Transcription Factors. In: Kliegman RM, Stanton BMD, St Geme J, Schor N, Behrman RE, editors. Nelson Textbook of Pediatrics. 2011. pp. 2431–2432. [Google Scholar]
- 2.Mundlos S. Cleidocranial dysplasia: clinical and molecular genetics. J Med Genet. 1999;36:177–182. [PMC free article] [PubMed] [Google Scholar]
- 3.Lee B, Thirunavukkarasu K, Zhou L, Pastore L, Baldini A, Hecht J, Geoffroy V, Ducy P, Karsenty G. Missense mutations abolishing DNA binding of the osteoblast-specific transcription factor OSF2/CBFA1 in cleidocranial dysplasia. Nat Genet. 1997;16:307–310. doi: 10.1038/ng0797-307. [DOI] [PubMed] [Google Scholar]
- 4.Kagoshima H, Shigesada K, Satake M, Ito Y, Miyoshi H, Ohki M, Pepling M, Gergen P. The Runt domain identifies a new family of heteromeric transcriptional regulators. Trends Genet. 1993;9:338–341. doi: 10.1016/0168-9525(93)90026-e. [DOI] [PubMed] [Google Scholar]
- 5.Ducy P. Cbfa1: a molecular switch in osteoblast biology. Dev Dyn. 2000;219:461–471. doi: 10.1002/1097-0177(2000)9999:9999<::AID-DVDY1074>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 6.Wang GX, Sun RP, Song FL. A novel RUNX2 mutation (T420I) in Chinese patients with cleidocranial dysplasia. Genet Mol Res. 2010;9:41–47. doi: 10.4238/vol9-1gmr685. [DOI] [PubMed] [Google Scholar]
- 7.Li YL, Xiao ZS. Advances in Runx2 regulation and its isoforms. Med Hypotheses. 2007;68:169–175. doi: 10.1016/j.mehy.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 8.Karsenty G. Transcriptional Control of Skeletogenesis. Annu Rev Genomics Hum Genet. 2008;9:183–196. doi: 10.1146/annurev.genom.9.081307.164437. [DOI] [PubMed] [Google Scholar]
- 9.Zelzer E, Olsen BR. The genetic basis for skeletal diseases. Nature. 2003;423:343–348. doi: 10.1038/nature01659. [DOI] [PubMed] [Google Scholar]
- 10.Komori T, Kishimoto T. Cbfa 1 in bone development. Curr Opin Genet Dev. 1998;8:494–499. doi: 10.1016/s0959-437x(98)80123-8. [DOI] [PubMed] [Google Scholar]
- 11.Mundlos S, Otto F, Mundlos C, Mulliken JB, Aylsworth AS, Albright S, Lindhout D, Cole WG, Henn W, Knoll JH, Owen MJ, Mertelsmann R, Zabel BU, Olsen BR. Mutations Involving the Transcription Factor CBFA1 Cause Cleidocranial Dysplasia. Cell. 1997;89:773–779. doi: 10.1016/s0092-8674(00)80260-3. [DOI] [PubMed] [Google Scholar]
- 12.Sakai N, Hasegawa H, Yamazaki Y, Ui K, Tokunaga K, Hirose R, Uchinuma E, Susami T, Takato T. A case of a Japanese patient with cleidocranial dysplasia possessing a mutation of CBFA1 gene. J Craniofac Surg. 2002;13:31–34. doi: 10.1097/00001665-200201000-00005. [DOI] [PubMed] [Google Scholar]
- 13.Zhang YW, Yasui N, Kakazu N, Abe T, Takada K, Imai S, Sato M, Nomura S, Ochi T, Okuzumi S, Nogami H, Nagai T, Ohashi H, Ito Y. PEBP2-alphaA/CBFA1 mutations in Japanese cleidocranial dysplasia patients. Gene. 2000;244:21–28. doi: 10.1016/s0378-1119(99)00558-2. [DOI] [PubMed] [Google Scholar]
- 14.Tsai FJ, Wu JY, Lin WD, Tsai CH. A stop codon mutation in the CBFA 1 gene causes cleidocranial dysplasia. Acta Paediatrica. 2000;89:1262–1265. doi: 10.1080/080352500750027673. [DOI] [PubMed] [Google Scholar]
- 15.Yokozeki M, Ohyama K, Tsuji M, Goseki-Sone M, Oida S, Orimo H, Moriyama K, Kuroda T. A case of Japanese cleidocranial dysplasia with a CBFA1 frameshift mutation. J Craniofac Genet Dev Biol. 2000;20:121–126. [PubMed] [Google Scholar]
- 16.Han MS, Kim HJ, Wee HJ, Lim KE, Park NR, Bae SC, van Wijnen AJ, Stein JL, Lian JB, Stein GS, Choi JY. The cleidocranial dysplasia-related R131G mutation in the Runt-related transcription factor RUNX2 disrupts binding to DNA but not CBF-beta. J Cell Biochem. 2010;110:97–103. doi: 10.1002/jcb.22516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang GX, Ma LX, Xu WF, Song FL, Sun RP. Clinical and image features, and identification of pathogenic gene mutation of two cleidocranial dysplasia families. Zhonghua Er Ke Za Zhi. 2010;48:834–838. [PubMed] [Google Scholar]
- 18.Zhang C, Zheng S, Wang Y, Zhao Y, Zhu J, Ge L. Mutational analysis of RUNX2 gene in Chinese patients with cleidocranial dysplasia. Mutagenesis. 2010;25:589–594. doi: 10.1093/mutage/geq044. [DOI] [PubMed] [Google Scholar]
- 19.Gao C, Wu L, Geng XJ, Song LJ, Luo Q. Two novel RUNX2 gene mutations in two Chinese families with cleidocranial dysplasia. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2010;27:140–143. doi: 10.3760/cma.j.issn.1003-9406.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 20.Li Y, Pan W, Xu W, He N, Chen X, Liu H, Darryl Quarles L, Zhou H, Xiao Z. RUNX2 mutations in Chinese patients with cleidocranial dysplasia. Mutagenesis. 2009;24:425–431. doi: 10.1093/mutage/gep025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xuan D, Li S, Zhang X, Hu F, Lin L, Wang C, Zhang J. Mutations in the RUNX2 gene in Chinese patients with cleidocranial dysplasia. Ann Clin Lab Sci. 2008;38:15–24. [PubMed] [Google Scholar]
- 22.Tang S, Xu Q, Xu X, Du J, Yang X, Jiang Y, Wang X, Speck N, Huang T. A novel RUNX2 missense mutation predicted to disrupt DNA binding causes cleidocranial dysplasia in a large Chinese family with hyperplastic nails. BMC Med Genet. 2007;8:82. doi: 10.1186/1471-2350-8-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Y, Wu H, Zhang XX, Zhao HS, Feng HL. Gene mutation detection in a cleidocranial dysplasia family. Zhonghua Kou Qiang Yi Xue Za Zhi. 2005;40:459–462. [PubMed] [Google Scholar]
- 24.Qiu ZQ, Tang AL, Yu W, Ao Y, Wilson HY, Wei M, Zhang X. A Chinese girl with cleidocranial dysplasia (CCD) caused by the recurrent R190W mutation in RUNX 2. Zhonghua Er Ke Za Zhi. 2004;42:759–761. [PubMed] [Google Scholar]
- 25.Lin WD, Lin SP, Wang CH, Tsai Y, Chen CP, Tsai FJ. RUNX2 mutations in Taiwanese patients with cleidocranial dysplasia. Genet Mol Biol. 2011;34:201–204. doi: 10.1590/S1415-47572011005000002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fang CY, Xue JJ, Tan L, Jiang CH, Gao QP, Liang DS, Wu LQ. A novel single-base deletion mutation of the RUNX2 gene in a Chinese family with cleidocranial dysplasia. Genet Mol Res. 2011;10:3539–3544. doi: 10.4238/2011.December.14.5. [DOI] [PubMed] [Google Scholar]
- 27.Qi Z, Yang W, Meng Y, Liu Y. Identification of three novel frameshift mutations in the RUNX2 gene in three sporadic Chinese cases with cleidocranial dysplasia. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2014;31:415–419. doi: 10.3760/cma.j.issn.1003-9406.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 28.Wu LZ, Su WQ, Liu YF, Ge X, Zhang Y, Wang XJ. Role of the RUNX2 p. R225Q mutation in cleidocranial dysplasia: a rare presentation and an analysis of the RUNX2 protein structure. Genet Mol Res. 2014;13:1187–1194. doi: 10.4238/2014.February.27.3. [DOI] [PubMed] [Google Scholar]
- 29.Jiang T, Jiang X, Zhang Y. Mutation analysis of the RUNX2 gene in a family with cleidocranial dysplasia. Hua Xi Kou Qiang Yi Xue Za Zhi. 2013;31:522–525. [PubMed] [Google Scholar]
- 30.Ding B, Li C, Xuan K, Liu N, Tang L, Liu Y, Guo W, Liu W, Jin Y. The effect of the cleidocranial dysplasia-related novel 1116_1119insC mutation in the RUNX2 gene on the biological function of mesenchymal cells. Eur J Med Genet. 2013;56:180–187. doi: 10.1016/j.ejmg.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 31.Chen T, Hou J, Hu LL, Gao J, Wu BL. A novel small deletion mutation in RUNX2 gene in one Chinese family with cleidocranial dysplasia. Int J Clin Exp Pathol. 2014;7:2490–2495. [PMC free article] [PubMed] [Google Scholar]
- 32.Anthonappa RP, Yan-Hui F, King NM, Rabie AB, You-Qiang S. Novel complex disease allele mutations in cleidocranial dysplasia patients. J Oral Pathol Med. 2014;43:798–800. doi: 10.1111/jop.12198. [DOI] [PubMed] [Google Scholar]
- 33.Huang Y, Song Y, Zhang C, Chen G, Wang S, Bian Z. Novel RUNX2 frameshift mutations in Chinese patients with cleidocranial dysplasia. Eur J Oral Sci. 2013;121:142–147. doi: 10.1111/eos.12048. [DOI] [PubMed] [Google Scholar]
- 34.Thirunavukkarasu K, Mahajan M, McLarren KW, Stifani S, Karsenty G. Two domains unique to osteoblast-specific transcription factor Osf2/Cbfa1 contribute to its transactivation function and its inability to heterodimerize with Cbfbeta. Mol Cell Biol. 1998;18:4197–4208. doi: 10.1128/mcb.18.7.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lou Y, Javed A, Hussain S, Colby J, Frederick D, Pratap J, Xie R, Gaur T, van Wijnen AJ, Jones SN, Stein GS, Lian JB, Stein JL. A Runx2 threshold for the cleidocranial dysplasia phenotype. Hum Mol Genet. 2009;18:556–568. doi: 10.1093/hmg/ddn383. [DOI] [PMC free article] [PubMed] [Google Scholar]