

Abstract
Elucidating the genomic diversity of CD209 gene promoter polymorphism could assist in clarifying disease pathophysiology as well as contribution to co-morbidities. CD209 gene promoter polymorphism has been shown to be associated with susceptibility to infection. We hypothesize that CD209 mutant variants occur at a higher frequency among Africans and in sickle cell disease. We analyzed the frequency of the CD209 gene (rs4804803) in healthy control and sickle cell disease (SCD) populations and determined association with disease. Genomic DNA was extracted from blood samples collected from 145 SCD and 231 control Africans (from Mali), 331 SCD and 379 control African Americans and 159 Caucasians. Comparative analysis among and between groups was carried out by polymerase chain reaction-restriction fragment length polymorphism (PCR-RFLP). Per ethnic diversification, we found significant disparity in genotypic (23.4% versus 16.9% versus 3.2%) and allelic frequencies (48.7% versus 42.1% versus 19.8%) of the homozygote mutant variant of the CD209 (snp 309A/G) gene promoter between Africans, African Americans and Caucasians respectively. Comparative evaluation between disease and control groups reveal a significant difference in genotypic (10.4% versus 23.4%; p = 0.002) and allelic frequencies (39.7% versus 48.7%; p = 0.02) of the homozygote mutant variant in African SCD and healthy controls respectively, an observation that is completely absent among Americans. Comparing disease groups, we found no difference in the genotypic (p = 0.19) or allelic (p = 0.72) frequencies of CD209 homozygote mutant variant between Africans and Americans with sickle cell disease. The higher frequency of CD209 homozygote mutant variants in the African control group reveals a potential impairment of the capacity to mount an immune response to infectious diseases, and possibly delineate susceptibility to or severity of infectious co-morbidities within and between groups.
Keywords: Sickle cell disease, Africa, African Americans, Caucasians, CD209, Diversity, Interethnic, Populations, Co-morbidities
Introduction
Sickle cell disease (SCD) is an inherited multisystem disorder, characterized by chronic hemolytic anemia, vaso-occlusive crises and several other disease outcomes such as acute chest syndrome, bacteremia, leg ulcers and priapism (Bunn, 1997; Benkerrou et al., 2002). SCD has shown marked variability in severity between individuals, with evidence of extensive differences in both clinical and disease haplotypes, with a global distribution, especially in sub-Saharan Africa, Middle East, parts of the Indian subcontinent, and Americans with an African or Caribbean descent (Hassell, 2010; Piel et al., 2013; Bandeira et al., 2014; Saraf et al., 2014; Thakur et al., 2014). SCD occurs in patients that are homozygous for the hemoglobin S gene, produced by a defective β-globin gene on chromosome 11 and has also been defined as resulting from compound heterozygosity for hemoglobin S and another β-globin chain abnormality (typically hemoglobin C or β-thalassemia), with α-thalassemia serving as a modifier of the clinical manifestations (Weatherall, 2010; Saraf et al., 2014). Patients commonly require red cell transfusions to manage complications, with alloimmunization a common occurrence (Charache, Bleecker & Bross, 1983; Rosse, Gallagher & Kinney, 1990; Tatari-Calderone et al., 2013) leaving such multiply transfused patients at risk for delayed hemolytic transfusion reactions (Piomelli, 1985; Petz et al., 1997; Taylor et al., 2008; Yazdanbakhsh, Ware & Noizat-Pirenne, 2012), development of autoimmune hemolytic anemia.
Infectious pathogens are a threat to those individuals with SCD, particularly children, that are prone to frequent and severe attacks (Overturf, 1999; Halasa et al., 2007; Szczepanek et al., 2013). For children in endemic countries, with very high circulating immune complexes due to constant exposure to multiple pathogenic stimuli, the added burden of these co-morbidities can severely impact immune response and survival (Thomas et al., 2012a). Recent reports showing high mortality rates post-vaccination in transgenic animals demonstrates that a dysregulated immune response might be responsible for such mortality and could be a major drawback to the current push to vaccinate (Adamkiewicz et al., 2003; McCavit et al., 2011; Szczepanek et al., 2013). In fact, other reports have shown that there is an over-stimulation of pro-inflammatory cytokines in sickle cell disease patients, which might be be related to vaso-occlusion (Makis, Hatzimichael & Bourantas, 2000; Pathare et al., 2004; Steinberg, 2006; Conran, Franco-Penteado & Costa, 2009; Qari, Dier & Mousa, 2012; Bandeira et al., 2014). In fact, this hyperstimulation has been associated with sickle cell haplotype in Brazil, and as such is the obvious consequence of worsening immune response to secondary infectious pathogens or co-morbidities of infection.
Recently published data have shown that there are wide differences in Plasmodium falciparum infection rates and multiplicity of infection between children who are carriers of the sickle cell trait (hemoglobin AS) and those patients that possess the normal hemoglobin (HbAA) gene (Williams et al., 2005; Kreuels et al., 2010; Gong et al., 2012; Taylor, Parobek & Fairhurst, 2012; Gong et al., 2013). In addition, extensive differences in genomic diversity of endothelial nitric oxide synthase (eNOS) genes, that had been reported to bear clinical significance on sickle cell pathogenesis, has been reported between Africans and African Americans (Thomas et al., 2013). These polymorphisms have been shown to be potential modifiers of clinical disease, with significant differences reported between Indian and African sickle cell disease patients (Nishank et al., 2013; Thakur et al., 2014), and these differences could be potentially linked to disease haplotype. These interethnic differences can be attributed to the introduction of single nucleotide polymorphisms over a very long period, which can ultimately influence gene expression, protein structure and potentially function. Therefore, single nucleotide polymorphisms located in certain promoter regions can affect transcription thereby altering variability in the immune response, and contributing to disease susceptibility or host resistance (Sakuntabhai et al., 2005). Despite the fact that African Americans can trace their ancestry to sub-Saharan Africa, recombination and genetic diversity in the African American gene pool has facilitated the introduction of single nucleotide polymorphisms leading to differing immune response to infectious pathogens, such as malaria and tuberculosis (Thomas et al., 2005; Jallow et al., 2009; Thomas et al., 2012b; Noumsi et al., 2011; Hansson et al., 2013), and demonstrated in an Afro-Brazilian population (Covas et al., 2007; Dettogni et al., 2013) sharing phenotypic and genotypic similarity with African Americans. In addition, they are exposed to different groups of infectious agents compared to their African counterparts, which in turn directs immune system development, as shown in complement receptor-1 (CR1) polymorphisms in malaria-endemic and non-endemic populations (Thomas et al., 2005). These phenomena would undergo a similar diversification in the sickle cell disease population as well.
One of the most common immunogenetic markers, usually evaluated for immune system response and susceptibility to infectious pathogens is dendritic cell-specific ICAM-3 grabbing non-integrin (DC-SIGN) encoded by CD209. It assists in the migration dendritic cells on endothelium as well as enabling the activation of signal transduction pathways (Rappocciolo, Jenkins & Hensler, 2006; Dettogni et al., 2013). They are targets for pathogens, seeking to impair the immune response in early infection, and are known to recognize diverse pathogens, with reports showing association between CD209 gene polymorphisms and infectious agents (Mummidi et al., 2001; Martin et al., 2004). The guanine (G) to adenine (A) transition within the gene promoter (SNP-336 A/G; rs4804803) polymorphism has shown the most significance, demonstrating association with susceptibility to HIV, tuberculosis, leishmaniasis and dengue (Tailleux, Schwartz & Herrmann, 2003; Tassaneetrithep, Burgess & Granelli-Piperno, 2003; Van Kooyk, Appelmelk & Geijtenbeek, 2003; Martin et al., 2004; Sakuntabhai et al., 2005; Barreiro, Neyrolles & Babb, 2006). Due to the interaction between malaria and sickle cell disease, the possibility of imposing selection pressures, leading to changes in allele frequencies that can exacerbate or ameliorate outcome of disease co-morbidities exists (Thomas et al., 2012a; Thomas et al., 2012b). We have shown that there is an extensive diversity in the ethnogenomic distribution of endothelial nitric oxide synthase (eNOS) polymorphisms (Thomas et al., 2013). Despite reports to the contrary, we have also demonstrated that endothelin-1 polymorphisms, rather than eNOS, are the most important in African SCD (Thakur et al., 2014). Therefore, since infections are common occurrences in SCD, there is a need to characterize the genomic diversity as well as haplotype frequency of immunogenetic markers, thereby clarifying their contributions to infectious disease susceptibility or co-morbidities. To this end, we examined the genotypic and allelic frequency of CD209 gene promoter polymorphism (SNP-336 A/G; rs4804803) in control groups (Africans versus African Americans versus Caucasians) and between sickle cell disease populations (African versus American). We conducted our analyses using a polymerase chain reaction-restriction fragment length polymorphism (PCR-RFLP) assay.
Materials and Methods
Subjects
This study encompasses sickle cell disease patients (cases) and control groups (Africans versus African Americans), as well as diverse ethnic groups (Africans, African Americans and Caucasians). The African portion was conducted at the Centre de Recherche et de Lutte contre la Drepanocytose (CRLD), a sickle cell disease treatment and referral center in Bamako, Mali. This study was approved by the Institutional Review Board (IRB), Rochester Institute of Technology in addition to the original approval granted by the National Ethical Review Board in Mali. Inclusion criteria include diagnosis with sickle cell disease and presentation during crisis or during regular follow-up. Sickle cell disease and control group demographic data has been described previously (Thakur et al., 2014). Briefly, African sickle cell disease group consists of 51.5% males and 48.4% females (mean age: 21 years; range: 1-51 years), and predominantly of the Bambaran tribe. Healthy population controls comprised of family members or those recruited by word of mouth, able to provide informed consent and without a diagnosis of sickle cell disease. In the United States, control groups are African American and Caucasian self-identified individuals, recruited from Shreveport, Louisiana. African American sickle cell disease patients were recruited as part of the National Institute of Health-funded Cooperative Study of Sickle Cell Disease (CSSCD).
Samples and genomic DNA extraction
Discarded EDTA-anticoagulated blood samples, from 376 subjects (145 sickle cell disease patients and 231 controls) were spotted onto filter papers (GE Healthcare Sciences, Piscataway, New Jersey, USA) and genomic DNA samples extracted from the dried, spotted samples with the Qiagen Blood Mini Kit (Qiagen Inc., Valencia, California, USA), with some changes to the manufacturer’s instruction (Thakur et al., 2014). Final elution volume was 100 µl and DNA samples were stored at −20 °C until further analysis. Genomic DNA samples from African American sickle cell disease patients as well as African American and Caucasian controls were gratefully provided (Betty Pace, Georgia Regents University and Joann Moulds, Grifols USA respectively).
Genotyping for CD209 single nucleotide polymorphism
To genotype for the single nucleotide polymorphisms of the CD209 gene promoter, we utilized a previously published mis-matched primer, designed to artificially introduce a restriction site (Sakuntabhai et al., 2005) and PCR assay (Dettogni et al., 2013), with a slight modification to the protocol. The primer sequences are 5′-GGATGGTCTGGGGTTGACAG-3 (forward reaction) and 5′-ACTGGGGGTGCTACCTGGC-3′ (reverse reaction). One µl of genomic DNA served as the template for PCR amplification, with conditions optimized to 25 µl final volume and amplified using the Lucigen EconoTaq Plus Green 2X Master Mix PCR system (Lucigen Corporation, Middleton WI), as described previously (Thomas et al., 2012a), and PCR cycling parameters as published (Sakuntabhai et al., 2005). Amplified PCR products (5 µl) was examined on a 2% (w/v) agarose gel and photographed. Positive amplification yielded products of 150 bp, with size estimated with a TriDye 100 bp DNA ladder (New England Biolabs, Boston, Massachusetts, USA).
Restriction fragment length polymorphism assay
We utilized the MscI (New England Biolabs, Boston, Massachusetts, USA) restriction endonuclease for restriction fragment length polymorphism analysis of CD209 (DC-SIGN 336A/G) gene promoter variants. 10 µl of PCR product was mixed with 0.5 µl of enzyme (5,000 U/ml), 5 µl of 1X CutSmart buffer and incubated at 37 °C for 1 h. Digested products were analyzed on an ethidium bromide-stained agarose gel, and band analysis carried out with a Doc-It LS Image Analysis Software (UVP Life Sciences, Upland, California, USA). Restriction analysis was conducted by two investigators anonymously and 50% of amplified products subjected to repeat digestion (3rd investigator), with 100% concordance. Homozygote wild type variants (−336A/A) were undigested (150 bp) while homozygote mutant variants (−336G/G) produced bands of 131 and 19 bp (Supplemental Information 1).
Statistical analysis
Genotypic and allelic frequencies were determined with a simple PERL script, as described previously (Thakur et al., 2014). Differences in genotype and allele frequencies between populations were assessed by chi-square tests, while differences between sickle cell disease and controls were assessed by odds ratio. Tests for deviation from Hardy-Weinberg equilibrium (HWE) were performed, with SNP’s rejected based on the recommended threshold of p < 0.05 in control individuals. Briefly, we calculated the number of alleles and observed genotypes, and compared observed numbers of genotypes with that expected under HWE, where the latter are computed on the basis of allele frequencies estimated from the genotype frequencies (null hypothesis, H0). For observed numbers, the relative cell frequencies are the estimates of the genotype probabilities (alternative hypothesis, H1). For the comparison between observed and expected numbers of genotypes, likelihood ratio chi-square is computed. Power calculations were computed using the Vanderbilt University PS program http://biostat.mc.vanderbilt.edu/wiki/Main/PowerSampleSize.
Results
We found a significant difference in the genetic diversity of the promoter variant of CD209 (DC-SIGN1-336A/G; rs4804803) gene polymorphism in different populations. Genotypic frequencies of 23.4%, 16.9% and 3.2% were observed for the homozygote mutant variant between Africans, African Americans and Caucasians respectively (Table 1). Similar findings were made for the allelic frequencies of the homozygote mutant variants (48.7%, 42.1% and 19.8% respectively), with a significant difference in the genotypic and allelic frequencies (P < 0.05) of CD209 gene promoter variant between all population groups. Surprisingly, the homozygote mutant variant (GG) is almost absent among Caucasians (3.2%; Fig. 1). The genotypic and allelic frequencies of the homozygote mutant variant (snp-336GG) had the highest frequency among Africans (23.4% and 48.7% respectively). The wild type and heterozygote variants (AA and AG), that are necessary to facilitate dendritic cell activation and function during immune response, occurred at higher frequencies among Caucasians (96.8%) and African Americans (83.1%), compared to the 76% among healthy African controls.
Table 1. Genotypic and allelic frequency of CD209 gene promoter polymorphism in diverse populations.
Genotypic and allelic frequencies of the CD209 gene promoter (rs4803803) polymorphism, determined among African (n = 244), African American (n = 379) and Caucasian (n = 159) healthy controls. Healthy control populations are individuals without sickle cell disease (HbAA). Africans were recruited from Bamako, Mali; African American and Caucasian populations were recruited from Shreveport, Louisiana. Odds ratio was calculated by Fisher’s two-tailed exact test. P value <0.05 was considered significant.
| Polymorphism | Genotype | Ethnic groups | Chi square | P-value | ||
|---|---|---|---|---|---|---|
| African n = 231 (%) |
African American n = 379 (%) |
Caucasian n = 159 (%) |
||||
| CD209 (rs4084803) | A/A | 60 (26.0) | 124 (32.7) | 101 (63.5) | 62.97 | 2.12E−14 |
| A/G | 117 (50.6) | 191 (50.4) | 53 (33.3) | 14.91 | 5.78E−04 | |
| G/G | 54 (23.4) | 64 (16.9) | 5 (3.2) | 29.13 | 4.72E−07 | |
| Allelic diversity | ||||||
| Allele | n = 462 (%) | n = 758 (%) | n = 318 (%) | Chi square | P-value | |
| CD209 (rs4804803) | A | 237 (51.3) | 439 (57.9) | 255 (80.2) | 70.09 | 6.03E−16 |
| G | 225 (48.7) | 319 (42.1) | 63 (19.8) | 70.09 | 6.03E−16 | |
Figure 1. Genotypic distribution of CD209 gene promoter polymorphism (SNP-336 A/G; rs4804803) in Caucasian, African American and African healthy controls.
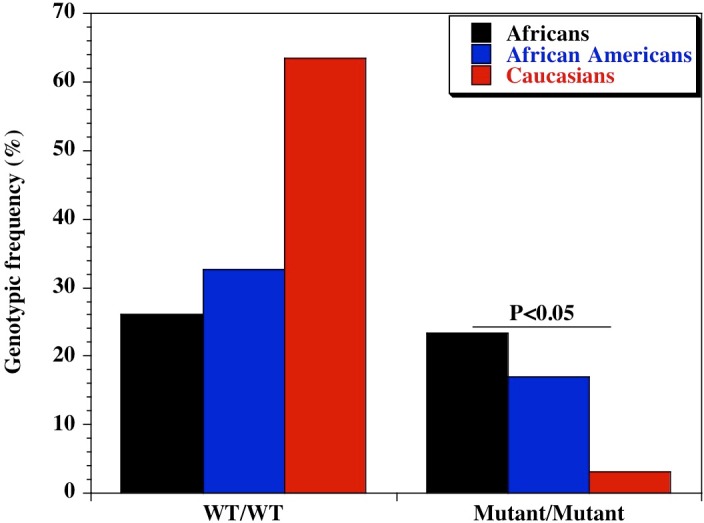
Amplified PCR products were digested with MscI restriction endonuclease (Fisher Scientific, Waltham, Massachusetts, USA), and expressed on a 2% ethidium bromide-stained agarose gel. Homozygote wild type variant (snp-336A) showed no digestion (150 bp); homozygote mutant variant (snp-336G) produced two bands (131 and 19 bp) on digestion (lower band size not shown). Marker: 100 bp ladder, where the 500 bp band stains most intensely (New England Biolabs, Ipswich, Massachusetts, USA). Black bars: Africans; blue bars: African Americans; red bars: Caucasians.
We also examined the diversity of CD209 (snp 336A/G) gene promoter polymorphisms between sickle cell disease and healthy control groups in Africa and United States. There was an extensive and significant difference in the genotypic (Fig. 2 and Table 2) frequency of the CD209 mutant variant (snp 336G/G) between sickle cell disease and control populations in Africa (P = 0.002). Surprisingly, this was not the case between sickle cell disease and control populations recruited from the United States (Fig. 3) (P = 0.54). In addition, the mutant variant has a higher frequency among healthy control groups than sickle cell patients (23.4% versus 10.4% respectively) in Africa, but no difference in the United States (16.9% versus 15.1% for controls and cases respectively). Similar observation was made for the allelic frequencies between controls and cases in Africa and United States (Table 3). The SNP effect is insignificant between American sickle cell disease and controls, but significant among Africans, with a 40% power from our analysis.
Figure 2. Genotypic frequency of CD209 gene promoter polymorphism (SNP-336 A/G; rs4804803) among African sickle cell disease and control groups.

Amplified PCR products were digested with MscI restriction endonuclease (Fisher Scientific, Waltham, Massachusetts, USA), and expressed on a 2% ethidium bromide-stained agarose gel. Homozygote wild type variant (snp-336A) showed no digestion (150 bp); homozygote mutant variant (snp-336G) produced two bands (131 and 19 bp) on digestion (lower band size not shown). Marker: 100 bp ladder, where the 500 bp band stains most intensely (New England Biolabs). Blue bars-sickle cell disease; red bars-control groups.
Table 2. Genotypic frequency of CD209 polymorphisms between sickle cell and control groups.
Genotypic frequencies of the CD209 gene promoter (rs4803803) polymorphism, determined among African and American sickle cell disease and control groups. Sickle cell disease (HbSS) populations were recruited from Bamako, Mali (African) and Augusta GA (American). Control populations are individuals without sickle cell disease, and were recruited from Mali (African) and Shreveport LA (American). A/G denotes the alleles at the CD209 locus. Odds ratio was calculated by Fisher’s two-tailed exact test. P value <0.05 was considered significant.
| African | |||||
|---|---|---|---|---|---|
| Polymorphism | Genotype | SCD: n = 145 (%) |
Controls: n = 231 (%) |
Odds ratio (95% CI) |
P-value |
| CD209 (rs4804803) | A/A | 45 (31.0) | 60 (26.0) | 1.28 (0.79-2.08) | 0.29 |
| A/G | 85 (58.6) | 117 (50.6) | 1.38 (0.89-2.15) | 0.14 | |
| G/G | 15 (10.4) | 54 (23.4) | 0.38 (0.19-0.72) | 0.002 | |
| African American | |||||
|
SCD: n = 331 (%) |
Controls: n = 379 (%) |
Odds ratio
(95% CI) |
P-value | ||
| CD209 (rs4804803) | A/A | 110 (33.2) | 124 (32.7) | 1.02 (0.74–1.42) | 0.94 |
| A/G | 171 (51.7) | 191 (50.4) | 1.05 (0.77–1.43) | 0.76 | |
| G/G | 50 (15.1) | 64 (16.9) | 0.88 (0.57–1.33) | 0.54 | |
Figure 3. Genotypic frequency of CD209 gene promoter polymorphisms (SNP-336 A/G; rs4804803) among African American sickle cell disease and control groups.

Table 3. Allelic frequency of CD209 polymorphisms between sickle cell and control groups.
Allelic frequencies of the CD209 gene promoter (rs4803803) polymorphism, determined among African and American sickle cell disease and control groups. Sickle cell disease (HbSS) populations were recruited from Bamako, Mali (African) and Augusta GA (American). Control populations are individuals without sickle cell disease, and were recruited from Mali (African) and Shreveport LA (American). A/G denotes the alleles at the CD209 locus. Odds ratio was calculated by Fisher’s two-tailed exact test. P value <0.05 was considered significant.
| African | |||||
|---|---|---|---|---|---|
| Polymorphism | Allele | SCD: n = 290 (%) |
Controls: n = 462 (%) |
Odds ratio (95% CI) |
P-value |
| CD209 (rs4804803) | A | 175 (60.3) | 237 (51.3) | 1.44 (1.60–1.97) | 0.02 |
| G | 115 (39.7) | 225 (48.7) | 0.69 (0.51–0.94) | 0.02 | |
| African American | |||||
|
SCD: n = 662 (%) |
Controls: n = 758 (%) |
Odds ratio (95% CI) |
P-value | ||
| CD209 (rs4804803) | A | 391 (59.1) | 439 (57.9) | 1.05 (0.84–1.30) | 0.67 |
| G | 271 (40.9) | 319 (42.1) | 0.95 (0.77–1.19) | 0.67 | |
Notes.
- SCD
- sickle cell disease
- CI
- confidence interval
Since clinical manifestation of sickle cell disease varies greatly within an individual, across individuals of the same population and those of different populations, we evaluated the diversity of CD209 (snp 336A/G) gene promoter polymorphisms between sickle cell groups recruited from Africa and United States. Surprisingly, there was no difference either in genotypic (P = 0.19) or allelic frequencies (P = 0.72) of mutant variants (snp 336G/G) between sickle cell disease groups (Table 4). The similarities in the genotypic and allelic frequencies (10.4% versus 15.1% and 39.7% versus 40.9% for genotypes and alleles respectively) of the homozygote mutant variants were statistically insignificant.
Table 4. Genotypic and allelic frequency of CD209 polymorphisms between sickle cell disease groups.
Genotypic and allelic frequencies of the CD209 gene promoter (rs4803803) polymorphism between African and American sickle cell disease groups. Sickle cell disease (HbSS) populations were recruited from Bamako, Mali (African) and Augusta GA (American). A/G denotes the alleles at the CD209 locus. Odds ratio was calculated by Fisher’s two-tailed exact test. P value <0.05 was considered significant.
| Genotypic frequency | |||||
|---|---|---|---|---|---|
| Polymorphism | Genotype | Mali: n = 145 (%) |
USA: n = 331 (%) |
Odds ratio (95% CI) |
P-value |
| CD209 (rs4804803) | A/A | 45 (31.0) | 110 (33.2) | 0.90 (0.59–1.40) | 0.67 |
| A/G | 85 (58.6) | 171 (51.7) | 1.32 (0.87–2.00) | 0.16 | |
| G/G | 15 (10.4) | 50 (15.1) | 0.65 (0.33–1.23) | 0.19 | |
| Allelic frequency | |||||
| Polymorphism | Allele |
Mali: n = 290 (%) |
USA: n = 662 (%) |
Odds ratio (95% CI) |
P-value |
| CD209 (rs4804803) | A | 175 (60.3) | 391 (59.1) | 1.05 (0.79–1.41) | 0.72 |
| G | 115 (39.7) | 271 (40.9) | 0.95 (0.71–1.27) | 0.72 | |
Notes.
- SCD
- sickle cell disease
- CI
- confidence interval
Discussion
Sickle cell disease is the most commonly inherited hemoglobinopathy with a worldwide distribution. It is a major disease represented in populations of sub-Saharan Africa, the Middle East and several parts of India; it remains a significant health burden borne by the African American population in the United States, as well as in several Caribbean island nations, whose populations are dominated by ethnicities of African origin. It has recently been classified as a disease that would create a global challenge to the population of three major countries (Piel et al., 2013), therefore requiring a need to clarify and decipher the various parameters contributing to its severity and diverse clinical pathophysiology among and between individuals from different populations. To our understanding, this is the first report to elucidate the genomic diversity of CD209 gene promoter (snp-336A/G) polymorphisms in sickle cell disease, with the potential to clarify its role or otherwise in susceptibility to infectious pathogens between sickle cell disease and control groups. We chose three ethnically distinct populations (Bambarans from Mali, African Americans from Shreveport, Louisiana and Augusta, Georgia, and Caucasians from Shreveport Louisiana), and as such permits conclusive inferences based on our finding. African case and controls were all from the Bambaran tribe in Mali, thereby facilitating analysis from an ethnically homogeneous population in comparison to the genetic heterogeneity of the African American group.
Our observation that the CD209 gene promoter homozygous wild-type variant (snp-336A/A) occurred at a lower frequency among Africans compared to African Americans and Caucasians is significant, though not unexpected considering the degree of genetic admixture in the African American population. This is similar to our previous finding while examining the genomic diversity of endothelial nitric oxide synthase gene polymorphisms in differing populations (Thomas et al., 2013). Though both populations share a common ancestry, the many years of genetic admixture and the legacies of slavery would affect the genetic contribution of African genes into the African American genome. The homozygote wild type variant is necessary for dendritic cell activation and initiation of adaptive immune response. Therefore, the reduced frequency of this allele among Africans might be a probable contributory factor to the susceptibility to infectious pathogens. On the other hand, selective pressure would favor those with adapting mechanisms, leading to them becoming adapted to other types of infections, diseases or conditions. A very good example is the hypothesis that sickle cell disease is highly prevalent in malaria endemic areas because of selection pressure that favors individuals with hemoglobin S, believed to be a contributor to malaria resistance in this group (Gong et al., 2012; Gong et al., 2013). Unfortunately, sub-Saharan Africa is blessed with a geographic and weather pattern that sustains the endemicity of many neglected diseases, and could potentially explain the often-encountered cases of multiple co-morbidities in a single host. This could be a disadvantage in the African continent, whereby immune response is limited, contributing to preponderance of infections. The possibility that these infectious agents might have contributed to the imposition of selection pressure (presenting as an advantage among sickle cell disease cases in Mali) is of potential significance and deserves further analysis. One approach to clarify this would be to generate monocyte-derived dendritic cells from peripheral blood mononuclear cells, collected from healthy controls that are CD209 homozygote wild type and homozygote mutant, and between sickle cell disease groups, and evaluate the differential expression of CD14 (monocyte/macrophage marker) or DC-SIGN (dendritic cell marker). A decrease in CD209 expression in the homozygote mutant variant of DC-SIGN in sickle cell disease patients resulting in lower susceptibility to infectious stimulus, with the reverse the case among control groups with the homozygote wild type variant would be a confirmatory outcome. Similar observation has been reported in dengue virus infection (Sakuntabhai et al., 2005; Wang et al., 2011).
An additional mechanism for our observation could be that there is an increased mortality of sickle cell disease patients that are also carriers of the CD209 ‘G’ allele, especially homozygotes, and are therefore missing from the Malian SCD cohort. Such phenomenon might not be the case in Western industrialized countries, where early sickle cell disease mortality is prevented by antibiotics and other prophylactic measures. An alternative approach to decipher the present observation would be to replicate this study in newborns or children with sickle cell disease recruited during the first year of life, before the expected mortality.
This observation in Africans is enhanced by the reverse observation in the Caucasian population of the United States. The wild type variants (AA, AG) allele is 97% among Caucasians and 83% among African Americans, with the mutant variant almost absent in both groups (3.2% among Caucasians and 16% among African Americans). This low genotypic frequency of the homozygote mutant variant is similar to results from previous reports, which showed 0%, 3% and 5% in a Taiwanese, general Brazilian and Sao Paulo populations respectively (Kashima et al., 2009; Wang et al., 2011; Dettogni et al., 2013). In a study conducted among three groups of healthy control populations in Thailand, a similar scenario was observed, with a genotypic frequency of 5%, 1% and 3% (Sakuntabhai et al., 2005). This observation confirms our hypothesis that this marker may have undergone evolutionary change in extant populations outside of Africa Miller, 1994; Gibbons, 2001; Zimmer, 2001; Thomas et al., 2005. Populations with the homozygote wild type variant are able to fight infections, hence the reduced prevalence of infectious agents, while the reverse may be the case in Africa. Further studies are imperative, before a definitive argument can be made, whereby other infectious diseases are examined viz-a-viz genotypic and allelic diversities of CD209 gene promoter polymorphism in the African population. The ancestral-susceptibility model, which states that disease susceptibility alleles are ancestral while derived variants are protective, has been proposed and validated (Di Rienzo & Hudson, 2005; Biswas & Akey, 2006), further emphasizing that ancestral alleles previously adapted might become maladaptive due to dispersal into new environmental niches (Biswas & Akey, 2006). Extensive reports of geographically restricted selection have been found in genome-wide studies of humans and human diseases (Carlson et al., 2005; Weir et al., 2005; Voight, Wen & Pritchard, 2006; Nakajima et al., 2004; Sakagami et al., 2004; Di Rienzo & Hudson, 2005; Young et al., 2005), and seems clear therefore, that local adaptation in extant populations is a major contributor (Fullerton et al., 2002; Rockman et al., 2004; Thompson et al., 2004).
The lack of differences in genotypic and allelic frequencies of homozygote mutant variants between sickle cell disease and control groups in the United States could be due to the low frequency of the mutant allele (small proportion of individuals with the mutant allele) in the US population. A proposed method to clarify this further would be to study sickle cell disease patients from other regions of the United States, considering the known reports of sub-continental regional population substructure in African American genetic makeup (Kayser et al., 2003; Lao et al., 2010) and different rates of Caucasian gene contribution to the genomic ancestry of African Americans.
Based on our present observation, we conclude that the sickle cell gene (as confirmed for malaria infection) probably confers protection against common infectious co-morbidities in Africa. The higher frequency of CD209 gene promoter homozygote mutants in the non-SCD group reveals an impaired capacity to mount an immune response to infectious diseases, potentially a contributor to the dominance of infectious co-morbidities in this population. The CD209 gene promoter polymorphism might be a major player in susceptibility to common infectious pathogens among Africans, and a contributor to diversity and severity of SCD that requires elucidation, while characterizing genetic risks imposed by locale-specific allele frequencies (Mtatiro et al., 2014). The implication of this finding for infectious co-morbidities or as modifiers of SCD pathophysiology, and its significance in African Americans with SCD deserves further deconvolution. Determining if this protection is regulated in any fashion by sickle cell disease haplotypes in Africa (Benin, Bantu, Cameroon, Senegal) and evaluating plasma levels of immunoglobulin E and eosinophilia, as markers of common helminthic infections, between disease and control groups, is needed.
Finally, it is important to clarify the synergistic or pathogenic role of the sickle cell gene in disparate disease and population groups. This report should be considered preliminary because of sample size limitations, thereby advocating for expansive studies in other population groups, as well as examination of other immunogenetic markers, especially as it relates to clinical endpoints in sickle cell disease. Genetic ancestry studies that might clarify the extent of admixture in the American sickle cell disease group and how this impact our current finding would be imperative. Analyzing American sickle cell disease groups, recruited from different regions (Northeast, Mid-Atlantic, Midwest etc.) under same conditions as this report, would be very important, considering the richness and diversity of the African American gene pool (Collins-Schramm et al., 2002; Kittles et al., 2002; Kayser et al., 2003; Lind et al., 2007).
Supplemental Information
Genomic DNA samples were amplified with standard primers (Dettogni et al., 2013, modified from Sakuntabhai et al.2008); amplified products were digested with MscI restriction endonuclease (New England Biolabs), and expressed on a 2% (w/v) ethidium bromide-stained agarose gel. Homozygous wild type variant showed no digestion (150 bp), while mutant variant produced two bands (131 and 19 bp) on digestion; lower band size not seen. Lane 1: 100 bp TriDye DNA ladder (New England Biolabs); lane 2–5: representative homozygous wild type (snp-336 A/A); lane 6–8: representative heterozygote (snp-336 A/G); lane 9–10: representative homozygous mutant (snp-336 G/G)
{kind=link}
Acknowledgments
We are grateful to Joann Moulds, Grifols USA for the African American and Caucasian control DNA samples. Special gratitude to Betty Pace, Georgia Regents University, who graciously provided the African American sickle cell DNA samples, collected as part of the Cooperative Study of Sickle Cell Disease. Special thanks to Iuri Drummond Louro, Universidad Federal do Espirito Santo, Brazil for technical assistance and Andrew Clark, Cornell University, for critical reading of the manuscript. This work was presented at the Annual Biomedical Research Conference for Minority Students (ABRCMS), San Antonio TX, November 12–15, 2014.
Abbreviations
- SCD
sickle cell disease
- HbAA
hemoglobin AA
- HbSS
hemoglobin SS;
- PCR-RFLP
polymerase chain reaction-restriction fragment length polymorphism
- OR
odds ratio
Funding Statement
College of Health Sciences and Technology, Rochester Institute of Technology provided the funding for this project (Bolaji Thomas). Additional support to Jenelle A Noble was provided by the Rochester Institute of Technology Louis Stokes Alliance for Minority Participation (NSF Award #25901-03200-S01) and the College of Health Sciences and Technology Summer Undergraduate Research Fellowship. Travel awards were supported by the Provost Fund (Kimberely C Duru) and the Federation of American Societies for Experimental Biology Maximizing Access To Research Careers (Jenelle A Noble, Bolaji Thomas). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Additional Information and Declarations
Competing Interests
The authors declare there are no competing interests.
Author Contributions
Jenelle A. Noble and Kimberley C. Duru performed the experiments, reviewed drafts of the paper.
Aldiouma Guindo and Dapa A. Diallo performed the experiments, contributed reagents/materials/analysis tools, reviewed drafts of the paper.
Li Yi and Ikhide G. Imumorin analyzed the data, wrote the paper, reviewed drafts of the paper.
Bolaji N. Thomas conceived and designed the experiments, performed the experiments, analyzed the data, contributed reagents/materials/analysis tools, wrote the paper, prepared figures and/or tables, reviewed drafts of the paper.
Human Ethics
The following information was supplied relating to ethical approvals (i.e., approving body and any reference numbers):
Approval was obtained from the Institutional Review Board, Rochester Institute of Technology (exempt).
References
- Adamkiewicz et al. (2003).Adamkiewicz TV, Sarnaik S, Buchanan GR, Iyer RV, Miller ST, Pegelow CH, Rogers ZR, Vichinsky E, Elliott J, Facklam RR, O’Brien KL, Schwartz B, Van Beneden CA, Cannon MJ, Eckman JR, Keyserling H, Sullivan K, Wong WY, Wang WC. Invasive pneumococcal infections in children with sickle cell disease in the era of penicillin prophylaxis, antibiotic resistance, and 23-valent pneumococcal polysaccharide vaccination. Journal of Pediatrics. 2003;143:438–444. doi: 10.1067/S0022-3476(03)00331-7. [DOI] [PubMed] [Google Scholar]
- Bandeira et al. (2014).Bandeira IC, Rocha LB, Barbosa MC, Elias DB, Querioz JA, Freitas MV, Gonçalves RP. Chronic inflammatory state in sickle cell anemia patients is associated with HBB(*)S haplotype. Cytokine. 2014;65:217–221. doi: 10.1016/j.cyto.2013.10.009. [DOI] [PubMed] [Google Scholar]
- Barreiro, Neyrolles & Babb (2006).Barreiro LB, Neyrolles O, Babb CL, Tailleux L, Quach H, McElreavey K, Van Helden PD, Hoal EG, Gicquel B, Quintana-Murci L. Promoter variation in the DC-SIGN-encoding gene CD209 is associated with tuberculosis. PLoS Medicine. 2006;3:e799. doi: 10.1371/journal.pmed.0030020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benkerrou et al. (2002).Benkerrou M, Delarche C, Brahimi L, Fay M, Vilmer E, Elion J, Gougerot-Pocidalo MA, Elbim C. Hydroxyurea corrects the dysregulated L-selectin expression and increased H2O2 production of polymorphonuclear neutrophils from patients with sickle cell anemia. Blood. 2002;99:2297–2303. doi: 10.1182/blood.V99.7.2297. [DOI] [PubMed] [Google Scholar]
- Biswas & Akey (2006).Biswas S, Akey JM. Genomic insights into positive selection. Trends in Genetics. 2006;22:437–446. doi: 10.1016/j.tig.2006.06.005. [DOI] [PubMed] [Google Scholar]
- Bunn (1997).Bunn HF. Pathogenesis and treatment of sickle cell disease. New England Journal of Medicine. 1997;337:762–769. doi: 10.1056/NEJM199709113371107. [DOI] [PubMed] [Google Scholar]
- Carlson et al. (2005).Carlson CS, Thomas DJ, Eberle MA, Swanson JE, Livingston RJ, Rieder MJ, Nickerson DA. Genomic regions exhibiting positive selection identified from dense genotype data. Genome Research. 2005;15:1553–1565. doi: 10.1101/gr.4326505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charache, Bleecker & Bross (1983).Charache S, Bleecker ER, Bross DS. Effects of blood transfusion on exercise capacity in patients with sickle-cell anemia. American Journal of Medicine. 1983;74:757–764. doi: 10.1016/0002-9343(83)91063-X. [DOI] [PubMed] [Google Scholar]
- Collins-Schramm et al. (2002).Collins-Schramm H, Phillips C, Operario D, Lee JL, Hanson R, Knowler W, Cooper R, Li H, Seldin M. Ethnic-difference markers for use in mapping by admixture linkage disequilibrium. American Journal of Human Genetics. 2002;70:737–750. doi: 10.1086/339368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conran, Franco-Penteado & Costa (2009).Conran N, Franco-Penteado CF, Costa FF. Newer aspects of the pathophysiology of sickle cell disease vaso-occlusion. Hemoglobin. 2009;33:1–16. doi: 10.1080/03630260802625709. [DOI] [PubMed] [Google Scholar]
- Covas et al. (2007).Covas DT, de Oliveira FS, Rodrigues ES, Abe-Sandes K, Silva WA, Jr, Fontes AM. Knops blood group haplotypes among distinct Brazilian populations. Transfusion. 2007;47:147–153. doi: 10.1111/j.1537-2995.2007.01077.x. [DOI] [PubMed] [Google Scholar]
- Dettogni et al. (2013).Dettogni RS, Sa RT, Tovar TT, Louro ID. Polymorphic genetic variation in immune system genes: a study of two populations of Espirito Santo, Brazil. Molecular Biology Reports. 2013;40:4843–4849. doi: 10.1007/s11033-013-2582-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Rienzo & Hudson (2005).Di Rienzo A, Hudson RR. An evolutionary framework for common diseases: the ancestral-susceptibility model. Trends in Genetics. 2005;21:596–601. doi: 10.1016/j.tig.2005.08.007. [DOI] [PubMed] [Google Scholar]
- Di Rienzo & Hudson (2006).Di Rienzo A, Hudson RR. Genomic insights into positive selection. Trends in Genetics. 2006;22:437–446. doi: 10.1016/j.tig.2006.06.005. [DOI] [PubMed] [Google Scholar]
- Fullerton et al. (2002).Fullerton SM, Bartoszewicz A, Ybazeta G, Horikawa Y, Bell GI, Kidd KK, Cox NJ, Hudson RR, Di Rienzo A. Geographic and haplotype structure of candidate type 2 diabetes susceptibility variants at the calpain-10 locus. American Journal of Human Genetics. 2002;70:1096–1106. doi: 10.1086/339930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons (2001).Gibbons A. Tools show humans reach Asia early. Science. 2001;293:2368–2369. doi: 10.1126/science.293.5539.2368b. [DOI] [PubMed] [Google Scholar]
- Gong et al. (2012).Gong L, Maiteki-Sebuguzi C, Rosenthal PJ, Hubbard AE, Drakeley CJ, Dorsey G, Greenhouse B. Evidence for both innate and acquired mechanisms of protection from Plasmodium falciparum in children with sickle cell trait. Blood. 2012;119:3808–3814. doi: 10.1182/blood-2011-08-371062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong et al. (2013).Gong L, Parikh S, Rosenthal PJ, Greenhouse B. Biochemical and immunological mechanisms by which sickle cell trait protects against malaria. Malaria Journal. 2013;12:317. doi: 10.1186/1475-2875-12-317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halasa et al. (2007).Halasa NB, Shankar SM, Talbot TR, Arbogast PG, Mitchel EF, Wang WC, Schaffner W, Craig AS, Griffin MR. Incidence of invasive pneumococcal disease among individuals with sickle cell disease before and after the introduction of the pneumococcal conjugate vaccine. Clinical Infectious Diseases. 2007;44:1428–1433. doi: 10.1086/516781. [DOI] [PubMed] [Google Scholar]
- Hansson et al. (2013).Hansson HH, Kurtzhals JA, Goka BQ, Rodriques OP, Nkrumah FN, Theander TG, Bygbjerg IC, Alifrangis M. Human genetic polymorphisms in the Knops blood group are not associated with a protective advantage against Plasmodium falciparum malaria in Southern Ghana. Malaria Journal. 2013;12:400. doi: 10.1186/1475-2875-12-400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassell (2010).Hassell KL. Populations estimates of sickle cell disease in the United States. American Journal of Preventive Medicine. 2010;38:S512–S521. doi: 10.1016/j.amepre.2009.12.022. [DOI] [PubMed] [Google Scholar]
- Jallow et al. (2009).Jallow M, Teo YY, Small KS, Rockett KA, Deloukas P, Clark TG, Kivinen K, Bojang KA, Conway DJ, Pinder M, Sirugo G, Sisay-Joof F, Usen S, Auburn S, Bumpstead SJ, Campino S, Coffey A, Dunham A, Fry AE, Green A, Gwilliam R, Hunt SE, Inouye M, Jeffreys AE, Mendy A, Palotie A, Potter S, Ragoussis J, Rogers J, Rowlands K. Genome-wide and fine-resolution association analysis of malaria in West Africa. Nature Genetics. 2009;41:657–665. doi: 10.1038/ng.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashima et al. (2009).Kashima S, Rodrigues ES, Azevedo R, da Cruz Castelli E, Mendes-Junior CT, Yoshioka FK, da Silva IT, Takayanagui OM, Covas DT. DC-SIGN (CD209) gene promoter polymorphisms in a Brazilian population and their association with human T-cell lymphotropic virus type 1 infection. Journal of General Virology. 2009;90:927–934. doi: 10.1099/vir.0.008367-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayser et al. (2003).Kayser M, Brauer S, Schädlich H, Prinz M, Batzer MA, Zimmerman PA, Boatin BA, Stoneking M. Y chromosome STR haplotypes and the genetic structure of US populations of African, European and Hispanic ancestry. Genome Research. 2003;13:624–634. doi: 10.1101/gr.463003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittles et al. (2002).Kittles RA, Chen W, Panguluri RK, Ahaghotu C, Jackson A, Adebamowo CA, Griffin R, Williams T, Ukoli F, Adams-Campbell L, Kwagyan J, Isaacs W, Freeman V, Dunston GM. CYP3A4-V and prostate cancer in African Americans: causal or confounding association because of population stratification? Human Genetics. 2002;110:553–560. doi: 10.1007/s00439-002-0731-5. [DOI] [PubMed] [Google Scholar]
- Kreuels et al. (2010).Kreuels B, Kreuzberg C, Kobbe R, Ayim-Akonor M, Apiah-Thompson P, Thompson B, Ehmen C, Adjei S, Langefeld I, Adjei O, May J. Differing effects of HbS and HbC traits on uncomplicated falciparum malaria, anemia, and child growth. Blood. 2010;115:4551–4558. doi: 10.1182/blood-2009-09-241844. [DOI] [PubMed] [Google Scholar]
- Lao et al. (2010).Lao O, Vallone PM, Coble MD, Diegoli TM, Van Oven M, Van der Gaag J, Pijpe J, de Knijff P, Kayser M. Evaluating self-declared ancestry of US American with autosomal, Y-chemosomal and mitochondrial DNA. Human Mutation. 2010;31:E1875–E1893. doi: 10.1002/humu.21366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lind et al. (2007).Lind JM, Hutcheson HB, Williams SM, Moore JH, Essex M, Ruiz-Pesini E, Wallace DC, Tishkoff SA, O’Brien SJ, Smith MW. Elevated make European and female African contributions to the genomes of African American individuals. Human Genetics. 2007;120:713–722. doi: 10.1007/s00439-006-0261-7. [DOI] [PubMed] [Google Scholar]
- Makis, Hatzimichael & Bourantas (2000).Makis AC, Hatzimichael EC, Bourantas KL. The role of cytokines in sickle cell disease. Annals of Hematology. 2000;79:407–413. doi: 10.1007/s002770000173. [DOI] [PubMed] [Google Scholar]
- Martin et al. (2004).Martin MP, Lederman MM, Hutcheson HB, Goedert JJ, Nelson GW, van Kooyk Y, Detels R, Buchbinder S, Hoots K, Vlahov D, O’Brien SJ, Carrington M. Association of DC-SIGN promoter polymorphism with increased risk for parenteral, but not mucosal, acquisition of human immunodeficiency virus type 1 infection. Journal of Virology. 2004;78:14053–14056. doi: 10.1128/JVI.78.24.14053-14056.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCavit et al. (2011).McCavit TL, Quinn CT, Techasaensiri C, Rogers ZR. Increase in invasive Streptococcus pneumoniae infections in children with sickle cell disease since pneumococcal conjugate vaccine licensure. Journal of Pediatrics. 2011;158:505–507. doi: 10.1016/j.jpeds.2010.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller (1994).Miller LH. Impact of malaria on genetic polymorphisms and genetic diseases in Africans and African-Americans. Proceedings of the National Academy of Science of the United States of America. 1994;91:2415–2419. doi: 10.1073/pnas.91.7.2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mtatiro et al. (2014).Mtatiro SN, Singh T, Rooks H, Mgaya J, Mariki H, Soka D, Mmbando B, Msaki E, Kolder I, Thein SL, Menzel S, Cox SE, Makani J, Barrett JC. Genome wide association study of fetal hemoglobin in sickle cell anemia in Tanzania. PLoS ONE. 2014;9:e799. doi: 10.1371/journal.pone.0111464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mummidi et al. (2001).Mummidi S, Catano G, Lam L, Hoefle A, Telles V, Begum K, Jimenez F, Ahuja SS, Ahuja AK. Extensive repertoire of membrane-bound and soluble dendritic cell-specific ICAM-3-grabbing nonintegrin 1 (DC-SIGN1) and DC-SIGN2 isoforms. Inter-individual variation in expression of DC-SIGN transcripts. Journal of Biological Chemistry. 2001;276:196–212. doi: 10.1074/jbc.M009807200. [DOI] [PubMed] [Google Scholar]
- Nakajima et al. (2004).Nakajima T, Woodling S, Sakagami T, Emi M, Tokunaga K, Tamiya G, Ishigami T, Umemura S, Munkhbat B, Jin F, Guan-Jun J, Hayasaka I, Ishida T, Saitou N, Pavelka K, Lalouel JM, Jorde LB, Inoue I. Natural selection and population history in the human angiotensinogen gene (AGT): 736 complete ATG sequences in chromosomes from around the world. American Journal of Human Genetics. 2004;74:898–916. doi: 10.1086/420793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishank et al. (2013).Nishank SS, Singh MP, Yadav R, Gupta RB, Gadge VS, Gwal A. Endothelial nitric oxide synthase gene polymorphism is associated with sickle cell disease patients in India. Journal of Human Genetics. 2013;58:775–779. doi: 10.1038/jhg.2013.99. [DOI] [PubMed] [Google Scholar]
- Noumsi et al. (2011).Noumsi GT, Tounkara A, Diallo H, Billingsley K, Moulds JJ, Moulds JM. Knops blood group polymorphism and susceptibility to Mycobacterium tuberculosis infection. Transfusion. 2011;51:2462–2469. doi: 10.1111/j.1537-2995.2011.03161.x. [DOI] [PubMed] [Google Scholar]
- Overturf (1999).Overturf GD. Infections and immunizations of children with sickle cell disease. Advances in Pediatrics Infectious Diseases. 1999;14:191–218. [PubMed] [Google Scholar]
- Pathare et al. (2004).Pathare A, Al Kindi S, Alnaqdy AA, Daar S, Knox-Macaulay H, Dennison D. Cytokine profile of sickle cell disease in Oman. American Journal of Hematology. 2004;77:323–328. doi: 10.1002/ajh.20196. [DOI] [PubMed] [Google Scholar]
- Petz et al. (1997).Petz LD, Calhoun L, Shulman IA, Johnson C, Herron RM. The sickle cell hemolytic transfusion reaction syndrome. Transfusion. 1997;37:382–392. doi: 10.1046/j.1537-2995.1997.37497265338.x. [DOI] [PubMed] [Google Scholar]
- Piel et al. (2013).Piel FB, Hay SI, Gupta S, Weatherall DJ, Williams TN. Global burden of sickle cell anemia in children under five, 2010–2050: modeling based on demographics, excess mortality, and interventions. PLoS Medicine. 2013;10:e799. doi: 10.1371/journal.pmed.1001484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piomelli (1985).Piomelli S. Chronic transfusions in patients with sickle cell disease. Indications and problems. American Journal of Pediatric Hematology/Oncology. 1985;7:51–55. [PubMed] [Google Scholar]
- Qari, Dier & Mousa (2012).Qari MH, Dier U, Mousa SA. Biomarkers of inflammation, growth factor, and coagulation activation in patients with sickle cell disease. Clinical and Applied Thrombosis/Hemostasis. 2012;18:195–200. doi: 10.1177/1076029611420992. [DOI] [PubMed] [Google Scholar]
- Rappocciolo, Jenkins & Hensler (2006).Rappocciolo G, Jenkins FJ, Hensler HR, Piazza P, Jais M, Borowski L, Watkins SC, Rinaldo CR. DCSIGN is a receptor for human herpes virus 8 on dendritic cells and macrophages. Journal of Immunology. 2006;176:1741–1749. doi: 10.4049/jimmunol.176.3.1741. [DOI] [PubMed] [Google Scholar]
- Rockman et al. (2004).Rockman MV, Hahn MW, Soranzo N, Loisel DA, Goldstein DB, Wray GA. Positive selection on MMP3 regulation has shaped heart disease risk. Current Biology. 2004;14:1531–1539. doi: 10.1016/j.cub.2004.08.051. [DOI] [PubMed] [Google Scholar]
- Rosse, Gallagher & Kinney (1990).Rosse WF, Gallagher D, Kinney TR, Castro O, Dosik H, Moohr J, Wang W, Levy PS. Transfusion and alloimmunization in sickle cell disease. Blood. 1990;76:1431–1437. [PubMed] [Google Scholar]
- Sakagami et al. (2004).Sakagami T, Witherspoon DJ, Nakajima T, Jinnai N, Wooding S, Jorde LB, Hasegawa T, Suzuki E, Gejyo F, Inoue I. Local adaptation and population differentiation at the interleukin 13 and interleukin 4 loci. Genes and Immunity. 2004;5:389–397. doi: 10.1038/sj.gene.6364109. [DOI] [PubMed] [Google Scholar]
- Sakuntabhai et al. (2005).Sakuntabhai A, Turbpaiboon C, Casadémont I, Chuansumrit A, Lowhnoo T, Kajaste-Rudnitski A, Kalayanarooj SM, Tangnararatchakit K, Tangthawornchaikul N, Vasanawathana S, Chaiyaratana W, Yenchitsomanus PT, Suriyaphol P, Avirutnan P, Chokephaibulkit K, Matsuda F, Yoksan S, Jacob Y, Lathrop GM, Malasit P, Desprès P, Julier C. A variant in the CD209 promoter is associated with severity of dengue disease. Nature Genetics. 2005;37:507–513. doi: 10.1038/ng1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraf et al. (2014).Saraf SL, Zhang X, Kanias T, Lash JP, Molokie RE, Oza B, Lai C, Rowe JH, Gowhari M, Hassan J, Desimone J, Machado RF, Gladwin MT, Little JA, Gordeuk VR. Haemoglobinuria is associated with chronic kidney disease and its progression in patients with sickle cell anemia. British Journal of Haemtology. 2014;164:729–739. doi: 10.1111/bjh.12690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg (2006).Steinberg MH. Pathophysiologically based drug treatment of sickle cell disease. Trends in Pharmacological Sciences. 2006;27:204–210. doi: 10.1016/j.tips.2006.02.007. [DOI] [PubMed] [Google Scholar]
- Szczepanek et al. (2013).Szczepanek SM, Secor ER, Jr, Bracken SJ, Guernsey L, Rafti E, Matson A, Thrall RS, Andemariam B. Transgenic sickle cell disease mice have high mortality and dysregulated immune responses after vaccination. Pediatric Research. 2013;74:141–147. doi: 10.1038/pr.2013.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tailleux, Schwartz & Herrmann (2003).Tailleux L, Schwartz O, Herrmann J-L, Pivert E, Jackson M, Amara A, Legres L, Dreher D, Nicod LP, Gluckman JC, Lagrange PH, Gicquel B, Neyrolles O. DC-SIGN is the major Mycobacterium tuberculosis receptor on human dendritic cells. Journal of Experimetnal Medicine. 2003;197:121–127. doi: 10.1084/jem.20021468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassaneetrithep, Burgess & Granelli-Piperno (2003).Tassaneetrithep B, Burgess TH, Granelli-Piperno A, Trumpfheller C, Finke J, Sun W, Eller MA, Pattanapanyasat K, Sarasombath S, Birx DL, Steinman RM, Schlesinger S, Marovich MA. DC-SIGN (CD209) mediates dengue virus infection of human dendritic cells. Journal of Experimetnal Medicine. 2003;197:823–829. doi: 10.1084/jem.20021840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatari-Calderone et al. (2013).Tatari-Calderone Z, Tamouza R, Le Bouder GP, Dewan R, Luban NLC, Lasserre J, Maury J, Lionnet F, Krishnamoorthy R, Girot R, Vukmanovic S. The association of CD81 polymorphisms with alloimmunization in sickle cell disease. Clinical and Developmental Immunology. 2013;2013:1–9. doi: 10.1155/2013/937846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor et al. (2008).Taylor JG, 6th, Nolan VG, Mendelsohn L, Kato GJ, Gladwin MT, Steinberg MH. Chronic hyper-hemolysis in sickle cell anemia: association of vascular complications and mortality with less frequent vasoocclusive pain. PLos ONE. 2008;3:e799. doi: 10.1371/journal.pone.0002095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, Parobek & Fairhurst (2012).Taylor SM, Parobek CM, Fairhurst RM. Hemoglobinopathies and the clinical epidemiology of malaria: a systematic review and meta-analysis. The Lancet Infectious Diseases. 2012;12:457–468. doi: 10.1016/S1473-3099(12)70055-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur et al. (2014).Thakur TJ, Guindo A, Cullifer LR, Li Y, Imumorin IG, Diallo DA, Thomas BN. Endothelin-1 but not endothelial nitric oxide synthase gene polymorphism is associated with sickle cell disease in Africa. Gene Regulation and Systems Biology. 2014;8:119–126. doi: 10.4137/GRSB.S14836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas et al. (2012a).Thomas BN, Diallo DA, Noumsi GT, Moulds JM. Circulating immune complex levels are associated with disease severity and seasonality in children with malaria from Mali. Biomarker Insights. 2012a;7:81–86. doi: 10.4137/BMI.S9624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas et al. (2005).Thomas BN, Donvito B, Cockburn I, Fandeur T, Rowe JA, Cohen JHM, Moulds JM. A complement receptor-1 polymorphisms with high frequency in malaria endemic regions of Asia but not Africa. Genes and Immunity. 2005;6:31–36. doi: 10.1038/sj.gene.6364150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas et al. (2012b).Thomas BN, Petrella CR, Thakur TJ, Crespo SR, Diallo DA. Genetic polymorphism of Plasmodium falciparum merozoite surface protein-1 and 2 and diversity of drug resistance genes in blood donors from Bamako, Mali. Infectious Diseases: Research and Treatment. 2012b;6:49–57. doi: 10.4137/IDRT.S10094. [DOI] [Google Scholar]
- Thomas et al. (2013).Thomas BN, Thakur TJ, Yi L, Guindo A, Diallo DA, Ott JG. Extensive ethnogenomic diversity of endothelial nitric oxide synthase (eNOS) polymorphisms. Gene Regulation and Systems Biology. 2013;7:1–10. doi: 10.4137/GRSB.S10857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson et al. (2004).Thompson EE, Kuttab-Boulos H, Witonskky D, Yang L, Roe BA, Di Rienzo A. CYP3A variation and the evolution of salt-sensitivity variants. American Journal of Human Genetics. 2004;75:1059–1069. doi: 10.1086/426406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Kooyk, Appelmelk & Geijtenbeek (2003).Van Kooyk Y, Appelmelk B, Geijtenbeek TB. A fatal attraction: mycobacterium tuberculosis and HIV-1 target DC-SIGN to escape immune surveillance. Trends in Molecular Medicine. 2003;9:153–159. doi: 10.1016/S1471-4914(03)00027-3. [DOI] [PubMed] [Google Scholar]
- Voight, Wen & Pritchard (2006).Voight BF, Kudaravalli S, Wen X, Pritchard JK. A map of recent positive selection in the human genome. PLoS Biology. 2006;4:e799. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang et al. (2011).Wang L, Chen RF, Liu JW, Lee IK, Lee CP, Kuo HC, Huang SK, Yang KD. DC-SIGN (CD209) Promoter-336 A/G polymorphism is associated with dengue hemorrhagic fever and correlated to DC-SIGN expression and immune augmentation. PLoS Neglected Tropical Diseases. 2011;5:e799. doi: 10.1371/journal.pntd.0000934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weatherall (2010).Weatherall DJ. The inherited diseases of hemoglobin are an emerging global health burden. Blood. 2010;115:4331–4336. doi: 10.1182/blood-2010-01-251348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir et al. (2005).Weir BS, Cardon LR, Anderson AD, Nielsen DM, Hill WG. Measures of human population structure show heterogeneity among genomic regions. Genome Research. 2005;15:1468–1476. doi: 10.1101/gr.4398405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams et al. (2005).Williams TN, Mwangi TW, Roberts DJ, Alexander ND, Weatherall DJ, Wambua S, Kortok M, Snow RW, Marsh K. An immune basis for malaria protection by the sickle cell trait. PLoS Medicine. 2005;2:e799. doi: 10.1371/journal.pmed.0020128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazdanbakhsh, Ware & Noizat-Pirenne (2012).Yazdanbakhsh K, Ware RE, Noizat-Pirenne F. Red blood cell alloimmunization in sickle cell disease: pathophysiology, risk factors, and transfusion management. Blood. 2012;120:528–537. doi: 10.1182/blood-2011-11-327361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young et al. (2005).Young JH, Hang YP, Kim JD, Chretien JP, Klag MJ, Levine MA, Ruff CB, Wang NY, Chakravarti A. Differential susceptibility to hypertension is due to selection during the out-of-Africa Expansion. PLoS Genetics. 2005;1:e799. doi: 10.1371/journal.pgen.0010082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer (2001).Zimmer C. Genetic trees reveal disease origins. Science. 2001;292:1090–1093. doi: 10.1126/science.292.5519.1090. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Genomic DNA samples were amplified with standard primers (Dettogni et al., 2013, modified from Sakuntabhai et al.2008); amplified products were digested with MscI restriction endonuclease (New England Biolabs), and expressed on a 2% (w/v) ethidium bromide-stained agarose gel. Homozygous wild type variant showed no digestion (150 bp), while mutant variant produced two bands (131 and 19 bp) on digestion; lower band size not seen. Lane 1: 100 bp TriDye DNA ladder (New England Biolabs); lane 2–5: representative homozygous wild type (snp-336 A/A); lane 6–8: representative heterozygote (snp-336 A/G); lane 9–10: representative homozygous mutant (snp-336 G/G)