

Abstract
Class IIa histone deacetylases (HDACs4, -5, -7, and -9) modulate the physiology of the human cardiovascular, musculoskeletal, nervous, and immune systems. The regulatory capacity of this family of enzymes stems from their ability to shuttle between nuclear and cytoplasmic compartments in response to signal-driven post-translational modification. Here, we review the current knowledge of modifications that control spatial and temporal histone deacetylase functions by regulating subcellular localization, transcriptional functions, and cell cycle-dependent activity, ultimately impacting on human disease. We discuss the contribution of these modifications to cardiac and vascular hypertrophy, myoblast differentiation, neuronal cell survival, and neurodegenerative disorders.
The enzymatic addition of acetyl moieties to lysine residues and the removal of these modifications via deacetylation were first documented over forty years ago (1, 2). As histones were the first substrates shown to be modified by acetylation (2), the enzymes responsible for adding and removing acetylations are commonly known as histone acetyltransferases and histone deacetylases (HDACs)1, respectively. HDACs have been shown to regulate a multitude of cellular processes, including cell cycle progression, differentiation, and apoptosis (3–5). In particular, HDACs have been intensively studied for their abilities to control gene transcription and to manipulate the epigenetic status of cells (6). HDACs are generally thought to promote transcriptional repression and gene silencing (7–9); however, nonhistone proteins are also targeted by these enzymes, and include cell receptors, chaperones, and cytoskeletal proteins (10). To date, 18 human HDACs have been identified and categorized into four classes based on homology to yeast deacetylases (11). Class I (HDAC1, -2, -3, and -8) are related to Rpd3, class IIa (HDAC4, -5, -7, and -9) and class IIb (HDAC6 and 10) are similar to Hda1, and class IV (HDAC11) has homology to both Rpd3 and Hda1 (11). These HDAC classes are zinc-dependent enzymes, in contrast to class III HDACs, commonly known as sirtuins (SIRT1–7), which require the cofactor NAD+ for activity (12, 13).
In recent years, the class IIa HDACs have been established as critical regulators of numerous cellular processes (14, 15), with misregulation being associated with a wide range of human diseases. Roles for class IIa HDACs have been reported in the development of cardiovascular disease (16), cancer (17), immune response to viral infection (18), epigenetic response to drug stimulus (19), diabetes (20), and neurodegenerative diseases, such as Huntington's disease (21). Intriguingly, class IIa HDACs have been shown to limit intrinsic enzymatic activity. Their repressive activity is proposed to depend largely on their association with class I enzymes, in particular HDAC3, and on their ability to repress transcription factors in a deacetylation-independent manner. Structurally, the limited activity of class IIa HDACs is the result of an amino acid substitution (His) at the catalytic Tyr present in the DAC domains of other eukaryotic and prokaryotic deacetylases. Indeed, His-to-Tyr mutation of HDAC4 resulted in elevated (1000×) enzymatic activity compared with wild type (22). Additionally, the identification of a zinc binding region, containing a CCHC motif, within class IIa HDACs suggests the possibility that this and other regions could be important for substrate recognition, binding, and regulation of HDAC catalytic activity because of proximity to the active site (23). However, in contrast to the better understanding of class I HDAC regulation and functions (24), the mechanisms governing class IIa HDAC functions in health and disease states still remain less well understood.
It has recently become increasingly clear that class IIa HDAC functions are significantly regulated by post-translational modifications (PTMs), including phosphorylations, acetylations, sumoylations, and ubiquitinations (25). PTMs provide important dynamic points of regulation for determining protein structure, protein interactions, and protein localizations, and additionally, in the case of HDACs, their transcriptional repressive functions (26–28). Given the recent expansion in the knowledge of numerous PTMs on class IIa HDACs, an ongoing challenge is to collate this meaningful information and identify key residues that elicit precise physiological functions. This review will first construct a comprehensive map of site-specific class IIa HDAC PTMs and the enzymes that regulate them. The functional significance of individual PTMs will then be discussed relative to their intracellular roles and their contributions to disease progression.
Site-specific Post-translational Modifications of Class IIa HDACs
Diversification of the human proteome can be achieved at the post-transcriptional level through mRNA splicing (29) and post-translational modification of polypeptides (30). A protein can be “decorated” with such chemical modifications that alter its activity and functions. For class IIa HDACs, the functions of these specific PTMs are critically associated with their location within the protein domains. HDAC4, -5, -7, and -9 possess a definitive bipartite structure (31). The C terminus contains a conserved deacetylase domain and a hydrophobic nuclear export sequence (NES), which promotes cytoplasmic accumulation (32) (Fig. 1). The N-terminal region, or adaptor domain, contains a lysine/arginine-rich nuclear localization sequence (NLS) and binding sites for transcription factors, such as the myocyte enhancer factor-2 (MEF2) (33, 34), and chaperone proteins (35–37). These domains serve as platforms for the formation of higher order protein complexes, such as by interactions with the C-terminal-binding protein and heterochromatin P1, through which HDACs perform specific enzymatic and functional activities (31). HDAC association with chaperone proteins enables their nuclear export and relieves repression of HDAC target genes (35–37) (discussed in “PTMs Regulate HDAC Subcellular Localization and Orchestrate Transcription”). This section will review the current knowledge of site-specific modifications, as integrated from comprehensive literature searches and from multiple databases, highlighting their functional relevance and regulation by distinct enzymes (Fig. 1).
Fig. 1.
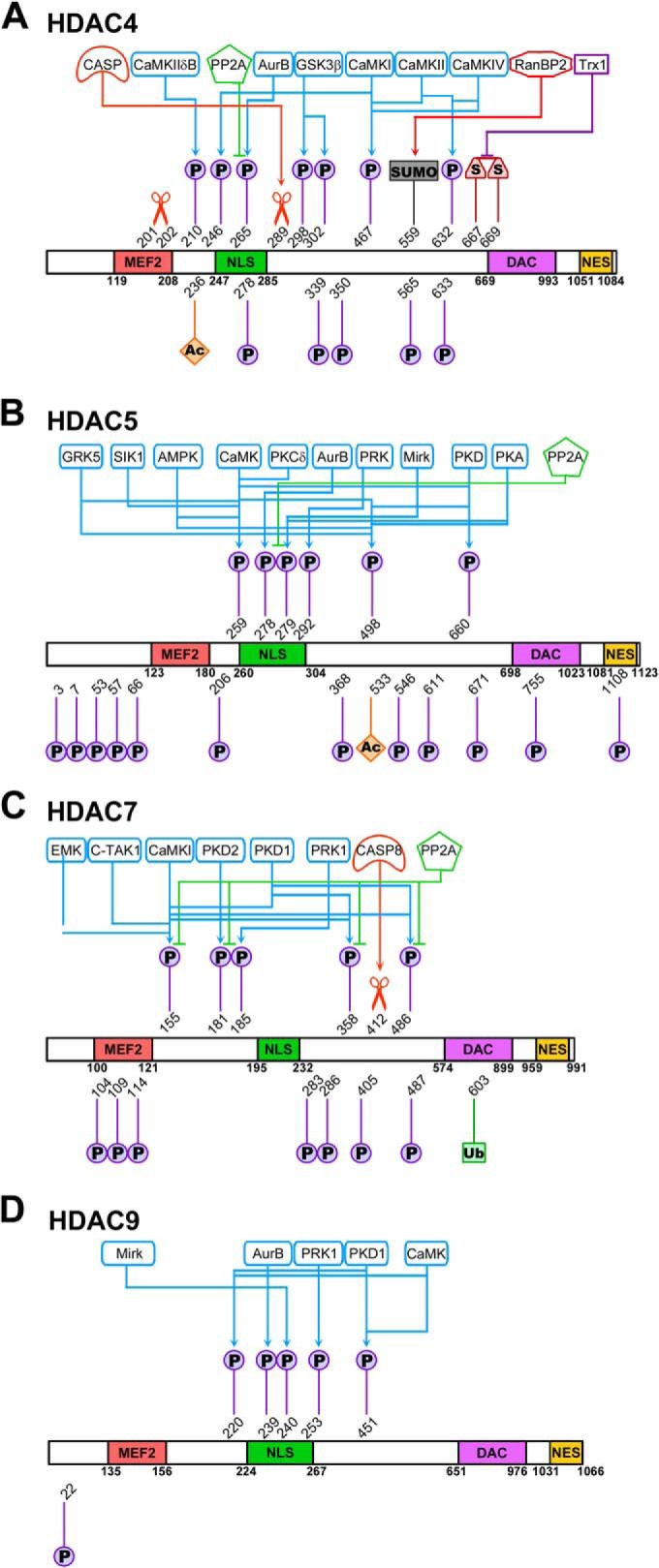
Numerous signaling pathways converge to post-translationally modify class IIa HDACs. Class IIa HDACs are modified by phosphorylation, sumoylation, disulfide bridges, acetylation, ubiquitinylation, and are cleaved by caspases. Sites with enzymes assigned to their modification are shown at the top of each protein schematic; uncharacterized sites are shown at the bottom. Functional domains of class IIa HDACs are highlighted: MEF, MEF2 binding domain; NLS, nuclear localization signal; DAC, deacetylation domain; NES, nuclear export sequence. A, HDAC4, B, HDAC5, C, HDAC7, D, HDAC9.
HDAC4
HDAC4 is the most well-characterized member of the class IIa family in terms of the number of identified PTMs, which include phosphorylation, acetylation, sumoylation, and disulfide bond formation (Fig. 1A). Furthermore, many of the enzymes regulating these modifications have been characterized. The numerous phosphorylations on HDAC4 provide a platform for finely tuned control of protein functions, as each modification can exert a function as a single signal, or can act in tandem with other modifications (supplemental Table S1). Phosphorylations at S246, S467, and S632 initiate binding of 14-3-3 chaperone proteins that promote shuttling of HDAC4 to the cytoplasm (35, 37). As HDAC localization is tightly connected to transcriptional regulatory function, loss of phosphorylation enhances MEF2 expression by spatially excluding HDAC4 from the nucleus rather than by limiting its intrinsic enzymatic activity (35, 37).
Several isoforms of the calcium/calmodulin-dependent kinase (CaMK) family phosphorylate HDAC4 (38–46) and regulate its ability to repress target genes in the nucleus (supplemental Table S1), suggesting a high degree of regulatory specificity with respect to individual sites. CaMKI preferentially phosphorylates S246 on HDAC4, having only limited activity toward S467 and S632 (40). The major regulatory kinase of S467 is CaMKII, which is also able to efficiently phosphorylate S632 (40) (Fig. 1A). S467 and S632 are also known to be phosphorylated by CaMKIV and can promote nuclear export by a mechanism independent of 14–3-3 binding (39). CaMKIIδB is also known to preferentially target S210 in cardiac cells (41), and this modification is associated with heart disease (see “The Post-translational Regulation of HDACs is Critically Linked to Human Disease”).
Apart from the CaMK superfamily, a diverse set of enzymes is involved in regulating HDAC4 modifications. Although the regulation of some sites remains unknown, the S265 and S266 NLS sites were shown to be phosphorylated by Aurora B and Mirk/dyrk1B, respectively (47, 48). S298 and S302 phosphorylation status are both under the control of the glycogen synthase kinase 3β (GSK3β), and the phospho-mimetic S298D mutation promotes polyubiquitination of HDAC4, suggesting a link between phosphorylation and ubiquitin PTMs (49). The proteins and pathways involved in the dephosphorylation of HDAC4 have also been investigated. Several studies have shown that the protein phosphatase 2A (PP2A) family can remove HDAC4 phosphorylation, promoting its nuclear accumulation (50–53).
Additional PTMs modulate the subcellular localization of HDAC4 independently of its phosphorylation status. Hypertrophic stimuli promote the oxidation of C667 and C669 and the formation of a disulfide bond that signals HDAC4 nuclear export (54). Conversely, sumoylation of HDAC4 by the E3 ligase RanBP2, which covalently attaches SUMO-1 to HDAC4 at K559, promotes nuclear retention (55). Caspase-mediated cleavage can also enhance HDAC4 nuclear accumulation. caspase 2- and caspase 3-dependent cleavage of HDAC4 at D289 generates an NLS-containing protein fragment that represses transcription factors and activates apoptotic pathways (56). HDAC4 cleavage is also linked to protein kinase A (PKA)-mediated processing that leads to the repression of MEF2 activity (57). Cleavage occurs between T201 and W202 to release a ∼28 kDa N-terminal fragment; however, as no PKA phosphorylation sites were determined for HDAC4, the association between PKA, HDAC4, and the protease requires further investigation (57).
HDAC5
Although numerous HDAC5 PTMs have been identified in recent years, fewer of these modifications have been functionally characterized. Those that have been investigated are clustered within and around the NLS (Fig. 1B, top). However, proteomic investigation of HDAC5 has also revealed extensive PTMs throughout the protein sequence, including within the DAC and NES domains (28) (Fig. 1B, bottom). Through a mechanism conserved across all class IIa HDACs, critical HDAC5 residues (S259 and S498) promote 14–3-3 chaperone protein binding (27, 36) and nuclear export upon phosphorylation by CaMK-I, -II, and -IV (27, 43, 58, 59). Distinct from analogous HDAC4 sites, these key residues are known to be phosphorylated by additional kinases (Fig. 1B, supplemental Table S1). Regulation of HDAC5 by protein kinase D (PKD) and its various isoforms has been widely reported (e.g. (60–65)). PKD also phosphorylates S660, an additional 14–3-3 binding site (66). PKC, an upstream regulator of PKD, can also phosphorylate S259 directly (67), whereas PKA can modify S498 (66). PKC-related kinases 1 and 2 (PRK1 and PRK2) also target T292 (68), highlighting the PKC signaling pathway as an important determinant of HDAC5 phosphorylation status. Other enzymes that target both S259 and S498 are AMP-activated protein kinase (69, 70), G protein-coupled receptor kinase-5 (GRK5) (71), whereas salt-inducible kinase 1 (SIK1) phosphorylates only S259 (72).
Recent investigation of phosphorylations within the NLS has uncovered new regulatory features of HDAC5. S279 phosphorylation has been shown to be critical for the nuclear HDAC5 localization (28) (discussed in “PTMs Regulate HDAC Subcellular Localization and Orchestrate Transcription”) (supplemental Table S1). Mirk/dyrk1B (48), PKA (73), and Cdk5 (74) have all been shown to phosphorylate S279, whereas PP2A has been reported to dephosphorylate it (74). Interestingly, the phosphorylation of the immediately adjacent NLS serine residue, S278, was shown to have a distinct spatial and temporal regulation, being controlled by Aurora B in a cell cycle-dependent manner (47). NLS phosphorylation by Aurora B is conserved in both HDAC4 and HDAC9.
HDAC7
Readers should note that HDAC7 amino acid numbering has changed considerably over the years as sequence information has been updated. To limit ambiguities, the sites we discuss are consistent with those reported in the PhosphoSitePlus database (Fig. 1C); please note that in some studies sites 179, 344, and 479 correspond to 155, 358, and 486 (supplemental Table S1). Similar to other class IIa HDACs, phosphorylation regulates the interaction between HDAC7 and chaperone proteins and protein stability (75). The HDAC7 residues critical for these processes are S181, S155, S358, and S486 (76). The latter three sites can be modified by CaMKI (77, 78), whereas PKD can phosphorylate all four residues (76, 79–83). In addition, hPar-1 kinases EMK and C-TAK1 phosphorylate S155 (84), and PRK1 targets S185 (68). The modification of individual sites by multiple distinct kinases demonstrates that phosphorylation is a finely tuned system for regulation of HDAC function. Furthermore, the ability of a diverse set of kinases to regulate the same sites may reflect an evolutionary flexibility that allows differential modulation of critical functional sites. Interestingly, dephosphorylation of the four 14–3-3 binding sites in HDAC7 is mediated by PP2A, which shows no preference for one site over the others (85). Although the precise sites of action remain unknown, protein phosphatase 1β and myosin phosphatase targeting subunit 1 promote nuclear accumulation of HDAC7 (86). Like HDAC4, the stability of HDAC7 can be mediated though caspase-dependent cleavage. Caspase-8 cleaves HDAC7 at D412 (87), generating an unstable N-terminal fragment that resides in the nucleus and a C-terminal fragment that is excluded from the nucleus (88).
HDAC9
HDAC9 remains the least well-characterized class IIa HDAC in terms of known PTMs and modifying enzymes (Fig. 1D). Information about HDAC9 has been derived based on its high degree of homology to the other class IIa family members. Prior to the cloning of HDAC9 (89), an isoform lacking the DAC domain and consisting of residues sharing homology with HDAC4 and HDAC5 was identified and named MEF2-interacting transcription repressor (MITR) (34). The sites regulating 14–3-3 binding in HDAC5 (27) were observed to be conserved in MITR at S220 and S451. Moreover, phosphorylation by CaMK disrupted MEF2 interaction and altered the localization of MITR (90). Later, PKD was also shown to phosphorylate these residues (81), modifications also detected in cortical neurons (91). Additionally, HDAC9 residues S239, S240, and S253 are phosphorylated by Aurora B (47), Mirk/dyrk1B (48), and PRK1 (68), respectively (Fig. 1D), similar to the analogous residues in other class IIa HDACs.
The Distribution of PTMs within HDAC Structure
The positioning of PTMs within different structural features of HDACs can significantly contribute to determining their functions. As detailed previously, the PTMs within or in the proximity of the NLS have been well documented to modulate HDAC localization and transcriptional repressive function. However, the presence of PTMs within multiple HDAC functional domains and regions with distinct structural properties must also be considered. To examine the distribution of HDAC PTMs (red stars, Fig. 2), disordered protein regions were predicted using the PrDOS software (92). Established domains, including the MEF binding, NLS, DAC, and NES, exhibit varying degrees of disorder, with the DAC domain representing the least disordered region of each HDAC. The highly structured prediction for the DAC domain is not surprising, given that the first crystal structure solved for class IIa HDACs was the catalytic domain of HDAC7 (23). Highly ordered regions may be favorable environments for structural PTMs, such as the disulfide bond that forms immediately adjacent to the DAC domain in HDAC4 (Figs. 1 and 2). The MEF domain itself harbors no currently known PTMs, but appears to have greater structural variation across the class IIa HDAC family than the NLS, suggesting a possible source of plasticity in transcription factor binding preferences among HDACs. Interestingly, phosphorylations found in the NLS appear to map to the more disordered portion of this region (Fig. 2).
Fig. 2.
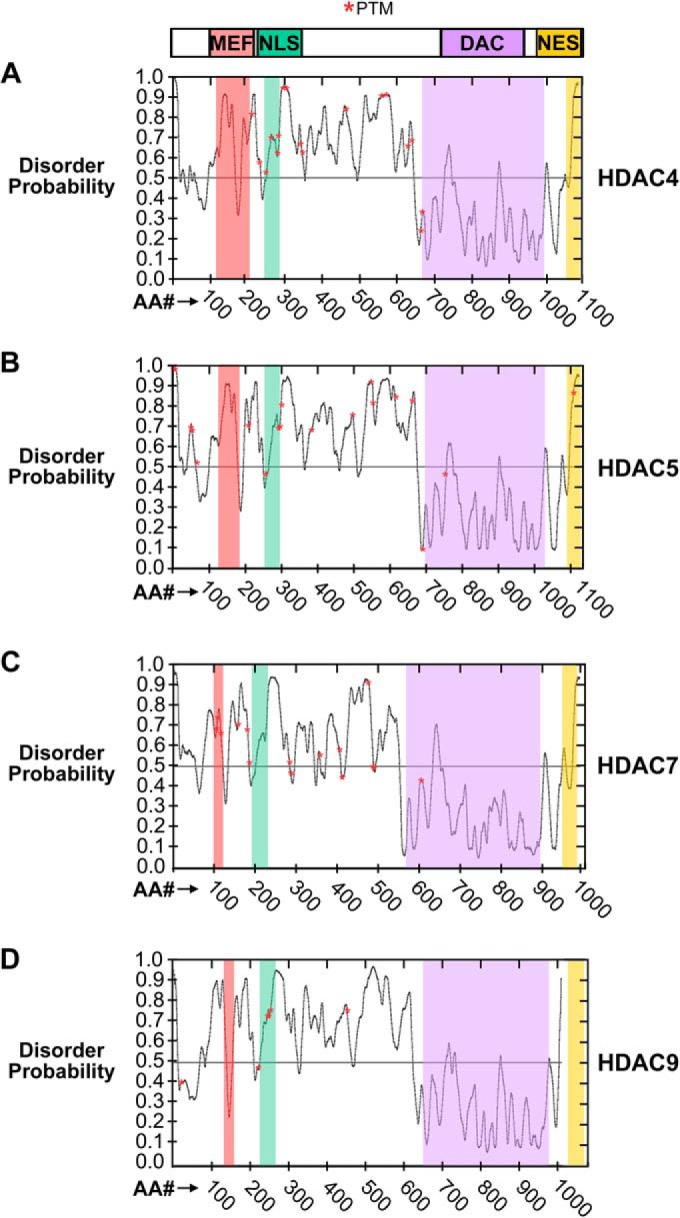
The distribution of class IIa HDAC post-translational modifications within ordered and disordered regions. The probability of disorder was determined for each HDAC amino acid sequence using the PrDOS prediction software. Known PTMS are represented as red stars. Regions falling within established HDAC functional domains are shaded: MEF, red; NLS, green; DAC, purple; NES, yellow. A, HDAC4 sequence, B, HDAC5 sequence, C, HDAC7 sequence, D, HDAC9 sequence.
Previous HDAC5 structure prediction indicated that the majority of phosphorylations map to the external surface of the protein, suggesting that these modifications may be primarily important for mediating protein interactions (12). Loss of phosphorylation within an acidic region, the DAC domain, and the NES was shown to modulate HDAC5 protein interactions, including associations necessary for transcriptional repression (12). Interestingly, mass spectrometry-based quantification of HDAC5 phosphorylations within these functional domains showed substantial variation in their relative abundances (12). Prominent modifications could point to sites necessary for protein folding or structural stability, whereas modifications with low abundances may reflect dynamic or transient events that are spatially and temporally modulated. It may be of interest to investigate possible correlations between the degree of order surrounding a PTM and its abundance.
Most HDAC PTMs seem to localize within regions predicted as naturally disordered (Fig. 2), in agreement with previous reports of abundant phosphorylation within disordered regions of other proteins (93). These regions are likely to represent more rapidly evolving sequences, and it is estimated that a significant percentage of S/T/Y sites (∼25%) within disordered regions are modified (29). The increased availability of these sites may also promote their regulation by multiple enzymes, thereby allowing flexible control over protein function without altering amino acid sequences and conferring an evolutionary advantage. Indeed, numerous sites on class IIa HDACs are targeted by multiple enzymes (Fig. 1) and under different biological conditions (supplemental Table S1).
Although it is clear that numerous PTMs play important roles in regulating protein functions, it is also possible that a subset of PTMs occur from random subcellular events. Although enzymatic post-translational modification does require energy input, additions could be energetically favorable within local reaction environments of the densely packed cell. Thus, expending additional energy to prevent their occurrence or to actively remove PTMs would be of no great benefit when these PTMs do not impact protein function. Another likely explanation for yet-uncharacterized PTMs is that the appropriate functional assays have not yet been discovered or employed. Individual PTMs may exert their functions at specific time points or locations. Currently, as little is known about the co-occurrence of multiple PTMs on HDACs at any given time, it would be of significant interest to examine the coordinated regulation of PTMs and the possibility of synergistic modification. Multiple sites await detailed site-specific investigation of their biological functions, which will no doubt be of tremendous value in building a more complete understanding the regulation of HDACs functions. For these yet functionally uncharacterized sites, we direct interested readers to the following papers that reported their discovery: HDAC4-K236 (94), T278 (95), S339 (96), S565 (97–99), S633 (97); HDAC5-(S3, S7, S53, S55, S66, S206, T234, S368, S755) (28), K533 (100), T546 (101), S671 (28, 102), S1108 (28, 103); HDAC7-T104 (104), S109 (97, 104), Y114 (105), S283 (97, 106), T286 (97, 106), S405 (102), S487 (107, 108), K603 (109), and HDAC9-S22 (96). The remainder of this review will focus on PTMs that have been functionally characterized and shown to regulate subcellular localization and transcription, thereby significantly contributing to maintenance of health and disease states.
PTMs Regulate HDAC Subcellular Localization and Orchestrate Transcription
Class IIa HDAC PTMs profoundly impact their capacity to modulate transcription of gene targets. Within the nucleus, HDACs carry out their transcriptional repressive functions as components of numerous multiprotein complexes (10), including the nuclear corepressor (NCoR) (110–112) and the Bcl-6 corepressor complexes (113). The nuclear association of class IIa HDACs to transcription factors through their N termini and to corepressors through their C termini provides a mechanism of targeting these specific functional protein complexes to particular genomic regions. The deacetylation of targets themselves is performed through association with various corepressor complexes that contain a class I HDAC (i.e. predominantly HDAC3), such as C-terminal-binding protein (114), NCoR/SMRT (110), and BCoR (115). For HDAC4, HDAC5, and HDAC9, the interaction with the E1A C-terminal binding protein occurs through a conserved N terminus PxDLS-like motif (114). The NCoR/SMRT complexes have been shown to associate with and regulate multiple transcription factors, including PLZF (116), BCL6 (117), MyoD (118), and Bach2 (119). The interaction with the SMRT complex was mapped to the C-terminal deacetylation domain of HDAC5 and HDAC7 and domains III and IV of SMRT (111, 120). NCoR and SMRT are also known to interact with HDAC3, as well as the HDAC1/HDAC2-containing Sin3 repressor complex (121). HDAC7 also interacts directly with mSin3A via an amphipathic helix in its N terminus (111), pointing to the complex regulation of HDAC associations with corepressors through both direct and indirect mechanisms. Another important nuclear interaction of HDAC4, HDAC5, and HDAC9 is with heterochromatin protein 1, which represses transcription via recruitment of histone methyltransferases, thus, linking the functional roles of deacetylation and methylation in regulating gene expression (122).
As mentioned previously, phosphorylation is a well-established regulator of HDAC protein localization, impacting the association with these distinct functional complexes (Fig. 3A). Phosphorylation of sites flanking the NLS in all class IIa HDACs promotes 14–3-3 chaperone protein binding and subsequent nuclear export in a CRM1-mediated fashion (27, 33, 78, 90, 123). Specifically, CRM1 binds a hydrophobic C-terminal NES motif conserved among class IIa HDACs (123). Association of 14–3-3 proteins was initially discovered for HDAC4 and HDAC5 using yeast two-hybrid and protein co-immunoprecipitation studies (35, 36). In the same study, HDAC4 and HDAC5 were also shown to associate with the Class I enzyme HDAC3 (35); recent quantitative proteomic studies have highlighted that this association occurs via binding to NCoR components (10). Although 14–3-3 simultaneously recognizes and binds to multiple phosphorylated sites, phosphorylation of a single site is sufficient for maintaining this interaction (35). 14–3-3 association restricts the binding of HDAC4 to importin-α, indicating that inhibition of HDAC nuclear import likely occurs via occlusion of the NLS (35). A similar regulatory mechanism has been reported for other proteins, including Cdc25 in Xenopus XTC cells and ATXN1 in neurons (21, 124).
Fig. 3.

Phosphorylations and functions of class IIa HDACs are spatially and temporally regulated. A, Nuclear HDACs repress transcription of target genes through associations with corepressor complexes. Phosphorylation of HDACs in the nucleus promotes binding of 14–3-3 chaperone proteins and nuclear export of all class IIa HDACs, whereas NLS phosphorylation promotes nuclear accumulation of HDAC5. B, In dividing cells, HDACs 4, 5, and 9 localize to the midzone within a mitotic midzone phosphorylation gradient and are phosphorylated by Aurora B within NLS.
The phosphorylation sites important for nuclear export represent convergence points for multiple signaling pathways, most notably the CaMK superfamily, PKC/PKD, and MARK 1 and 2 (84). Ca2+-mediated activation of the CaMK pathway promotes nuclear export of HDAC5 by stimulating phosphorylation of 14–3-3 binding sites (36). This change in localization triggers the dissociation of the MEF2/HDAC complex and subsequent activation of MEF2 transcriptional targets (36, 58, 125), a process observed to be essential for proper cardiac function (92, 126) (discussed in “The Post-translational Regulation of HDACs is Critically Linked to Human Disease”). In agreement, overexpression of HDAC4, HDAC5, and HDAC9 suppresses MEF2 activity in cardiomyocytes (16).
Similarly, phosphorylation of HDAC7 at its 14–3-3 binding sites by PKD promotes dissociation of HDAC7 from the transcriptional targets Nur77 (78, 80, 127), RUNX2 (82), and RCAN2 (81). Moreover, retention of HDAC7 in the nucleus results in repression of the matrix metalloproteinase MTI-matrix metalloproteinase (MTI-MMP) and MMP10, which are important for microvessel sprouting in angiogenesis (79). In addition to PKD signaling, the MARK kinases C-TAK1 and EMK also serve to regulate HDAC4-, HDAC5-, and HDAC7-mediated repression of Nur77 and c-Jun (84). Interestingly, in HDAC7, the initial phosphorylation of one 14–3-3 binding site promotes the subsequent phosphorylation of the other chaperone binding sites (84), suggesting the cooperative regulation of PTMs within HDACs.
In addition to the PTMs flanking the NLS region, phosphorylation within the NLS at S279 is also critical for nuclear localization of HDAC5. Specifically, phosphorylation was shown to be important for efficient nuclear import in HEK293 and U2OS cells (28), whereas found to prevent PKD/CaMK-induced nuclear export in Cos7 cells (73).
Thus far, understanding of the PTM-induced changes in HDAC localization have been limited to the modification of the NLS region and 14–3-3 binding sites; however, recent studies have suggested that other PTMs can also trigger changes in HDAC5 protein association with important transcriptional regulators. Specifically, S611 mutation led to changes in the association of HDAC5 with the transcription factor MEF2D (12). Therefore, future studies investigating additional PTMs could shed light on their impact on HDAC localizations and interactions within different regions of the same subcellular compartments (e.g. subnuclear). Furthermore, it is worth noting that the aforementioned HDAC-mediated mechanisms are restricted to the effects of the presence or absence of HDACs on their nuclear roles. To date, the cytoplasmic functions of class IIa HDACs remain insufficiently explored, and are likely to be relevant in numerous cellular processes.
Cell Cycle-dependent Regulation of HDACs
As transcription is carefully regulated throughout cell cycle progression, it is expected that HDAC and histone acetyltransferases functions are also dynamic during different stages of the cell cycle. Along with changes in chromosome condensation status, histone acetylation levels have been shown to decrease early in mitosis, reaching their lowest levels during metaphase and anaphase, and rising at the late telophase/interphase transition (128). Additionally, global inhibition of HDACs using small molecules has been shown to inhibit mitotic progression by inducing G2-phase checkpoint response (129, 130). HDAC inhibition by Trichostatin A results in spindle and kinetochore attachment defects in mitotic cell populations, indicating that HDAC-containing complexes are important in properly maintaining mitotic structures (131). Additional HDAC inhibitors have been shown to promote cell cycle arrest at G0/G1 and at G2/M, and to be associated with increased apoptosis (132, 133). The acetylation status of spindle components has been proposed to be important for proper mitotic progression (131), and the class IIb enzyme HDAC6 is directly responsible for deacetylation of tubulin (17).
Changes in the distribution of HDAC4, HDAC5, and HDAC7 have been observed during mitosis, including the exclusion of HDAC4 from condensed chromatin regions (128). Moreover, HDAC5 was shown to localize to the spindle midzone during mitosis and to the midbody during cytokinesis, where it interacts with the mitotic kinase Aurora B and is phosphorylated at S278 within the NLS (47) (Fig. 3B). Phosphorylation of this site is conserved in HDAC4 and HDAC9, suggesting a shared point of regulation; however, in HDAC7 this NLS serine residue is replaced by lysine, indicating divergent regulation of HDAC7 from the rest of the class IIa family (47). Although phosphorylation of S278 coincides with HDAC localization to the mitotic midzone, whether S278 phosphorylation determines this redistribution, or whether the presence of HDAC5 at the midzone promotes its phosphorylation remains to be explored. Consistent with the midzone sequestration of HDAC5 during mitosis, interactions of HDAC5 with members of the NCoR complex were significantly diminished in a G2/M-arrested population of HEK293 cells, with a concomitant decrease in in vitro deacetylation activity (47). It is clear that histone acetylation profiles are dynamic during cell cycle progression, contributing to the regulation of gene expression during mitosis; therefore, further investigation of the mechanisms governing HDAC and histone acetyltransferase localization and activity during specific cell cycle stages will be an important resource for gaining a better understanding of transcriptional control throughout the cell cycle.
The Post-translational Regulation of HDACs is Critically Linked to Human Disease
Although class I HDACs have previously stirred significant interest for their roles in human disease progression and their therapeutic potential, in particular in cancers, studies in recent years have established unique functions for class IIa HDACs in different tissues and organs that can critically impact the development of disease. Given the extensive PTMs and localization-dependent functions of class IIa HDACs, and their intricate regulation by numerous signaling pathways, it is not surprising that these enzymes impact a wide range of cellular processes relevant to human health. Class IIa HDACs are expressed abundantly in the heart, brain, and musculoskeletal tissues, and known to regulate physiological processes in a tissue-specific manner (Fig. 4 and supplemental Table S1). The misregulation of class IIa HDAC levels, PTMs, localization, and transcriptional regulatory functions have been critically linked to significant human diseases, including cardiac, neurodegenerative, and musculoskeletal disorders. As such, these HDACs have emerged as potential novel targets for therapeutic interventions. This section presents an overview of the regulation and functions of class IIa HDACs in the context of human disease, as specifically connected to their PTM status.
Fig. 4.
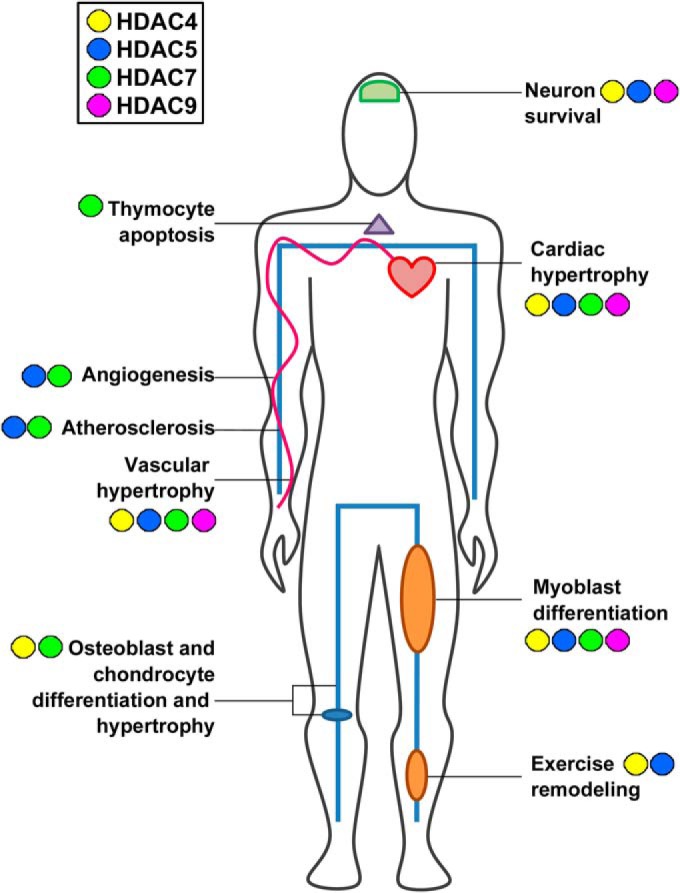
Class IIa HDACs have broad impact on the development of human disease. HDAC4, 5, 7, and 9 have shared roles in cardiac development and muscle differentiation; however, specific roles for individual HDACs have been reported in certain organ systems and pathologies.
Cardiovascular System and Cardiac Hypertrophy
One of the best established roles of class IIa HDACs in human disease is their impact on the cardiovascular system, in particular on the development of cardiac hypertrophy. The constant response of heart tissue to physiological and pathological stimuli can lead to increased cardiac cell size, also known as cardiac hypertrophy. Hypertrophy strengthens the cardiac wall in response to increased stress arising from hypertension, cardiac injury, valve disease, and myocardial infarction (134, 135); however, prolonged hypertrophy can lead to cardiomyopathy, fibrosis, and heart failure (136). Cardiac remodeling results from the activation of the “fetal gene program” that mediates the expression of proteins involved in muscle contraction and calcium absorption (137). In the absence of injury, this program is repressed by nuclear MEF2-interacting HDACs (138, 139). Hypertrophic stimuli activate a cardiac kinase that relieves HDAC-mediated repression thorough phosphorylation-dependent nuclear export (16). HDAC mutants lacking phosphorylation sites were refractory to hypertrophic stimuli, whereas HDAC9 knock-out mice displayed enhanced sensitivity to hypertrophic stimuli (16). Aberrant calcium signaling has also been implicated in the pathological remodeling of the heart in response to stress signaling (140) and the downstream phosphorylation of HDAC4 via CaMKI and CaMKII (40). Although some level of redundancy exists with regard to enzymes targeting multiple sites for phosphorylation (Fig. 1), several kinases only modify specific HDACs and/or sites. For example, CaMKIIδ-null mice display reduced kinase activity toward HDAC4, whereas HDAC5 was unaffected (41, 141). Additional specificity is conferred via the alternate splicing of CaMKIIδ, which yields localization-dependent phosphorylation of HDAC4 by a nuclear δB isoform activated by phenylephrine, and a cytoplasmic δC isoform activated by caffeine (27). Both isoforms can phosphorylate HDAC4 and induce similar MEF2 gene expression, and CaMKIIδC can also phosphorylate calcium regulatory proteins to modulate Ca2+ levels (42). Several other HDAC-modifying enzymes have been shown to induce cardiac hypertrophy via transmission of calcium-independent signals. PKD, a downstream effector of PKC signaling, can directly phosphorylate HDAC5 to stimulate its nuclear export (64, 142). Cardiac A-kinase anchoring proteins serve as scaffolds for PKA- and PKC-dependent activation of PKD1, thus facilitating nuclear export of HDAC5 (66). Export of HDAC5, and subsequent MEF2 activation, can also be triggered by nuclear GRK5 under hypertensive conditions, leading to ventricular hypertrophy (124, 143). Independently of blood pressure, high salt intake has also been known to be associated with left ventricle hypertrophy (128, 144). More recently, this mechanism was shown to be phosphorylation-dependent, as increased sodium levels activate the critical cardiac kinase SIK1, leading to HDAC5 phosphorylation and enhanced MEF2 and NFAT transcriptional activity (72). Interestingly, a recent study examined the mechanisms through which clinically relevant neurohormonal stimuli regulate HDAC phosphorylation in adult cardiomyocytes (62). Although PKD was found to be a critical mediator of endothelin-1-induced phosphorylation and nuclear export of HDAC5, β1-adrenergic receptor (β1-AR) also stimulated nuclear export and MEF2 activation through a phosphorylation-independent mechanism (62). Therefore, more research is needed to understand how these multiple pathways are integrated to achieve a common end point, such as fetal gene program activation.
Mechanisms that act to inhibit cardiac hypertrophy through nuclear retention of HDACs and repression of MEF2 activity further contribute to the challenge of fully understanding the regulation of HDACs during cardiac disease (supplemental Table S1). Such mechanisms are of particular interest as they can potentially be harnessed for therapeutic benefit. Phenylephrine signaling was shown to promote the formation of disulfide bonds between cysteine 667 and 669 of HDAC4 and nuclear export (54). Importantly, reduction of these disulfide bonds by thioredoxin 1 has the potential to limit cardiac hypertrophy (54). Specific phosphorylations are also capable of limiting HDAC nuclear export. PKA-mediated phosphorylation of HDAC5 at S279 impairs 14–3-3 binding, ultimately leading to nuclear retention, repression of MEF2-dependent transcription, and inhibition of cardiac hypertrophy (73). Activation of PKA by β1-AR also promotes HDAC5 nuclear accumulation, most likely through a PP2A-dependent mechanism (145). Another cardioprotective function of PKA involves the proteolytic cleavage of HDAC4, which releases an N-terminal fragment that translocates to the nucleus and represses MEF2 (57). Also inhibiting nuclear export is the direct binding of the transcription factor YY1 to HDAC5 that prevents its phosphorylation (146).
Vascularization
Like cardiac muscle cells, vascular smooth muscle cells can also undergo pathological remodeling leading to hypertrophy via similar molecular mechanisms. Class IIa HDAC PTMs are also modulated under conditions of vascular hypertrophy. Phosphorylation of HDAC4 and HDAC5 in vascular smooth muscle cells is modulated by stimulation of PKD1 (65) and CaMKII (147). Formation of new blood vessels (angiogenesis) involves recruitment of both vascular smooth muscle cells and mature endothelial cells 126). During angiogenesis, VEGF induces phosphorylation of 14–3-3 binding sites and nuclear export of HDAC5 (60) and HDAC7 (79, 81). These phosphorylation events are mediated by PKD signaling, and result in the expression of VEGF-responsive genes (supplemental Table S1) that promote cell proliferation and migration (81), tube formation, and microvessel sprouting (60, 79). HDAC5 phosphorylation and localization are similarly regulated by CaMK during atherosclerosis, triggering enhanced expression of Kruppel-like factor 2 and endothelial nitric oxide synthase in endothelial cells (148). It has also been reported that nitric oxide, a product of endothelial nitric oxide synthase, induces PP2A-dependent dephosphorylation of HDAC4 (50). Lastly, PKD phosphorylation of HDAC5 has been shown to regulate erythropoiesis (149), suggesting that HDACs have roles in the development of both cardiovascular structures and blood cells.
Musculoskeletal System
Phosphorylation is also responsible for controlling the ability of class IIa HDACs to modulate gene programs associated with muscle and bone development, implicating them in diseases of the musculoskeletal system, including myopathies and osteoarthritis.
Muscle Development
Myogenesis encompasses the conversion of myoblasts into differentiated myotubes and involves the activation of hundreds of muscle-specific genes and concomitant repression of cell proliferation genes (14). In undifferentiated cells, MEF2 transcription factors are repressed when in a complex with HDAC4 or HDAC5 (133), which undergo nuclear export upon phosphorylation of 14–3-3 binding sites by GIT1 and CaMKs (27, 36). Although class IIa HDACs have a characteristic dual-compartment localization, individual HDACs exhibit different localization patterns upon myoblast differentiation. The pool of HDAC4 sequestered in the cytoplasm returns to the nucleus following myoblast fusion; however, CaMKIV can inhibit HDAC4 nuclear relocalization (38). Interestingly, HDAC7 is exclusively cytoplasmic in differentiated myotubes (150). The HDAC9 splice variant MITR inhibits the myogenic program, and is also subject to CaMK and Mirk/dyrk1B-dependent phosphorylation-dependent subcellular localization (48, 90). Mirk targets residues within the NLS domains of HDAC5 and MITR, rather than the 14–3-3 binding sites targeted by CaMKs (Fig. 1).
Bone Development
Skeletal biology, including the differentiation of osteoblasts and chondrocytes, is regulated by bone morphogenic proteins (BMPs) that establish bone formation and growth (151, 152). BMP2 induces nuclear export of HDAC7, but not of HDAC5, via PKD-mediated signal-dependent phosphorylation (82). This process relieves the HDAC7-mediated repression of Runx2, the master transcriptional regulator of the skeletal system (153). Differentiation of proliferating chondrocytes into prehypertrophic chondrocytes is also induced by CaMKIV-mediated nuclear exclusion of HDAC4 and elevated Runx2 activity (46), whereas HDAC4-null mice exhibit premature bone ossification (154), implicating these regulatory mechanisms in the development of osteoarthritis. This maturation process can be reversed by expression of the parathyroid hormone-related peptide, which represses Runx2 via PP2A-dependent dephosphorylation of HDAC4 (52), suggesting that modulation of HDAC phosphorylation may represent a promising therapeutic for osteoarthritis and bone calcification disorders.
The Nervous System and Neurodegenerative Diseases
The cytoplasmic localization of class IIa HDACs in neurons allows MEF2 and CREB to express numerous genes involved in neuronal differentiation (155). Neuronal activity itself specifies HDAC subcellular localization; spontaneous synaptic activity is sufficient to induce nuclear export of HDAC4, whereas export of HDAC5 requires CaMK inhibition and calcium flux (156). Similarly, phosphorylation-dependent HDAC9 translocation occurs following spontaneous firing in cortical neurons (91), and subsequent expression of c-fos promotes dendritic growth. Knowledge of the cytoplasmic functions of these HDACs is currently limited; however, multiple studies indicate that the roles and regulatory mechanisms of HDACs in the cytoplasm are highly relevant to understanding their contribution to human disease progression.
In the cytoplasm of peripheral neurons, HDAC5 deacetylates tubulin, promoting neuro-regenerative growth (157). HDAC5 also regulates axon regeneration when activated by PKC (157), further indicating that phosphorylation is an important determinant of HDAC function during neuronal development and in the presence of neuronal injury. Cocaine treatment induces the nuclear export of HDAC5 and activation of MEF2C via SIK1-dependent phosphorylation (158). Interestingly, a separate study has demonstrated that cocaine can alternatively induce nuclear accumulation of HDAC5 through cAMP signaling and PP2A-mediated dephosphorylation of S279 (74). Thus, it is possible that multiple signaling pathways may direct differentially phosphorylated HDAC species to the relevant subcellular compartments under such stress conditions.
HDAC4 has a substantial cytoplasmic localization in neurons (159). Although the nuclear accumulation of HDAC4 because of increased PP2A activity is associated with ataxia telangiectasia (53), the cytoplasmic functions of HDAC4 may also be involved in neurodegenerative diseases. Huntington's disease is caused by the mis-folding of the huntingtin protein, and its pathology is associated with the formation of both nuclear and cytoplasmic aggregates (160). HDAC4 has emerged as a possible promising target for the treatment of Huntington's disease because of its association with the huntingtin protein and its ability to influence the formation of cytoplasmic aggregates (21). As HDAC4 localization is determined by its phosphorylation status, these signaling pathways could be exploited to promote the nuclear retention of HDAC4 in order to limit the formation of cytoplasmic aggregates associated with Huntington's disease pathogenesis. This observation is especially noteworthy as the cytoplasmic roles of class IIa HDACs remain poorly characterized, and increased understanding of such roles will likely be of significant clinical relevance.
Emerging Roles for Class IIa HDACs in Cancer and Diabetes
Although the PTM status of class IIa HDACs has been established to modulate their functions in cardiovascular and neurodegenerative disease, recent studies indicate that their roles in human disease may be far reaching.
It is well-established that HDACs have roles in cancer development. The broad HDAC inhibitor Vorinostat is currently approved for treatment of T-cell lymphomas, indicating that HDACs represent important targets for limiting cancer progression. Thus far, class I HDACs have been the major focus of cancer studies, as their loss of function is linked to cancer development (132). However, class II HDACs may also represent important regulators of tumorigenesis. In gastric cancer cells, PKD2 accumulates in the nucleus and induces phosphorylation of HDAC7, ultimately resulting in HDAC7 nuclear export and expression of downstream targets including Nur77 (83). Conversely, HDAC4 localizes to the nucleus in cancerous colon cells, where it interacts with Sp1 and represses transcription of p21, promoting cancer cell growth (161). Additional investigation of class IIa HDAC localizations, post-translational modifications, and functions in multiple cancer types will provide important insight into their roles in tumor development.
The pathogenesis of diabetes is also linked to HDAC function and involves resistance to insulin in tissues such as skeletal muscle. The fasting hormone glucagon induces dephosphorylation and nuclear retention of HDAC4, HDAC5, and HDAC7 (20). Surprisingly, however, the expression of gluconeogenic genes, such as the catalytic subunit of G6Pase, requires class IIa HDACs to deacetylate and activate FOXO transcription factors (20), indicating that HDACs are able to regulate the production of hormones that can control blood glucose levels. Additionally, HDAC5 modulates the expression of GLUT4, a key regulator of insulin resistance, via phosphorylation-dependent nuclear export and derepression (69). Interestingly, HDAC5 phosphorylation levels have been shown to be elevated during aerobic exercise (162), which could indicate that HDACs respond to change in blood oxygen levels. The broader family of human deacetylases is known to respond to changes in cellular metabolic environments. It is well established that the class III deacetylase (SIRT) activities are regulated by NAD+, and their enzymatic activities can be modulated by glucose and glutamine availability (163–166). Moreover, acetyl-CoA and other CoA derivatives, along with NADPH, have been shown to allosterically activate class I enzymes, HDAC1 and HDAC2 (167). Additional metabolic regulators of HDACs include short chain fatty acids (e.g. butyrate), which formed the basis for the design of chemotherapeutic treatments (168). Increased abundance of short chain fatty acids and butyrate have been proposed to contribute to the cardioprotective and antidiabetic roles of dietary whole grains (169). It is possible that alterations in butyrate concentrations could contribute to modulating HDAC activities in diabetic patients.
Perspectives
Class IIa HDACs are comprehensively controlled by numerous signaling pathways and enzymes that mediate their post-translational modification (Fig. 1). These modifications are found in multiple protein domains and areas of disordered structure (Fig. 2), and not only alter HDAC subcellular localization, but fundamentally impact the ability of HDACs to repress/activate targets. Accumulating evidence indicates that site-specific PTMs control distinct spatial and temporal HDAC regulatory mechanisms, such as via mediating protein interactions, dynamic localization, and cell cycle-dependent effects. Nuclear export of class IIa HDACs has been generally investigated in the context of its ability to regulate transcription. However, the cytoplasmic functions of HDACs are now starting to be more widely explored, particularly with respect to their impacts on human diseases, including neurodegenerative disorders, cardiac disease, and virus-induced pathologies.
The HDAC regulatory network is highly complex, and is responsive to both tissue context and environmental cues. Proteomic studies and advances in mass spectrometry-based methods provide valuable tools for further expanding the identification of PTMs within these different biological contexts. Characterizing these modifications, their regulation by specific enzymes, and their possible coordinated functions in different tissues and under physiological conditions will be critical for gaining a deeper understanding of organ- and disease-specific HDAC functions (Fig. 4). Furthermore, multidisciplinary approaches integrating molecular, clinical, and mass spectrometry-based methods, promise to generate a systems-wide understanding of the dynamic regulation of HDACs. Given the critical links starting to be established between HDAC PTM status and human diseases, the quantification of site-specific PTMs by targeted mass spectrometry may provide accurate and sensitive detection of disease biomarkers in clinical samples.
HDACs have been exploited for their therapeutic benefit given their involvement in conditions such as cardiac and vascular hypertrophy, atherosclerosis, cancer, and diabetes. With several HDAC inhibitors in phase I and II clinical trials, and the development of small molecule inhibitors that block the MEF2 interaction, it is only a matter of time before the full potential of HDAC regulatory mechanisms are realized and used to treat human diseases and disorders. As numerous small molecules can simultaneously inhibit multiple HDACs, the use of such drugs can alter numerous gene programs and lead to cytotoxicity. Gaining a better understanding of HDAC regulation promises to provide targets with increased specificity that can selectively modulate a single HDAC or a given context-dependent function. Such targets may include kinases/phosphatases or other enzymes regulating a certain PTM site or factors influencing the formation of HDAC protein interactions.
Supplementary Material
Footnotes
Author contributions: R.A.M., A.J.G., and I.M.C. wrote the paper.
* This work was supported by NIH grants DP1DA026192, R21AI102187, and R21HD073044 to I.M.C., National Health & Medical Research Council of Australia Early Career CJ Martin Fellowship #APP1037043 to R.A.M., and an NSF graduate fellowship to A.J.G.
 This article contains supplemental Table S1.
This article contains supplemental Table S1.
1 The abbreviations used are:
- HDAC
- histone deacetylase
- PTM
- post-translational modification
- NES
- nuclear export sequence
- NLS
- nuclear localization sequence
- MEF2
- myocyte enhancement factor
- CaMK
- calcium/calmodulin dependent kinase
- PP2A
- protein phosphatase2A
- PKA
- protein kinase A
- PKD
- protein kinase D
- MITR
- MEF2 interacting transcription repressor
- NCoR
- nuclear co-repressor.
REFERENCES
- 1. Bantscheff M., Hopf C., Savitski M. M., Dittmann A., Grandi P., Michon A. M., Schlegl J., Abraham Y., Becher I., Bergamini G., Boesche M., Delling M., Duempelfeld B., Eberhard D., Huthmacher C., Mathieson T., Poeckel D., Reader V., Strunk K., Sweetman G., Kruse U., Neubauer G., Ramsden N. G., Drewes G. (2011) Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat. Biotechnol. 29, 255–265 [DOI] [PubMed] [Google Scholar]
- 2. Choi H., Larsen B., Lin Z. Y., Breitkreutz A., Mellacheruvu D., Fermin D., Qin Z. S., Tyers M., Gingras A. C., Nesvizhskii A. I. (2011) SAINT: probabilistic scoring of affinity purification-mass spectrometry data. Nat. Methods 8, 70–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Berger I., Bieniossek C., Schaffitzel C., Hassler M., Santelli E., Richmond T. J. (2003) Direct interaction of Ca2+/calmodulin inhibits histone deacetylase 5 repressor core binding to myocyte enhancer factor 2. J. Biol. Chem. 278, 17625–17635 [DOI] [PubMed] [Google Scholar]
- 4. Sefton B. M. (2001) Overview of protein phosphorylation. Curr. Protoc. Cell Biol. Chapter 14, Unit 14.1 [DOI] [PubMed] [Google Scholar]
- 5. Melo-Braga M. N., Schulz M., Liu Q., Swistowski A., Palmisano G., Engholm-Keller K., Jakobsen L., Zeng X., Larsen M. R. (2014) Comprehensive quantitative comparison of the membrane proteome, phosphoproteome, and sialiome of human embryonic and neural stem cells. Mol. Cell. Proteomics 13, 311–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Goldberg A. D., Allis C. D., Bernstein E. (2007) Epigenetics: a landscape takes shape. Cell 128, 635–638 [DOI] [PubMed] [Google Scholar]
- 7. Cress W. D., Seto E. (2000) Histone deacetylases, transcriptional control, and cancer. J. Cell. Physiol. 184, 1–16 [DOI] [PubMed] [Google Scholar]
- 8. Grozinger C. M., Schreiber S. L. (2002) Deacetylase enzymes: biological functions and the use of small-molecule inhibitors. Chem. Biol. 9, 3–16 [DOI] [PubMed] [Google Scholar]
- 9. Verdin E., Dequiedt F., Kasler H. G. (2003) Class II histone deacetylases: versatile regulators. Trends Genet. 19, 286–293 [DOI] [PubMed] [Google Scholar]
- 10. Joshi P., Greco T. M., Guise A. J., Luo Y., Yu F., Nesvizhskii A. I., Cristea I. M. (2013) The functional interactome landscape of the human histone deacetylase family. Mol. Syst. Biol. 9, 672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yang X. J., Seto E. (2008) The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat. Rev. Mol. Cell Biol. 9, 206–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Guise A. J., Mathias R. A., Rowland E. A., Yu F., Cristea I. M. (2014) Probing phosphorylation-dependent protein interactions within functional domains of histone deacetylase 5 (HDAC5). Proteomics 14, 2156–2166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cohen P. (2000) The regulation of protein function by multisite phosphorylation—a 25 year update. Trends Biochem. Sci. 25, 596–601 [DOI] [PubMed] [Google Scholar]
- 14. Martin M., Kettmann R., Dequiedt F. (2009) Class IIa histone deacetylases: conducting development and differentiation. Int. J. Dev. Biol. 53, 291–301 [DOI] [PubMed] [Google Scholar]
- 15. Carvalho A. S., Ribeiro H., Voabil P., Penque D., Jensen O. N., Molina H., Matthiesen R. (2014) Global mass spectrometry and transcriptomics array based drug profiling provides novel insight into glucosamine induced ER stress. Mol. Cell. Proteomics 13, 3294–3307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang C. L., McKinsey T. A., Chang S., Antos C. L., Hill J. A., Olson E. N. (2002) Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell 110, 479–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Clocchiatti A., Florean C., Brancolini C. (2011) Class IIa HDACs: from important roles in differentiation to possible implications in tumourigenesis. J. Cell. Mol. Med. 15, 1833–1846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Keedy K. S., Archin N. M., Gates A. T., Espeseth A., Hazuda D. J., Margolis D. M. (2009) A limited group of class I histone deacetylases acts to repress human immunodeficiency virus type 1 expression. J. Virol. 83, 4749–4756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Renthal W., Maze I., Krishnan V., Covington H. E., 3rd, Xiao G., Kumar A., Russo S. J., Graham A., Tsankova N., Kippin T. E., Kerstetter K. A., Neve R. L., Haggarty S. J., McKinsey T. A., Bassel-Duby R., Olson E. N., Nestler E. J. (2007) Histone deacetylase 5 epigenetically controls behavioral adaptations to chronic emotional stimuli. Neuron 56, 517–529 [DOI] [PubMed] [Google Scholar]
- 20. Mihaylova M. M., Vasquez D. S., Ravnskjaer K., Denechaud P. D., Yu R. T., Alvarez J. G., Downes M., Evans R. M., Montminy M., Shaw R. J. (2011) Class IIa histone deacetylases are hormone-activated regulators of FOXO and mammalian glucose homeostasis. Cell 145, 607–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mielcarek M., Landles C., Weiss A., Bradaia A., Seredenina T., Inuabasi L., Osborne G. F., Wadel K., Touller C., Butler R., Robertson J., Franklin S. A., Smith D. L., Park L., Marks P. A., Wanker E. E., Olson E. N., Luthi-Carter R., van der Putten H., Beaumont V., Bates G. P. (2013) HDAC4 reduction: a novel therapeutic strategy to target cytoplasmic huntingtin and ameliorate neurodegeneration. Plos Biol. 11, e1001717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lahm A., Paolini C., Pallaoro M., Nardi M. C., Jones P., Neddermann P., Sambucini S., Bottomley M. J., Lo Surdo P., Carfí A., Koch U., De Francesco R., Steinkühler C., Gallinari P. (2007) Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc. Natl. Acad. Sci. U.S.A. 104, 17335–17340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schuetz A., Min J., Allali-Hassani A., Schapira M., Shuen M., Loppnau P., Mazitschek R., Kwiatkowski N. P., Lewis T. A., Maglathin R. L., McLean T. H., Bochkarev A., Plotnikov A. N., Vedadi M., Arrowsmith C. H. (2008) Human HDAC7 harbors a class IIa histone deacetylase-specific zinc binding motif and cryptic deacetylase activity. J. Biol. Chem. 283, 11355–11363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Henningsen K., Palmfeldt J., Christiansen S., Baiges I., Bak S., Jensen O. N., Gregersen N., Wiborg O. (2012) Candidate hippocampal biomarkers of susceptibility and resilience to stress in a rat model of depression. Mol. Cell. Proteomics 11, doi: 10.1074/mcp.M111.016428. Epub 2012 Feb 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang F., Melo-Braga M. N., Larsen M. R., Jørgensen H. J., Palmisano G. (2013) Battle through signaling between wheat and the fungal pathogen septoria tritici revealed by proteomics and phosphoproteomics. Mol. Cell. Proteomics 12, 2497–2508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wright P. E., Dyson H. J. (1999) Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J. Mol. Biol. 293, 321–331 [DOI] [PubMed] [Google Scholar]
- 27. McKinsey T. A., Zhang C. L., Lu J., Olson E. N. (2000) Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature 408, 106–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Greco T. M., Yu F., Guise A. J., Cristea I. M. (2011) Nuclear import of histone deacetylase 5 by requisite nuclear localization signal phosphorylation. Mol. Cell. Proteomics 10, doi: 10.1074/mcp.M110.004317. Epub 2010 Nov 16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nishi H., Fong J. H., Chang C., Teichmann S. A., Panchenko A. R. (2013) Regulation of protein–protein binding by coupling between phosphorylation and intrinsic disorder: analysis of human protein complexes. Mol. Biosyst. 9, 1620–1626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schuetz A., Min J., Allali-Hassani A., Schapira M., Shuen M., Loppnau P., Mazitschek R., Kwiatkowski N. P., Lewis T. A., Maglathin R. L., McLean T. H., Bochkarev A., Plotnikov A. N., Vedadi M., Arrowsmith C. H. (2008) Human HDAC7 harbors a class IIa histone deacetylase-specific zinc binding motif and cryptic deacetylase activity. J. Biol. Chem. 283, 11355–11363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Martin M., Kettmann R., Dequiedt F. (2007) Class IIa histone deacetylases: regulating the regulators. Oncogene 26, 5450–5467 [DOI] [PubMed] [Google Scholar]
- 32. Parker B. L., Shepherd N. E., Trefely S., Hoffman N. J., White M. Y., Engholm-Keller K., Hambly B. D., Larsen M. R., James D. E., Cordwell S. J. (2014) Structural basis for phosphorylation and lysine acetylation cross-talk in a kinase motif associated with myocardial ischemia and cardioprotection. J. Biol. Chem. 289, 25890–25906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Miska E. A., Karlsson C., Langley E., Nielsen S. J., Pines J., Kouzarides T. (1999) HDAC4 deacetylase associates with and represses the MEF2 transcription factor. EMBO J. 18, 5099–5107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sparrow D. B., Miska E. A., Langley E., Reynaud-Deonauth S., Kotecha S., Towers N., Spohr G., Kouzarides T., Mohun T. J. (1999) MEF-2 function is modified by a novel corepressor, MITR. EMBO J. 18, 5085–5098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Grozinger C. M., Schreiber S. L. (2000) Regulation of histone deacetylase 4 and 5 and transcriptional activity by 14-3-3-dependent cellular localization. Proc. Natl. Acad. Sci. U.S.A. 97, 7835–7840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McKinsey T. A., Zhang C. L., Olson E. N. (2000) Activation of the myocyte enhancer factor-2 transcription factor by calcium/calmodulin-dependent protein kinase-stimulated binding of 14–3-3 to histone deacetylase 5. Proc. Natl. Acad. Sci. U.S.A. 97, 14400–14405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang A. H., Kruhlak M. J., Wu J., Bertos N. R., Vezmar M., Posner B. I., Bazett-Jones D. P., Yang X. J. (2000) Regulation of histone deacetylase 4 by binding of 14–3-3 proteins. Mol. Cell. Biol. 20, 6904–6912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Miska E. A., Langley E., Wolf D., Karlsson C., Pines J., Kouzarides T. (2001) Differential localization of HDAC4 orchestrates muscle differentiation. Nucleic Acids Res. 29, 3439–3447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhao X., Ito A., Kane C. D., Liao T. S., Bolger T. A., Lemrow S. M., Means A. R., Yao T. P. (2001) The modular nature of histone deacetylase HDAC4 confers phosphorylation-dependent intracellular trafficking. J. Biol. Chem. 276, 35042–35048 [DOI] [PubMed] [Google Scholar]
- 40. Backs J., Song K., Bezprozvannaya S., Chang S., Olson E. N. (2006) CaM kinase II selectively signals to histone deacetylase 4 during cardiornyocyte hypertrophy. J. Clin. Investig. 116, 1853–1864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Little G. H., Bai Y., Williams T., Poizat C. (2007) Nuclear calcium/calmodulin-dependent protein kinase II delta preferentially transmits signals to histone deacetylase 4 in cardiac cells. J. Biol. Chem. 282, 7219–7231 [DOI] [PubMed] [Google Scholar]
- 42. Zhang T., Kohlhaas M., Backs J., Mishra S., Phillips W., Dybkova N., Chang S., Ling H., Bers D. M., Maier L. S., Olson E. N., Brown J. H. (2007) CaMKIIdelta isoforms differentially affect calcium handling but similarly regulate HDAC/MEF2 transcriptional responses. J. Biol. Chem. 282, 35078–35087 [DOI] [PubMed] [Google Scholar]
- 43. Backs J., Backs T., Bezprozvannaya S., McKinsey T. A., Olson E. N. (2008) Histone deacetylase 5 acquires calcium/calmodulin-dependent kinase II responsiveness by oligomerization with histone deacetylase 4. Mol. Cell. Biol. 28, 3437–3445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McGee S. L., Fairlie E., Garnham A. P., Hargreaves M. (2009) Exercise-induced histone modifications in human skeletal muscle. J. Physiol. 587, 5951–5958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ginnan R., Sun L. Y., Schwarz J. J., Singer H. A. (2012) MEF2 is regulated by CaMKIIdelta2 and a HDAC4-HDAC5 heterodimer in vascular smooth muscle cells. Biochem. J. 444, 105–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Guan Y., Chen Q., Yang X., Haines P., Pei M., Terek R., Wei X., Zhao T., Wei L. (2012) Subcellular relocation of histone deacetylase 4 regulates growth plate chondrocyte differentiation through Ca2+/calmodulin-dependent kinase IV. Am J. Physiol. Cell Physiol. 303, C33–C40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Guise A. J., Greco T. M., Zhang I. Y., Yu F., Cristea I. M. (2012) Aurora B-dependent regulation of class IIa histone deacetylases by mitotic nuclear localization signal phosphorylation. Mol. Cell. Proteomics 11, 1220–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Deng X., Ewton D. Z., Mercer S. E., Friedman E. (2005) Mirk/dyrk1B decreases the nuclear accumulation of class II histone deacetylases during skeletal muscle differentiation. J. Biol. Chem. 280, 4894–4905 [DOI] [PubMed] [Google Scholar]
- 49. Cernotta N., Clocchiatti A., Florean C., Brancolini C. (2011) Ubiquitin-dependent degradation of HDAC4, a new regulator of random cell motility. Mol. Biol. Cell 22, 278–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Illi B., Dello Russo C., Colussi C., Rosati J., Pallaoro M., Spallotta F., Rotili D., Valente S., Ragone G., Martelli F., Biglioli P., Steinkuhler C., Gallinari P., Mai A., Capogrossi M. C., Gaetano C. (2008) Nitric oxide modulates chromatin folding in human endothelial cells via protein phosphatase 2A activation and class II histone deacetylases nuclear shuttling. Circ. Res. 102, 51–58 [DOI] [PubMed] [Google Scholar]
- 51. Paroni G., Cernotta N., Dello Russo C., Gallinari P., Pallaoro M., Foti C., Talamo F., Orsatti L., Steinkühler C., Brancolini C. (2008) PP2A regulates HDAC4 nuclear import. Mol. Biol. Cell 19, 655–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kozhemyakina E., Cohen T., Yao T. P., Lassar A. B. (2009) Parathyroid hormone-related peptide represses chondrocyte hypertrophy through a protein phosphatase 2A/histone deacetylase 4/MEF2 pathway. Mol. Cell. Biol. 29, 5751–5762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Li J., Chen J., Ricupero C. L., Hart R. P., Schwartz M. S., Kusnecov A., Herrup K. (2012) Nuclear accumulation of HDAC4 in ATM deficiency promotes neurodegeneration in ataxia telangiectasia. Nat. Med. 18, 783–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ago T., Liu T., Zhai P., Chen W., Li H., Molkentin J. D., Vatner S. F., Sadoshima J. (2008) A redox-dependent pathway for regulating class II HDACs and cardiac hypertrophy. Cell 133, 978–993 [DOI] [PubMed] [Google Scholar]
- 55. Kirsh O., Seeler J. S., Pichler A., Gast A., Müller S., Miska E., Mathieu M., Harel-Bellan A., Kouzarides T., Melchior F., Dejean A. (2002) The SUMO E3 ligase RanBP2 promotes modification of the HDAC4 deacetylase. EMBO J. 21, 2682–2691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Paroni G., Fontanini A., Cernotta N., Foti C., Gupta M. P., Yang X. J., Fasino D., Brancolini C. (2007) Dephosphorylation and caspase processing generate distinct nuclear pools of histone deacetylase 4. Mol. Cell. Biol. 27, 6718–6732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Backs J., Worst B. C., Lehmann L. H., Patrick D. M., Jebessa Z., Kreusser M. M., Sun Q., Chen L., Heft C., Katus H. A., Olson E. N. (2011) Selective repression of MEF2 activity by PKA-dependent proteolysis of HDAC4. J. Cell Biol. 195, 403–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wu X., Zhang T., Bossuyt J., Li X., McKinsey T. A., Dedman J. R., Olson E. N., Chen J., Brown J. H., Bers D. M. (2006) Local InsP3-dependent perinuclear Ca2+ signaling in cardiac myocyte excitation-transcription coupling. J. Clin. Investig. 116, 675–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mishra S., Gray C. B., Miyamoto S., Bers D. M., Brown J. H. (2011) Location matters: clarifying the concept of nuclear and cytosolic CaMKII subtypes. Circ. Res. 109, 1354–1362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ha C. H., Wang W., Jhun B. S., Wong C., Hausser A., Pfizenmaier K., McKinsey T. A., Olson E. N., Jin Z. G. (2008) Protein kinase D-dependent phosphorylation and nuclear export of histone deacetylase 5 mediates vascular endothelial growth factor-induced gene expression and angiogenesis. J. Biol. Chem. 283, 14590–14599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Harrison B. C., Kim M. S., van Rooij E., Plato C. F., Papst P. J., Vega R. B., McAnally J. A., Richardson J. A., Bassel-Duby R., Olson E. N., McKinsey T. A. (2006) Regulation of cardiac stress signaling by protein kinase D1. Mol. Cell. Biol. 26, 3875–3888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Haworth R. S., Stathopoulou K., Candasamy A. J., Avkiran M. (2012) Neurohormonal regulation of cardiac histone deacetylase 5 nuclear localization by phosphorylation-dependent and phosphorylation-independent mechanisms. Circ. Res. 110, 1585–1595 [DOI] [PubMed] [Google Scholar]
- 63. Papazyan R., Doche M., Waldron R. T., Rozengurt E., Moyer M. P., Rey O. (2008) Protein kinase D isozymes activation and localization during mitosis. Exp. Cell Res. 314, 3057–3068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Vega R. B., Harrison B. C., Meadows E., Roberts C. R., Papst P. J., Olson E. N., McKinsey T. A. (2004) Protein kinases C and D mediate agonist-dependent cardiac hypertrophy through nuclear export of histone deacetylase 5. Mol. Cell. Biol. 24, 8374–8385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Xu X., Ha C. H., Wong C., Wang W., Hausser A., Pfizenmaier K., Olson E. N., McKinsey T. A., Jin Z. G. (2007) Angiotensin II stimulates protein kinase D-dependent histone deacetylase 5 phosphorylation and nuclear export leading to vascular smooth muscle cell hypertrophy. Arterioscler. Thromb. Vasc. Biol. 27, 2355–2362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Carnegie G. K., Soughayer J., Smith F. D., Pedroja B. S., Zhang F., Diviani D., Bristow M. R., Kunkel M. T., Newton A. C., Langeberg L. K., Scott J. D. (2008) AKAP-Lbc mobilizes a cardiac hypertrophy signaling pathway. Mol. Cell 32, 169–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Huynh Q. K. (2011) Evidence for the phosphorylation of serine259 of histone deacetylase 5 by protein kinase Cdelta. Arch. Biochem. Biophys. 506, 173–180 [DOI] [PubMed] [Google Scholar]
- 68. Harrison B. C., Huynh K., Lundgaard G. L., Helmke S. M., Perryman M. B., McKinsey T. A. (2010) Protein kinase C-related kinase targets nuclear localization signals in a subset of class IIa histone deacetylases. FEBS Lett. 584, 1103–1110 [DOI] [PubMed] [Google Scholar]
- 69. McGee S. L., van Denderen B. J., Howlett K. F., Mollica J., Schertzer J. D., Kemp B. E., Hargreaves M. (2008) AMP-activated protein kinase regulates GLUT4 transcription by phosphorylating histone deacetylase 5. Diabetes 57, 860–867 [DOI] [PubMed] [Google Scholar]
- 70. Zhao J. X., Yue W. F., Zhu M. J., Du M. (2011) AMP-activated protein kinase regulates beta-catenin transcription via histone deacetylase 5. J. Biol. Chem. 286, 16426–16434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Martini J. S., Raake P., Vinge L. E., DeGeorge B. R., Jr., DeGeorge B., Chuprun J. K., Harris D. M., Gao E., Eckhart A. D., Pitcher J. A., Koch W. J. (2008) Uncovering G protein-coupled receptor kinase-5 as a histone deacetylase kinase in the nucleus of cardiomyocytes. Proc. Natl. Acad. Sci. U.S.A. 105, 12457–12462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Popov S., Venetsanou K., Chedrese P. J., Pinto V., Takemori H., Franco-Cereceda A., Eriksson P., Mochizuki N., Soares-da-Silva P., Bertorello A. M. (2012) Increases in intracellular sodium activate transcription and gene expression via the salt-inducible kinase 1 network in an atrial myocyte cell line. Am. J. Physiol. Heart Circ. Physiol. 303, H57-H65 [DOI] [PubMed] [Google Scholar]
- 73. Ha C. H., Kim J. Y., Zhao J., Wang W., Jhun B. S., Wong C., Jin Z. G. (2010) PKA phosphorylates histone deacetylase 5 and prevents its nuclear export, leading to the inhibition of gene transcription and cardiomyocyte hypertrophy. Proc. Natl. Acad. Sci. U.S.A. 107, 15467–15472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Taniguchi M., Carreira M. B., Smith L. N., Zirlin B. C., Neve R. L., Cowan C. W. (2012) Histone deacetylase 5 limits cocaine reward through cAMP-induced nuclear import. Neuron 73, 108–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Li X., Song S., Liu Y., Ko S. H., Kao H. Y. (2004) Phosphorylation of the histone deacetylase 7 modulates its stability and association with 14–3-3 proteins. J. Biol. Chem. 279, 34201–34208 [DOI] [PubMed] [Google Scholar]
- 76. Dequiedt F., Van Lint J., Lecomte E., Van Duppen V., Seufferlein T., Vandenheede J. R., Wattiez R., Kettmann R. (2005) Phosphorylation of histone deacetylase 7 by protein kinase D mediates T cell receptor-induced Nur77 expression and apoptosis. J. Exp. Med. 201, 793–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Gao C., Li X., Lam M., Liu Y., Chakraborty S., Kao H. Y. (2006) CRM1 mediates nuclear export of HDAC7 independently of HDAC7 phosphorylation and association with 14–3-3s. FEBS Lett. 580, 5096–5104 [DOI] [PubMed] [Google Scholar]
- 78. Kao H. Y., Verdel A., Tsai C. C., Simon C., Juguilon H., Khochbin S. (2001) Mechanism for nucleocytoplasmic shuttling of histone deacetylase 7. J. Biol. Chem. 276, 47496–47507 [DOI] [PubMed] [Google Scholar]
- 79. Ha C. H., Jhun B. S., Kao H. Y., Jin Z. G. (2008) VEGF stimulates HDAC7 phosphorylation and cytoplasmic accumulation modulating matrix metalloproteinase expression and angiogenesis. Arterioscler. Thromb. Vasc. Biol. 28, 1782–1788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Parra M., Kasler H., McKinsey T. A., Olson E. N., Verdin E. (2005) Protein kinase D1 phosphorylates HDAC7 and induces its nuclear export after T-cell receptor activation. J. Biol. Chem. 280, 13762–13770 [DOI] [PubMed] [Google Scholar]
- 81. Wang S., Li X., Parra M., Verdin E., Bassel-Duby R., Olson E. N. (2008) Control of endothelial cell proliferation and migration by VEGF signaling to histone deacetylase 7. Proc. Natl. Acad. Sci. U.S.A. 105, 7738–7743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Jensen E. D., Gopalakrishnan R., Westendorf J. J. (2009) Bone morphogenic protein 2 activates protein kinase D to regulate histone deacetylase 7 localization and repression of Runx2. J. Biol. Chem. 284, 2225–2234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. von Blume J., Knippschild U., Dequiedt F., Giamas G., Beck A., Auer A., Van Lint J., Adler G., Seufferlein T. (2007) Phosphorylation at Ser244 by CK1 determines nuclear localization and substrate targeting of PKD2. EMBO J. 26, 4619–4633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Dequiedt F., Martin M., Von Blume J., Vertommen D., Lecomte E., Mari N., Heinen M. F., Bachmann M., Twizere J. C., Huang M. C., Rider M. H., Piwnica-Worms H., Seufferlein T., Kettmann R. (2006) New role for hPar-1 kinases EMK and C-TAK1 in regulating localization and activity of class IIa histone deacetylases. Mol. Cell. Biol. 26, 7086–7102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Martin M., Potente M., Janssens V., Vertommen D., Twizere J. C., Rider M. H., Goris J., Dimmeler S., Kettmann R., Dequiedt F. (2008) Protein phosphatase 2A controls the activity of histone deacetylase 7 during T cell apoptosis and angiogenesis. Proc. Natl. Acad. Sci. U.S.A. 105, 4727–4732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Parra M., Mahmoudi T., Verdin E. (2007) Myosin phosphatase dephosphorylates HDAC7, controls its nucleocytoplasmic shuttling, and inhibits apoptosis in thymocytes. Genes Dev. 21, 638–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Mahrus S., Trinidad J. C., Barkan D. T., Sali A., Burlingame A. L., Wells J. A. (2008) Global sequencing of proteolytic cleavage sites in apoptosis by specific labeling of protein N termini. Cell 134, 866–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Scott F. L., Fuchs G. J., Boyd S. E., Denault J. B., Hawkins C. J., Dequiedt F., Salvesen G. S. (2008) Caspase-8 cleaves histone deacetylase 7 and abolishes its transcription repressor function. J. Biol. Chem. 283, 19499–19510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Zhou X., Marks P. A., Rifkind R. A., Richon V. M. (2001) Cloning and characterization of a histone deacetylase, HDAC9. Proc. Natl. Acad. Sci. U.S.A. 98, 10572–10577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Zhang C. L., McKinsey T. A., Olson E. N. (2001) The transcriptional corepressor MITR is a signal-responsive inhibitor of myogenesis. Proc. Natl. Acad. Sci. U.S.A. 98, 7354–7359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Sugo N., Oshiro H., Takemura M., Kobayashi T., Kohno Y., Uesaka N., Song W. J., Yamamoto N. (2010) Nucleocytoplasmic translocation of HDAC9 regulates gene expression and dendritic growth in developing cortical neurons. Eur. J. Neurosci. 31, 1521–1532 [DOI] [PubMed] [Google Scholar]
- 92. Ishida T., Kinoshita K. (2007) PrDOS: prediction of disordered protein regions from amino acid sequence. Nucleic Acids Res. 35, W460–W464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Holt L. J., Tuch B. B., Villén J., Johnson A. D., Gygi S. P., Morgan D. O. (2009) Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science 325, 1682–1686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Zhao S., Xu W., Jiang W., Yu W., Lin Y., Zhang T., Yao J., Zhou L., Zeng Y., Li H., Li Y., Shi J., An W., Hancock S. M., He F., Qin L., Chin J., Yang P., Chen X., Lei Q., Xiong Y., Guan K. L. (2010) Regulation of cellular metabolism by protein lysine acetylation. Science 327, 1000–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Sui S., Wang J., Yang B., Song L., Zhang J., Chen M., Liu J., Lu Z., Cai Y., Chen S., Bi W., Zhu Y., He F., Qian X. (2008) Phosphoproteome analysis of the human Chang liver cells using SCX and a complementary mass spectrometric strategy. Proteomics 8, 2024–2034 [DOI] [PubMed] [Google Scholar]
- 96. Mayya V., Lundgren D. H., Hwang S. I., Rezaul K., Wu L., Eng J. K., Rodionov V., Han D. K. (2009) Quantitative phosphoproteomic analysis of T cell receptor signaling reveals system-wide modulation of protein–protein interactions. Sci. Signal. 2, ra46. [DOI] [PubMed] [Google Scholar]
- 97. Dephoure N., Zhou C., Villén J., Beausoleil S. A., Bakalarski C. E., Elledge S. J., Gygi S. P. (2008) A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 105, 10762–10767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Old W. M., Shabb J. B., Houel S., Wang H., Couts K. L., Yen C. Y., Litman E. S., Croy C. H., Meyer-Arendt K., Miranda J. G., Brown R. A., Witze E. S., Schweppe R. E., Resing K. A., Ahn N. G. (2009) Functional proteomics identifies targets of phosphorylation by B-Raf signaling in melanoma. Mol. Cell 34, 115–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Zanivan S., Gnad F., Wickström S. A., Geiger T., Macek B., Cox J., Fässler R., Mann M. (2008) Solid tumor proteome and phosphoproteome analysis by high resolution mass spectrometry. J. Proteome Res. 7, 5314–5326 [DOI] [PubMed] [Google Scholar]
- 100. Choudhary C., Kumar C., Gnad F., Nielsen M. L., Rehman M., Walther T. C., Olsen J. V., Mann M. (2009) Lysine acetylation targets protein complexes and coregulates major cellular functions. Science 325, 834–840 [DOI] [PubMed] [Google Scholar]
- 101. Moritz A., Li Y., Guo A., Villén J., Wang Y., MacNeill J., Kornhauser J., Sprott K., Zhou J., Possemato A., Ren J. M., Hornbeck P., Cantley L. C., Gygi S. P., Rush J., Comb M. J. (2010) Akt-RSK-S6 kinase signaling networks activated by oncogenic receptor tyrosine kinases. Sci. Signal. 3, ra64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Huttlin E. L., Jedrychowski M. P., Elias J. E., Goswami T., Rad R., Beausoleil S. A., Villén J., Haas W., Sowa M. E., Gygi S. P. (2010) A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Christensen G. L., Kelstrup C. D., Lyngsø C., Sarwar U., Bøgebo R., Sheikh S. P., Gammeltoft S., Olsen J. V., Hansen J. L. (2010) Quantitative phosphoproteomics dissection of seven-transmembrane receptor signaling using full and biased agonists. Mol. Cell. Proteomics 9, 1540–1553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Chen R. Q., Yang Q. K., Lu B. W., Yi W., Cantin G., Chen Y. L., Fearns C., Yates J. R., 3rd, Lee J. D. (2009) CDC25B mediates rapamycin-induced oncogenic responses in cancer cells. Cancer Res. 69, 2663–2668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Cantin G. T., Yi W., Lu B., Park S. K., Xu T., Lee J. D., Yates J. R., 3rd (2008) Combining protein-based IMAC, peptide-based IMAC, and MudPIT for efficient phosphoproteomic analysis. J. Proteome Res. 7, 1346–1351 [DOI] [PubMed] [Google Scholar]
- 106. Olsen J. V., Vermeulen M., Santamaria A., Kumar C., Miller M. L., Jensen L. J., Gnad F., Cox J., Jensen T. S., Nigg E. A., Brunak S., Mann M. (2010) Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 3, ra3. [DOI] [PubMed] [Google Scholar]
- 107. Beausoleil S. A., Jedrychowski M., Schwartz D., Elias J. E., Villén J., Li J., Cohn M. A., Cantley L. C., Gygi S. P. (2004) Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc. Natl. Acad. Sci. U.S.A. 101, 12130–12135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Raijmakers R., Kraiczek K., de Jong A. P., Mohammed S., Heck A. J. (2010) Exploring the human leukocyte phosphoproteome using a microfluidic reversed-phase-TiO2-reversed-phase high-performance liquid chromatography phosphochip coupled to a quadrupole time-of-flight mass spectrometer. Anal. Chem. 82, 824–832 [DOI] [PubMed] [Google Scholar]
- 109. Wagner S. A., Beli P., Weinert B. T., Nielsen M. L., Cox J., Mann M., Choudhary C. (2011) A proteome-wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol. Cell. Proteomics 10, M111.013284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Fischle W., Dequiedt F., Hendzel M. J., Guenther M. G., Lazar M. A., Voelter W., Verdin E. (2002) Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol. Cell 9, 45–57 [DOI] [PubMed] [Google Scholar]
- 111. Kao H. Y., Downes M., Ordentlich P., Evans R. M. (2000) Isolation of a novel histone deacetylase reveals that class I and class II deacetylases promote SMRT-mediated repression. Genes Dev. 14, 55–66 [PMC free article] [PubMed] [Google Scholar]
- 112. Huang E. Y., Zhang J., Miska E. A., Guenther M. G., Kouzarides T., Lazar M. A. (2000) Nuclear receptor corepressors partner with class II histone deacetylases in a Sin3-independent repression pathway. Genes Dev. 14, 45–54 [PMC free article] [PubMed] [Google Scholar]
- 113. Lemercier C., Brocard M. P., Puvion-Dutilleul F., Kao H. Y., Albagli O., Khochbin S. (2002) Class II histone deacetylases are directly recruited by BCL6 transcriptional repressor. J. Biol. Chem. 277, 22045–22052 [DOI] [PubMed] [Google Scholar]
- 114. Zhang C. L., McKinsey T. A., Lu J. R., Olson E. N. (2001) Association of COOH-terminal-binding protein (CtBP) and MEF2-interacting transcription repressor (MITR) contributes to transcriptional repression of the MEF2 transcription factor. J. Biol. Chem. 276, 35–39 [DOI] [PubMed] [Google Scholar]
- 115. Huynh K. D., Fischle W., Verdin E., Bardwell V. J. (2000) BCoR, a novel corepressor involved in BCL-6 repression. Genes Dev. 14, 1810–1823 [PMC free article] [PubMed] [Google Scholar]
- 116. Tsao P. S., Buitrago R., Chan J. R., Cooke J. P. (1996) Fluid flow inhibits endothelial adhesiveness—nitric oxide and transcriptional regulation of VCAM-1. Circulation 94, 1682–1689 [DOI] [PubMed] [Google Scholar]
- 117. Huynh K. D., Bardwell V. J. (1998) The BCL-6 POZ domain and other POZ domains interact with the corepressors N-CoR and SMRT. Oncogene 17, 2473–2484 [DOI] [PubMed] [Google Scholar]
- 118. Tsao T. S., Burcelin R., Katz E. B., Huang L., Charron M. J. (1996) Enhanced insulin action due to targeted GLUT4 overexpression exclusively in muscle. Diabetes 45, 28–36 [DOI] [PubMed] [Google Scholar]
- 119. Ren J. M., Marshall B. A., Mueckler M. M., McCaleb M., Amatruda J. M., Shulman G. I. (1995) Overexpression of glut4 protein in muscle increases basal and insulin-stimulated whole-body glucose disposal in conscious mice. J. Clin. Investig. 95, 429–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Huang E. Y., Zhang J., Miska E. A., Guenther M. G., Kouzarides T., Lazar M. A. (2000) Nuclear receptor corepressors partner with class II histone deacetylases in a Sin3-independent repression pathway. Genes Dev. 14, 45–54 [PMC free article] [PubMed] [Google Scholar]
- 121. Morishita R., Gibbons G. H., Ellison K. E., Lee W., Zhang L., Yu H., Kaneda Y., Ogihara T., Dzau V. J. (1994) Evidence for direct local effect of angiotensin in vascular hypertrophy—in-vivo gene-transfer of angiotensin-converting enzyme. J. Clin. Investig. 94, 978–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Zhang C. L., McKinsey T. A., Olson E. N. (2002) Association of class II histone deacetylases with heterochromatin protein 1: Potential role for histone methylation in control of muscle differentiation. Mol. Cell. Biol. 22, 7302–7312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Wang A. H., Yang X. J. (2001) Histone deacetylase 4 possesses intrinsic nuclear import and export signals. Mol. Cell. Biol. 21, 5992–6005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Akhter S. A., Luttrell L. M., Rockman H. A., Iaccarino G., Lefkowitz R. J., Koch W. J. (1998) Targeting the receptor-G(q) interface to inhibit in vivo pressure overload myocardial hypertrophy. Science 280, 574–577 [DOI] [PubMed] [Google Scholar]
- 125. Linseman D. A., Bartley C. M., Le S. S., Laessig T. A., Bouchard R. J., Meintzer M. K., Li M., Heidenreich K. A. (2003) Inactivation of the myocyte enhancer factor-2 repressor histone deacetylase-5 by endogenous Ca(2+) //calmodulin-dependent kinase II promotes depolarization-mediated cerebellar granule neuron survival. J. Biol. Chem. 278, 41472–41481 [DOI] [PubMed] [Google Scholar]
- 126. Risau W. (1997) Mechanisms of angiogenesis. Nature 386, 671–674 [DOI] [PubMed] [Google Scholar]
- 127. Dequiedt F., Kasler H., Fischle W., Kiermer V., Weinstein M., Herndier B. G., Verdin E. (2003) HDAC7, a thymus-specific class II histone deacetylase, regulates Nur77 transcription and TCR-mediated apoptosis. Immunity 18, 687–698 [DOI] [PubMed] [Google Scholar]
- 128. Frohlich E. D., Chien Y., Sesoko S., Pegram B. L. (1993) Relationship between dietary-sodium intake, hemodynamics, and cardiac mass in shr and wky rats. Am. J. Physiol. 264, R30–R34 [DOI] [PubMed] [Google Scholar]
- 129. Shin H. J., Baek K. H., Jeon A. H., Kim S. J., Jang K. L., Sung Y. C., Kim C. M., Lee C. W. (2003) Inhibition of histone deacetylase activity increases chromosomal instability by the aberrant regulation of mitotic checkpoint activation. Oncogene 22, 3853–3858 [DOI] [PubMed] [Google Scholar]
- 130. Noh E. L., Lim D. S., Jeong G., Lee J. S. (2009) An HDAC inhibitor, trichostatin A, induces a delay at G(2)/M transition, slippage of spindle checkpoint, and cell death in a transcription-dependent manner. Biochem. Biophys. Res. Commun. 378, 326–331 [DOI] [PubMed] [Google Scholar]
- 131. Ishii S., Kurasawa Y., Wong J., Yu-Lee L. Y. (2008) Histone deacetylase 3 localizes to the mitotic spindle and is required for kinetochore-microtubule attachment. Proc. Natl. Acad. Sci. U.S.A. 105, 4179–4184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Ropero S., Esteller M. (2007) The role of histone deacetylases (HDACs) in human cancer. Mol. Oncol. 1, 19–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Lu J., McKinsey T. A., Nicol R. L., Olson E. N. (2000) Signal-dependent activation of the MEF2 transcription factor by dissociation from histone deacetylases. Proc. Natl. Acad. Sci. U.S.A. 97, 4070–4075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Heineke J., Molkentin J. D. (2006) Regulation of cardiac hypertrophy by intracellular signaling pathways. Nat. Rev. Mol. Cell Biol. 7, 589–600 [DOI] [PubMed] [Google Scholar]
- 135. McKinsey T. A., Olson E. N. (2005) Toward transcriptional therapies for the failing heart: chemical screens to modulate genes. J. Clin. Investig. 115, 538–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Chien K. R. (1999) Stress pathways and heart failure. Cell 98, 555–558 [DOI] [PubMed] [Google Scholar]
- 137. Frey N., Olson E. N. (2003) Cardiac hypertrophy: the good, the bad, and the ugly. Annu. Rev. Physiol. 65, 45–79 [DOI] [PubMed] [Google Scholar]
- 138. Black B. L., Olson E. N. (1998) Transcriptional control of muscle development by myocyte enhancer factor-2 (MEF2) proteins. Annu. Rev. Cell Dev. Biol. 14, 167–196 [DOI] [PubMed] [Google Scholar]
- 139. McKinsey T. A., Zhang C. L., Olson E. N. (2002) MEF2: a calcium-dependent regulator of cell division, differentiation, and death. Trends Biochem. Sci. 27, 40–47 [DOI] [PubMed] [Google Scholar]
- 140. Frey N., McKinsey T. A., Olson E. N. (2000) Decoding calcium signals involved in cardiac growth and function. Nat. Med. 6, 1221–1227 [DOI] [PubMed] [Google Scholar]
- 141. Backs J., Backs T., Neef S., Kreusser M. M., Lehmann L. H., Patrick D. M., Grueter C. E., Qi X., Richardson J. A., Hill J. A., Katus H. A., Bassel-Duby R., Maier L. S., Olson E. N. (2009) The delta isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload. Proc. Natl. Acad. Sci. U.S.A. 106, 2342–2347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Fielitz J., Kim M. S., Shelton J. M., Qi X., Hill J. A., Richardson J. A., Bassel-Duby R., Olson E. N. (2008) Requirement of protein kinase D1 for pathological cardiac remodeling. Proc. Natl. Acad. Sci. U.S.A. 105, 3059–3063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Gold J. I., Gao E., Shang X., Premont R. T., Koch W. J. (2012) Determining the absolute requirement of G protein-coupled receptor kinase 5 for pathological cardiac hypertrophy: short communication. Circ. Res. 111, 1048–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Schmieder R. E., Messerli F. H., Garavaglia G. E., Nunez B. D. (1988) Dietary salt intake—a determinant of cardiac involvement in essential-hypertension. Circulation 78, 951–956 [DOI] [PubMed] [Google Scholar]
- 145. Sucharov C. C., Dockstader K., Nunley K., McKinsey T. A., Bristow M. (2011) beta-Adrenergic receptor stimulation and activation of protein kinase A protect against alpha1-adrenergic-mediated phosphorylation of protein kinase D and histone deacetylase 5. J. Card. Fail. 17, 592–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Sucharov C. C., Dockstader K., McKinsey T. A. (2008) YY1 protects cardiac myocytes from pathologic hypertrophy by interacting with HDAC5. Mol. Biol. Cell 19, 4141–4153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Pang J., Yan C., Natarajan K., Cavet M. E., Massett M. P., Yin G., Berk B. C. (2008) GIT1 mediates HDAC5 activation by angiotensin II in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 28, 892–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Wang W., Ha C. H., Jhun B. S., Wong C., Jain M. K., Jin Z. G. (2010) Fluid shear stress stimulates phosphorylation-dependent nuclear export of HDAC5 and mediates expression of KLF2 and eNOS. Blood 115, 2971–2979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Delehanty L. L., Bullock G. C., Goldfarb A. N. (2012) Protein kinase D-HDAC5 signaling regulates erythropoiesis and contributes to erythropoietin cross-talk with GATA1. Blood 120, 4219–4228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Gao C., Liu Y., Lam M., Kao H. Y. (2010) Histone deacetylase 7 (HDAC7) regulates myocyte migration and differentiation. Biochim. Biophys. Acta 1803, 1186–1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Cao X., Chen D. (2005) The BMP signaling and in vivo bone formation. Gene 357, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Wan M., Cao X. (2005) BMP signaling in skeletal development. Biochem. Biophys. Res. Commun. 328, 651–657 [DOI] [PubMed] [Google Scholar]
- 153. Jensen E. D., Schroeder T. M., Bailey J., Gopalakrishnan R., Westendorf J. J. (2008) Histone deacetylase 7 associates with Runx2 and represses its activity during osteoblast maturation in a deacetylation-independent manner. J. Bone Min. Res. 23, 361–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Vega R. B., Matsuda K., Oh J., Barbosa A. C., Yang X. L., Meadows E., McAnally J., Pomajzl C., Shelton J. M., Richardson J. A., Karsenty G., Olson E. N. (2004) Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell 119, 555–566 [DOI] [PubMed] [Google Scholar]
- 155. West A. E., Griffith E. C., Greenberg M. E. (2002) Regulation of transcription factors by neuronal activity. Nat. Rev. Neurosci. 3, 921–931 [DOI] [PubMed] [Google Scholar]
- 156. Chawla S., Vanhoutte P., Arnold F. J., Huang C. L., Bading H. (2003) Neuronal activity-dependent nucleocytoplasmic shuttling of HDAC4 and HDAC5. J. Neurochem. 85, 151–159 [DOI] [PubMed] [Google Scholar]
- 157. Cho Y., Cavalli V. (2012) HDAC5 is a novel injury-regulated tubulin deacetylase controlling axon regeneration. EMBO J. 31, 3063–3078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Dietrich J. B., Takemori H., Grosch-Dirrig S., Bertorello A., Zwiller J. (2012) Cocaine induces the expression of MEF2C transcription factor in rat striatum through activation of SIK1 and phosphorylation of the histone deacetylase HDAC5. Synapse 66, 61–70 [DOI] [PubMed] [Google Scholar]
- 159. Bolger T. A., Yao T. P. (2005) Intracellular trafficking of histone deacetylase 4 regulates neuronal cell death. J. Neurosci. 25, 9544–9553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Davies S. W., Turmaine M., Cozens B. A., DiFiglia M., Sharp A. H., Ross C. A., Scherzinger E., Wanker E. E., Mangiarini L., Bates G. P. (1997) Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 90, 537–548 [DOI] [PubMed] [Google Scholar]
- 161. Wilson A. J., Byun D. S., Nasser S., Murray L. B., Ayyanar K., Arango D., Figueroa M., Melnick A., Kao G. D., Augenlicht L. H., Mariadason J. M. (2008) HDAC4 promotes growth of colon cancer cells via repression of p21. Mol. Biol. Cell 19, 4062–4075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Vissing K., McGee S. L., Roepstorff C., Schjerling P., Hargreaves M., Kiens B. (2008) Effect of sex differences on human MEF2 regulation during endurance exercise. Am. J. Physiol. Endocrinol. Metab. 294, E408-E415 [DOI] [PubMed] [Google Scholar]
- 163. Pirinen E., Lo Sasso G., Auwerx J. (2012) Mitochondrial sirtuins and metabolic homeostasis. Best Pract. Res. Clin. Endocrinol. Metab. 26, 759–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Zhu Y., Yan Y., Gius D. R., Vassilopoulos A. (2013) Metabolic regulation of sirtuins upon fasting and the implication for cancer. Curr. Opin. Oncol. 25, 630–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Poulsen R. C., Knowles H. J., Carr A. J., Hulley P. A. (2014) Cell differentiation versus cell death: extracellular glucose is a key determinant of cell fate following oxidative stress exposure. Cell Death Dis. 5, e1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Mathias R. A., Greco T. M., Oberstein A., Budayeva H. G., Chakrabarti R., Rowland E. A., Kang Y., Shenk T., Cristea I. M. (2014) Sirtuin 4 is a lipoamidase regulating pyruvate dehydrogenase complex activity. Cell 159, 1615–1625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Vogelauer M., Krall A. S., McBrian M. A., Li J. Y., Kurdistani S. K. (2012) Stimulation of histone deacetylase activity by metabolites of intermediary metabolism. J. Biol. Chem. 287, 32006–32016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Rajendran P., Williams D. E., Ho E., Dashwood R. H. (2011) Metabolism as a key to histone deacetylase inhibition. Crit. Rev. Biochem. Mol. Biol. 46, 181–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Nilsson A. C., Östman E. M., Knudsen K. E., Holst J. J., Bjorck I. M. (2010) A cereal-based evening meal rich in indigestible carbohydrates increases plasma butyrate the next morning. J. Nutr. 140, 1932–1936 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.