

Abstract
β-catenin is the key component of the canonical Wnt pathway and plays a crucial role in a multitude of developmental and homeostatic processes. The different tasks of β-catenin are orchestrated by its subcellular localization and participation in multiprotein complexes. To gain a better understanding of β-catenin's role in living cells we have generated a new set of single domain antibodies, referred to as nanobodies, derived from heavy chain antibodies of camelids. We selected nanobodies recognizing the N-terminal, core or C-terminal domain of β-catenin and applied these new high-affinity binders as capture molecules in sandwich immunoassays and co-immunoprecipitations of endogenous β-catenin complexes. In addition, we engineered intracellularly functional anti-β-catenin chromobodies by combining the binding moieties of the nanobodies with fluorescent proteins. For the first time, we were able to visualize the subcellular localization and nuclear translocation of endogenous β-catenin in living cells using these chromobodies. Moreover, the chromobody signal allowed us to trace the accumulation of diffusible, hypo-phosphorylated β-catenin in response to compound treatment in real time using High Content Imaging. The anti-β-catenin nanobodies and chromobodies characterized in this study are versatile tools that enable a novel and unique approach to monitor the dynamics of subcellular β-catenin in biochemical and cell biological assays.
Wnt signaling regulates cell proliferation, differentiation, and tissue homeostasis during metazoan development ranging from embryogenesis to the adult organism. β-catenin is the key effector molecule of the canonical Wnt pathway and it exerts two crucial roles within the cell. Firstly, it functions in cell adhesion at the plasma membrane where it connects cadherins via α-catenin to the cytoskeleton (1) and secondly, it mediates the expression of genes controlled by Wnt-responsive elements as a transcriptional co-activator (2, 3).
To fulfill these different tasks properly, well-balanced intracellular levels of β-catenin are required. The cellular concentration of β-catenin is tightly controlled by a destruction complex consisting of the scaffold protein Axin, Adenomatous Polyposis Coli protein, protein phosphatase 2A, casein kinase 1, and glycogen synthase kinase 3β (GSK3β), which constitutively phosphorylates newly synthesized β-catenin at key amino-terminal Ser and Thr residues (Ser33, Ser37, Thr41, Ser45 – the so called “SSTS-motif”) flagging it for proteasome-mediated degradation (4–7). Upon extrinsic activation of the Wnt receptors the destruction complex is functionally inactivated (8–12). This leads to the accumulation of hypo-phosphorylated β-catenin in the cytoplasm followed by its translocation into the nucleus where it interacts with members of the Lymphoid enhancer factor/T-cell factor (LEF/TCF)1 family to activate transcription of Wnt-responsive genes (13–17).
In pathological conditions, β-catenin is enriched when key components of the destruction complex are defective or the Ser and Thr residues of the N-terminal SSTS-motif are mutated. Consequently, increased nuclear and global levels of β-catenin are found in many types of human epithelial cancers including breast, colorectal and hepatocellular carcinoma (3, 18–22). The emerging role as a mediator of transcription of numerous genes involved in cell proliferation, epithelial-mesenchymal transition, and tumor progression converts β-catenin and its interactors into interesting targets for therapeutic intervention (reviewed in (23)). Hence, there is an ongoing need for reliable tools to follow the dynamics of β-catenin in living cells. The most widespread approach to study the composition of β-catenin-containing multiprotein complexes (MPCs) are biochemical assays. For such analyses, recombinant β-catenin is either applied as a bait protein or antibodies targeting endogenous β-catenin are used in immunoprecipitation studies. Interacting components can then be identified by immunodetection or mass spectrometry analysis. Besides biochemical analyses, the dynamic subcellular redistribution of β-catenin in response to extrinsic or intrinsic signals are of particular interest. Numerous studies report the use of GFP- or Yellow Fluorescent Protein-fusions of β-catenin in this context (24–27). However, because of its complex regulation β-catenin is not a suitable target to be ectopically expressed as a fluorescently labeled fusion protein because even minor changes of cellular levels can have dramatic effects on the subcellular distribution and transcriptional activity (25).
We generated anti-β-catenin nanobodies to follow the dynamics of β-catenin using biochemical and cell biological assays. The advantage of nanobodies lies in their single domain character, ease of generation, stability, and small size (28–30). Their simplicity in structure and availability of their sequence makes nanobodies amendable to genetic modification and intracellular expression (31). In our screen, we identified five nanobodies specific for the N-terminal, core or the C-terminal domain of β-catenin. We demonstrate the application of these novel binding molecules in various biochemical approaches including SPR measurements, sandwich immunoassays and co-immunoprecipitation followed by MPC analysis of endogenous β-catenin. For visualization in living cells, we genetically fused the nanobodies to fluorescent proteins generating so-called “chromobodies” (31–33). Following the chromobody signal, we were able to trace subcellular localization and nuclear translocation of endogenous β-catenin for the first time in living cells. Additionally, we traced the accumulation of diffusible, hypophosphorylated β-catenin in response to compound treatment in real time by monitoring changes in the intensity of chromobody fluorescence using High Content Imaging. We propose that changes in fluorescence are mediated by an antigen-dependent stabilization of the chromobody protein levels in living cells.
MATERIALS AND METHODS
VHH Libraries
Alpaca immunizations with purified β-catenin protein and VHH-library construction were carried out as described previously (31). Animal immunization has been approved by the government of Upper Bavaria (Permit number: 55.2-1-54-2531.6-9-06). In brief, 6 weeks after immunization of two animals (Vicugna pacos) with either GST-β-catenin or C-terminal histidine-tagged β-catenin (β-catenin-His6), ∼100 ml blood were collected and lymphocytes were isolated by Ficoll gradient centrifugation using the Lymphocyte Separation Medium (PAA Laboratories GmbH, Pasching, Austria). Total RNA was extracted using TRIzol (Invitrogen, Carlsbad, CA) and mRNA was reverse transcribed to cDNA using a First-Strand cDNA Synthesis Kit (GE Healthcare, Freiburg, Germany). The VHH repertoire was isolated in three subsequent PCR reactions using following primer combinations: 1) CALL001 (5′-GTC CTG GCT GCT CTT CTA CA A GG-3′) and CALL002 (5′-GGT ACG TGC TGT TGA ACT GTT CC-3′), 2) SM017 and SM018 (5′-CCA GCC GGC CAT GGC TCA GGT GCA GCT GGT GGA GTC TGG-3′ and 5′-CCA GCC GGC CAT GGC TGA TGT GCA GCT GGT GGA GTC TGG-3′, respectively) and reverse primer CALL002, and 3) A4short (5′-CAT GCC ATG ACT CGC GGC CAC GCC GGC CAT GGC-3′) and reverse Primer 38 (5′-GGA CTA GTG CGG CCG CTG GAG ACG GTG ACC TGG GT-3′) introducing SfiI and NotI restriction sites. The VHH library was subcloned into the SfiI/NotI sites of the pHEN4 phagemid vector (34).
VHH Screening
The VHH domains were expressed on phages after infecting the cells of the “immune” library in pHEN4 with M13K07 helper phages. VHH domains with specificity for β-catenin were enriched by two consecutive rounds of in vitro selection using full-length β-catenin coated on microtiter plates (10 μg/well). Bound phages were eluted with 100 mm tri-ethylamine, TEA (pH 10.0). The eluate was immediately neutralized with 1 m Tris/HCl (pH 7.4) and used to infect exponentially growing TG1 cells. The enrichment of phage particles carrying the antigen-specific VHH domains was monitored by comparing the number of phages eluted from wells with captured versus noncaptured antigen. Following panning 96 individual clones of each antigen were screened by standard ELISA procedures using a horseradish peroxidase-labeled anti-M13 monoclonal antibody (GE Healthcare).
Expression Plasmids
For bacterial expression of VHH domains (nanobodies, Nbs), sequences were cloned into the pHEN6 vector (34), thereby adding a C-terminal 6xHis-tag for IMAC purification as described previously (35). The expression plasmids for the β-catenin fusions have been described previously (36, 37). For protein production, E. coli BL21(DE3) CodonPlus-RIL cells (Stratagene, Amsterdam, The Netherlands) were used. Expression and purification of β-catenin-specific Nbs was carried out as described previously (35). For mammalian expression, translational fusions of β-catenin-specific nanobodies (BCs) with TagGFP (Evrogen, Moscow, Russia) or eGFP were constructed by introducing BglII/HindIII restriction sites in the target backbone vector (CMV pEGFP-C1). BglII and HindIII restriction sites were introduced by PCR using following set of forward primers comprising a BglII restriction site: 5′-GGG GAG ATC TCC GGC CAT GGC TCA GGT GCA GCT GGT GGA GTC TGG-3′, 5′-GGG GAG TTC TCC GGC CAT GGC TCA GGT GCA GCT GCA GGA GTC TGG-3′, 5′-GGG GAG ATC TCC GGC CAT GGC TCA TGT GCA GCT GCA GGA GTC TGG-3′ in combination with following set of reverse primers including a HindIII site: 5′-GGG GGA AGC TTC TTG AGG AGA CGG TGA CCT GCA T-3′, 5′-GGG GGA AGC TTC TTG AGG AGA CGG TGA CCT GGG-3′, 5′-GGG GGA AGC TTC TGC TGG AGA CGG TGA CCT GGG T-3′. The PCR products were purified, digested with BglII and HindIII, and ligated in the BglII/HindIII sites of the indicated vector.
The TOPflash luciferase reporter assay was carried out with 8xTCF/LEF-driven luciferase reporters. Constructs coding for the SuperTop-flash and SuperFop-flash have been described by Braeuning et al. (38). For constitutive expression of renilla luciferase in the reporter assay a pRL-CMV control vector (Promega, Madison, WI) was used. The plasmid for transient GFP-β-catenin expression was constructed by using BamHI/KpnI restriction sites in the target backbone vector CMV pEGFP-C1. The restriction sites were introduced into CTNNB1 cDNA using PCR with following primers: forward primer comprising a KpnI restriction site 5′-CCC CGG ATC CTC ACA GGT CAG TAT CAA AC-3′ and reverse primer 5′-CCC CGG TAC C GCT ACT CAA GCT GAT TT -3′ comprising a BamHI restriction site. The lac operator corresponding mCherry-lacI-β-catenin construct was generated by PCR-amplification of CTNNB1 cDNA using the following primers comprising restriction sites SalI/BamHI: forward primer comprising SalI restriction site 5′-GGG TCG ACG ATC CCA TCT ACA CAG TTT GAT G-3′ and reverse primer comprising BamHI restriction site 5′-CCG GAT CCT CAC AGG TCA GT A TCA AAC CAG GC-3′. Subsequently, the PCR-product was cloned into mCherry-C1-lacI-NLS vector (ChromoTek GmbH, Martinsried, Germany) using SalI/BamHI restriction sites. All resulting constructs were sequenced and tested for expression in HEK293T cells followed by immunoblot analysis.
SDS-PAGE and Immune Blotting
Denaturing polyacrylamid gel electrophoresis (SDS-PAGE) was performed according to standard procedures. Protein samples were boiled in 2×SDS-sample buffer containing 60 mm Tris/HCl, pH 6.8; 2% (w/v) SDS; 5% (v/v) 2-mercaptoethanol, 10% (v/v) glycerol, and 0.02% bromphenole blue. For immunoblotting proteins were transferred on nitrocellulose membrane (Bio-Rad Laboratories, Munich, Germany).
Antibodies
For immunoblotting the following primary antibodies were used: anti-GFP clone 3H9 or clone 3H5, anti-RFP clone 3F5 or clone 5F8, anti-GST clone 6G9 (ChromoTek), anti-TagCGY (Evrogen), anti-β-catenin, clone 14 (BD-Biosciences, Heidelberg, Germany), anti-active β-catenin (anti-ABC) clone 8E7 (MerckMillipore, Darmstadt, Germany), anti-nonphospho (active) β-catenin (Ser33/37/Thr41) clone D13A1 (Cell Signaling, Leiden, The Netherlands), anti-E-cadherin, clone 24E10 (Cell Signaling), anti-TCF4, clone 6H5–3 (Upstate, Darmstadt, Germany), anti-GSK3β, clone 27C10 (Cell Signaling), anti-α-catenin, clone 6D202 (US Biological, Salem, MA), anti-TCF1, clone C46C7 (Cell Signaling), anti-Axin1, clone C7B12 (Cell Signaling), and anti-GAPDH (Abcam, Cambridge, UK). For detection fluorophore-labeled species-specific secondary antibodies (Alexa-647, Alexa-546, Alexa-488; goat-anti-mouse, goat-anti-rabbit, goat-anti-rat, Invitrogen; IRDye 680CW donkey-anti-mouse, IRDye 800CW donkey-anti-rabbit, LI-COR) were used. Blots were scanned on the Typhoon-Trio laser scanner (GE Healthcare) or the Odyssey CLx (LI-COR, Lincoln, NE) and quantitatively analyzed using Image Quant TL 7.0 (GE Healthcare) or Image Studio Software for Odyssey 4.0 (LI-COR).
Cells Culture and Transfection
HEK293T, HeLa, SW480, HCT116, and BHK cells were cultivated according to standard protocols. Briefly, growth media contained DMEM (high glucose, pyruvate, 10% fetal calf serum (FCS)), supplemented with l-Glutamine and antibiotics if not stated differently. Cells were trypsinized for passaging and cultivated at 37 °C in a humidified chamber with a 5% CO2 atmosphere. Plasmid-DNA was transfected with Lipofectamine 2000 (Invitrogen) or polyethylenimine, PEI (Sigma Aldrich). Stable HEK293T_BC1-CB and HeLa_BC1-CB cells were maintained in medium supplemented with 3 μg/ml Blasticidine (Sigma Aldrich), HeLa_GFP cells in medium containing 500 μg/ml G418 (Sigma Aldrich, St. Louis, MO). To generate DNA/PEI complexes for transfection in 6-well plates, 4 μg DNA were mixed with 16 μl PEI in 150 μl DMEM per well, incubated for 15 min, and added to the cells. Lipofectamine 2000 were used according to the manufacturer′s protocol.
Production of Lentivirus and Generation of Stable Cell Lines
The BC1-CB sequence was transferred into pLenti6/V5-DEST by Gateway recombination and lentiviral particles were produced using Virapower lentiviral packaging mix according to manufacturer′s protocols (Invitrogen) with a yield of ∼5 × 107 transducing units/ml. Twenty-four hours after transduction, HeLa or HEK293T cells we subjected to a 2-week selection period with 3 μg/ml Blasticidine. HeLa_GFP cells were generated by plasmid transfection and subsequent selection with 500 μg/ml G418.
Immunoprecipitation
1x10E6 - 1x10E7 HEK293T or HeLa cells were either left untreated or incubated with CHIR-99021 dissolved in H2O (GSK3β inhibitor, Tocris Bioscience, Bristol, UK) for 24 h. Cells were washed and harvested in phosphate buffered saline (PBS), snap-frozen in liquid nitrogen and stored at −20 °C. Cell pellets were homogenized in 200 μl lysis buffer (10 mm Tris/Cl, pH 7.5, 150 mm NaCl, 0.5% Nonidet P-40, 1 μg DNaseI, 2 mm MgCl2, 2 mm PMSF, 1×phosSTOP phosphatase inhibitor (Roche, Mannheim, Germany), 1×protease inhibitor mix M (Serva) by repeated pipetting for 40 min on ice. After a centrifugation step (10 min at 18,000 × g) the protein concentration of each lysate was determined using Coomassie Plus according to manufacturer's protocol (Thermo Scientific, Darmstadt, Germany) and the protein solutions were adjusted with dilution buffer (10 mm Tris/Cl, pH 7.5, 150 mm NaCl, 2 mm PMSF) to equal concentrations. Two percent of the lysate were added to SDS-containing sample buffer (referred to as total). To deplete β-catenin associated with the plasma membrane 200–1000 μl ConA (Concanavalin A)-Sepharose beads (GE Healthcare) were added followed by incubation for 2–12 h on an end-over-end rotor at 4 °C. After a centrifugation step (2 min, 2500 × g) the precleared supernatant was transferred to a new cup. Two percent were added to SDS-containing sample buffer (referred to as input). ConA bound proteins were eluted by boiling the ConA-beads in 0.5 ml SDS-sample buffer. For immunoprecipitation 50–200 μl of the agarose-coupled nanobodies and 5 μg of anti-β-catenin antibodies precoupled to 50 μl of an equilibrated mixture of protein A/G Sepharose (GE Healthcare) were added and incubated for 4–12 h on an end-over-end rotor at 4 °C. As a negative control a nonrelated nanobody (specific for BSA) coupled to agarose was used. The bead pellet was washed two times in 0.5 ml dilution buffer. After the last washing step the beads were transferred to a new cup, resuspended in 2×SDS-containing sample buffer and boiled for 10 min at 95 °C. Samples (0.5% total, 2% ConA-bound, 0.5% input, 10% bound) were analyzed by SDS-PAGE followed by Western blotting. Immunoblots were probed with indicated anti-β-catenin and anti-GAPDH antibodies for normalization of the input fractions and as negative control to detect unspecific binding to Nb-matrix.
Immunofluorescent Staining
For immunofluorescence HeLa cells expressing BC1-CB, BC6-CB, BC9-CB, or GFP were grown to near confluence in 96-well format (Greiner Bio One, Frickenhausen, Germany), fixed with 4% formaldehyde/PBS, permeabilized with 3% BSA/PBS plus 0.1% Triton X-100 and incubated with anti-β-catenin antibody (clone H102, Santa Cruz Inc, Santa Cruz, CA) overnight at 4 °C. For detection, an Alexa-488 tagged, goat anti-rabbit secondary antibody (Invitrogen) was used. Subsequently, nuclei were stained with DAPI (Invitrogen) and fluorescent imaging was performed using ImageXpress MicroXL (Molecular Devices, Biberach, Germany).
Co-immunoprecipitation
Co-immunoprecipitations were carried out as described above. Immunostaining of interacting partner was done with indicated antibodies. Signal of GAPDH antibody was used for normalization of the input fractions and as negative control to detect unspecific binding to Nb-matrix.
Intracellular IP Experiments
1x10E6 - 1x10E7 HEK293T or HeLa cells were transiently transfected with equal amount of expression vectors encoding for the β-catenin chromobodies BC1-CB, BC2-CB, BC6-CB, BC9-CB, BC13-CB, or eGFP using Lipofectamine 2000 (Invitrogen) or polyethylenimine (Sigma Aldrich). Transfection efficiency was monitored on the next day by fluorescence microscopy and cells were split 1:2 in p100 cell culture dishes and grown for additional 24 h. Subsequently, cells were either mock treated or incubated with 10 μm CHIR-99021 dissolved in H2O (GSK3β inhibitor, Tocris Bioscience) for 24 h in growth-media. Cell lysates were generated as described and β-catenin chromobodies or eGFP were precipitated using the GFP-Trap (ChromoTek) according to manufacturer's instructions. Input (I) and bound (B) fractions were subjected to SDS-PAGE followed by immunoblotting for β-catenin and tagGFP (chromobody).
Fluorescent 2-Hybrid Analysis
The fluorescent 2-hybrid assay was carried out as described previously (39, 40). In brief, transgenic BHK cells (clone #2) containing lac operator repeats were cultured under selective conditions in DMEM supplemented with 10% fetal calf serum and 150 μg/ml hygromycin B (PAA Laboratories GmbH). For microscopy, cells were grown to 50–70% confluence in 96-well microplates (Greiner) and then cotransfected with mCherry-lacI-β-catenin and the indicated chromobody expression constructs. Twenty-four hours after transfection cells were imaged with ImageXpress Micro XL. Immobilized mCherry-lacI-β-catenin appears as a defined spot in the nucleus. Colocalization of the chromobody with this defined spot indicates the interaction of the two proteins.
Resazurin Assay
24 and 48 h post transfection with BC1-, BC6-, BC9-, or EGFP-coding plasmids HeLa cells were incubated for one hour in cell culture medium containing resazurin (Sigma Aldrich) at a final concentration of 10 μg/ml. Fluorescence was measured with a PHERAstar plate reader (BMG Labtech).
Microsphere-based Sandwich Immunoassays
Nbs were covalently immobilized on microspheres using a modified manufacturer's protocol described in Poetz et al. (41). Immobilized Nbs were incubated with purified proteins representing full-length β-catenin or the following domains of β-catenin: N terminus amino acid (aa) 1–119, armadillo domain aa 120–683, and C terminus aa 683–781. Purified Glutathion-S-transferase (GST) was used as a negative control. Concentrations range from 0.25 μg/ml to 2 μg/ml. Bound proteins were detected with domain-specific antibodies including ABC antibody clone 8E7 (Merck Millipore) specific for the N terminus, anti-β-catenin 9G10 (Merck Millipore) targeting the armadillo domain and anti-β-catenin clone 14 (Becton Dickinson) specifically recognizing the C terminus. To detect unspecific binding of the Nbs to GST an antibody against GST (6G9, ChromoTek) was used. Secondary species-specific antibodies (anti-mouse, anti-rat, anti-rabbit) tagged with phycoerythrin (PE) (Dianova, Hamburg, Germany) were used for immunodetection of antibody/protein complexes. The microspheres were measured in a FlexMap 3D instrument (Luminex, Austin, United States). The evaluation of the data was done with Excel (Microsoft).
Surface Plasmon Resonance
The affinity measurement of the nanobodies was performed using surface plasmon resonance spectroscopy with a Biacore 3000 instrument (GE Healthcare). Recombinant β-catenin was covalently coupled on dextran fibers of a CM5-chip (GE Healthcare) according to the manufacturer's protocol. β-catenin was coupled to a response ranging from 1200 to 3000 RU. One flow cells was activated and blocked in the absence of protein to determine background. For kinetic measurement five concentrations ranging from 0.625 nm to 5 μm of the Nbs were injected. Each measurement was done in duplicates. As running/dilution buffer 10 mm HEPES, pH 7.4, 150 mm NaCl, 0.5% surfactant P20 was used. For the kinetics of BC13, 3 mm EDTA was added to the running and dilution buffer. Measurements were performed at 25 °C: For the association of the Nbs a flow rate of 30 μl/min for 3 min and for the dissociation a flow rate of 60 μl/min for 5 min was applied. The dissociation was induced by two injections of regeneration solution of 15 μl each at the identical flow rates. Different regeneration solutions were used: for BC1: 10 mm glycine, pH 2.0; BC2: 100 mm H3PO4; BC6, BC9: 10 mm glycine, pH 2.5; BC13: 3 m MgCl2. The data was evaluated using the software Bia evaluation 4.1 and the 1:1 Langmuir binding model.
Immobilization of Peptides onto OVA-microspheres
MagPlex microspheres (Luminex), with varying IDs per peptide (n = 29), were coated with Imject ovalbumin (OVA) (Thermo Fisher Scientific). Coating procedure was done according to a modified manufacturer's protocol (41). Subsequently, 250,000 microspheres of each ID were washed one time with 1×PBS, the OVA was activated with 1660 μm sulfosuccinimidyl 4-[p-maleimidophenyl] butyrate (sulfo-SMPB) (Thermo Fisher Scientific) dissolved in dimethyl sulfoxide (Roth, Karlsruhe, Germany) and diluted to the final concentration with 1×PBS. The activation of OVA-microspheres was done for 1 h at RT on a plate incubator (Eppendorf AG, Hamburg, Germany) at 650 rpm. In parallel, the 29 peptides were reduced with tris(2-carboxyethyl)phosphine (TCEP). Each peptide was diluted in 1×PBS containing 40% (v/v) acetonitrile to a final concentration of 600 μm. An equimolar solution of TCEP in PBS was prepared and one volume of each the peptide solutions and the TCEP-solution were mixed and incubated for 20 min at RT at 250 rpm on a plate incubator. After activation, the OVA-microspheres were washed two times with 1×PBS 0.005% Triton X-100 to remove excess sulfo-SMPB and solubilized in reduced peptide solutions. Coupling procedure of the peptides to OVA was done for 1 h at RT with continuous shaking at 650 rpm. Afterward, the microspheres were again washed two times with 1×PBS 0.005% Triton X-100 and transferred into a blocking buffer containing 10 mg/ml BSA dissolved in 1×PBS to block all free activated OVA. The blocking of OVA was done for 10 min at room temperature at 850 rpm followed by an additional washing step with 1×PBS containing 0.005% Triton and transferred into 1×Roche-buffer (Roche) containing 0.05% sodium azide for storage.
Epitope Mapping
For the epitope mapping Nbs (BC1 and BC2) were biotinylated with sulfo-NHS-LC-biotin (Thermo Fisher Scientific) using manufacturer's protocol. The biotinylated Nbs were then used as detection reagent in a sandwich-immunoassay similar to the domain mapping procedure using the peptide coated OVA-microspheres to screen for specific epitopes. As negative controls, microspheres comprising Ovalbumin and the Myc-peptide (EQKLISEEDL) covalently coupled to OVA-microspheres were used. The biotinylated Nbs were applied at a concentration of 1 μg/ml. Peptide-bound Nbs were detected with 2.5 μg/ml streptavidin-phycoerythrin (PE) solution (Prozyme, Hayward, CA) dissolved in 1×Roche buffer 0.05% Tween. Evaluation of the PE-signals were done in a Flexmap 3D (Luminex).
β-catenin Suspension Bead Array-Based Assay
Cell lysates (10 μg protein extract) either total or membrane protein-depleted fractions were incubated with immobilized nanobodies BC1, BC2, or nonrelated nanobody overnight at 4 °C. After removal of the bound fraction, remaining supernatants were analyzed using bead array-based sandwich immunoassays for total and hypo-phosphorylated β-catenin (nonphospho Ser33/Ser37/Thr41), as described earlier (37). E-cadherin/β-catenin complex was measured by miniaturized co-immunoprecipitation as described previously (41).
TOP-Flash Assay
The transcriptional activity of β-catenin was measured with a TOPflash luciferase reporter assay (2). A reporter construct containing eight TCF-Promoter-luciferase-reporter sites (SuperTOPflash/TOP) and a corresponding control construct with mutated TCF-binding sites (SuperFopflash/FOP) was used. Top/Fop-constructs were kindly provided by Braeuning (38). HEK293T cells were transfected with TOP or FOP and CMV-Renilla luciferase (pRen) in a stoichiometric ratio of 7:1. Additionally, BC-chromobodies were transfected using a ratio of 1:1 to TOP or FOP. Twenty-four hours after transfection, cells were incubated with 10 mm LiCl. Four hours and 24 h after LiCl treatment a Dual Glo luciferase-assay was performed according to manufacturer's protocol (Promega).
Image Acquisition, Segmentation, and Analysis
2500 HeLa_BC1-CB cells/well were plated in a black μClear 96-well plate (Greiner). Twenty-four hours after plating, cells were either left untreated or were treated with 20 mm NaCl or LiCl, 10 μm CHIR, 1 μm LH846, or a combination of 10 μm CHIR and 1 μm LH846. In order to allow automated nucleocytoplasmic segmentation, nuclei of live cells were stained by the addition of 1 μg/ml Hoechst33258 (Sigma Aldrich) to the cell culture medium. Two hours after stimulation time-lapse image acquisition was started with an Image Xpress micro XL system and analyzed by MetaXpress software (64 bit, 5.1.0.41). Several images were acquired for each condition comprising a statistically relevant number of cells (∼500). An algorithm calculating the intensity above local background was used to segment entire cells and nuclei. The average fluorescence intensities in the cytoplasm and the nuclei were determined for each image and background fluorescence was subtracted. From these values the average cytoplasmic and nuclear fluorescence and standard errors were calculated for three biological replicates.
Quantitative Real-Time PCR
From inhibitor-treated or transfected HeLa_BC1-CB cells RNA was prepared using the RNeasy Mini Kit (Qiagen, Hilden, Germany). Subsequently, RNA samples were subjected to a DNase digest according to manufacturer's protocol (RQ1-DNase, Promega) to remove any residual genomic DNA. Using M-MuLV reverse transcriptase (New England Biolabs, Frankfurt, Germany) and hexanucleotide primers (dN6-primers, Sigma Aldrich) cDNA was synthesized. Real-time PCR analysis was conducted with a StepOne Plus system (Applied Biosystems, Invitrogen) using standard predesigned TaqMan probes for GAPDH and β-catenin and a custom-made probe for TagGFP (Invitrogen). Relative mRNA expression levels were determined by the 2(−ΔΔC(T)) method (42).
RESULTS
Identification of β-catenin-specific Nanobodies
To generate β-catenin-specific nanobodies, we immunized two alpacas (Vicugna pacos) with purified recombinant human β-catenin. A phagemid library was generated comprising ∼2 × 107 clones representing the full repertoire of the variable heavy chains of heavy chain antibodies (VHHs or nanobodies, Nbs) derived from one animal. The library was subjected to phage display and biopanning was performed using full-length β-catenin. Two subsequent phage display cycles revealed an enrichment of 14 unique nanobody sequences (Fig. 1A) that were positively tested for antigen binding in a solid-phase phage ELISA (data not shown).
Fig. 1.

Selection of nanobodies (Nbs) against β-catenin. A, Amino acid sequences of the complementarity determining region 3 from unique Nbs selected after two rounds of biopanning are shown. B, Recombinant expression and purification of Nbs. Coomassie staining of 1–2 mg of purified Nbs is shown. C, Precipitation of recombinant β-catenin with Nbs. Soluble lysate of β-catenin-expressing bacteria was incubated with Nbs immobilized on Sepharose beads. Bound fractions were subjected to SDS-PAGE followed by immunoblot analysis using a β-catenin-specific antibody, CTR: pulldown with nonrelated Nb. D, Microsphere-based sandwich immunoassay using β-catenin-specific Nbs as capture molecules. Nbs were immobilized on microspheres and incubated with increasing β-catenin concentrations ranging from 0.25 μg/ml to 2 μg/ml. Bound protein was detected with an anti-β-catenin antibody. Background level of control Nb was set to one. Shown are mean signal intensities of three independent replicates ± stds. Binding values of the five best Nbs are highlighted in dark gray.
Individual Nbs were cloned with a C-terminal 6xHis-tag, expressed in Eschericha coli (E. coli) and purified using immobilized metal ion affinity chromatography (IMAC) followed by size exclusion chromatography (35, 43) (Fig. 1B). To test the binding capabilities, we covalently immobilized the purified Nbs on agarose beads and used them to pull down recombinant β-catenin. Immunoblot analysis showed that all selected Nbs precipitate their corresponding antigen (Fig. 1C). Next, we tested whether the β-catenin-specific Nbs are functional as capture molecules in a microsphere-based sandwich immunoassay system. To this end, we immobilized the Nbs on magnetic microspheres (MagPlex) and incubated them with decreasing amounts of β-catenin. Bound β-catenin was detected with a C terminus-specific antibody (clone 14/BD). The results showed that the Nbs have different capabilities to capture β-catenin (Fig. 1D). For further studies, we chose the five best-performing nanobodies: BC1, BC2, BC6, BC9, and BC13. In combination with the C-terminal β-catenin-specific antibody as detector, bound β-catenin was detectable down to ∼1 ng/ml when using BC1 and BC2 as capture molecules, whereas applying BC6, BC9, or BC13 lower sensitivities (∼50 ng/ml) for the detection of β-catenin were observed (supplemental Fig. S1).
Affinity Measurements
For further characterization of the selected nanobodies, we determined their affinities to recombinant β-catenin using surface plasmon resonance (SPR). After immobilizing β-catenin, we measured the association/dissociation rates by injecting serial dilutions of five different concentrations for each binder. For BC1 and BC2, we determined affinities (KD values) in the low nanomolar range of ∼1.9 nm and ∼3.1 nm, respectively, which is in accordance with the strong binding signals observed in the microsphere-based sandwich immunoassay experiments. The C-terminal-specific BC13 shows a slightly lower affinity of ∼44 nm. For BC6 and BC9 we detected affinities in the low μm range (Fig. 2, supplemental Fig. S2).
Fig. 2.

Nanobodies bind β-catenin with high affinities. For surface plasmon resonance spectroscopy (SPR)-based affinity measurements, β-catenin was covalently coupled on a CM5-chip. Kinetic measurements were performed by injecting five concentrations of purified Nbs ranging from 0.625 nm to 5 μm. The obtained data sets were evaluated using the 1:1 Langmuir binding model. As an example the sensogram of the BC1 nanobody at indicted concentrations is shown. The table summarizes affinities (KD), association (Kon) and dissociation constants (Koff) determined for individual Nbs.
Domain and Epitope Mapping of β-catenin Binders
Structural analysis of β-catenin revealed a tripartite structure: a negatively charged N terminus (aa 1–140), the core domain (aa 141–664) composed of 12 Armadillo repeats and a short C terminus (aa 665–781) (36, 44, 45). To test whether the selected binders recognize these individual domains of β-catenin, we performed a microsphere-based sandwich immunoassay capturing either full-length β-catenin or the indicated domains with the selected Nbs. With this approach, we could show that BC1 and BC2 exclusively recognize the N-terminal domain (aa 1–119) whereas BC6 and BC9 specifically capture the isolated armadillo domain (aa 120–683). With BC13, we identified one binder, which recognizes the C terminus (aa 684–781) of β-catenin (Fig. 3A). We did not detect any domain cross-specificity of the selected Nbs suggesting a strict domain-specific binding.
Fig. 3.

Nanobodies bind distinct domains of β-catenin. A, Microsphere-immobilized Nbs were incubated with GST-fusion constructs comprising full-length β-catenin or indicated domains. Captured β-catenin constructs were detected with domain-specific antibodies. For direct comparison, fluorescence intensities obtained with domain-specific antibodies were normalized to signals obtained with an anti-GST-antibody. B, Identification of the epitope recognized by BC2. In a peptide screen 29 overlapping 15-mer peptides covering the N terminus from aa 1–127 of β-catenin were immobilized on microspheres with varying IDs per peptide and incubated with biotinylated BC2. Peptide-bound BC2 was detected with streptavidin-phycoerythrin (PE) solution. The Myc-peptide (EKLISEEDL) was used as negative control (neg CTR). C, Phosphorylation of the epitope abolishes binding by BC2. Binding analysis of BC2 to a peptide representing aa 15–29 with (+) or without (−) a phosphorylated Ser23 (phos Ser23) was performed as described in B. Columns represent mean signal intensities of three independent experiments ± stds.
In order to study endogenous β-catenin in a physiologically relevant context, interference with interaction partners, physiological conformation and subcellular dynamics should be as low as possible. In contrast to the armadillo domain and the C terminus, for which a large number of interaction partners have been described (reviewed in (16)), to our knowledge the N terminus of β-catenin is only addressed by very few and transient interactions. On this basis, we speculate that the N terminus-specific nanobodies BC1 and BC2 might be suitable to study β-catenin with minimal bias regarding competition with endogenous interaction partners or subcellular localization. Therefore, we further analyzed the binding properties of the two N terminus-specific nanobodies BC1 and BC2. To determine the minimal linear epitope of BC1 and BC2 we performed a pepscan analysis. For this purpose, we used 29 synthetic 15-mer peptides with a 11 amino acids overlap between consecutive peptides representing aa 1–127 of the β-catenin N terminus. All peptides were immobilized via an additional cysteine at the N terminus of the peptide on individual microsphere particles and incubated with biotinylated BC1 or BC2 in increasing concentrations (46).
For BC1, we received binding signals for the consecutive peptides representing the residues QVLYEWEQGFSQSFTQEQVADIDGQYA (aa 61–87) (supplemental Fig. S3A). With more than 25 amino acid residues the epitope is quite large and the detectable binding signals were rather low and do not reflect the high affinity determined for this binder. Therefore, it might be possible that the indicated peptide is part of an extended three-dimensional epitope and only partially bound by BC1 in the pepscan assay. For BC2, the analysis showed strong binding to two consecutive peptides comprising the residues 13–31 (Fig. 3B and supplemental Fig. S3B). Further truncation revealed that BC2 recognizes the amino acid sequence PDRKAAVSHWQQ (aa 16–27) (supplemental Fig. 3B). This observation is quite notable because only very few nanobodies are known to bind short linear peptide sequences (30). The recognized epitope is evolutionarily highly conserved and it harbors a central serine residue (Ser23) that is described to be post-translationally modified either upon targeted phosphorylation mediated by GSK3β in the same fashion as the SSTS-motif or by addition of O-linked-beta-N-acetylglucosamine (O-GlcNAc) (47, 48). For further analyses of the BC2 epitope specificity, we performed binding studies using the identified peptide with a phosphorylated Ser23 residue. This modification completely abolishes binding of BC2 suggesting that this nanobody recognizes β-catenin only when Ser23 is not phosphorylated (Fig. 3C). In summary, our data shows that BC1 and BC2 recognize two distinct, non-overlapping epitopes within the N-terminal domain of β-catenin. Although BC1 seems to recognize a three-dimensional epitope close to the SSTS-motif, BC2 binds a linear peptide at the very N terminus of β-catenin in a phosphorylation-dependent manner.
Precipitation of Endogenous β-catenin
Next, we investigated the potential of the selected nanobodies to precipitate endogenous β-catenin from human cell extracts. To modulate levels of β-catenin HEK293T cells were incubated with the GSK3β inhibitor CHIR (CHIR-99021) (49). After cell lysis soluble protein fractions were incubated with Concanavalin A coupled to agarose (ConA-beads) to remove membrane-associated cadherin-bound β-catenin (50). Membrane-depleted lysates were subjected to immunoprecipitation using all β-catenin-specific nanobodies. Bound fractions were analyzed by SDS-PAGE and immunoblotting. The analysis showed that all nanobodies precipitate endogenous β-catenin but with different efficiencies (Fig. 4A). Although only minor amounts of β-catenin are detectable in the bound fraction of BC6 and BC9, BC2, and BC13 efficiently precipitate endogenous β-catenin in the absence of GSK3β inhibitor and show comparable pulldown efficiencies to conventional antibodies (Fig. 4A, supplemental Fig. S4). Upon elevation of total β-catenin by GSK3β inhibition, increased amounts of precipitated β-catenin were detected for all binders. Notably, for BC1, which precipitates only a minor amount of β-catenin from nontreated cells, the inhibition of GSK3β increases the levels of bound β-catenin by an extend that exceeds the elevation in total β-catenin (Fig. 4A).
Fig. 4.

Nanobodies bind endogenous β-catenin. A, HEK293T cells were either left untreated (nt) or were incubated with CHIR (GSK3β-inhibitor) for 24 h. Cells were lysed under native conditions and membrane-bound fraction was depleted using Concanavalin A (ConA)-beads. Depleted supernatants were adjusted to 2 mg/ml and incubated with equal amounts of immobilized Nbs. Bound protein fractions were subjected to SDS-PAGE followed by immunoblot analysis using antibodies specific for β-catenin and GAPDH. Total: 0.5% of total lysate; ConA: 2.5% of ConA-bound fraction; input: 0.5% of ConA-depleted supernatant; BC1–BC13: 10% of Nb-bound fraction, CTR: 10% of bound fraction of a nonrelated Nb. Shown are representative blots of three independent experiments. B, Precipitation of β-catenin with immobilized Nbs as described in A from membrane-depleted lysates derived from HCT116 cells (upper panel) or SW480 cells (lower panel). Cells were either left untreated (nt) or were incubated with CHIR for 24 h. Immunoblot analysis of input and bound fraction with anti-β-catenin antibody is shown.
This could be explained either by an altered accessibility of the epitope or an increased affinity induced by posttranslational changes of the epitope. Because GSK3β inhibition induces high levels of hypo-phosphorylated β-catenin in the cytoplasm we suspected, that BC1 might have a preference for β-catenin when the SSTS-motif is not phosphorylated. To test this, we applied these nanobodies to precipitate β-catenin from human cell lines comprising different levels of hypo-phosphorylated β-catenin. We used human colorectal cancer HCT116 cells that harbor a mono-allelic deletion at residue Ser45 leading to an increased level of hypo-phosphorylated β-catenin and extracts derived from the colorectal adenocarcinoma cell line SW480 comprising mutations in the Adenomatous polyposis coli gene (51). Immunoblot analysis of the membrane-depleted lysate showed that the global level of diffusible β-catenin is only slightly increased in HCT116 cells after induction with CHIR whereas no induction was observed in SW480 cells (Fig. 4B). Unlike as previously observed in HEK293T cells the increase in β-catenin precipitated by BC1 is comparable to the increase of the diffusible β-catenin fraction in HCT116 cells upon induction with CHIR. A similar increase could be detected for β-catenin precipitated by BC2 or BC13 (Fig. 4B, upper panel). In SW480 cells BC1 and BC2 seem to precipitate more β-catenin from the membrane-depleted lysate upon treatment with CHIR although the global level of β-catenin was not affected. Such an increase in binding was not observed for the other nanobodies (Fig. 4B, lower panel).
To further dissect possible binding preferences of BC1 and BC2, we performed an immunoassay comparing the amount of global and differentially phosphorylated forms of β-catenin in the supernatant of (1) total lysate, (2) after removal of membrane-bound β-catenin, (3) after precipitation with BC1, (4) BC2, or a (4) nonrelated nanobody. Cellular levels of β-catenin were modulated by incubation with CHIR as described. For a multiplex immunoassay we took advantage of a binding panel comprising antibodies recognizing either total (Fig. 5A), Ser33-Ser37-Thr41 nonphosphorylated β-catenin (Fig. 5B), or E-cadherin-associated β-catenin (Fig. 5C) (37). Measuring the total fraction of β-catenin, we detected a ∼twofold increase upon induction with CHIR. Depletion with ConA beads led to a slight decrease of detected β-catenin in the lysate of both, nontreated and CHIR-treated cells. After precipitation with BC1 we observed a further reduction of β-catenin by ∼20% and with BC2 by ∼45% in nontreated and CHIR-treated cells. Notably, this is independent of the amount of applied nanobody matrix. However, the observed reduction is caused by specific binding of the nanobodies because we could not detect any depletion precipitating with a nonrelated nanobody matrix (Fig. 5A). Measuring the amount of nonphosphorylated β-catenin we detected a ∼10-fold increase upon induction in the total and the membrane-depleted fraction. After precipitation with the nanobodies, we observed a significant reduction of nonphosphorylated β-catenin by ∼80% for BC1 and ∼95% for BC2. As shown before, no depletion of β-catenin was observed after precipitation with a nonrelated nanobody (Fig. 5B). This indicates a preferential binding of BC1 and BC2 to N-terminally nonphosphorylated β-catenin. Capturing with an E-cadherin-specific antibody showed a similar reduction of E-cadherin-bound β-catenin in untreated and treated cells by ∼60% after incubation with ConA-beads (Fig. 5C). E-cadherin-bound β-catenin could not be further depleted using either more ConA-beads or longer incubation times (data not shown). This indicates that there is a residual fraction of cytoplasmic or nuclear E-cadherin (52, 53). Precipitation with BC1 does not deplete E-cadherin-bound β-catenin, whereas precipitation with BC2 leads to a further reduction by ∼50% (Fig. 5C). This suggests that BC2 but not BC1 binds to β-catenin associated with E-cadherin.
Fig. 5.

BC1 and BC2 preferentially bind nonphosphorylated β-catenin. HEK293T cells were treated with (bars in gray) and without CHIR for 24 h (bars in dark gray). Cells were lysed under native conditions. Soluble cell lysates (1 mg/ml) were incubated with ConA (ii), ConA and BC1 (iii), ConA and BC2 (iv), and ConA and a nonrelated Nb (v) to deplete different fractions of β-catenin. Ten micrograms of depleted fractions were subjected to a multiplex bead-based immunoassay capturing total A, Ser33/-Ser37/-Thr41 nonphosphorylated β-catenin, B, or E-cadherin/β-catenin complex C. Bound β-catenin in the total lysate (i) and the individual supernatants of the different depleted fractions (ii–v) was detected with an anti-β-catenin antibody. Shown are mean signal intensities of three independent replicates ± stds.
Coprecipitation of Multiprotein Complexes Comprising β-catenin with Nanobodies
According to its central role in multiple cellular processes β-catenin cooperates with a plethora of interaction partners in a spatially and temporally coordinated manner. One of the most widespread methods to reveal these interactions is co-immunoprecipitation. For such an approach it is important to use high-quality monoclonal antibodies against well-defined epitopes to dissect real interactions from unspecific binding and to avoid interference with potential binding partners. Although numerous antibodies against β-catenin are available, they do not always fulfill those criteria properly because of steric hindrances, large binding interfaces and subcellular accessibility. Alternatively, many studies are using ectopically expressed β-catenin fusion constructs (e.g. Flag- or Myc-tagged β-catenin) in combination with tag-specific antibodies to identify potential interacting partners. Considering the complex regulation of cellular β-catenin levels, such constructs do not necessarily reflect the behavior of the endogenous protein. Therefore, results from interaction studies have to be taken with caution. To overcome these limitations, we tested the N terminus-specific nanobodies to coprecipitate interaction partners of endogenous β-catenin from cellular lysates. We incubated total soluble protein fractions of HEK293T cells that were either left untreated or after inhibition of GSK3β, with immobilized BC1 and BC2. As a control we used an immobilized nonrelated nanobody. Bound fractions were subjected to SDS-PAGE followed by immunoblot analysis using antibodies directed against known or potential interaction partners of β-catenin. The Western blot analysis showed that both nanobodies are able to coprecipitate numerous interacting proteins that are described to bind to different domains of β-catenin (Fig. 6). With E-cadherin and α-catenin we detected two proteins that are described to interact with β-catenin at the plasma membrane. Notably, BC2 seems to coprecipitates more E-cadherin in comparison to BC1 irrespective of the induction of β-catenin. Because the membrane fraction was not removed in these experiments, it might be possible that BC2 precipitates β-catenin bound to E-cadherin located at the plasma membrane. However, analyzing the bound fractions for α-catenin no differences in precipitation efficiency of both binders were detectable.
Fig. 6.

Nanobodies coprecipitate interaction partners of β-catenin. HEK293T cells were either left untreated or were incubated with CHIR for 24 h. Cells were lysed under native conditions and soluble protein fractions were incubated with immobilized BC1, BC2, or a nonrelated Nb (CTR). Input and bound fractions (BC1, BC2) were subjected to SDS-PAGE and immunoblot analysis using antibodies against indicated proteins. Shown are representative results of three independent experiments.
Monitoring the interaction of β-catenin with components of the destruction complex Axin1 and GSK3β showed that both proteins are significantly enriched in the bound fractions after inhibition of GSK3β (Fig. 6). These results suggest that binding of either nanobody does not interfere or compete with the interaction between β-catenin and Axin1. Moreover, the coprecipitation of GSK3β, which does not stably interact with β-catenin, but with Axin1, indicates that both nanobodies are suitable to precipitate the intact destruction complex.
We also tested the interaction of β-catenin with transcription factors. Although we did not detect TCF7L2 (TCF4) or LEF in the lysate of HEK293T cells (data not shown), we obtained a strong signal for TCF1 in the bound fractions of both binders almost exclusively after induction of β-catenin. Compared with BC1, BC2 more efficiently coprecipitated TCF1 (Fig. 6). Together with the finding that BC2 also binds β-catenin interacting with E-cadherin, we conclude that BC2 might address at least two different fractions of β-catenin: a β-catenin complex mediating the formation of adherens junctions at the plasma membrane and a nuclear complex involved in gene regulation. In summary, the results show that BC1 and BC2 that bind to different epitopes located in the N terminus of β-catenin efficiently precipitate their antigen comprising multiprotein complexes (MPCs) on endogenous levels and in response to compound treatment.
Intracellular Application of β-catenin Chromobodies
Recently, we and others have shown that VHH domains derived from heavy chain antibodies of camelids can be selected as intrabodies for functional interference and visualization of target structures (31, 33, 54, 55). To detect endogenous β-catenin within living cells, we fused the coding sequences of the nanobodies to the fluorescent protein tagGFP generating so-called chromobodies (BC-chromobodies) and introduced them on DNA-level either by transfection or viral transduction in living cells. Upon cellular expression chromobodies become visible and can be analyzed using fluorescence microscopy (Fig. 7A).
Fig. 7.

Chromobodies bind to β-catenin upon intracellular expression. A, Schematic illustration of a BC-chromobody and its introduction into cells on DNA level. B, Intracellular immunoprecipitation (IC-IP) of β-catenin. HEK293T cells expressing indicated chromobodies or GFP were either left untreated or were incubated with CHIR for 24 h. Cells were lysed under native conditions and chromobodies and GFP were precipitated using the GFP-Trap. Input (I) and bound fraction of the GFP-Trap (B) were subjected to SDS-PAGE followed by immunoblotting using an anti-β-catenin antibody (upper panel). Chromobodies or GFP were detected using an anti-GFP antibody (lower panel). C, Relative increase of precipitated β-catenin from cells after inhibition of GSK3β. Input reflects the relative increase of total β-catenin derived from the “input”-samples normalized to GAPDH. Columns represent the ratios of three independent experiments ± stds.
In a first step, we investigated whether the individual BC-chromobodies constitute functional intrabodies recognizing β-catenin. To this end, we performed a Fluorescent Two-Hybrid (F2H) assay (39, 40). By expression of a mCherry-β-catenin construct coupled to the lac inhibitor (lacI) we generated a fluorescently labeled bait protein. After transfection of BHK-2 cells containing a stably integrated lac-operator array mCherry-lacI-β-catenin is recruited to the nuclear interaction platform and becomes visible as a bright red spot (supplemental Fig. S5). To analyze chromobody binding, we used a microscopic read-out based on the colocalization of red- (β-catenin) and green- (chromobody) fluorescence at the interaction platform. Applying the F2H assay, we observed that all BC-chromobodies colocalize with anchored β-catenin whereas GFP alone shows a diffuse distribution (supplemental Fig. S5). In addition, we tested intracellular binding of the BC-chromobodies biochemically by performing intracellular immunoprecipitations (IC-IPs). HEK293T cells expressing the indicated chromobodies were either left untreated or β-catenin levels were induced by treatment with CHIR. After generation of soluble cell extracts, chromobodies were precipitated using the fluorescent moiety as an affinity tag and the formation of complexes was analyzed by immunoblotting using antibodies directed against tagGFP and β-catenin. As a negative control we used HEK293T cells expressing GFP only (Fig. 7B). In densitometric analysis, we compared the normalized signals of β-catenin in the bound fraction after induction of β-catenin (Fig. 7C). The results show that BC1-, BC6-, and BC9-chromobodies efficiently bind endogenous β-catenin derived from CHIR-treated cells upon intracellular expression, whereas BC2- and BC13-chromobodies only bind minor fractions of β-catenin (Fig. 7B, C). Immunoblot analysis revealed a very weak intracellular expression of both chromobodies whereas BC1-, BC6-, and BC9-chromobodies are highly abundant after transient transfection (supplemental Fig. S6). We were not able to increase the expression levels of BC2- and BC13-chromobodies neither by changing the transfection conditions nor the cell type used for transfection (data not shown). Next, we investigated whether expression of the chromobodies affects cell viability. To this end, we performed a Resazurin assay (AlamarBlue) 24 h and 48 h after transfection of the readily expressed BC1-, BC6-, or BC9-chromobodies in comparison to EGFP (supplemental Fig. S7). Our results revealed that the tested chromobodies have no effect on cellular viability.
Fluorescent microscopic analysis showed that none of these chromobodies localizes to the plasma membrane (supplemental Fig. S8A) indicating that they do not bind to membrane-associated β-catenin upon expression in living cells. For a more detailed analysis, we performed pulldown studies from human cells constitutively expressing the BC1-chromobody using ConA beads and analyzed the association of the BC1-chromobody with the membrane fraction. The results show, that the BC1-chromobody is predominantly detectable in the nonbound fraction and only negligible amounts coprecipitate with membrane-associated β-catenin irrespective of an induction of β-catenin upon treatment with CHIR (supplemental Fig. S8B). This result further underlines our hypothesis that BC1 binds to diffusible, nonphosphorylated β-catenin also upon intracellular expression. In summary, from the colocalization of the chromobody-signal in the F2H assay and the intracellular coprecipitation studies we concluded that the chromobodies bind to the diffusible fraction of endogenous β-catenin within living cells.
Next, we asked whether binding of the chromobodies has an impact on the transcriptional activity of β-catenin. To this end, we analyzed the effect of intracellular chromobody expression in a TOPflash luciferase assay upon treatment with NaCl (control) or LiCl-mediated induction of β-catenin after 24 h. As controls, we used HEK293T cells expressing either EGFP or a GFP-β-catenin construct. In cells expressing GFP-β-catenin, we observed a strong transcriptional activation independent of induction with LiCl compared with GFP (supplemental Fig. S9) whereas we did not detected any differences between expression of GFP and chromobodies in noninduced cells (Fig. 8). Although we detected similar transcriptional activity of β-catenin in cells expressing BC1-CB compared with GFP, a significant reduction in reporter fluorescence by ∼35% for BC6- and BC9-expressing cells was observable after induction with LiCl. From that, we concluded that in contrast to BC6 and BC9 the BC1-chromobody does not affect the transcriptional activity of β-catenin.
Fig. 8.

Chromobodies binding the armadillo domain affect transcriptional activity of β-catenin. HEK293T cells were either cotransfected with GFP or the indicated BC-chromobodies in combination with reporter constructs containing TCF-Promoter-luciferase-reporter sites (TOP-flash) or a corresponding control construct with mutated TCF-binding sites (FOP-flash). Reporter activity of NaCl-treated cells is shown in light gray bars and LiCl-induced reporter activity is shown in dark gray bars. All values are normalized to mean luminescence values of LiCl-treated GFP control. Reporter induction after 24 h upon LiCl treatment is shown. Columns represent the results of three independent experiments ± stds.
Chromobody as a Sensor for β-catenin Localization and Expression
Current technologies to study subcellular localization of β-catenin have significant limitations. Immunostaining of fixed cells with specific antibodies provide snap shots in an end point measurement that makes it difficult to trace dynamics of β-catenin in living cells. In order to visualize β-catenin in real time, fluorescent β-catenin fusion constructs have been artificially over-expressed (25–27). However, as shown in our gene reporter assay, transient expression of fluorescently labeled β-catenin dramatically increases the cellular levels of β-catenin and the transcription of Wnt-responsive elements (supplemental Fig. S9). Here, intracellular expression of the BC1-chromobody may provide a unique opportunity to visualize endogenous β-catenin in living cells and thereby overcome the most significant limitations of current imaging approaches.
In this context, we analyzed whether the BC1-chromobody (BC1-CB) is able to trace dynamic changes in the localization of β-catenin. As shown above, BC1-CB is well expressed, nontoxic and does not interfere with the function of β-catenin as transcriptional co-activator. Importantly, the results of the co-immunoprecipitation and IC-IP experiments indicate that BC1-CB preferentially binds the diffusible, nonphosphorylated β-catenin. Hence, we chose BC1-CB as the best candidate to trace dynamic relocalization of β-catenin in living cells.
Because transient chromobody expression displays a large intercellular heterogeneity, we generated stable HeLa cell lines by lentiviral transduction of a BC1-chromobody expression construct. In order to minimize background fluorescence because of an excess of unbound chromobody, we selected a monoclonal cell line (HeLa_BC1-CB) with low BC1-CB expression levels and high intercellular homogeneity. As a control, we established a HeLa cell line stably expressing GFP (HeLa_GFP).
The common model of Wnt activation suggests, that β-catenin, after it accumulates in the cytoplasm, translocates to the nucleus (56–58). This nuclear translocation of β-catenin might lead to a concomitant redistribution of the BC1-CB signal. To test this, we induced β-catenin in HeLa_BC1-CB and performed live cell imaging. Microscopic analysis after 18 h of either control-treated (NaCl) or GSK3β-inhibited (LiCl) cells revealed an increase of chromobody signal in the cytoplasm and the nucleus. In HeLa_GFP cells no accumulation of the fluorescent signal was detectable upon LiCl treatment (Fig. 9A, supplemental Movie S1 (NaCl) and supplemental Movie S2 (LiCl)). In order to detect a possible relocalization of β-catenin, we quantified the fluorescence intensities in the cytoplasm and the nucleus in a statistically relevant number of cells. The analysis shows a slight increase of the ratio of nuclear to cytoplasmic fluorescence from ∼1.3 in the control treated cells to ∼1.6 for LiCl treatment (Fig. 9B). These data do not support a model in which cytoplasmic enrichment and nuclear accumulation are regarded as two consecutive events. Instead, it suggests a simultaneous accumulation of β-catenin in both compartments.
Fig. 9.
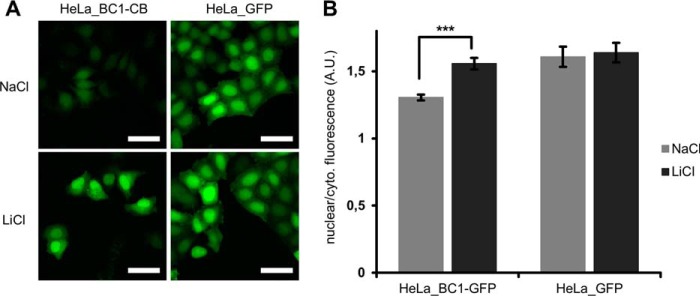
Tracing nuclear translocation of β-catenin with the BC1-chromobody. A, HeLa_BC1-CB and HeLa_GFP cells were incubated with NaCl or LiCl. Representative images of cells after 18 h of compound treatment are shown. Scale bar = 50 μm. B, After image segmentation, mean fluorescence in cytoplasm und nuclei was determined and the ratio of nuclear to cytoplasmic fluorescence was calculated. Columns represent the mean ratio and standard deviation from five biological replicates (∼500 cells each). Highly significant increase in nuclear/cytoplasmic ratio is indicated by *** (p < 0.001, t test).
Based on the observed global elevation in chromobody fluorescence upon induction of β-catenin, we speculate that the BC1-CB may provide a fluorescent read-out to monitor alterations of the diffusible β-catenin fraction over time in living cells. To test this, we determined the average fluorescence in the cytoplasm and nucleus of HeLa_BC1-CB cells in a time-lapse analysis for various treatments (Fig. 10A). For LiCl we detected a ∼2.5-fold increase of chromobody signal after 12 h in the cytoplasm and the nucleus, which was not further increased at later time points (Fig. 10B, 10C). In contrast, using the more potent GSK3β inhibitor CHIR, a plateau in cytoplasmic fluorescence with a ∼4.5-fold increase was reached after ∼18 h (Fig. 10B). However, chromobody fluorescence in nuclei is further increased and reached a ∼sixfold elevation after 23 h compared to starting level (Fig. 10C). After incubation with the casein kinase 1-inhibitor LH846 we did not observe any increase of the chromobody signal. Interestingly, a combinatorial treatment with LH846 and CHIR further increased the fluorescence compared with CHIR alone. For this treatment, we observed a cytoplasmic elevation of ∼5.5-fold (Fig. 10B) and a nuclear increase of ∼sevenfold after 23 h (Fig. 10C).
Fig. 10.

Time-lapse microscopy of compound-treated HeLa_BC1-CB cells. A, HeLa_BC1-CB cells were imaged in hourly intervals for 24 h with control treatment (H2O) or in the presence of NaCl, LiCl, LH846, CHIR, or a combination of CHIR and LH846. Time-lapse imaging of HeLa_BC1-CBs is shown for each condition starting 5 h after compound treatment. After image segmentation mean fluorescence intensity in the cytoplasm, B, and nuclei C, was determined and values of three biological replicates (∼500 cells each) were plotted against time ± standard errors.
In order to find hints regarding the molecular mechanism responsible for the observed changes in chromobody fluorescence, we investigated whether compound treatment affects chromobody expression on mRNA level. Therefore, we performed qRT-PCR analysis of cells after 24 h treatment with indicated compounds (supplemental Fig. S10). Only minor changes in mRNA expression are detectable, which do not correspond to the observed changes of the chromobody fluorescence. Because the increase of chromobody signal is not caused by an induction at transcriptional level, we hypothesized that an elevation in β-catenin might lead to a concomitant stabilization of the chromobody on protein level. To evaluate this possibility we performed an immunoblot analysis of induced HeLa_BC1-CB cells at different time points. We observed a significant increase of endogenous β-catenin after induction with CHIR at different time points (Fig. 11A). In addition, we detected a comparable accumulation of the BC1-CB protein over time. Most interestingly, the densitometric analysis reveals a correlation of the increase of β-catenin with the elevation of the chromobody at different time points after induction (Fig. 11B). This strongly suggests that the chromobody becomes stabilized upon binding to β-catenin, which is reflected by an increased fluorescent chromobody signal (Fig. 10). In summary, these results show that the chromobody signal is suitable to trace the diffusible β-catenin fraction in living cells. Moreover, we demonstrate that the observable changes of chromobody fluorescence are directly linked to the cellular levels of β-catenin.
Fig. 11.

Cellular level of BC1-chromobody correlates with endogenous β-catenin. A, Immunoblot analysis of β-catenin, BC1-CB and GAPDH of CHIR-treated HeLa_BC1-CB cells at indicated time points. B, Densitometric analysis of immunoblot analysis. Values represent the mean of four replicates ± stds.
DISCUSSION
Aberrant regulation of the Wnt/β-catenin signaling pathway is associated with numerous diseases (15) including neurodegenerative disorders (59, 60), metabolic diseases (61), or different types of cancer (3, 62, 63). β-catenin as the key effector of the Wnt-pathway has been shown to modulate numerous processes including gene expression, cell proliferation, epithelial-mesenchymal transition, and tumor progression (15, 27, 64, 65). Although the role of β-catenin in many cellular processes has been known for many years, we are still at the beginning of understanding how its multiple dynamic and interdependent functions are orchestrated in a spatial and temporal manner. The development of new research tools has always proven to advance the knowledge of β-catenin. This includes the generation and application of specific antibodies recognizing different post-translationally modified versions of β-catenin (47, 66–68), reliable immunoassays to study protein dynamics upon compound treatment (37) or the development of fluorescently labeled constructs to trace the dynamics of β-catenin in different cellular models (26, 27).
In this study, we have generated a set of single domain antibodies (so-called nanobodies) derived from heavy chain only antibodies of camelids to target and trace endogenous β-catenin, its participation in multiprotein complexes and the dynamic of its translocation and stabilization in cellular models. Starting with a set of 14 β-catenin-specific nanobodies we selected five specific binding molecules that recognize distinct domains of β-catenin with affinities down to the low nanomolar range. These nanobodies can be easily produced in high yields in bacteria and functionalized as stable and efficient capture molecules for sandwich immunoassays e.g. to perform high throughput screening of compound libraries on protein–protein interactions of β-catenin with binding partners including TCF4, α-catenin, or components of the destruction complex. One of the most widespread methods to reveal protein–protein interactions is co-immunoprecipitation. The usage of nanobodies for such analyses has been recently shown for ectopically expressed proteins including GFP- or mCherry-fusion constructs (35, 69, 70). However, the analysis of interaction partners of endogenous β-catenin is more challenging, because the accessibility of binding epitopes might be sterically blocked because of its participation in multiprotein complexes in various subcellular compartments. Based on our detailed domain and epitope analysis, we identified N- and C terminus-specific nanobodies, which efficiently precipitate endogenous β-catenin. For BC2, we were able to narrow down the epitope to a linear stretch of 12 aa residues located at the very N terminus of β-catenin. This observation is quite notable because only very few nanobodies are known to bind short linear peptide sequences (30). Moreover, to our knowledge, there are no interaction partners known that block this epitope upon binding to β-catenin. Therefore, we postulate that BC2 is a very potent binder to precipitate various cellular fractions of β-catenin. This is supported by the results of our coprecipitation experiments showing that BC2 precipitates β-catenin in complex with binding partners of the plasma membrane, the cytoplasm and the nucleus. With BC1 we have identified a second N-terminal binder. In contrast to BC2, this nanobody does not seem to bind the β-catenin fraction associated with the plasma membrane but appears to preferentially bind the diffusible, nonphosphorylated and therefore transcriptionally active β-catenin. This turns BC1 into a powerful tool to study the changes of this specific subfraction of β-catenin upon Wnt-pathway engagement.
Recent studies have demonstrated that single domain antibody fragments can be functionally expressed in living cells. Depending on their binding properties they can be used to visualize subcellular antigen location or to modulate their target structure and function (31, 33, 43, 55, 71). Here, we have demonstrated that all selected nanobodies are able to bind β-catenin in living cells. However, although BC1, BC6, and BC9 were well detectable, BC2 and BC13 only showed minor expression. The molecular basis for the different protein levels is not clear but as BC2 comprises an additional disulfide bond, which is not properly formed in the reducing environment of the cell, it can be speculated that this binder shows misfolding or aggregation and is subjected to a faster degradation.
With BC6 and BC9 we selected two binding molecules addressing the armadillo domain. Numerous interaction partners of β-catenin have been shown to bind to this domain including E-cadherin, Adenomatous polyposis coli, Axin, or TCF4 (reviewed in (15, 16)). As these interactions are present under various cellular conditions the poor pulldown efficiency of both nanobodies applied as extracellular binding molecules could be explained by domain occupation and hence an inaccessibility of BC6 and BC9 binding epitopes. However, intracellular binding studies suggest, that both binding molecules can compete with endogenous interaction partners and bind to β-catenin within living cells. This hypothesis is further supported by the observation that both chromobodies negatively affect the transcriptional activity of β-catenin. We are currently developing cellular models where we induce the expression of either chromobody and follow changes in gene regulation and cellular morphology. Such models could provide a new intracellular proteome-based approach for target validation, dissecting the functional relevance of the armadillo domain for transcriptional activation.
Engineering an intracellularly functional binding molecule based on the BC1 domain (BC1-chromobody), we were able to visualize for the first time dynamic changes of endogenous β-catenin in living cells. Using the BC1-chromobody in time-lapse analysis we visualized the enrichment of nonphosphorylated β-catenin after compound treatment. Automated image segmentation and analysis shows, that upon induction the global enrichment of endogenous β-catenin is accompanied only by a slightly stronger accumulation in the nucleus. This is in accordance with previous findings describing that the absolute number of β-catenin molecules entering and exiting the nucleus per time increases proportionally when the β-catenin concentration is increased (26, 72). Thereby, our data supports a model wherein a global increase of β-catenin rather than a subcompartment-specific accumulation occurs after interference with β-catenin degradation.
Most interestingly, we detected a significant increase in global chromobody signal after inhibiting the destruction complex. This increase is not caused by higher mRNA expression levels of the chromobodies initiated by the compounds but rather correlates with increased cellular levels of β-catenin. Therefore, we speculate about a mechanism leading to an antigen-mediated stabilization of the chromobody in living cells. Recent reports have described intrabodies modified by the addition of a degradation promoting domain. Upon ectopical expression of their antigens these intrabodies become stabilized (73). In our approach we observed an enrichment of the chromobody independently of the introduction of a destabilizing domain. This suggests that antigen-dependent stabilization is an intrinsic property of intracellularly expressed chromobodies. This constitutes a novel and flexible approach to follow dynamic changes of endogenous protein levels using chromobody fluorescence. We currently test this for a number of different intracellular antigens that we have recently generated chromobodies against.
In conclusion, the nanobodies presented in this study offer a robust and versatile tool box to study β-catenin. Their applicability in biochemical and cell biological assays enable a fast and efficient integration of data obtained for protein interactions in vitro with subcellular translocation and dynamic changes of protein stability upon compound treatment in cellular systems.
Using the chromobody fluorescence as an indicator for changes in endogenous β-catenin levels offers a new approach for an imaging-based high-throughput screening of β-catenin modulating compounds in end point and live cell assays.
Supplementary Material
Footnotes
Author contributions: B.T., F.E., R.A., O.P., and U.R. designed research; B.T., F.E., R.A., O.P., R.S.H., J.M., P.D.K., A.B., T.R., and U.R. performed research; A.M.S. and S.N. contributed new reagents or analytic tools; B.T., F.E., O.P., and U.R. analyzed data; B.T., F.E., and U.R. wrote the paper.
* This work was supported by the Ministry of Science, Research and the Arts of Baden-Württemberg (V.1.4.-H3-1403-74) and the GO-Bio Program of the Bundesministerium fuer Bildung und Forschung (BMBF) (ChromoTek GmbH).
 This article contains supplemental Figs. S1 to S10 and Movies S1 and S2.
This article contains supplemental Figs. S1 to S10 and Movies S1 and S2.
Competing interests: U.R. is shareholder of the commercial company ChromoTek GmbH, which develops and market research reagents based on the chromobody-technology. A.B. and T.R. are employees of the commercial company ChromoTek GmbH.
1 The abbreviations used are:
- LEF/TCF
- Lymphoid enhancer factor/T-cell factor
- BC1-CB
- Beta-catenin chromobody 1
- CB
- Chromobody
- CMV
- Cytomegalo virus
- F2H
- Fluorescent-2 Hybrid
- GFP
- Green fluorescent protein
- GSK3β
- Glycogen synthase kinase 3 beta
- GST
- Glutathione S-transferase
- ICIP
- Intracellular immunoprecipitation
- MPC
- Multiprotein complexes
- Nb
- Nanobody
- sdAbs
- Single domain antibodies
- SPR
- Surface plasmon resonance
- VHH
- Variable heavy chain of heavy chain antibody.
REFERENCES
- 1. Takeichi M. (1995) Morphogenetic roles of classic cadherins. Curr. Opin. Cell Biol. 7, 619–627 [DOI] [PubMed] [Google Scholar]
- 2. Korinek V., Barker N., Morin P. J., van Wichen D., de Weger R., Kinzler K. W., Vogelstein B., Clevers H. (1997) Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC−/− colon carcinoma. Science 275, 1784–1787 [DOI] [PubMed] [Google Scholar]
- 3. Behrens J., Lustig B. (2004) The Wnt connection to tumorigenesis. Int. J. Dev. Biol. 48, 477–487 [DOI] [PubMed] [Google Scholar]
- 4. Aberle H., Bauer A., Stappert J., Kispert A., Kemler R. (1997) beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 16, 3797–3804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hart M., Concordet J. P., Lassot I., Albert I., del los Santos R., Durand H., Perret C., Rubinfeld B., Margottin F., Benarous R., Polakis P. (1999) The F-box protein beta-TrCP associates with phosphorylated beta-catenin and regulates its activity in the cell. Curr. Biol. 9, 207–210 [DOI] [PubMed] [Google Scholar]
- 6. Marikawa Y., Elinson R. P. (1998) beta-TrCP is a negative regulator of Wnt/beta-catenin signaling pathway and dorsal axis formation in Xenopus embryos. Mech. Dev. 77, 75–80 [DOI] [PubMed] [Google Scholar]
- 7. Liu C., Li Y., Semenov M., Han C., Baeg G. H., Tan Y., Zhang Z., Lin X., He X. (2002) Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 108, 837–847 [DOI] [PubMed] [Google Scholar]
- 8. Huang H., He X. (2008) Wnt/beta-catenin signaling: new (and old) players and new insights. Curr. Opin. Cell Biol. 20, 119–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kimelman D., Xu W. (2006) beta-catenin destruction complex: insights and questions from a structural perspective. Oncogene 25, 7482–7491 [DOI] [PubMed] [Google Scholar]
- 10. Bilic J., Huang Y. L., Davidson G., Zimmermann T., Cruciat C. M., Bienz M., Niehrs C. (2007) Wnt induces LRP6 signalosomes and promotes dishevelled-dependent LRP6 phosphorylation. Science 316, 1619–1622 [DOI] [PubMed] [Google Scholar]
- 11. MacDonald B. T., Tamai K., He X. (2009) Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev. Cell 17, 9–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li V. S., Ng S. S., Boersema P. J., Low T. Y., Karthaus W. R., Gerlach J. P., Mohammed S., Heck A. J., Maurice M. M., Mahmoudi T., Clevers H. (2012) Wnt signaling through inhibition of beta-catenin degradation in an intact Axin1 complex. Cell 149, 1245–1256 [DOI] [PubMed] [Google Scholar]
- 13. Nusse R. (1999) WNT targets. Repression and activation. Trends Genet. 15, 1–3 [DOI] [PubMed] [Google Scholar]
- 14. Staal F. J., Clevers H. (2000) Tcf/Lef transcription factors during T-cell development: unique and overlapping functions. Hematol. J. 1, 3–6 [DOI] [PubMed] [Google Scholar]
- 15. Clevers H. (2006) Wnt/beta-catenin signaling in development and disease. Cell 127, 469–480 [DOI] [PubMed] [Google Scholar]
- 16. Valenta T., Hausmann G., Basler K. (2012) The many faces and functions of beta-catenin. EMBO J. 31, 2714–2736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mosimann C., Hausmann G., Basler K. (2009) Beta-catenin hits chromatin: regulation of Wnt target gene activation. Nat. Rev. Mol. Cell Biol. 10, 276–286 [DOI] [PubMed] [Google Scholar]
- 18. Kinzler K. W., Vogelstein B. (1996) Lessons from hereditary colorectal cancer. Cell 87, 159–170 [DOI] [PubMed] [Google Scholar]
- 19. Morin P. J., Sparks A. B., Korinek V., Barker N., Clevers H., Vogelstein B., Kinzler K. W. (1997) Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science 275, 1787–1790 [DOI] [PubMed] [Google Scholar]
- 20. Logan C. Y., Nusse R. (2004) The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 20, 781–810 [DOI] [PubMed] [Google Scholar]
- 21. Reya T., Clevers H. (2005) Wnt signaling in stem cells and cancer. Nature 434, 843–850 [DOI] [PubMed] [Google Scholar]
- 22. Miyoshi K., Hennighausen L. (2003) Beta-catenin: a transforming actor on many stages. Breast Cancer Res. 5, 63–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kahn M. (2014) Can we safely target the WNT pathway? Nat. Rev. Drug Discov. 13, 513–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Damalas A., Ben-Ze'ev A., Simcha I., Shtutman M., Leal J. F., Zhurinsky J., Geiger B., Oren M. (1999) Excess beta-catenin promotes accumulation of transcriptionally active p53. EMBO J. 18, 3054–3063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Giannini A. L., Vivanco M. M., Kypta R. M. (2000) Analysis of beta-catenin aggregation and localization using GFP fusion proteins: nuclear import of alpha-catenin by the beta-catenin/Tcf complex. Exp. Cell Res. 255, 207–220 [DOI] [PubMed] [Google Scholar]
- 26. Krieghoff E., Behrens J., Mayr B. (2006) Nucleo-cytoplasmic distribution of beta-catenin is regulated by retention. J. Cell Sci. 119, 1453–1463 [DOI] [PubMed] [Google Scholar]
- 27. Johnson M., Sharma M., Jamieson C., Henderson J. M., Mok M. T., Bendall L., Henderson B. R. (2009) Regulation of beta-catenin trafficking to the membrane in living cells. Cell. Signal. 21, 339–348 [DOI] [PubMed] [Google Scholar]
- 28. Hamers-Casterman C., Atarhouch T., Muyldermans S., Robinson G., Hamers C., Songa E. B., Bendahman N., Hamers R. (1993) Naturally occurring antibodies devoid of light chains. Nature 363, 446–448 [DOI] [PubMed] [Google Scholar]
- 29. Muyldermans S., Baral T. N., Retamozzo V. C., De Baetselier P., De Genst E., Kinne J., Leonhardt H., Magez S., Nguyen V. K., Revets H., Rothbauer U., Stijlemans B., Tillib S., Wernery U., Wyns L., Hassanzadeh-Ghassabeh G., Saerens D. (2009) Camelid immunoglobulins and nanobody technology. Vet. Immunol. Immunopathol. 128, 178–183 [DOI] [PubMed] [Google Scholar]
- 30. Muyldermans S. (2013) Nanobodies: natural single-domain antibodies. Annu. Rev. Biochem. 82, 775–797 [DOI] [PubMed] [Google Scholar]
- 31. Rothbauer U., Zolghadr K., Tillib S., Nowak D., Schermelleh L., Gahl A., Backmann N., Conrath K., Muyldermans S., Cardoso M. C., Leonhardt H. (2006) Targeting and tracing antigens in live cells with fluorescent nanobodies. Nat. Methods 3, 887–889 [DOI] [PubMed] [Google Scholar]
- 32. Zolghadr K., Gregor J., Leonhardt H., Rothbauer U. (2012) Case study on live cell apoptosis-assay using lamin-chromobody cell-lines for high-content analysis. Methods Mol. Biol. 911, 569–575 [DOI] [PubMed] [Google Scholar]
- 33. Helma J., Schmidthals K., Lux V., Nuske S., Scholz A. M., Krausslich H. G., Rothbauer U., Leonhardt H. (2012) Direct and dynamic detection of HIV-1 in living cells. PloS One 7, e50026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Arbabi Ghahroudi M., Desmyter A., Wyns L., Hamers R., Muyldermans S. (1997) Selection and identification of single domain antibody fragments from camel heavy-chain antibodies. FEBS Lett. 414, 521–526 [DOI] [PubMed] [Google Scholar]
- 35. Rothbauer U., Zolghadr K., Muyldermans S., Schepers A., Cardoso M. C., Leonhardt H. (2008) A versatile nanotrap for biochemical and functional studies with fluorescent fusion proteins. Mol. Cell. Proteomics 7, 282–289 [DOI] [PubMed] [Google Scholar]
- 36. Aberle H., Butz S., Stappert J., Weissig H., Kemler R., Hoschuetzky H. (1994) Assembly of the cadherin-catenin complex in vitro with recombinant proteins. J. Cell Sci. 107, 3655–3663 [DOI] [PubMed] [Google Scholar]
- 37. Luckert K., Gotschel F., Sorger P. K., Hecht A., Joos T. O., Potz O. (2011) Snapshots of protein dynamics and post-translational modifications in one experiment–beta-catenin and its functions. Mol. Cell. Proteomics 10, M110 007377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Braeuning A., Menzel M., Kleinschnitz E. M., Harada N., Tamai Y., Kohle C., Buchmann A., Schwarz M. (2007) Serum components and activated Ha-ras antagonize expression of perivenous marker genes stimulated by beta-catenin signaling in mouse hepatocytes. FEBS J. 274, 4766–4777 [DOI] [PubMed] [Google Scholar]
- 39. Zolghadr K., Mortusewicz O., Rothbauer U., Kleinhans R., Goehler H., Wanker E. E., Cardoso M. C., Leonhardt H. (2008) A fluorescent two-hybrid assay for direct visualization of protein interactions in living cells. Mol. Cell. Proteomics 7, 2279–2287 [DOI] [PubMed] [Google Scholar]
- 40. Zolghadr K., Rothbauer U., Leonhardt H. (2012) The fluorescent two-hybrid (F2H) assay for direct analysis of protein–protein interactions in living cells. Methods Mol. Biol. 812, 275–282 [DOI] [PubMed] [Google Scholar]
- 41. Poetz O., Luckert K., Herget T., Joos T. O. (2009) Microsphere-based co-immunoprecipitation in multiplex. Anal. Biochem. 395, 244–248 [DOI] [PubMed] [Google Scholar]
- 42. Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 43. Kirchhofer A., Helma J., Schmidthals K., Frauer C., Cui S., Karcher A., Pellis M., Muyldermans S., Casas-Delucchi C. S., Cardoso M. C., Leonhardt H., Hopfner K. P., Rothbauer U. (2010) Modulation of protein properties in living cells using nanobodies. Nat. Struct. Mol. Biol. 17, 133–138 [DOI] [PubMed] [Google Scholar]
- 44. Xu W., Kimelman D. (2007) Mechanistic insights from structural studies of beta-catenin and its binding partners. J. Cell Sci. 120, 3337–3344 [DOI] [PubMed] [Google Scholar]
- 45. Huber A. H., Nelson W. J., Weis W. I. (1997) Three-dimensional structure of the armadillo repeat region of beta-catenin. Cell 90, 871–882 [DOI] [PubMed] [Google Scholar]
- 46. Bauer M., Chicca A., Tamborrini M., Eisen D., Lerner R., Lutz B., Poetz O., Pluschke G., Gertsch J. (2012) Identification and quantification of a new family of peptide endocannabinoids (Pepcans) showing negative allosteric modulation at CB1 receptors. J. Biol. Chem. 287, 36944–36967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. van Noort M., Meeldijk J., van der Zee R., Destree O., Clevers H. (2002) Wnt signaling controls the phosphorylation status of beta-catenin. J. Biol. Chem. 277, 17901–17905 [DOI] [PubMed] [Google Scholar]
- 48. Olivier-Van Stichelen S., Dehennaut V., Buzy A., Zachayus J. L., Guinez C., Mir A. M., El Yazidi-Belkoura I., Copin M. C., Boureme D., Loyaux D., Ferrara P., Lefebvre T. (2014) O-GlcNAcylation stabilizes beta-catenin through direct competition with phosphorylation at threonine 41. FASEB J. 28, 3325–3338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bennett C. N., Ross S. E., Longo K. A., Bajnok L., Hemati N., Johnson K. W., Harrison S. D., MacDougald O. A. (2002) Regulation of Wnt signaling during adipogenesis. J. Biol. Chem. 277, 30998–31004 [DOI] [PubMed] [Google Scholar]
- 50. Miller J. R., Moon R. T. (1997) Analysis of the signaling activities of localization mutants of beta-catenin during axis specification in Xenopus. J. Cell Biol. 139, 229–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ilyas M., Tomlinson I. P., Rowan A., Pignatelli M., Bodmer W. F. (1997) Beta-catenin mutations in cell lines established from human colorectal cancers. Proc. Natl. Acad. Sci. U.S.A. 94, 10330–10334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chetty R., Serra S., Asa S. L. (2008) Loss of membrane localization and aberrant nuclear E-cadherin expression correlates with invasion in pancreatic endocrine tumors. Am. J. Surg. Pathol. 32, 413–419 [DOI] [PubMed] [Google Scholar]
- 53. Ferber E. C., Kajita M., Wadlow A., Tobiansky L., Niessen C., Ariga H., Daniel J., Fujita Y. (2008) A role for the cleaved cytoplasmic domain of E-cadherin in the nucleus. J. Biol. Chem. 283, 12691–12700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Burgess A., Lorca T., Castro A. (2012) Quantitative live imaging of endogenous DNA replication in mammalian cells. PloS One 7, e45726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Van Audenhove I., Van Impe K., Ruano-Gallego D., De Clercq S., De Muynck K., Vanloo B., Verstraete H., Fernandez L. A., Gettemans J. (2013) Mapping cytoskeletal protein function in cells by means of nanobodies. Cytoskeleton 70, 604–622 [DOI] [PubMed] [Google Scholar]
- 56. Behrens J., von Kries J. P., Kuhl M., Bruhn L., Wedlich D., Grosschedl R., Birchmeier W. (1996) Functional interaction of beta-catenin with the transcription factor LEF-1. Nature 382, 638–642 [DOI] [PubMed] [Google Scholar]
- 57. Huber O., Korn R., McLaughlin J., Ohsugi M., Herrmann B. G., Kemler R. (1996) Nuclear localization of beta-catenin by interaction with transcription factor LEF-1. Mech. Dev. 59, 3–10 [DOI] [PubMed] [Google Scholar]
- 58. Monga S. P., Mars W. M., Pediaditakis P., Bell A., Mule K., Bowen W. C., Wang X., Zarnegar R., Michalopoulos G. K. (2002) Hepatocyte growth factor induces Wnt-independent nuclear translocation of beta-catenin after Met-beta-catenin dissociation in hepatocytes. Cancer Res. 62, 2064–2071 [PubMed] [Google Scholar]
- 59. Okerlund N. D., Cheyette B. N. (2011) Synaptic Wnt signaling-a contributor to major psychiatric disorders? J. Neurodev. Disorders 3, 162–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Inestrosa N. C., Montecinos-Oliva C., Fuenzalida M. (2012) Wnt signaling: role in Alzheimer disease and schizophrenia. J. Neuroimmune Pharmacol. 7, 788–807 [DOI] [PubMed] [Google Scholar]
- 61. Schinner S., Willenberg H. S., Schott M., Scherbaum W. A. (2009) Pathophysiological aspects of Wnt-signaling in endocrine disease. Eur. J. Endocrinol. 160, 731–737 [DOI] [PubMed] [Google Scholar]
- 62. Giles R. H., van Es J. H., Clevers H. (2003) Caught up in a Wnt storm: Wnt signaling in cancer. Biochim. Biophys. Acta 1653, 1–24 [DOI] [PubMed] [Google Scholar]
- 63. Cui J., Jiang W., Wang S., Wang L., Xie K. (2012) Role of Wnt/beta-catenin signaling in drug resistance of pancreatic cancer. Curr. Pharm. Des. 18, 2464–2471 [DOI] [PubMed] [Google Scholar]
- 64. Mokkapati S., Niopek K., Huang L., Cunniff K. J., Ruteshouser E. C., Finegold M. J., Huff V. (2014) β-catenin activation in a novel liver progenitor cell type is sufficient to cause hepatocellular carcinoma and hepatoblastoma. Cancer Res. 3275.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Santoro A., Pannone G., Papagerakis S., McGuff H. S., Cafarelli B., Lepore S., De Maria S., Rubini C., Mattoni M., Staibano S. (2014) Beta-Catenin and Epithelial Tumors: A Study Based on 374 Oropharyngeal Cancers. BioMed Res. Int. 2014, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. van Noort M., Weerkamp F., Clevers H. C., Staal F. J. (2007) Wnt signaling and phosphorylation status of beta-catenin: importance of the correct antibody tools. Blood 110, 2778–2779 [DOI] [PubMed] [Google Scholar]
- 67. Fang D., Hawke D., Zheng Y., Xia Y., Meisenhelder J., Nika H., Mills G. B., Kobayashi R., Hunter T., Lu Z. (2007) Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. J. Biol. Chem. 282, 11221–11229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zhu G., Wang Y., Huang B., Liang J., Ding Y., Xu A., Wu W. (2012) A Rac1/PAK1 cascade controls beta-catenin activation in colon cancer cells. Oncogene 31, 1001–1012 [DOI] [PubMed] [Google Scholar]
- 69. Trinkle-Mulcahy L., Boulon S., Lam Y. W., Urcia R., Boisvert F. M., Vandermoere F., Morrice N. A., Swift S., Rothbauer U., Leonhardt H., Lamond A. (2008) Identifying specific protein interaction partners using quantitative mass spectrometry and bead proteomes. J. Cell Biol. 183, 223–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Fridy P. C., Li Y., Keegan S., Thompson M. K., Nudelman I., Scheid J. F., Oeffinger M., Nussenzweig M. C., Fenyo D., Chait B. T., Rout M. P. (2014) A robust pipeline for rapid production of versatile nanobody repertoires. Nat. Methods 11, 1253–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Delanote V., Vanloo B., Catillon M., Friederich E., Vandekerckhove J., Gettemans J. (2010) An alpaca single-domain antibody blocks filopodia formation by obstructing L-plastin-mediated F-actin bundling. FASEB J. 24, 105–118 [DOI] [PubMed] [Google Scholar]
- 72. Tan C. W., Gardiner B. S., Hirokawa Y., Smith D. W., Burgess A. W. (2014) Analysis of Wnt signaling beta-catenin spatial dynamics in HEK293T cells. BMC Sys. Biol. 8, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Sibler A. P., Courtete J., Muller C. D., Zeder-Lutz G., Weiss E. (2005) Extended half-life upon binding of destabilized intrabodies allows specific detection of antigen in mammalian cells. FEBS J. 272, 2878–2891 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
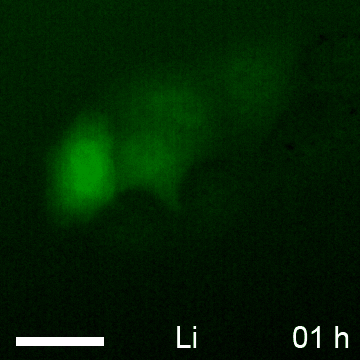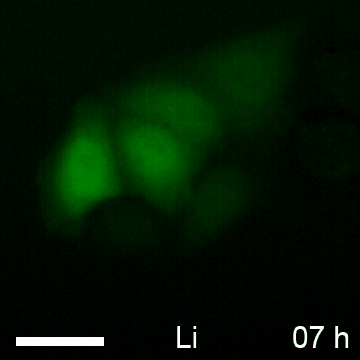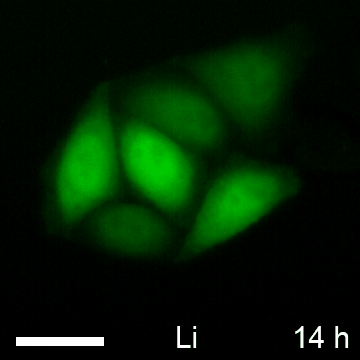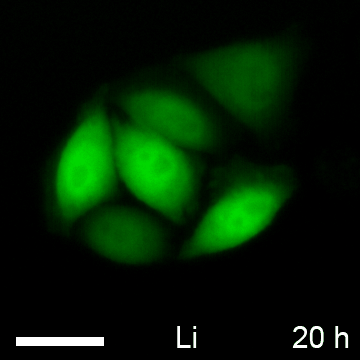{kind=link}