

Abstract
The challenge in high-quality membrane proteomics is all about sample preparation prior to HPLC, and the cell-to-protein step poses a long-standing bottleneck. Traditional protein extraction methods apply ionic or poly-disperse detergents, harsh denaturation, and repeated protein/peptide precipitation/resolubilization afterward, but suffer low yield, low reproducibility, and low sequence coverage. Contrary to attempts to subdue, we resolved this challenge by providing proteins nature-and-activity-promoting conditions throughout preparation. Using 285-kDa hetero-pentameric human GABA type A receptor overexpressed in HEK293 as a model, we describe a n-dodecyl-β-d-maltopyranoside/cholesteryl hemisuccinate (DDM/CHS)-based affinity purification method, that produced active receptors, supported protease activity, and allowed high performance with both in-gel and direct gel-free proteomic analyses—without detergent removal. Unlike conventional belief that detergents must be removed before HPLC MS, the high-purity low-dose nonionic detergent DDM did not interfere with peptides, and obviated removal or desalting. Sonication or dropwise addition of detergent robustly solubilized over 90% of membrane pellets. The purification conditions were comparable to those applied in successful crystallizations of most membrane proteins. These results enabled streamlined proteomics of human synaptic membrane proteins, and more importantly, allowed directly coupling proteomics with crystallography to characterize both static and dynamic structures of membrane proteins in crystallization pipelines.
Transmembrane (TM)1 proteins are abundant and play critical roles in nearly all biological processes. About 25% of the 29,375 unique protein sequences in human proteome contain at least one TM alpha-helix, and 13% contain at least two (1). TM proteins such as ligand-gated ion channels (LGICs) and G protein-coupled receptors (GPCRs) are cell gate-keepers that convert external signals into cellular activities throughout the central nervous system (CNS), and predominate as desired drug targets. Ion channels already represent 10% of current drug targets before the structure-function mechanisms of CNS LGICs become clear (1). Formed by five homologous TM subunits (main form in brain (α1)2(β2/β3)2(γ2L)1) (2, 3), Cys-loop LGIC gamma-aminobutyric acid type A receptor (GABAAR) is the major inhibitory (Cl̄) ion channel that balances excitation in CNS. Genomic studies have pinpointed several single mutations in GABAAR as the causes—and potential drug targets—of epilepsy, a devastating disorder that afflicts 2.2 million Americans and 65 million people worldwide at tremendous health and economic costs (4–12). For decades, antiepileptic drugs remain limited, control only 2/3 of epilepsies, and incur serious side effects including sedation, addiction, dizziness, and suicidality, indicating urgency for more effective mechanism-driven pharmacotherapy. However, no atomic structure of full-length mammalian Cys-loop LGICs is yet available (crystallography structures of two truncated mutated homo-pentamers appeared during the review of this manuscript (13, 14)), and their structure-function mechanisms in native forms remain unclear.
Current structure-function relationship studies of Cys-loop LGIC rely on relating ion flux function to genomic sequence-based alignment with homomeric bacterial or Caenorhabditis elegans structural analogs, and offer little explanation for the high diversity and complicacy of subunit-specific drug selectivity of mammalian LGIC (2, 15). Arguably most post-translational modifications (PTMs) on mammalian CNS membrane proteins have high functional priority, and increasingly, PTMs are emerging as the key to decipher their function and regulation mechanisms (16, 17). However, direct protein sequence and PTM are rarely considered in structure-function relationship studies of mammalian Cys-loop LGIC (18, 19), largely for shortage of such robust data. Membrane proteins are hydrophobic and technically difficult to study at all levels (20)—including global proteomic analysis by MS: discrepant with their abundance and importance, they are typically underrepresented by peptide spectral matches (PSMs), unique peptides, and sequence coverage, compared with soluble proteins. Benchmark sequence coverage for membrane proteins in global studies remains 20–30%, after improvements using cleavable detergents or extreme 3-dimension 8-day HPLC separations (21, 22). Such limitations impede robust analysis of their modifications on a comprehensive or quantitative scale.
We reason that the challenge in proteomic analysis of membrane proteins is all about the cell-protein-peptide sample preparation prior to HPLC. Cell-to-protein conversion is the critical first step, because it directly—and often irreversibly—steers downstream method design and impacts the quality of the entire study. The goal for this step is to achieve sufficient protein solubility and structural integrity to promote protease access for the next digestion. Current protein extraction methods have the problem of low yield and low reproducibility for membrane proteins, and have remained an outstanding impediment over the past decade, despite rapid revolution in downstream HPLC and MS capacities (23–25). After purification in Triton X-100, current sequencing methods for LGIC take daunting 10 days (in-gel) or 2 days (gel-free) and combine complicated 6–10 overnight digestions (both sequential and parallel) to prepare peptide sample alone for each analysis (26–30), and yield limited information in both depth and width. Therefore, more efficient cell-to-protein preparation method is in urgent need to facilitate high-performance proteomic studies of these important membrane proteins.
Recent improvements subjected membrane proteins to various extremely harsh denaturing conditions, such as boiling the cell lysate in up to 4–7.5% SDS, SDS detergent removal using dia-filtration, multiple rounds of precipitation, and re-solubilizing proteins or peptides into detergent-free solutions such as 8 m urea (31–34). Only up to 50% of the total proteins in cell lysate prepared as such could be converted into peptides, and 50% resisted digestion (32, 33). These difficulty and complicacy stem from the use of SDS. SDS has to be removed prior to digestion and HPLC MS analysis, because it inactivates proteases such as trypsin and severely interferes with peptide separation and ionization in HPLC and MS. However, SDS removal from membrane proteins by dia-filtration using 30 kDa filters is rarely clean, and membrane proteins aggregate upon detergent depletion. Membrane proteins also aggregate upon boiling in SDS (35, 36), even though no precipitation is visible in sample tubes. Conventional denaturants such as high urea are successful to denature and spread soluble proteins for more effective protease digestion, but they tend to denature and aggregate membrane proteins upon contact, preventing protease access.
As the primary tool to extract proteins from cell membranes by forming protein-detergent co-micelles, detergent application is key to the cell-to-protein step. Selecting the suitable detergent is critical for membrane protein to maintain solubility, stability, structural integrity, and activity (35–37). Most of the recent over 400 different membrane proteins with resolved atomic structures in Protein Data Bank were enabled by purification using detergent n-dodecyl-β-d-maltopyranoside (DDM) (37–43) and its derivatives (44–47), whereas other common detergents including SDS, CHAPS, cholate, Tween 20, and Triton X-100 failed despite decades of efforts (48, 49). Detergents interfere with ionization of peptides in ESI and MALDI, though nonionic detergents such as DDM are the least detrimental (50, 51). Ionic and poly-disperse detergents also overlap with peptide elution throughout reversed-phase (RP) HPLC, and some inactivate proteases. Therefore, detergent removal and consequent complicated sample processing are so far considered mandatory prior to digestion or HPLC analysis (34). However, our recent GPCR study suggested high-purity nonionic DDM, with low critical micelle concentration (cmc), can be tolerated by RP-HPLC MS (52). Other tools to keep membrane proteins soluble include TM-barrel-forming peptides (53) and multi-lysine lipopeptides (54) that were developed for nuclear magnetic resonance and electron microscopy (EM) studies, but they might introduce chemical noise that skews digestion and quantitation of proteins and peptides.
Here using full-length hetero-pentameric (FLAG-α1)2(β3)2(γ2L)1 human GABAAR overexpressed in HEK293 cells as a model, we describe a DDM/cholesteryl hemisuccinate (DDM/CHS)-based affinity purification method of human CNS membrane proteins for high-performance proteomic analyses. Contrary to attempts to destroy membrane proteins, we harnessed them by providing the most nature-and-activity-promoting conditions. Sonication or dropwise addition of detergent robustly solubilized over 90% of membrane pellets. Besides producing active receptors, the method afforded near 97% MS/MS peptide sequence coverage (α1) by in-gel analysis following a concise trypsin-chymotrypsin tandem overnight digestion—contrasting earlier 10-day, 3-protease, 6- or 10-digestion in-gel methods—and further allowed direct gel-free digestion and HPLC MS analysis, without detergent removal or protein/peptide precipitation. The purification conditions are comparable to those applied in successful crystallization of most membrane proteins reported to date (37, 39–41, 43, 55). With the subunits at unequal stoichiometry ratios and copurified proteins, this system serves as simple model of protein mixture and suggests that, the method less the stringent enrichment can be broadly applicable to prepare complex samples such as whole cell lysates. These results paved the way for streamlined high-performance proteomics of GABAAR, and more importantly allowed directly coupling proteomics with crystallography to map both static and dynamic structures of membrane proteins in crystallization pipelines.
EXPERIMENTAL PROCEDURES
Materials
Tetracycline-inducible human embryonic kidney (HEK293-TetR) cells were provided by the late Dr. H. G. Khorana's lab at the Massachusetts Institute of Technology. Dulbecco's Modified Eagle Medium containing nutrient mixture F-12 (DMEM/F-12) was from ATCC (Manassas, VA). Fetal bovine serum (FBS) premium was from Atlanta Biologicals (Lawrenceville, GA). Glycerol-free flavobacterium meningosepticum-sereted PNGase F solution was purchased from New England Biolabs (Ipswich, MA). M2 monoclonal anti-FLAG antibody-coupled agarose beads (average binding capacity 1.0 mg/ml FLAG-BAP), FLAG peptide bromide, γ-aminobutyric acid (GABA), protease inhibitors leupeptin, aprotinin, chymostatin, pepstatin (LACP), phenylmethylsulfonyl fluoride (PMSF), soybean asolectin, porcine pepsin, 50% (m/v, 15 m) hydroxylamine (NH2OH), 2-mercaptoethanol, Tris-2-carboxyethylphosphine (TCEP), dithiolthreitol (DTT), and iodoacetamide (iodoaa) were from Sigma-Aldrich (St. Louis, MO). Glass column cartridge was from GE Healthcare Bio-Sciences (Piscataway, NJ). Dia-filtration units with molecular weight cutoff (MWCO) 30 kDa, 50 kDa, or 100 kDa were from Millipore (EMD Millipore Corporation, Billerica, MA). N-dodecyl-β-d-maltopyranoside (DDM, Anagrade), cholesteryl hemisuccinate Trizma salt (CHS, Anagrade) and CHAPS (Anagrade) were from Anatrace-Affymetrix (Santa Clara, CA). Phosphate buffered saline (PBS, 10x, final pH 7.4), POROS AL beads (bead size 20 μm), BCATM protein assay kit, EZ-RUN BP3603 (10–170 kDa) protein molecular weight markers for SDS-PAGE, and precast NuPAGE 4–12% (m/v) Bis-Tris gels with MOPS-SDS and MES-SDS running buffers were from Thermo Fisher Scientific (Rockford, IL). Sequence-grade porcine trypsin and chymotrypsin were from Promega (Madison, WI). Radioactive [3H]muscimol (3-hydroxy-5-aminomethylisoxazole, [methylene-3H(N)], 35.6 Ci/mmol) was from Perkin Elmer (Waltham, MA).
Cell Growth, Membrane Isolation and GABAAR Purification
HEK293-TetR cells stably expressing human (N)-FLAG-α1β3γ2L-(GGS)3GK-1D4 GABAAR were obtained as recently described (56). Briefly, the genes coding for GABAAR subunits were individually cloned using independent antibiotic selections and introduced to HEK293-TetR at α1 (bovine): β3 (human): γ2L (human) subunit plasmid copy ratio of 2:2:1. Full-length genomic sequences of the subunits were used, except for an N-terminal FLAG tag inserted after the signaling peptide in α1, and a C-terminal -(GGS)3GK-1D4 tag in γ2L. The cells were grown in a 5% CO2 incubator at 37 °C on poly-d-lysine-coated 15-cm cell culture plates. 72 h after cell seeding in the DMEM/F-12 media supplemented with 10% (v/v) FBS, 0.2% (m/v) penicillin/streptomycin and subunit expression suppressors to reach typical 90% confluence, the cells were induced with 1 μg/ml tetracycline to start the 24-hour GABAAR expression. The cells were then washed with 1xPBS and harvested into lysis buffer composed of 10 mm HEPES (pH 7.5) and protease inhibitors (LACP, PMSF), and frozen at −80 °C. One batch of growth typically produced 60 plates of cells at passage numbers below 25.
Cell lysis and protein purification were all performed at 4 °C or on ice. The cells were lysed with sonic probe at 9500 rpm for durations of 5–7 s, for three cycles at 2–3 min intervals. The cell lysate was homogenized with a hand-held glass homogenizer for 20 strokes, and centrifuged at 20,000 rpm for 30 min to collect membrane pellets. Crude membrane pellets were resuspended in fresh lysis buffer, again homogenized, and centrifuged. Final membrane pellets were resuspended in lysis buffer at a typical protein concentration of 6 mg/ml (BCA assay), further homogenized by inspiration and expiration through 24- and 27-gauge needles, and flash-frozen in liquid nitrogen for storage at −80 °C.
Expressed (N)-FLAG-α1β3γ2L-(GGS)3GK-1D4 GABAAR was affinity-purified on a house-packed anti-FLAG column (M2 monoclonal anti-FLAG agarose beads, 7 mm i.d. x 40 mm) connected to a T valve and a syringe pump operated at a constant flow rate of 0.3 ml/min. A Tris buffer composed of 50 mm Tris-HCl (pH 7.4), 150 mm NaCl, 2 mm CaCl2, 5 mm KCl, 5 mm MgCl2, 4 mm EDTA, and 10% (v/v) glycerol served as the base buffer throughout the purification. The membrane pellets were thawed and solubilized by dropwise addition (∼0.8 ml/min) of DDM/CHS stock to final concentrations of 20 mm (1%, m/v) DDM/4 mm (0.25% Trizma salt, m/v, equivalent to 0.2% free acid form) CHS against 2 mg/ml protein over vigorous stirring for 2.5 h. The 1.2∼2x DDM/CHS stock was prepared in the base buffer. The solubilized proteins were collected by centrifugation at 20,000 rpm for 30 min (unsolubilized pellets were small and typically white), and applied immediately to preconditioned anti-FLAG column. Two batches (60 cell places ×2) of membranes were sequentially solubilized and loaded to the column in a typical purification. The column was then washed with 11 column volumes (CV, 1 CV was 4 ml) of the protein buffer composed of 1 mm (0.05% m/v) DDM and 0.2 mm (0.0125% m/v) CHS prepared in the base buffer, and bound receptors were eluted with 0.15 mg/ml (150 μm) FLAG peptide. Fractions of eluted GABAAR were pooled, cleaned and concentrated by dia-filtration with 100 kDa MWCO filters at 5000–8000 rpm unless specified otherwise, to obtain final product of 25 μm (7 mg/ml) purified GABAAR in the 0.05% DDM/0.0125% CHS/10% glycerol protein buffer. The 120 plates of cells could yield 500 μl of 25 μm purified GABAAR. Flowthroughs throughout the purification at 4 ml, 2 ml, or 1 ml fraction size were monitored on time with UV-Vis over wavelength 200–700 nm, and all steps were sampled and measured with SDS-PAGE afterward. Earlier method development also compared purifying DDM-solubilized membranes into 0.05% DDM versus 5 mm CHAPS/asolectin buffer, and applying anti-FLAG beads in slurry mode versus column mode. Filter sizes of 30 kDa, 50 kDa, and 100 kDa for dia-filtration were compared by subjecting blank DDM/CHS protein buffer to the same dia-filtration and SDS-PAGE processes as applied to proteins.
Detergent Selection
Detergents were screened both for membrane solubilization efficiency by using BCA protein assay and visual inspection of insoluble pellets, and for ligand-binding activity of GABAAR in solubilized membrane by using [3H]muscimol radioligand binding assay. Different detergents (DDM, DDM+CHS, and MNG-2) and mixing methods (simple stirring, grinding, dropwise additions, and brief sonication on ice for 5–7 s x 3–5 times) were examined for solubilization efficiency, using membrane pellet suspension at final concentration of 1 mg/ml. Default solubilization condition represented 1% DDM added all at once to final 6 ml of 1 mg/ml protein followed by vigorous stirring for 2 h, and was included in each set of experiments. Solubilized supernatants were collected by centrifugation as described above. The effect of de-palmitoylation by NH2OH and alkylation by iodoacetamide on solubilization was also tested. The % solubilization efficiency was presented as the % ratio of protein concentration of supernatant over that of membrane suspension before solubilization. The % solubilization improvement was calculated as the % ratio of the solubilization efficiency of specified method over that of the default. Statistical significance in solubilization efficiency difference was evaluated with unpaired t test (two-tailed distribution, two-sample unequal variance) in Excel.
Effects of asolectin, CHAPS and CHS on receptor activity were evaluated by including 0.1 mm soy asolectin, 5 mm CHAPS, or 4 mm CHS (0.25%, m/v) in diluted DDM/CHS-solubilized membrane sample (final DDM 0.05%, 1 mm, CHS 0.015%, 0.24 mm, and total protein 0.15 mg/ml) during protein–ligand incubation, followed by measurement of the specific binding of 2 nm [3H]muscimol and its enhancement by 10 μm etomidate. Each reaction was measured by four filters from single incubation, and each filter was loaded with 0.7 mg membrane protein. The effect of CHAPS on GABAAR ligand binding activity was further examined by comparing the full modulation curves of [3H]muscimol binding by R and S barbiturate enantiomers, in the presence and absence of 5 mm CHAPS using membrane pellet suspensions (Supplemental Note S1). Structures of relevant detergents and additives are shown in supplemental Fig. S1.
Protein Characterization
Purified protein composition was analyzed by SDS-PAGE, and by mass spectrometry following in-gel digestion of multiple gel segments from the full lane. Samples were resolved by NuPAGE 4–12% or 10% Bis-Tris precast gels (1.5 mm thick) over MES-SDS (pH 7.3) or MOPS-SDS (pH 7.7) running buffer, and stained with Coomassie blue in 10% (v/v) acetic acid/50% (v/v) methanol solution. Concentration of purified protein was measured by BCA protein assay using an estimated molecular weight of 285 kDa for the pentameric GABAAR. BCA reaction was performed in triplicates at 60 °C for 10 min using BSA as calibration standard, FLAG elution buffer as blank control and quartz cuvette. Receptor activity of purified protein was characterized by the abundance of specific binding for GABA-mimetic agonist [3H]muscimol and its sensitivity toward 10 μm etomidate modulation, using classical radioligand filtration assay (Supplemental Note S1). Protein dispersion or aggregation was monitored using negative-stain EM.
Negative-stain Electron Microscopy of Purified GABAAR
Negative-stain EM analysis of purified GABAAR was performed at the Microscopy Center at MGH using standard protocols. Nine μl of GABAAR at 25 μm, or at 2.5 μm after 10-fold H2O dilution, was applied to formvar-coated gold grid under room temperature, incubated for 2 min, and rinsed with H2O. The grid was then stained for 2 min on drops of 2% (m/v) uranyl acetate aqueous solution (pH 3.5–4.0), rinsed with H2O again, and dried in air. Protein buffer was used as control. The grids were examined in a JEOL JEM 1011 TEM at 80 kV and images were collected using an AMT digital imaging system (Advanced Microscopy Techniques, Danvers, MA).
PNGase F Deglycosylation of Purified GABAAR
N-glycosylation modification of purified GABAAR was probed by deglycosylation treatment using PNGase F. Various enzyme final concentration (0.5, 1, or 50 units (U)/μl, over 1–2 μm GABAAR), reaction time (1, 2, 4, or 6 h) and temperature (4, 21, or 37 °C) were screened. Reactions were measured with SDS-PAGE (10% Bis-Tris) developed in MOPS-SDS buffer to expand the 40–70 kDa region, using 5–10 μl of 1 μm GABAAR per lane for clear images.
In-gel and Gel-free Digestions of Purified GABAAR
SDS-PAGE gel bands were subjected to in-gel digestion and proteomic analysis at the Taplin Biological Mass Spectrometry Facility. Typically, 20 μl of 6 μm purified GABAAR was applied to each gel lane for in-gel digestion. The gel bands at ∼57 kDa or multiple segments of a full lane were excised, diced into 1 mm squares, de-stained in 50 mm NH4HCO3 and 50% acetonitrile, dehydrated, reduced with DTT, alkylated with iodoacetamide in the dark, and each applied to sequential overnight trypsin (12.5 μg/ml) and chymotrypsin (12.5 μg/ml) digestions at 37 °C. Overnight digestion with only trypsin or chymotrypsin was also tested. The digestion was quenched with 0.4% trifluoroacetic acid, peptides were dried and redissolved in formic acid buffer (0.1%, v/v) for HPLC injection. Direct gel-free digestion of purified GABAAR was tested by using porcine pepsin solution, pepsin-immobilized POROS AL beads slurry, or pepsin-immobilized column. Preparation of pepsin beads and columns was described in another manuscript (57). Purified GABAAR was adjusted to pH 2.5 by mixing with pepsin digestion buffer (0.1 m NaPi-HCl, pH 2.4, 15 mm TCEP, and 0.02% DDM) (52) at 1:1 (v/v) at final GABAAR concentration less than 0.1 μm or 1–2 μm, and incubated with pepsin solution (100 μg/ml, 15 min 4 °C), pepsin beads slurry (20 μl net beads, 30 min 37 °C) over mixing every 5–10 min, or pepsin column (1 mm i.d. × 20 mm, 20–22 °C, GABAAR solution kept on ice) at flow rate 25 μl/min. The digests were directly loaded to HPLC or frozen in liquid nitrogen until use. Digestion efficiency was measured with SDS-PAGE.
Mass Spectrometry
Peptide digests at pH 2–2.5 were analyzed by RP HPLC MS and CID MS/MS on an LTQ-Orbitrap XL mass spectrometer equipped with nanoelectrospray ionization (nESI) source, Agilent 1100 binary pump (Agilent Technologies, Palo Alto, CA) with nanoliter flow splitters, and Famos autosampler (LC Packings, San Francisco, CA) operated in direct injection mode. About 6 μl of peptide sample was loaded in each run. HPLC buffer A contained 0.1% (v/v) formic acid (FA), and buffer B contained 0.1% (v/v) FA and 60% (v/v) acetonitrile. Peptides were separated on a C18 fused silica microcapillary column (3 μm 200 Å Maccel AQ C18 beads, 125 μm i.d. × 18 cm, hand-pulled tip i.d. 5 μm) over a 75-min 0–40% linear gradient of buffer B. The total HPLC analysis time including loading, separation, and column equilibration was 85 min. A high-low top10 data-dependent method was used for MS and CID MS/MS acquisition, where one high-resolution full MS scan over m/z 370–1600 in the orbitrap was followed by up to 10 MS/MS scans of the most abundant MS ions in the linear ion trap under normal scan mode, using isolation width 2.0 Da, normalized collision energy 35%, activation q 0.25, and activation time 50 ms. Dynamic exclusion of precursor ion was applied with a 0.2 Da width for durations of 20 s. Ion maximum injection time was 125 ms for MS and MS/MS.
Proteomic Data Processing
The CID MS/MS spectra were assigned by searching against the GABAAR protein sequences supplied for overexpression or the human proteome containing both forward and reversed sequences (Taplin), using the SEQUEST algorithm (b and y ions), with a fixed modification for cysteine (Cys, C) carbamidomethylation (+57.02146 Da, C-Cb) and dynamic modifications for methionine (Met, M) oxidation (+15.99492 Da, Mox), and N(Q) deamidation (+0.98402 Da, when PNGase F-specific, defined as N(Q)-deglycosylation), at mass variation tolerance of 5 or 10 ppm for MS and 0.8 Da for MS/MS, unless specified otherwise. Common contaminations such as human keratin and trypsin were excluded for in-gel data, and porcine pepsin sequence was added for the gel-free pepsin digest data for test. In-gel trypsin data were searched with trypsin enzyme specificity and up to two missed cleavages; In-gel chymotrypsin, trypsin-chymotrypsin, and gel-free pepsin digests were searched without enzyme specification. Unless specified otherwise, peptide assignments were filtered to a peptide false discovery rate (FDR) below 1% by applying a target-decoy database search strategy and a linear discrimination algorithm that integrated multiple parameters (16); For human proteome search of gel bands, the resulting MS/MS data were further filtered to obtain protein FDR below 1% (16). Gel-free MS/MS spectra were also searched against human proteome (downloaded May 23, 2013, Ensembl) or GABAAR sequences (dynamic M oxidation) with SEQUEST, and filtered to peptide FDR<1% using Percolator (by q-value) in Proteome Discoverer version 1.3 (PD 1.3). A protein was considered identified only when three or more unique peptides were detected. Peptide coverage maps over protein sequence were prepared in part using the HDX workbench software (58), and other graphics were prepared using Excel.
N-glycosylation sites were validated by decoy deamidation (N, Q) search on PNGase F nontreated protein samples as a control for false positive, and by applying a strict 0.2 Da mass tolerance to CID MS/MS spectra (same 10 ppm for MS) of both versions of samples (PD 1.3, peptide FDR <1% by Percolator). Only those sites identified as deamidated in PNGase F-treated samples and not as deamidated in nontreated samples were decided to be real N-glycosylation sites. Those identified as deamidated in both were considered false positive and their peptides were discarded, assuming that there was no natural occurrence of both amide and acid forms of the residues and that PNGase F-deamidation equaled N(Q)-glycosylation.
For absence of cells with empty vector, to screen for putative GABAAR-specific associated proteins, we searched identified proteins against CRAPome (version 1.0, 343 entries), a database of known contaminants in affinity-purifications from human cell lines (59), and categorized them with arbitrary percent occurrence boundaries as nonspecific (>30.1%), likely real (10.1–30.0%) or real GABAAR-specific proteins (≤10.0%).
RESULTS
Cell-to-Protein Method Workflow
We developed a DDM/CHS-based cell-to-protein affinity purification method, suitable for deep and direct proteomic analysis for human GABAAR from HEK293 cells (Fig. 1). Our method featured the uses of: DDM/CHS throughout solubilization and purification, anti-FLAG beads as a column operated at constant low flow rate, and 100 kDa filters for dia-filtration in protein concentrating. We included CHS with DDM in all solubilization and protein buffers, to mimic the natural ∼20–30% (w/w) cholesterol composition in total lipids of CNS cell membranes (60), to therefore promote less-biased membrane protein extraction and maintain protein structure/activity—based on earlier findings that addition of CHS to DDM stabilized the structure and activity of human GPCRs (39). DDM/CHS at 1%/0.25% added dropwise using 1.2–2x detergent stock afforded robust membrane solubilization efficiency at over 90% (Fig. 2A), and DDM/CHS at 0.05%/0.0125% in the protein buffer was well tolerated by subsequent HPLC and MS. CHS facilitated the receptor activity in ligand binding and modulation measured using solubilized membranes (Fig. 2B).
Fig. 1.
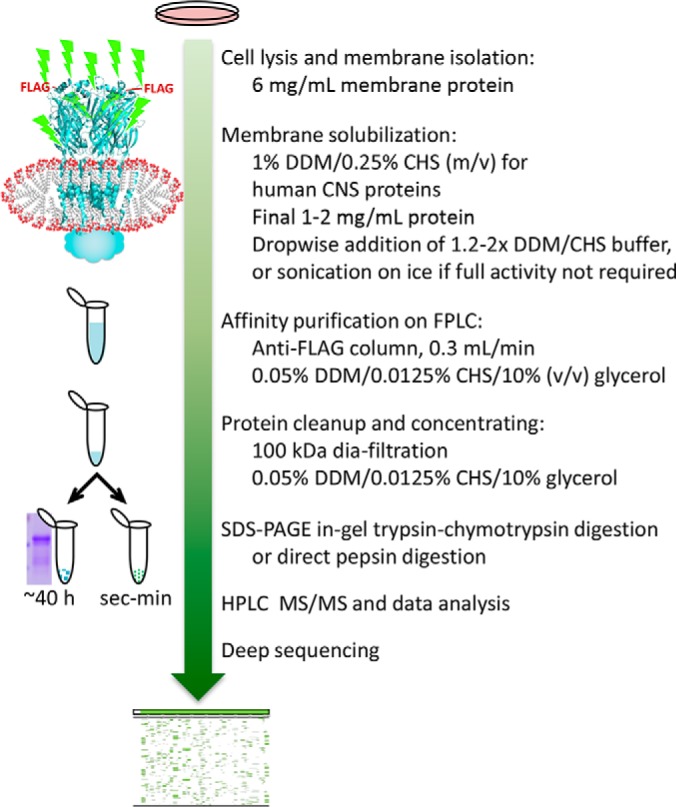
Method workflow of protein purification and subsequent validation with in-gel and gel-free sequencing analyses.
Fig. 2.

GABAAR protein purification and characterization. A, Membrane solubilization methods screened by protein concentration (BCA) and imaging of unsolubilized pellets. *** designates statistically significant difference at 95% confidence by unpaired t test (p = 0.029); *, p = 0.105. B, The effects of detergents and additives on receptor activities, represented by [3H] muscimol (2 nm) binding and etomidate (10 μm) modulation using solubilized membranes (0.15 mg/ml), and the binding and modulation activities of DDM/CHS-purified GABAAR (0.03 μm in reaction). C, SDS-PAGE profile of a representative purification. Lane 1, solubilized membrane (2 mg/ml) before loading to the anti-FLAG column; lane 2, flowthrough during column loading at 0.3 ml/min; lanes 3 and 4, flowthroughs from column washing after loading; lane 5, flowthrough after loading ¼ CV of the eluting buffer containing 150 μm FLAG peptide; lanes 6, 7, 8, 9, and 10, flowthroughs from 11 CV eluting buffer: lanes 6 and 7 were pooled and concentrated to obtain final GABAAR product; lanes 11 and 12, final concentrated GABAAR (25 μm) applied at 1 μl/well, incubated for 10 min with SDS sample buffer at 100 °C, and with 0.3 fold more SDS at 60 °C, respectively; lanes 13 and 14, flowthroughs during protein concentrating using dia-filtration with 100 kDa filters; lane 15, protein molecular weight markers (3 μl). D, SDS-PAGE of purified GABAAR before and after complete PNGase F reaction revealed extensive N-glycosylation.
To improve separation efficiency and consistency in lipid retention control across purifications, we applied anti-FLAG beads as a column with small internal diameter (7 mm i.d. × 40 mm) at estimated site occupancy of 60–80%, over slurry mode. To facilitate affinity binding of protein or FLAG peptide during column loading or eluting, we critically applied a constant low flow rate of 0.3 ml/min, about 10% of averaged flow rate by gravity, throughout the purification. Immediately eluted proteins were low in GABAAR concentration, high in FLAG peptide, and not practical for downstream analyses; they were thus cleaned and concentrated via dia-filtration at low centrifugation speed using 100 kDa filters (instead of precipitation). Earlier use of 30 kDa filters caused accumulation in DDM/CHS, and CHS overwhelmed half of the HPLC and MS spectra in direct gel-free digest analysis, although receptor protein composition by SDS-PAGE showed no difference.
Using DDM/CHS protein buffer without proteins, comparison of filter sizes showed that both 30 kDa and 50 kDa filters caused accumulation of DDM/CHS micelles, manifested as a bright blue spot below 11 kDa in SDS-PAGE upon Coomassie blue staining, whereas 100 kDa filter did not (supplemental Fig. S2B). No protein stain was observed in the filter-through of protein dia-filtration using 100 kDa filters either (Fig. 2C lanes 13 and 14). The molecular weight of 510 Da and micelle aggregation number of 98 lead to DDM micelles of 50 kDa, and incorporating CHS is expected to further increase the micelle size, whereas DDM/CHS/protein co-micelle should be much larger than 100 kDa. This result suggested that, in the purification buffer, most proteins likely remained as complexes wrapped in micelles. The 100 kDa filter was also applied in successful purifications of 36 kDa one-subunit GPCRs (39, 52), cytochrome c oxidase containing multiple subunits at sizes of 4–60 kDa (55, 61), and membrane protein super-complexes (51).
Detergent Selection and Validation
Both detergent chemical structure and method of addition affected membrane solubilization efficiency (Fig. 2A). Default condition using 1% DDM (∼115 cmc), added all at once to final 1 mg/ml membrane protein followed by vigorous stirring for 2 h (four replicates), gave 61 ± 9% solubilization efficiency by BCA. Addition of CHS appeared to maintain if not increase efficiency. MNG-2, recently developed to extend GPCR activity over time and to promote complex stability for crystallization (44), did not increase solubilization efficiency per se here. Grinding solubilization mixture throughout 2 h made little difference in solubilization efficiency. By contrast, brief sonication—of 5–7 s for 3–5 times on ice during the 2-hour stirring—led to strikingly clearer mixtures and smaller pellets following centrifugation, consistent with the higher solubilized protein level measured by BCA. Likewise, gradual addition of 1.2–2x detergent stock over 80 min led to higher solubilization efficiency (93%), but dropwise addition of 10x detergent stock at similar flow rate over 30 min didn't show such an advantage.
These results highlighted the mechanism of membrane protein solubilization via forming detergent-protein co-micelles, and suggested that promoting protein-detergent co-micelle formation solves the challenge of efficient membrane solubilization. Although fast addition of detergent at high cmc represented a process of protein insertion into existing detergent micelles, slow addition of detergent allowed the protein and detergent sufficient time to fully disperse, thus facilitated formation of co-micelles once the detergents reached the critical micelle concentration. Sonication also aided micelle rearrangement and co-micelle formation. Attempts to address solubilization by removing palmitoylation anchors using NH2OH and iodoacetamide treatments turned out impractical, as they led to sample loss because of multiple separations and the reagents interfered with BCA assay (Fig. 2A).
Detergents for protein buffer were screened by the ability to maintain receptor ligand binding activity (Fig. 2B). Adding 4 mm CHS to DDM/CHS-solubilized membrane maintained receptor activity and modulation sensitivity to etomidate. Addition of 0.1 mm soy asolectin showed no advantage, whereas CHAPS removed over 80% of the binding sites, despite an apparent 200% relative modulation by etomidate. Because CHAPS is a common detergent used for membrane protein extraction and reconstitution, we further examined the influence of CHAPS on receptor activity by comparing the full modulation curves over a pair of barbiturate enantiomers (62), using membranes (supplemental Fig. S2C), and observed similar effects.
Negative-stain EM analysis of purified GABAAR in 0.05% DDM/0.0125% CHS/10% glycerol buffer showed that GABAAR were well dispersed particles with diameters around 25 μm, but upon a 10-fold depletion of detergent (0.005% DDM/0.00125% CHS, 0.57 cmc), the protein aggregated instantly (supplemental Fig. S2A). This stark contrast emphasized that sufficient detergent is essential in maintaining the integrity of membrane proteins. Further EM method optimizations such as buffers for membrane proteins might improve the image. Taken together, DDM/CHS allowed high-efficiency membrane solubilization, receptor activity, and structural integrity.
Characterization of Purified Protein
The purified GABAAR maintained agonist-binding activity and displayed sensitivity toward etomidate modulation of [3H]agonist binding (Fig. 2B). SDS-PAGE of the purified protein showed band density predominantly at 40–57 kDa, the expected MW range for GABAAR subunits (Fig. 2C); proteomic analysis following in-gel digestion found that each of these bands contained all three types of α1, β3, and γ2L subunits as predominant components, based on PSMs. Low amounts of other proteins were also copurified in the lane, most of which were reported as common contaminants from nucleus and mitochondria (59). PNGase F treatment lowered all GABAAR subunit bands to 35–55 kDa, but still each band contained all three types of subunits (Fig. 2D). NH2OH incubation did not induce any band shift, suggesting palmitoylation did not cause the multi-band distribution. Similar formation of multi-bands for glyco-proteins previously occurred in soluble Aplysia acetylcholine binding protein (AChBP) produced from the same HEK293-TetR, and displayed dependence on host cell line, affinity tag type and tag position (63).
Comparison of a range of reaction temperature, time and PNGase F enzyme concentration confirmed that, 50 U/μl of PNGase F at 4 °C over 1 h against 1–2 μm GABAAR reached completion of reaction, whereas 1 U/μl enzyme, even at 37 °C over 4 h, was too low to finish deglycosylation (supplemental Fig. S3). Intriguingly, such requirements for longer time and more enzymes contrasted an earlier observation that 0.05 U/μl (50 U/ml) PNGase F at 4 °C within 1 h completely deglycosylated 15 μm of a human GPCR β2AR construct containing two adjacent N-glycans on its extracellular surface (52)—suggesting more intricate glycosylations in GABAAR. Compared with boiling (Fig. 2C lane 11), 60 °C incubation (Fig. 2C lane 12) of protein in 0.3-fold more SDS alleviated protein aggregation as shown in the upper part of the gel.
In-gel Tandem Digestion Achieved High Sequence Coverage of GABAAR
Compared with trypsin (t) or chymotrypsin (c) overnight in-gel digestion, tandem trypsin-chymotrypsin (tc) consistently generated higher MS/MS sequence coverage, more unique peptides and more PSMs, using gel bands from multiple batches of purified GABAAR (Fig. 3A). Protein concentrating further increased the number of unique peptides (4–5 fold), PSMs and sequence coverage, and decreased subunit-bias (Fig. 3A), indicating concentrating crude elutes is necessary to obtain samples useful for sequencing, even with in-gel method.
Fig. 3.

Tandem trypsin-chymotrypsin in-gel digestion of DDM/CHS-purified GABAAR achieved high sequence coverage. A, Comparison of sequencing metrics—% sequence coverage, number of unique peptides and number of peptide spectral matches (PSMs)—of in-gel digestions using trypsin (t), chymotrypsin (c), or tandem trypsin-chymotrypsin (tc) on GABAAR gel bands from various purifications. Gel band indicates equivalent migration position. B, Gel segments used for deep overall protein composition profiling by tc digestion, in the absence (lane 2) and presence (lane 4) of complete PNGase F treatment. C, Subunit distribution within major GABAAR-containing bands, before and after PNGase F treatment: one aggregation band above 170 kDa (a and a′), two bands at 40–72 kDa that shifted to 40–55 kDa after PNGase F (e and e′; f and f′), and two replicates of the whole segment of 40–72 kDa (equivalent to e+f; gel not shown). D, Reproducibility and E, length distribution of unique peptide ions identified for GABAAR, from replicate tc analyses of the e+f whole bands. Oxidized peptides were included as unique.
Using the whole gel bands at 40–57 kDa from 20–40 μl of 6 μm purified GABAAR, our succinct tandem overnight tc digestion reproducibly achieved robust over 90% sequence coverage for α1, over 80% for β3, and about 60% for γ2L, and more than 500 unique peptides for GABAAR, with peptide lengths predominantly in the useful range of 4–20 residues (64) (Fig. 3A experiments 8 and 9, Fig. 3E, and Fig. 4). PNGase F treatment of protein further increased the coverage to 96.8% for α1, and 87.0% for β3 (Fig. 4). Both sequence coverage and useful unique peptides greatly improved in our tc method, contrasting previous reports that needed either 10-day 3-protease 10-digestion in-gel, or 2-day 3-protease 6-digestion gel-free methods—using Triton X-100-purified GABAAR from rat brain or Sf9 cells (27–30). However, common to in-gel and previous gel-free methods that spanned long hours and dried products, nearly every M-containing peptide had an additional Mox version (orange lines in Fig. 4A), further complicating the digest mixture.
Fig. 4.

Peptide coverage map of GABAAR from one typical experiment of in-gel trypsin-chymotrypsin digestion using the whole 40–72 kDa band (e+f in Fig. 3B) combined with PNGase F-specific deamidated peptides (e' and f' bands in Fig. 3B). A, Unique peptides mapped against protein sequence. % sequence coverage was calculated after excluding expected signaling residues (black triangle). Black line represents one unique peptide sequence; orange, Mox version; dented front, PNGase F-specific deamidated peptide ions. Green NXX, validated N-glycosylation site. Bold lines indicate domains: disulfide Cys-loop (cyan); TM helix (TM, blue); intracellular loop (ICL, red dash line). B, and C, Representative MS/MS spectra for deamidation identification: β3-NMS and β3-NLT, respectively. Spectra for other sites are shown in supplemental Fig. S4. N#, N-deamidation; M*, Mox.
In-gel tc Digestion and PNGase F Identified New N-glycosylation Sites
Our in-gel tandem tc digestion method significantly improved sequence coverage and number of unique peptides for GABAAR, and when coupled with PNGase F treatment, further identified multiple N-glycosylation sites that complied to canonical NX(!P)S/T motif on the α1 and β3 subunits (Fig. 4 lines with dented fronts); only two of these sites were previously reported in mouse or rat (α1 subunit) (28, 65). We defined N-glycosylation sites by identifying a deamidation change (N to D, or Q to E) specific to PNGase F treatment using narrow MS/MS mass tolerance (0.2 Da) in spectra search. All the validated glycosylation sites occurred on N and none on Q. We found two sites (N46TT and N146MT) in α1 extracellular domain (ECD), consistent with two previous sequencing reports in mouse and rat (28, 65), and three new sites (N33MS, N105LT, and N174CT) in β3 ECD. We saw no sites on γ2L for its limited sequence coverage, but solved this problem in another study (57).
Remarkably, the β3 N174CT site occurred in its Cys loop (Fig. 4), a name-defining structural feature of Cys-loop family LGICs. However, only two unique peptides were detected to harbor this site, largely because intertwined C-C bond and bulky glycosylation obstructed effective protease access, a well-established challenge (23) also addressed in another study (57). Strikingly, during the review of this manuscript, crystallography of a truncated construct of (β3)5 hGABAAR expressed in HEK293S-GnTĪ resolved three saccharides for the β3 N174CT glycan, confirming it exists and interacts with adjacent β-sheet arms of loop C (14). Most N-glycosylation sites on ECD were covered only after PNGase F treatment, indicating near-full site occupancy. The exception was β3-N33MS: abundant unique peptides identified both before and after PNGase F, suggested its partial occupancy (Fig. 4). In addition, we found two sites (N373NT and N422SV) in the intracellular domain of α1 at low site-occupancy; they met the data filtering criteria, and the NXV motif was noticed in previous studies (65, 66), but their true modified structures and physiological implications remained unclear.
GABAAR and Copurified Proteins Profiled by Deep In-gel tc Analysis
We profiled the proteins co-purified with GABAAR from HEK293 by dissecting the full gel lanes of purified GABAAR into multiple segments (Fig. 3B) for individual tc digestion, HPLC CID-MS/MS, and data search against human proteome. The top 10 most abundant proteins in each segment before and after PNGase F treatment were summarized in supplemental Tables S1 and S2, respectively. We found that boiling protein in SDS, despite high concentration of 2-mercaptoethanol, caused a marked aggregation beyond 170 kDa (Fig. 3B lane 2), whereas using 60 °C and a 0.3-fold more SDS alleviated such aggregations (Fig. 3B lane 4 and supplemental Fig. S3). Typical to long in-gel and gel-free methods, many peptides were generated from keratin contamination, thus the sequence of keratin was removed from the database for in-gel analysis.
We identified three gel segments that predominantly contained all three subunits of GABAAR: the aggregation band above 170 KDa, and the two most intense bands at 50–57 kDa and 40–50 kDa. Compared with expected assembly stoichiometry and plasmid copy ratio of α1:β3:γ2L at 2:2:1, far more peptides were identified for α1 and β3 than for γ2L in the 40–57 kDa bands, whereas the aggregation band above 170 kDa displayed high content of γ2L peptides, with α1, β3, and γ2L PSM ratio at 102:159:104 and unique peptide ratio at 20:21:18. Deglycosylation shifted the relative quantity of α1 and β3 subunits within bands (Fig. 3C bands e and f versus e' and f'), suggesting subunit-biased N-glycan contents. Further NH2OH treatment of deglycosylated GABAAR (aiming to remove Cys-palmitoylation) slightly increased density in the aggregation band (data not shown). These results suggested that γ2L differs from α1 and β3 by being more prone to aggregation upon heating, consistent with the higher Cys content of γ2 in its intracellular and TM domains. Besides α1, β3, and γ2L, several other GABAAR subunits were also assigned by search, because of shared sequence.
Although purification conditions (such as extensive washing after column loading) were aimed to obtain high-purity GABAAR, some other proteins still copurified, though at estimated total gel stain density less than about 15% of that for total GABAAR subunits (Fig. 2C, D, Fig. 3B, and supplemental Fig. S3). CRAPome screening highlighted potentially real and physiologically meaningful GABAAR-specific proteins (supplemental Tables S1 and S2). Those found at above 30.1% occurrence (“nonspecific”) were typically from nucleus and mitochondria introduced during whole cell lysis. In about 50 “real” or “likely real” GABAAR-specific proteins identified at various PSMs across the gel lane, calnexin was found in the 70–100 kDa gel segment (supplemental Table S1). Most proteins known implicated in GABAAR endocytosis (15) were not found in the top10 lists by in-gel analysis with HEK293. Other putatively real associated proteins included UDP-glucosyltransferase (UGCGL1) and mannosyl-oligosaccharide glucosidase (MOGS).
DDM/CHS-purification Supported Direct Gel-free Digestion and Proteomic Analysis
Most importantly, the DDM/CHS-purified GABAAR demonstrated full compatibility with direct protease digestion within seconds-minutes, and with direct HPLC-ESI MS/MS analysis, using much less protein sample without requiring detergent removal (Fig. 5). The HPLC MS total ion count (TIC) trace showed that DDM eluted only in the end of the gradient at high %B at modest ion count intensity without interfering with peptides, and that the most intense chromatography peaks at 16 and 18 min were peptide ions mostly at charge state 2+ or 3+ (Fig. 5B). Most of the remaining unidentified sequence gaps contained Cys and NXS/T sites, and likely attributed to unaddressed PTMs. Thus the DDM/CHS-purification entirely eliminated the needs for protein/peptide precipitation/resolubilization, high urea, desalting, dia-filtration, and centrifugation, and thereby cleared the barrier toward a sensitive uninterrupted proteomic streamline amenable for complete automation. CHAPS-purified GABAAR failed to produce spectra with quality worth searching: by forming strong fixed positive ions and adducts at acidic pH, CHAPS overwhelmed HPLC leaving few MS/MS spectra legible for peptides.
Fig. 5.

DDM/CHS-purified GABAAR allowed direct gel-free pepsin digestion and HPLC CID MS/MS analysis without detergent removal or protein/peptide precipitation. A, Comparison of the sequencing metrics—% sequence coverage, numbers of unique peptides and PSMs—of direct digestion methods using pepsin solution, pepsin beads, or pepsin column, on GABAAR purified in various conditions. B, Representative HPLC MS chromatography (top) of a direct HPLC MS/MS analysis, and averaged MS scans for retention time 16–17 min (middle) and 18–19.5 min (bottom) showing mostly 2+ and 3+ charged peptide precursor ions. Relative abundance represented % normalized ion counts over the highest (marked on each panel). C, Peptide coverage map of GABAAR from one typical experiment of direct pepsin-column digestion at 25 μl/min followed by HPLC CID MS/MS analysis using less than 10 μl x1 μm DDM/CHS-purified GABAAR. Peptides were not deglycosylated or alkylated in this experiment. The % sequence coverage was calculated after excluding expected signaling residues (black triangle). Bold lines represent domains: Cys-loop (cyan); TM helix (blue); ICL (red dash line).
Comparison of the sequencing metrics of direct digestions of purified GABAAR using pepsin solution, pepsin-immobilized bead slurry, and pepsin column demonstrated a clear advantage of using concentrated GABAAR and pepsin column (Fig. 5A). Seconds of digestion time through pepsin column led to complete digestion of GABAAR (gel not shown), high sequence coverage (84% for α1, 80.1% for β3, and 61.6% for γ2L), over 350 unique peptides and over 1100 PSMs for GABAAR with subunit distributions closer to the 2:2:1 stoichiometry (Fig. 5), and few artificial modifications: only a few peptides contained Mox (supplemental Table S4C).
When searched against the human proteome (Fig. 6A and supplemental Table S3), GABAAR α1, β3, and γ2L subunits were the predominant top three components by PSMs, at sequence coverage of 45%, 45%, and 26%, unique peptides at 81, 49, and 18, and PSMs at 201, 141, and 59, respectively. Sufficient peptides and coverage in γ2 intracellular loop confirmed that the γ2 subunit in purified GABAAR assumed the long form (NP-LLRMFSFK-AP) and not the short form (NPAP) (Figs. 5C and 6A). Further, contaminations from sample processing such as keratin (two unique peptides, PSM 4) and collagen (one unique peptide, PSM 1) were remarkably low—below the designated 3-unique-peptide detection criteria (supplemental Table S3). Only a small number of peptides were generated by porcine pepsin using pepsin column, in contrast to the overwhelming (over 100-fold) pepsin auto-digested peptides when using pepsin as a solution.
Fig. 6.

Direct gel-free profiling of DDM/CHS-purified GABAAR against human proteome and CRAPome suggested tentative associated proteins. A, Proteins identified by direct analysis and searching against human proteome (May 23, 2013 Ensembl) and CRAPome (version 1.0, total 343 experiments). Keys: * Spectra were searched without modification, and identification required three or more unique peptides at peptide FDR<1%. ** Relative % occurrence in CRAPome was defined as % found/total (343) entries in database. Occurrence in CRAPome: nonspecific (blue), sample-specific (red). # Specificity was defined with arbitrary boundaries of % occurrence: >30.1% nonspecific; 10.1–30.0%, likely real; 0–10.0%, real. B, Speculated presence of GABAAR-calnexin super-complex that survived DDM/CHS-purification. Cartoon, GABAAR (adapted from C. elegans GluCl 3RHW), glycans not shown; Surface, calnexin extracellular domain (PDB: 1JHN), colored from N (blue) to C terminus (red).
Minimal protein contaminants or artifacts from sample processing greatly enhanced the sensitivity and accuracy in the identification and label-free quantitation of low-abundance species in shotgun proteomics. Among the myriad proteins in membrane lysates subjected to purification, CRAPome screening revealed only two “real” or “likely real” specific associated proteins at PSM levels beyond 20% of GABAAR γ2 subunit (PSM 59): calnexin (PSM 40), and pyrroline-S-carboxylate reductase (PYCR2, PSM 18) (Fig. 6A and supplemental Table S3). Nonspecific and lower-abundance proteins, such as heat shock proteins (Fig. 6A), may be ubiquitous in cells but assume important functions upon cell stress.
Intriguingly, calnexin, a Ca2+-sensing 67-kDa one-TM-helix chaperone, now emerged as the most abundant associated protein, immediately following γ2 subunit (Fig. 6A). It was identified in multiple batches of purifications by both in-gel and gel-free analyses. Although traditionally located in endoplasmic reticulum (ER) to assist folding and assembly of nascent glyco-proteins including nicotinic acetylcholine receptor (nAChR) (67–69), calnexin was recently found abundant on neuron plasma membrane in rat brain by immunohistochemistry, and its recruitment increased with N-methyl-d-aspartate (NMDA) receptor activation (70). The comparable quantities of calnexin (PSM 40) and GABAAR γ2 subunit (PSM 59) led us to speculate the presence of GABAAR-calnexin super-complex that survived the DDM/CHS-purification (Fig. 6B), which can be examined further by quantifying changes with ligand stimulation.
Thus, DDM/CHS-purification, followed by gel-free analysis of all components as one sample, provided proteomic evidence for calnexin (Ca2+) associated with GABAAR (Cl̄) in HEK293, possibly on plasma membrane, and suggested calnexin may function for GABAAR beyond ER. A clear view of potential interacting candidates was masked by overwhelming other components in earlier deep in-gel analysis of separate bands. Interactome studies typically prepared protein samples using HPLC/MS-noncompatible detergents such as SDS, deoxycholate, Tween 20, and Triton X-100 (or no detergent but high urea), often followed by in-gel analysis, protein/peptide precipitation or overnight RapiGest detergent exchange. Here we showed that beyond convenience and cost-effectiveness, DDM/CHS-purification greatly simplified accurate identification of potential interacting proteins, by enabling direct gel-free as well as deep in-gel analyses.
DISCUSSION
In summary, compared with previous reports on Triton X-100-purified GABAAR (27–30), our DDM/CHS-based purification produced active GABAAR at high purity amenable for deep in-gel analyses; our simpler tandem trypsin-chymotrypsin in-gel digestion method afforded higher sequence coverage of all GABAAR subunits, and revealed new N-glycosylation sites (Fig. 7). The purification also fully supported direct gel-free digestion and instant HPLC-ESI MS/MS analysis, including TM domains (Fig. 7A), with minimal contamination or artifacts from sample processing. We achieved this progress by providing membrane proteins with nature and activity-promoting conditions throughout the preparation, rather than harsh denaturation.
Fig. 7.

Merits of DDM/CHS-purification for deep in-gel and direct gel-free proteomic sequencing of GABAAR. A, Peptides identified for the transmembrane region (red), from direct gel-free MS/MS analyses of purified GABAAR (extracted from Fig. 5C). B, Comparison with current literatures that used Triton X-100. (2012 gel-free Ref. (28): protein consumed in one experiment was based on compromised radioligand binding, but total protein concentration was not reported.)
This DDM/CHS-purification method, developed using heteromeric hGABAAR/HEK293 and compatible with most membrane proteins purified for crystallization (37, 39–41, 43, 55), is likely applicable to prepare other mammalian membrane proteins or cell lysates—with either more or less strict enrichment—for high-performance proteomics. These results not only paved the way for streamlined proteomic analysis of human synaptic membrane proteins, but more importantly allowed directly coupling proteomics with crystallography to map both dynamic and static structures, for essentially all membrane proteins in crystallization pipelines.
Supplementary Material
Acknowledgments
We thank Ross Tomaino and Dr. Steven P. Gygi at the Taplin Biological Mass Spectrometry Facility of Harvard Medical School for in-gel digestion, HPLC MS/MS data acquisition, data search and helpful discussions on experimental design and result analysis. We thank Mary McKee and Dr. Dennis Brown at the Microscopy Core of the Program in Membrane Biology at MGH for electron microscopy data acquisition and result discussions. We thank Bruce Pascal and Dr. Patrick R. Griffin at Scripps Florida for providing the HDX workbench program. We thank Dr. Shelagh M. Ferguson-Miller for reviewing the manuscript. The late Khorana lab at MIT generously provided the HEK293-TetR cell line, and the Miller lab provided technical support in maintaining GABAAR/HEK293-TetR cells.
Footnotes
Author contributions: X.Z. designed research; X.Z. performed research; X.Z. and K.W.M. contributed new reagents or analytic tools; X.Z. analyzed data; X.Z. wrote the paper; K.W.M. read manuscript.
* This work was supported in part by the National Institute of General Medical Sciences (GM 58448, K.W.M.) and by the Department of Anesthesia, Critical Care and Pain Medicine, Massachusetts General Hospital (K.W.M.).
 This article contains supplemental Figs. S1 to S4, Tables S1 to S4, and Note S1.
This article contains supplemental Figs. S1 to S4, Tables S1 to S4, and Note S1.
1 The abbreviations used are:
- TM
- transmembrane, or transmembrane helix
- GABAAR
- gamma-aminobutyric acid type A receptor
- LGIC
- ligand-gated ion channel
- CNS
- central nervous system
- GPCR
- G protein-coupled receptor
- β2AR
- beta-2 adrenergic receptor
- HEK293
- human embryonic kidney cell line 293
- TetR
- tetracycline repressor
- PTM
- post-translational modification
- ICL
- intracellular loop
- ECD
- extracellular domain
- C-C loop
- disulfide-bonded Cys-loop
- Mox
- Methionine oxidation
- DDM
- n-dodecyl-β-D-maltopyranoside
- CHS
- cholesteryl hemisuccinate Trizma salt
- MNG-2
- maltose-neopentyl glycol type 2
- iodoaa
- iodoacetamide
- TCEP
- Tris-2-carboxyethylphosphine
- cmc
- critical micelle concentration
- FPLC
- fast protein liquid chromatography
- CV
- column volume
- tc
- trypsin-chymotrypsin
- t
- trypsin
- c
- chymotrypsin
- U
- units
- RP
- reversed-phase
- FDR
- false discovery rate
- PSM
- peptide spectral match
- TIC
- total ion count
- MWCO
- molecular weight cutoff
- EM
- electron microscopy
- ER
- endoplasmic reticulum.
REFERENCES
- 1. Pieper U., Schlessinger A., Kloppmann E., Chang G. A., Chou J. J., Dumont M. E., Fox B. G., Fromme P., Hendrickson W. A., Malkowski M. G., Rees D. C., Stokes D. L., Stowell M. H., Wiener M. C., Rost B., Stroud R. M., Stevens R. C., Sali A. (2013) Coordinating the impact of structural genomics on the human alpha-helical transmembrane proteome. Nat. Struct. Mol. Biol. 20, 135–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Olsen R. W., Sieghart W. (2009) GABA A receptors: subtypes provide diversity of function and pharmacology. Neuropharmacology 56, 141–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Forman S. A., Miller K. W. (2012) Anesthetic sites and allosteric mechanisms of action on Cys-loop ligand-gated ion channels. Can. J. Anaesth. 58, 191–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Baulac S., Huberfeld G., Gourfinkel-An I., Mitropoulou G., Beranger A., Prud'homme J. F., Baulac M., Brice A., Bruzzone R., LeGuern E. (2001) First genetic evidence of GABA(A) receptor dysfunction in epilepsy: a mutation in the gamma2-subunit gene. Nat. Genet. 28, 46–48 [DOI] [PubMed] [Google Scholar]
- 5. Wallace R. H., Marini C., Petrou S., Harkin L. A., Bowser D. N., Panchal R. G., Williams D. A., Sutherland G. R., Mulley J. C., Scheffer I. E., Berkovic S. F. (2001) Mutant GABA(A) receptor gamma2-subunit in childhood absence epilepsy and febrile seizures. Nat. Genet. 28, 49–52 [DOI] [PubMed] [Google Scholar]
- 6. Macdonald R. L., Gallagher M. J., Feng H. J., Kang J. (2004) GABA(A) receptor epilepsy mutations. Biochem. Pharmacol. 68, 1497–1506 [DOI] [PubMed] [Google Scholar]
- 7. Brooks-Kayal A. R., Shumate M. D., Jin H., Rikhter T. Y., Coulter D. A. (1998) Selective changes in single cell GABA(A) receptor subunit expression and function in temporal lobe epilepsy. Nat. Med. 4, 1166–1172 [DOI] [PubMed] [Google Scholar]
- 8. Marini C., Harkin L. A., Wallace R. H., Mulley J. C., Scheffer I. E., Berkovic S. F. (2003) Childhood absence epilepsy and febrile seizures: a family with a GABA(A) receptor mutation. Brain 126, 230–240 [DOI] [PubMed] [Google Scholar]
- 9. Hernandez C. C., Gurba K. N., Hu N., Macdonald R. L. (2011) The GABRA6 mutation, R46W, associated with childhood absence epilepsy, alters 6beta22 and 6beta2 GABA(A) receptor channel gating and expression. J. Physiol. 589, 5857–5878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ding L., Feng H. J., Macdonald R. L., Botzolakis E. J., Hu N., Gallagher M. J. (2010) GABA(A) receptor alpha1 subunit mutation A322D associated with autosomal dominant juvenile myoclonic epilepsy reduces the expression and alters the composition of wild type GABA(A) receptors. J. Biol. Chem. 285, 26390–26405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jones-Davis D. M., Macdonald R. L. (2003) GABA(A) receptor function and pharmacology in epilepsy and status epilepticus. Curr. Opin. Pharmacol. 3, 12–18 [DOI] [PubMed] [Google Scholar]
- 12. Gurba K. N., Hernandez C. C., Hu N., Macdonald R. L. (2012) GABRB3 mutation, G32R, associated with childhood absence epilepsy alters alpha1beta3gamma2L gamma-aminobutyric acid type A (GABAA) receptor expression and channel gating. J. Biol. Chem. 287, 12083–12097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hassaine G., Deluz C., Grasso L., Wyss R., Tol M. B., Hovius R., Graff A., Stahlberg H., Tomizaki T., Desmyter A., Moreau C., Li X. D., Poitevin F., Vogel H., Nury H. (2014) X-ray structure of the mouse serotonin 5-HT3 receptor. Nature 512, 276–281 [DOI] [PubMed] [Google Scholar]
- 14. Miller P. S., Aricescu A. R. (2014) Crystal structure of a human GABAA receptor. Nature 512, 270–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Luscher B., Fuchs T., Kilpatrick C. L. (2011) GABAA receptor trafficking-mediated plasticity of inhibitory synapses. Neuron 70, 385–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huttlin E. L., Jedrychowski M. P., Elias J. E., Goswami T., Rad R., Beausoleil S. A., Villen J., Haas W., Sowa M. E., Gygi S. P. (2010) A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nobles K. N., Xiao K., Ahn S., Shukla A. K., Lam C. M., Rajagopal S., Strachan R. T., Huang T. Y., Bressler E. A., Hara M. R., Shenoy S. K., Gygi S. P., Lefkowitz R. J. (2011) Distinct phosphorylation sites on the beta(2)-adrenergic receptor establish a barcode that encodes differential functions of beta-arrestin. Sci. Signal. 4, ra51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Corringer P. J., Poitevin F., Prevost M. S., Sauguet L., Delarue M., Changeux J. P. (2012) Structure and pharmacology of pentameric receptor channels: from bacteria to brain. Structure 20, 941–956 [DOI] [PubMed] [Google Scholar]
- 19. Bergmann R., Kongsbak K., Sorensen P. L., Sander T., Balle T. (2013) A unified model of the GABA(A) receptor comprising agonist and benzodiazepine binding sites. PLoS One 8, e52323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Baker M. (2010) Making membrane proteins for structures: a trillion tiny tweaks. Nat. Methods 7, 429–434 [DOI] [PubMed] [Google Scholar]
- 21. Chen E. I., Cociorva D., Norris J. L., Yates J. R., 3rd (2007) Optimization of mass spectrometry-compatible surfactants for shotgun proteomics. J. Proteome Res. 6, 2529–2538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhou F., Lu Y., Ficarro S. B., Adelmant G., Jiang W., Luckey C. J., Marto J. A. (2013) Genome-scale proteome quantification by DEEP SEQ mass spectrometry. Nat. Commun. 4, 2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yates J. R., 3rd (2013) The revolution and evolution of shotgun proteomics for large-scale proteome analysis. J. Am. Chem. Soc. 135, 1629–1640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wu C. C., Yates J. R., 3rd (2003) The application of mass spectrometry to membrane proteomics. Nat. Biotechnol. 21, 262–267 [DOI] [PubMed] [Google Scholar]
- 25. Whitelegge J. P. (2013) Integral membrane proteins and bilayer proteomics. Anal. Chem. 85, 2558–2568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yip G. M., Chen Z. W., Edge C. J., Smith E. H., Dickinson R., Hohenester E., Townsend R. R., Fuchs K., Sieghart W., Evers A. S., Franks N. P. (2013) A propofol binding site on mammalian GABA receptors identified by photolabeling. Nat. Chem. Biol. 9, 715–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kang S. U., Heo S., Lubec G. (2011) Mass spectrometric analysis of GABAA receptor subtypes and phosphorylations from mouse hippocampus. Proteomics 11, 2171–2181 [DOI] [PubMed] [Google Scholar]
- 28. Chen Z. W., Fuchs K., Sieghart W., Townsend R. R., Evers A. S. (2012) Deep amino acid sequencing of native brain GABAA receptors using high-resolution mass spectrometry. Mol. Cell. Proteomics 11, M111 011445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kang S. U., Fuchs K., Sieghart W., Pollak A., Csaszar E., Lubec G. (2009) Gel-based mass spectrometric analysis of a strongly hydrophobic GABAA-receptor subunit containing four transmembrane domains. Nat. Protoc. 4, 1093–1102 [DOI] [PubMed] [Google Scholar]
- 30. Kang S. U., Fuchs K., Sieghart W., Lubec G. (2008) Gel-based mass spectrometric analysis of recombinant GABA(A) receptor subunits representing strongly hydrophobic transmembrane proteins. J. Proteome Res. 7, 3498–3506 [DOI] [PubMed] [Google Scholar]
- 31. Manza L. L., Stamer S. L., Ham A. J., Codreanu S. G., Liebler D. C. (2005) Sample preparation and digestion for proteomic analyses using spin filters. Proteomics 5, 1742–1745 [DOI] [PubMed] [Google Scholar]
- 32. Wisniewski J. R., Zougman A., Nagaraj N., Mann M. (2009) Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362 [DOI] [PubMed] [Google Scholar]
- 33. Wisniewski J. R., Mann M. (2012) Consecutive proteolytic digestion in an enzyme reactor increases depth of proteomic and phosphoproteomic analysis. Anal. Chem. 84, 2631–2637 [DOI] [PubMed] [Google Scholar]
- 34. Nagaraj N., Lu A., Mann M., Wisniewski J. R. (2008) Detergent-based but gel-free method allows identification of several hundred membrane proteins in single LC-MS runs. J. Proteome Res. 7, 5028–5032 [DOI] [PubMed] [Google Scholar]
- 35. Newby Z. E., O'Connell J. D., 3rd, Gruswitz F., Hays F. A., Harries W. E., Harwood I. M., Ho J. D., Lee J. K., Savage D. F., Miercke L. J., Stroud R. M. (2009) A general protocol for the crystallization of membrane proteins for X-ray structural investigation. Nat. Protoc. 4, 619–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Drew D., Newstead S., Sonoda Y., Kim H., von Heijne G., Iwata S. (2008) GFP-based optimization scheme for the overexpression and purification of eukaryotic membrane proteins in Saccharomyces cerevisiae. Nat. Protoc. 3, 784–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Garavito R. M., Ferguson-Miller S. (2001) Detergents as tools in membrane biochemistry. J. Biol. Chem. 276, 32403–32406 [DOI] [PubMed] [Google Scholar]
- 38. Rosevear P., VanAken T., Baxter J., Ferguson-Miller S. (1980) Alkyl glycoside detergents: a simpler synthesis and their effects on kinetic and physical properties of cytochrome c oxidase. Biochemistry 19, 4108–4115 [DOI] [PubMed] [Google Scholar]
- 39. Cherezov V., Rosenbaum D. M., Hanson M. A., Rasmussen S. G., Thian F. S., Kobilka T. S., Choi H. J., Kuhn P., Weis W. I., Kobilka B. K., Stevens R. C. (2007) High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 318, 1258–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hilf R. J., Dutzler R. (2008) X-ray structure of a prokaryotic pentameric ligand-gated ion channel. Nature 452, 375–379 [DOI] [PubMed] [Google Scholar]
- 41. Hibbs R. E., Gouaux E. (2011) Principles of activation and permeation in an anion-selective Cys-loop receptor. Nature 474, 54–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liu W., Chun E., Thompson A. A., Chubukov P., Xu F., Katritch V., Han G. W., Roth C. B., Heitman L. H., AP I. J., Cherezov V., Stevens R. C. (2012) Structural basis for allosteric regulation of GPCRs by sodium ions. Science 337, 232–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Brohawn S. G., del Marmol J., MacKinnon R. (2012) Crystal structure of the human K2P TRAAK, a lipid- and mechano-sensitive K+ ion channel. Science 335, 436–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chae P. S., Rasmussen S. G., Rana R. R., Gotfryd K., Chandra R., Goren M. A., Kruse A. C., Nurva S., Loland C. J., Pierre Y., Drew D., Popot J. L., Picot D., Fox B. G., Guan L., Gether U., Byrne B., Kobilka B., Gellman S. H. (2010) Maltose-neopentyl glycol (MNG) amphiphiles for solubilization, stabilization, and crystallization of membrane proteins. Nat. Methods 7, 1003–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rasmussen S. G., DeVree B. T., Zou Y., Kruse A. C., Chung K. Y., Kobilka T. S., Thian F. S., Chae P. S., Pardon E., Calinski D., Mathiesen J. M., Shah S. T., Lyons J. A., Caffrey M., Gellman S. H., Steyaert J., Skiniotis G., Weis W. I., Sunahara R. K., Kobilka B. K. (2011) Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature 477, 549–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chung K. Y., Kim T. H., Manglik A., Alvares R., Kobilka B. K., Prosser R. S. (2012) Role of detergents in conformational exchange of a G protein-coupled receptor. J. Biol. Chem. 287, 36305–36311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tao H., Fu Y., Thompson A., Lee S. C., Mahoney N., Stevens R. C., Zhang Q. (2012) Synthesis and properties of dodecyl trehaloside detergents for membrane protein studies. Langmuir 28, 11173–11181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lee S. C., Bennett B. C., Hong W. X., Fu Y., Baker K. A., Marcoux J., Robinson C. V., Ward A. B., Halpert J. R., Stevens R. C., Stout C. D., Yeager M. J., Zhang Q. (2013) Steroid-based facial amphiphiles for stabilization and crystallization of membrane proteins. Proc. Natl. Acad. Sci. U.S.A. 110, E1203–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Thompson D. A., Suarez-Villafane M., Ferguson-Miller S. (1982) The active form of cytochrome c oxidase: effects of detergent, the intact membrane, and radiation inactivation. Biophys. J. 37, 285–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cristoni S., Bernardi L. R. (2003) Development of new methodologies for the mass spectrometry study of bioorganic macromolecules. Mass Spectrom. Rev. 22, 369–406 [DOI] [PubMed] [Google Scholar]
- 51. Laganowsky A., Reading E., Hopper J. T., Robinson C. V. (2013) Mass spectrometry of intact membrane protein complexes. Nat. Protoc. 8, 639–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhang X., Chien E. Y., Chalmers M. J., Pascal B. D., Gatchalian J., Stevens R. C., Griffin P. R. (2010) Dynamics of the beta2-adrenergic G-protein coupled receptor revealed by hydrogen-deuterium exchange. Anal. Chem. 82, 1100–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tao H., Lee S. C., Moeller A., Roy R. S., Siu F. Y., Zimmermann J., Stevens R. C., Potter C. S., Carragher B., Zhang Q. (2013) Engineered nanostructured beta-sheet peptides protect membrane proteins. Nat. Methods 10, 759–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. McGregor C. L., Chen L., Pomroy N. C., Hwang P., Go S., Chakrabartty A., Prive G. G. (2003) Lipopeptide detergents designed for the structural study of membrane proteins. Nat. Biotechnol. 21, 171–176 [DOI] [PubMed] [Google Scholar]
- 55. Zhang X., Tamot B., Hiser C., Reid G. E., Benning C., Ferguson-Miller S. (2011) Cardiolipin deficiency in Rhodobacter sphaeroides alters the lipid profile of membranes and of crystallized cytochrome oxidase, but structure and function are maintained. Biochemistry 50, 3879–3890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dostalova Z., Zhou X., Liu A., Zhang X., Zhang Y., Desai R., Forman S. A., Miller K. W. (2014) Human alpha1beta3gamma2L gamma-aminobutyric acid type A receptors: High-level production and purification in a functional state. Protein Sci. 23, 157–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhang X., Miller K. W. (2014) Novel integrated ultra-deep quantitative-structural membrane proteomics discovered PTM signatures for human Cys-loop receptor subunit bias. Submitted. (under review at MCP) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pascal B. D., Willis S., Lauer J. L., Landgraf R. R., West G. M., Marciano D., Novick S., Goswami D., Chalmers M. J., Griffin P. R. (2012) HDX workbench: software for the analysis of H/D exchange MS data. J. Am. Soc. Mass Spectrom. 23, 1512–1521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mellacheruvu D., Wright Z., Couzens A. L., Lambert J. P., St-Denis N. A., Li T., Miteva Y. V., Hauri S., Sardiu M. E., Low T. Y., Halim V. A., Bagshaw R. D., Hubner N. C., Al-Hakim A., Bouchard A., Faubert D., Fermin D., Dunham W. H., Goudreault M., Lin Z. Y., Badillo B. G., Pawson T., Durocher D., Coulombe B., Aebersold R., Superti-Furga G., Colinge J., Heck A. J., Choi H., Gstaiger M., Mohammed S., Cristea I. M., Bennett K. L., Washburn M. P., Raught B., Ewing R. M., Gingras A. C., Nesvizhskii A. I. (2013) The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat. Methods 10, 730–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Norton W. T., Abe T., Poduslo S. E., DeVries G. H. (1975) The lipid composition of isolated brain cells and axons. J. Neurosci. Res. 1, 57–75 [DOI] [PubMed] [Google Scholar]
- 61. Zhang X., Hiser C., Tamot B., Benning C., Reid G. E., Ferguson-Miller S. M. (2011) Combined genetic and metabolic manipulation of lipids in Rhodobacter sphaeroides reveals non-phospholipid substitutions in fully active cytochrome c oxidase. Biochemistry 50, 3891–3902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Savechenkov P. Y., Zhang X., Chiara D. C., Stewart D. S., Ge R., Zhou X., Raines D. E., Cohen J. B., Forman S. A., Miller K. W., Bruzik K. S. (2012) Allyl m-trifluoromethyldiazirine mephobarbital: an unusually potent enantioselective and photoreactive barbiturate general anesthetic. J. Med. Chem. 55, 6554–6565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hansen S. B., Talley T. T., Radic Z., Taylor P. (2004) Structural and ligand recognition characteristics of an acetylcholine-binding protein from Aplysia californica. J. Biol. Chem. 279, 24197–24202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Dephoure N., Gould K. L., Gygi S. P., Kellogg D. R. (2013) Mapping and analysis of phosphorylation sites: a quick guide for cell biologists. Mol. Biol. Cell 24, 535–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zielinska D. F., Gnad F., Wisniewski J. R., Mann M. (2010) Precision mapping of an in vivo N-glycoproteome reveals rigid topological and sequence constraints. Cell 141, 897–907 [DOI] [PubMed] [Google Scholar]
- 66. Zielinska D. F., Gnad F., Schropp K., Wisniewski J. R., Mann M. (2012) Mapping N-glycosylation sites across seven evolutionarily distant species reveals a divergent substrate proteome despite a common core machinery. Mol. Cell 46, 542–548 [DOI] [PubMed] [Google Scholar]
- 67. Chang W., Gelman M. S., Prives J. M. (1997) Calnexin-dependent enhancement of nicotinic acetylcholine receptor assembly and surface expression. J. Biol. Chem. 272, 28925–28932 [DOI] [PubMed] [Google Scholar]
- 68. Keller S. H., Lindstrom J., Taylor P. (1996) Involvement of the chaperone protein calnexin and the acetylcholine receptor beta-subunit in the assembly and cell surface expression of the receptor. J. Biol. Chem. 271, 22871–22877 [DOI] [PubMed] [Google Scholar]
- 69. Keller S. H., Lindstrom J., Taylor P. (1998) Inhibition of glucose trimming with castanospermine reduces calnexin association and promotes proteasome degradation of the alpha-subunit of the nicotinic acetylcholine receptor. J. Biol. Chem. 273, 17064–17072 [DOI] [PubMed] [Google Scholar]
- 70. Itakura M., Tsujimura J., Yamamori S., Ohkido T., Takahashi M. (2013) NMDA receptor-dependent recruitment of calnexin to the neuronal plasma membrane. Neurosci. Lett. 550, 173–178 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.