

Abstract
The function of a large percentage of proteins is modulated by post-translational modifications (PTMs). Currently, mass spectrometry (MS) is the only proteome-wide technology that can identify PTMs. Unfortunately, the inability to detect a PTM by MS is not proof that the modification is not present. The detectability of peptides varies significantly making MS potentially blind to a large fraction of peptides. Learning from published algorithms that generally focus on predicting the most detectable peptides we developed a tool that incorporates protein abundance into the peptide prediction algorithm with the aim to determine the detectability of every peptide within a protein. We tested our tool, “Peptide Prediction with Abundance” (PPA), on in-house acquired as well as published data sets from other groups acquired on different instrument platforms. Incorporation of protein abundance into the prediction allows us to assess not only the detectability of all peptides but also whether a peptide of interest is likely to become detectable upon enrichment. We validated the ability of our tool to predict changes in protein detectability with a dilution series of 31 purified proteins at several different concentrations. PPA predicted the concentration dependent peptide detectability in 78% of the cases correctly, demonstrating its utility for predicting the protein enrichment needed to observe a peptide of interest in targeted experiments. This is especially important in the analysis of PTMs. PPA is available as a web-based or executable package that can work with generally applicable defaults or retrained from a pilot MS data set.
Post-translational modification (PTM)1 of proteins is a key regulatory mechanism in the vast majority of biological processes. Historically, to follow PTMs, site-specific antibodies had to be generated in a time-consuming and laborious process associated with high failure rates. Mass spectrometry (MS) holds enormous promise in PTM analysis as it is currently the only technique that has the ability to both discover, localize, and quantify proteome-wide modifications (1). Recent advances in instrumentation and method optimization makes it possible to detect the complete yeast proteome within one hour (2), an ever increasing proportion of the human proteome (3–6), and more than 10,000 phosphorylation sites in a single MS experiment (7, 8). As a result one of the major publicly available databases (www.phosphosite.org (9)) has curated >200,000 phosphorylation sites.
Although the number of proteins and PTMs that can be identified is impressive, many modifications have still not been identified in any MS-based experiment. The identification and quantification of biologically relevant modifications is challenging for three reasons: (1) many proteins of interest are of very low abundance rendering them difficult to detect and quantify; (2) many modifications sites are present at substoichiometric quantities, further reducing their detectability; and (3) as large scale proteomics is based on the detection of peptides after a proteolytic digest, and the detectability of a peptide is determined by its physiochemical properties (10), many peptides from highly abundant proteins are never detected. This is particularly important, as there is a shift in the use of MS-based proteomics from large scale, unbiased, discovery-focused experiments toward directed experiments for accurate and precise quantification of biologically relevant PTMs. Protein and peptide enrichment strategies and/or targeted MS experiments like single reaction monitoring (SRM) (11) have increased the number of detectable peptides; however, both of these methods are laborious, and often not successful, that is, the peptide carrying the modification of interest is still not observed as it is fundamentally very difficult to detect.
Protein enrichment is the method choice for most experimentalists, but there is no current way to determine whether this is likely to succeed prior to engaging in lengthy biochemical and/or analytical experiments. In an effort to gauge the chances of success for detecting a particular peptide we sought to develop an algorithm that can predict both the chances of detecting a particular peptide and, more importantly, what enrichment it would take to detect a particular peptide that is not easily detected. Here we present such a tool that predicts the detectability and estimates an enrichment factor, i.e. an increase in signal over the background that is necessary to actually detect a particular peptide. Our algorithm development was motivated by two premises: (1) In silico methods have been developed that focus on the prediction of easily detectable “proteotypic” peptides (peptides that are likely to provide the best detection sensitivity) with good accuracy (12–15). (2) Comprehensive proteome studies have shown that the number of detected peptides per protein, and thus the sequence coverage, varies with protein abundance (which is the basis for spectral counting-based protein quantification (16, 17)). We find that incorporation of protein abundance in a peptide classification tool improves the accuracy of the prediction of peptide detectability allowing us to predict the detectability of all peptides within a protein as well as the amount of enrichment needed to detect a peptide of interest.
We used a set of 120 purified in vitro expressed proteins as a training set to develop a prediction tool. We deliver this in the form of a web-based interface that provides information about: (1) the probability of detecting the different tryptic peptides of a protein, and (2) the fold enrichment that would be required to bring a peptide of interest into the detectable range. This tool will help guide researchers in their efforts to monitor particular peptides and their modified cognates by MS, specifically, in prioritizing their efforts toward enriching proteins where they would be likely to be able to detect a peptide or modification of interest.
MATERIALS AND METHODS
Cloning and In Vitro Expression of Full-length Stable Isotope Labeled Proteins
The pEU-E01-His-N1-FlexII in vitro expression vector is similar to the vector described by Singh et al. (18) with some modifications to create three FLEXII-peptides (supplemental Fig. S1). These peptides, TVLLFLEISK, TVLYFSEISK, and TSLYFSEISK, were synthesized by New England Peptide (Gardner, MA) and quantified by amino acid analysis (Molecular Biology Core Facilities, Dana-Farber Cancer Institute (DFCI), Boston, MA). A subset of the human ORFeome (846 genes) was generously provided by the Center for Cancer Systems Biology (DFCI).
The human ORFs were moved into the FLEXII-Tag-vector using Gateway Cloning (Invitrogen, Carlsbad, CA). Briefly, 100 ng of entry vector and 100 ng of destination vector were mixed in a 3 μl reaction mix using LR Clonase II Enzyme Mix (Invitrogen) for 18 h at 25 °C followed by 10min digestion with 1 μg proteinase K at 37 °C. Cells were then transformed into E. coli and liquid selection was performed. Spot sequencing was performed on one column from each of nine 96 well plates (Genewiz, South Plainfield, NJ).
The in vitro transcription and translation reactions were carried out as described by the manufacturer (Cell free sciences, Ehime, Japan, Wheat Germ Expression H Kit-NA) and Singh et al. (18) modified to 96 well format. Transcription was performed at 37 °C for 8 h and immediately used in translation in presence of heavy isotope labeled lysine (K8) and arginine (R10) for 17–20 h at 16 °C. Expressed proteins were purified with a His MultiTrap HP 96-well plate according to the manufacturers instruction (GE Healthcare) using a binding/wash buffer of 20 mm sodium phosphate, pH 7.4, 500 mm NaCl, 30 mm imidazole, and eluted two times with 70 μl of 20 mm sodium phosphate, pH 7.4, 500 mm NaCl, 500 mm imidazole, and 5% glycerol. The flowthrough from the initial binding to the His MultiTrap plate was rebound to the same plate and a third 70 μl elution was performed. Eluent was stored at −80 °C in 20 μl aliquots. Expression was checked with a 15 μl aliquot by 1D-SDS-PAGE (4–12% Bis-Tris, NuPage, Invitrogen). Expression was scored by visual assessment of Coomassie staining intensity with proteins sorted into five categories from zero (no expression) to five (highest expression).
In-solution Digestion
A 20 μl aliquot of expressed protein was mixed with an equal volume of a 0.2% (w/v) RapiGest SF (Waters, Milford, MA) solution and incubated at 37 °C for 30 min. The samples were then reduced with the addition of 5 μl of 200 mm DTT and 15 μl of 100 mm ammonium bicarbonate (ABC), pH 8.0, and heated to 60 °C for 30 min then cooled to room temperature for 5 min. The samples were alkylated by the addition of 4 μl of 40% pure acrylamide (Calbiochem, San Diego, CA) in the dark for 30 min. The reaction was quenched by the addition of 20 μl of 200 mm DTT for at least 30 min. Trypsin was added in a 20 μl aliquot of ABC to a total of 100 ng per sample. After digestions for 17–20 h at 37 °C 8 samples were pooled and acidified with the addition of a fresh vial of 0.1% trifluoroacetic acid (TFA, Pierce). For pooled samples, the samples were mixed after digestion and were then dried by speed-vac to a volume between 100 to 200 μl. Additionally, a subset of proteins was digested and subsequent analyzed singly.
Peptides were desalted using a C18 Silica Column (Nest Group #SS18V) spinning on a bench top centrifuge at 50 × g. Columns were washed two times with 200 μl 70% acetonitrile (ACN) 0.1% TFA and equilibrated two times with 200 μl 0.1% TFA. Then the samples were loaded and passed two times over the column. The columns were then washed three times with 100 μl of 0.1% TFA. Samples were eluted over three 75 μl elutions; 30% ACN, 0.1% TFA; 50% ACN, 0.1% TFA; 70% ACN, and 0.1% TFA. The final elution was passed over the column a second time. The samples were then dried down and resuspended in sample buffer containing 5% formic acid (FA), 5% ACN. For absolute quantification 10 fmol/μl of unlabeled FLEXII-peptides were added.
Additionally, commercially available tryptic digests of a human cell lysate (K562) and of a yeast cell lysate were analyzed (both from Promega, Fitchburg, WI).
LC-MS/MS Analysis and Data Processing
Samples were analyzed by a nanoLC system (Eksigent, Dublin, CA) equipped with a LC-chip system (cHiPLC nanoflex, Eksigent, trapping column: Nano cHiPLC Trap column 200 μm x 0.5 mm ChromXP C18-CL 3 μm 120 Å, analytical column: Nano cHiPLC column 75 μm x 15 cm ChromXP C18-CL 3 μm 120 Å) coupled online to a Q-Exactive mass spectrometer (Thermo Scientific). Peptides were separated by a linear gradient from 95% buffer A (0.2% FA in water)/5% buffer B (0.2% FA in ACN) to 65% buffer A/35% buffer B. The gradient length was altered between 10 min (single protein digest), 30 min (pools of eight) and 60 min (human and yeast digests). The MS was operated in data-dependent TOP10 mode with the following settings: mass range 300–1500 Th; resolution for MS1 scan 70,000 @ 200 Th; lock mass: 445.120025 Th; resolution for MS2 scan 17,500 @ 200 Th; isolation width 2 m/z; NCE 27; underfill ratio 0.1%; charge state exclusion: unassigned, 1; dynamic exclusion 15 s. Prior database search RAW-files were converted into MGF-files using the ProteoWizard software tool (19).
Additionally the yeast digest was analyzed with the same LC setup on a Q-ToF MS (AB Sciex TripleToF 5600). The MS was operated in data-dependent TOP25 mode with following settings: MS1 mass range 400–1000 Th with 200 ms acc. time; MS2 mass range 150–1400 Th with 60 ms acc. time and following MS2 selection criteria: UNIT resolution, intensity threshold 200 cts; charge states 2–4.
MGF- and WIFF-files were searched in ProteinPilot 4.5 (AB Sciex, Framingham, MA) with the implemented Paragon algorithm using following settings – sample type: identification or SILAC (K+8, R+10); Cys alkylation acrylamide; digestion: trypsin; instrument: Orbi MS, Orbi MS/MS; search effort: rapid ID. ProteinPilot does not allow it to choose the mass tolerances and number of missed cleavages. After database search (spectral library samples: combined database of human proteins in clone library (based on Uniprot May 2011) plus wheat database (Triticum aestivum, PlantGDB Aug 2011), 5169 entries; human sample: Uniprot database (May 2011), 35,807 entries; yeast sample: SGD database downloaded on Sept 2011, 6751 entries; all databases contained common laboratory contaminants). The search results were exported and based on the peptide confidence filtered by a global FDR of 1%. MaxQuant 1.5.0.0 (20) was used to generate the iBAQ abundance values (21) (same databases as mentioned above, trypsin with up to two missed cleavages, mass tolerances set to 20 ppm for first search and 4.5 ppm for main search, other settings were set to defaults). The search results were filtered based on a 1% FDR on peptide and protein level. Peak areas for peptide quantification were determined using a MS1-filtering experiment in Skyline (22).
Mass spectrometry data and search results for the enriched protein data sets are available at Peptideatlas.org (23) (identifier PASS00449, http://www.peptideatlas.org/PASS/PASS00449).
Peptide Classification
Training Data
The LC-MS/MS data from the 120 singly analyzed proteins were utilized as training data (2842 peptides, supplemental Table S1). Tryptic in silico digestion of these proteins was performed with the following settings (http://prospector.ucsf.edu/prospector/cgi-bin/msform.cgi?form = msdigest): minimum five amino acids, MW between 600 to 6400 Da. Any in silico peptide that was not identified in the LC-MS/MS experiments was labeled as not detected. To assess the data quality for model building/training the data have been 10× cross validated.
Calculation of the Physicochemical Feature Values of the Peptides
A total of 544 amino acid physicochemical features were considered as initial features in this study (10). Each physicochemical feature contains 20 numeric values (continuous or discrete) indicating its relative weights for the 20 amino acids. For each peptide the feature's aggregated value was computed by summing the feature weights of the amino acids contained in the peptides. Therefore, the peptide sequences in training data were transformed to a two-dimensional feature-value matrix containing N*544 (N being the number of peptides used for training) explanatory values, and a binary vector of dimension N*1 indicating whether each peptide from the in silico digestions was actually detected or not. Because different physicochemical features such as molecular weight, contain values orders of magnitude larger than other features, the gradient descent back-propagation algorithm adjusted weights for some features more than for others. To reduce the bias toward features with larger values the original input data was standardized—the mean and variance of each feature was set to zero and one, respectively.
Artificial Neural Network Backpropagation
Input to the Neural Network
For the neural network classification (24, 25), the input data consisted of a complete standardized feature list, xi (i.e. the maximum number of inputs could be 544), where i is an index to a specific physicochemical feature (input to first-order neurons, i = 1,2,…, 544). The number of intermediate neurons and layer were varied (see below). The network computed a single output value, o, for each input peptides standardized aggregated feature values. This model value will be compared with t, a binary value indicating the peptide was detected by (t = 1) and not detected (t = 0). The training process iteratively updated the model coefficients until the differences between all target values t⃗ and original data o⃗ were minimized.
Neural Network Structure
After multiple tests with different numbers of nodes and hidden layers on our initial test set (supplemental Table S4 and below), we determined that one hidden layer with two neurons recapitulated more complicated networks and hence used this simpler design for all further analysis. Two sets of network classifier parameters were estimated, w•j, the weight of the j th hidden neuron to output, j = 1,2, and wji, the weight of the i input to j hidden neuron, i = 1,2,…,15, and j = 1,2.
Each neuron in the network first calculates the weighted linear combination of the input nodes (1.3 first layer, 1.4 s layer) and then passes this value through a sigmoid function (1.5) to help force the network to categorize the outputs likely detected or likely not detected.
 |
 |
 |
Objective Function
The weights in the neural network were “trained” by minimizing the objective function (4) – the difference between the target vector, t⃗, and output of the neural network for all peptides, o⃗, from the input data
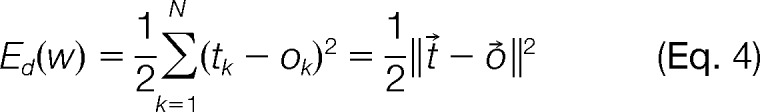 |
Minimizing the Objective Function
The objective function was minimized by iteratively testing small deviation from the current weights (5), input-to-hidden weight wji and hidden-to-output weight w•j, and updating w any time the objective function was smaller than the previous attempt.
Given the network learning rate η, the model learning rule for weight updates was a series of computations to find the steepest descent in the error surface defined by the partial derivatives of error surface with respect to weight vectors.
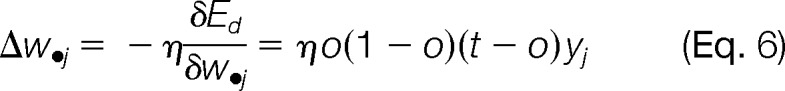 |
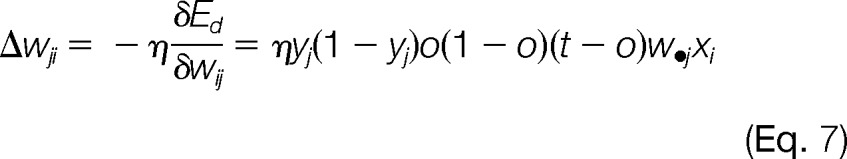 |
Backpropagation was known for the weight updates by starting from the hidden-to-output layer first and then propagated to the input-to-hidden layer. This process was iterative; weights for the hidden-to-output were updated first, followed by the input-to-hidden layer weights, which also utilized weight updates in the downstream layer.
Artificial Neural Network Model Training Procedures
Because many physicochemical features are highly correlated with others, feature selection methods such as back-selection or forward-selection were applied to reduce the number of redundant features.
During the training process, 90% of the data were randomly selected as training data and the remaining 10% as validation data. This procedure was repeated until every theoretical peptide was included in the validation set. For any selected feature set, sensitivity rate, specificity rate, and accuracy was computed to evaluate the model performance (supplemental Table S4). Results from the performance evaluations suggested that a considerable number of false positives came from a small number of proteins (30) that were expressed at low amounts (7 fmol/μl) and/or were identified with a low sequence coverage (< 20%). These proteins were removed from the training data.
After training with the experimental data, a neural network model containing 15 outer layer physicochemical features and a hidden layer with two neurons performed best (scheme in Fig. 2A, supplemental Table S5) and was utilized as basis for PPA.
Fig. 2.
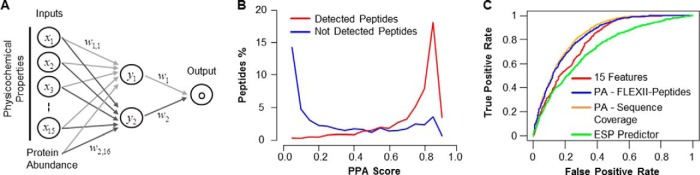
Incorporation of Protein Abundance in Peptide Detection Classifier Improves Performance. A, Scheme of the neural network model used for classification of peptide detection resulting in 16 features including 15 physicochemical properties and protein abundance, resulting in the Peptide Prediction with Abundance (PPA) score. The model was trained with 10-fold cross-validation. B, Distribution of PPA scores for detected versus not detected peptides of the 120 singly analyzed protein digests. C, The Receiver Operating Characteristic (ROC) was calculated for three models and compared with the previously published ESP predictor (15): red: 15 neural network model physicochemical features only; blue: PPA including protein abundance (PA) as FLEXII-peptide intensity; orange: PPA including protein abundance (PA) as sequence coverage; green: ESP predictor on the 10-fold cross-validated data set.
In addition to the physicochemical properties that were used for this model, our neural network was extended in one novel way by including an extra feature to represent the protein abundance. Four versions of the extended neural network full model have been developed: (1) absolute amount of protein (in fmol) determined using the FLEXII-peptides, (2) the sequence coverage, (3) Top-3 abundance (average signal intensity of the three highest intense tryptic peptides (26)), and (4) iBAQ abundance (intensity of identified peptides divided by theoretically observable peptides (21), supplemental Fig. S3). Using the identification result from the training data, the peptide's PPA were matched to its corresponding observation (detected/not detected) and the models were compared based on Receiver Operating Characteristic (ROC) and corresponding areas under the curve were computed (AUROC, R package: ROCR by Tobias Sing and www.vassarstats.net/roc_comp.html, supplemental Table S6, neural network feature weights in supplemental Table S8).
Artificial Neuron Network Validation
For validation, in silico digestion was performed on the identified proteins of the data set under study as described for the training data (identification data from this study in Supplemental Tables S2, S9–S11, additionally used data: Wisniewski et al. (27) and Hebert et al. (2)). Peptides were flagged whether they have been detected or not detected and the peptide score based on the 15-feature model was calculated. Afterward, two different sets of sequence coverage were applied to calculate the PPA score of the peptides:
Sequence coverage from another experiment (external sequence coverage, eSC): for the pool of eight and the Wisniewski et al. samples the sequence coverage data from the human lysates acquired on the Q-Exactive (K562 cells) were used and the Wisniewski et al. data for the K562 data; for all yeast samples sequence coverage information were used from de Godoy et al. (28).
Sequence coverage from the data itself (internal sequence coverage, iSC).
Similarly, a probability score was also computed for the same set of peptides using ESP Predictor (15). Finally, the peptide's PPA and ESP score were matched to its corresponding observation (detected/not detected) and the models were compared based on AUROC and a modified z-test (29) was applied to compute the significance of the difference between the models (supplemental Tables S6–S7).
Calculation of the Protein Enrichment/Prediction of Sequence Coverage Based on PPA Scores
To enable the calculation of protein enrichment, the protein abundance has to be transferred on a linear scale (intensity scale) as the sequence coverage does not correlate linearly with the protein abundance and reaches a maximum at 100%. To convert the sequence coverage based input onto an intensity scale, the PPA model was trained with Top-3 abundances (26) from the initial training data set (performance comparison in supplemental Fig. S6). Starting with an average protein abundance (log2 transformed intensity) from the training data, the PPA scores of each peptides per protein were calculated and a peptide length weighted average PPA score per protein was computed. This weighted average weighted PPA score can be seen as prediction of protein sequence coverage. Afterward the intensity was iterated to match the weighted PPA score average with the experimental sequence coverage, which is the input value for most PPA applications. For enrichment prediction, the intensity was increased so that peptides reach a predefined PPA score (likelihood of peptide detection, default value: 0.65).
For the prediction of sequence coverage of the dilution series data (Fig. 3B, supplemental Fig. S4C), PPA scores were calculated based on the 15 features and the FLEXII-Tag abundance. The FLEXII-Tag abundance was normalized by the ratio of the heavy FLEXII-Tag versus the spiked in light FLEXII-peptides to correct for run-to-run variation in MS signal intensity. Peptides with a score > 0.5 were assumed to be detected, and therefore used to compute the predicted sequence coverage.
Fig. 3.

Validation of PPA Enrichment Prediction. A, Each line represents the abundance depended PPA score for all individual peptides from DTX3 at different protein concentrations. These predictions were compared with the outcome of LC-MS/MS analyses of DTX3 digests at three different concentrations. Black: not-detected; green: detected at 4 fmol; blue: detected at 7.6 fmol; red: detected at 65.2 fmol. B, Prediction of protein abundance-dependent sequence coverage (blue curve) for DTX3. For a sequence coverage prediction, peptides with a PPA score > 0.5 were considered to be detected. Inset highlights our two evaluation metrics – ΔSC and ΔPA. The pie charts represent the experimental data points. Green color: correct prediction; red color: peptides were not detected, but predicted to be detected; white color: peptides were detected, but not predicted to be detected. Each data point (supplemental Fig. S4C) contributes to C and D. C, This histogram uses all 110 data points. D, This histogram uses 95 data points (all 110 data points are shown in supplemental Fig. S4D). We eliminate 15 data points where the maximal sequence coverage of our prediction was less than the actual observed sequence coverage. This results when a single peptide from a protein that has a very low PPA score is actually detected.
RESULTS AND DISCUSSION
Generation of a Spectral Library for Development of an Abundance-based Classifier
Previous classifiers (12–15) were developed and optimized for the prediction of the most easily detectable “proteotypic” peptides for the purpose of quantifying protein abundance. By focusing only on the most detectable peptides, it was possible to develop these classifiers using widely available LC-MS/MS data sets from complex protein samples such as whole cell lysates (14, 15). In contrast, in our study, we aimed at predicting the likelihood of detecting every peptide of a tryptic digest, and understanding how abundance quantitatively effects this prediction.
To this end, we used MS to analyze tryptic peptides derived from proteins of known abundance. To achieve this, we adapted our FLEXIQuant strategy (18, 30) (see Fig. 1A) to create a large number of peptides of known abundance. This strategy entails in vitro expression and purification of a large number of proteins for which we designed an improved FLEXII-plasmid to be used with wheat germ expression systems (pEU-E01-His-N1-FlexII, supplemental Fig. S1A). Expression from this improved plasmid results in recombinant proteins with an N-terminal His6-tag for efficient purification and a MS quantification tag, the FLEXII-Tag. The FLEXII-Tag proteolyzes into several reporter peptides upon digestion with trypsin, LysC, LysN, and/or GluC, thereby facilitating quick isotope dilution-based MS based quantification of the expressed protein (supplemental Fig. S1B). These reporter peptides (here called FLEXII-peptides) have artificial sequences that do not occur in any of the proteomes of any currently used common model organism.
Fig. 1.

Workflow of Spectral Library Generation. A, Workflow: 846 genes from the human ORFeome bank were introduced via Gateway cloning into a wheat germ expression vector (vector map in supplemental Fig. S1A) containing a 5′-FLEXII-Tag (sequence in supplemental Fig. S1B) resulting in a clone library with 801 of the selected 846 ORFs (95%). FLEXII-tagged proteins were expressed in cell-free wheat germ expression system and enriched via Ni-NTA beads. Eighty-four percent of the expressed clones yielded visible bands on SDS-PAGE (details in supplemental Fig. S1C). The purified proteins were in solution trypsinized and the peptide mixtures were analyzed in pools of eight by LC-MS/MS on a Q-Exactive mass spectrometer. Database searches were performed by the ProteinPilot software and a global FDR cutoff of 1% was chosen for peptide/protein identification resulting in 735 identified proteins, that is an identification rate of 90%; this included proteins not visible by SDS-PAGE. Absolute quantification was carried out using synthetic peptide-based isotope dilution mass spectrometry when necessary. MS data are publicly available at Peptide Atlas (www.peptideatlas.org). B, Sequence coverage distribution for the 735 identified proteins. C, Distribution of the observed number of peptides per protein for the 735 identified proteins.
Our efforts to clone 846 genes into our improved FLEXII-plasmid resulted in a clone library consisting of 801 gene constructs. After in vitro transcription and translation, 703 of the 801 expected proteins could be detected by SDS-PAGE and Coomassie staining. Spot checks showed that missing proteins typically resulted from stop codons, missing plasmids, or wrong clones. As determined by isotope dilution MS, proteins were expressed between 0.5 and 62 pmol of protein (95 percentile range reported here and below) for a single 240 μl in vitro transcription/translation reaction (median of 5.5 pmol). Digestion of 10–15% of each sample resulted in concentrations between 1.2 and 110 nm per protein digest (median 4.5 nm, supplemental Fig. S1C–E) as determined by MS-based quantification with our FLEXII-peptides. For absolute quantification by our FLEXIQuant approach and as training data set for the development of the classifier, we individually analyzed 120 of these purified proteins by LC-MS/MS. As expected, higher protein abundance results in higher sequence coverage (supplemental Fig. S1F, supplemental Table S1). To create a test set to validate our classification tool, 801 proteins were run in pools of eight proteins resulting in the identification of 735 proteins (supplemental Table S2). On average, we obtained a sequence coverage of 49% (16 to 77%) (Fig. 1B) with 21 peptides identified per protein (10 to 37, Fig. 1C), thereby providing an excellent basis for future FLEXIQuant experiments.
To validate our system, replicates of the same digest (“technical replicates”) as well as replicates of the entire workflow (“workflow replicates”) were analyzed. Using the binary read-out of being identified or not identified in a data dependent acquisition (DDA) routine, 89% of all peptides were detected in the technical replicates and 86% of the peptides in the workflow replicates (supplemental Fig. S2A). The two relevant FLEXII-peptides used for quantification purposes were detected in all replicates. Not surprisingly, peptides that were identified in only one of the two replicate were on average 4-fold lower in intensity than peptides identified in both replicates (supplemental Fig. S2B). A more detailed analysis of the data showed that for those peptides only identified in a single replicate, only for 12%, i.e. a minor fraction, the peptide signal was clearly absent in the MS1 spectra. Of the remaining 88% of the peptides identified only in a single replicate, MS/MS data were not acquired for ∼50% of the peptides suggesting that the precursor intensity was too low to trigger MS/MS experiments; for the other 50%, MS/MS spectra were acquired, but the quality of the spectra was too low to generate a positive database hit (within the 1% FDR threshold).
In summary, these experiments confirmed the notion that the reproducibility of peptide identification using DDA routines highly depends on the peptide signal intensities. To be able to create an abundance-based tool for predicting detectability it was also necessary to validate the quantitative reproducibility of our intensity measurements. To this end, we determined the reproducibility of our FLEXII-peptides as these peptides were the initial basis of the abundance measurement for our peptide classifier; one peptide had a correlation coefficient of 0.89 and the other 0.96, i.e. the average fold changes between the replicates were within 10% highlighting the precision of our approach (supplemental Fig. S2C).
A Peptide Classifier That Incorporates Protein Abundance
Following the example of previously published tools for predicting the easily detectable “proteotypic” peptides14,15, we started out using a set of 544 different amino acid features (10). A 10x-cross validation approach was used to train and test our classification model based on the LC-MS/MS data of the 120 individually analyzed protein digests. We evaluated our classifier not only on its ability to predict whether a peptide is detected, but also on its ability to predict whether a peptide is not detected.
Three different classification methods were tested: Support Vector Machine (SVM) (31), Random Forest (32), and neural network approaches (25). The initial evaluation of the classification methods suggested that the neural network provided slightly better results than Random Forest, which in turn gave better results than SVM. Thus, we used the former for furthering the development of our classification model (Fig. 2A and supplemental Table S3). Our classifier assigns each peptide a detectability classification score between zero and one that represents our confidence in detecting or not detecting a peptide. A score of one signifies 100% confidence that a peptide will be detected, whereas a score of zero signifies 100% confidence that a peptide will not be detected. We calculated the score for each detected and not detected peptides from the individually analyzed protein digests and plotted the densities (Fig. 2B). Our neural network model is able to separate between detected (Fig. 2B - red curve) and not detected peptides (Fig. 2B - blue curve). Importantly, ∼60% of peptides receive scores above 0.75 or below 0.15 underscoring the notion that for the majority of peptides we can predict with reasonable confidence that it will or will not be detected.
We first developed a classifier that incorporated all 544 different amino acid features similar to previously published studies (15). Because of the risk of overfitting, we sought to determine if the number of features could be reduced without significant reduction in classification performance. In the end, a classifier with only 15-features had comparable performance to a classifier with all 544 features (see Materials and Methods for details, supplemental Tables S4–S5 for the final set of 15 features) on the same training set. Our 15-feature classifier performed at least as well as the ESP predictor (Fig. 2C, Materials and Methods for details, and supplemental Table S6), also demonstrating that 2842 peptides from 120 proteins is a sufficiently large training set for peptide classification.
Next, abundance was integrated into our 15-feature classifier. Using the average signal intensity of our FLEXII-peptides as a measure of abundance significantly improved the area under the receiver operating characteristic curve (AUROC) from 0.79 to 0.83 (Fig 2C, p value <1e6 by modified z-test (29), supplemental Tables S6–S8). Although this significant improvement clearly shows that abundance information is a highly relevant feature for predicting the detectability of peptides, in practice, researchers will most often not have exogenously expressed tagged proteins available as a standard. Thus, we tested whether sequence coverage could be used as an easily available protein-specific abundance estimate instead of FLEXII-peptide intensity. Indeed, sequence coverage and signal intensity of the FLEXII-peptides resulted in similar performance improvements over our 15-feature classifier (Fig. 2C - AUROC 0.84 versus 0.83).
Although sequence coverage is an easily available protein abundance estimate, other studies have shown that it is only an estimate for protein abundance, i.e. it is not very precise (33, 34). Therefore, we tested whether the use of the iBAQ as a quantitative measure of protein abundance would improve our classifier; iBAQ is defined as the intensities of all observed peptides divided by theoretically observable peptides (21), which has been shown to be a reliable absolute protein quantification method (33). To ensure comparability of the iBAQ values independent of the mass spectrometer used for the analysis, we calculated normalized iBAQ values by dividing all iBAQ values by the highest iBAQ value in the data set of interest. Using these normalized iBAQ values we generated an iBAQ-based classifier and applied it to two different data sets: one acquired on a Q-Exactive (Orbitrap), the other on a TripleTOF 5600 (quadrupole TOF). Comparing the performances of an iBAQ based or sequence coverage based classifier, we obtained AUROCs of 0.85 versus 0.83 (Q-Exactive) and 0.82 versus 0.82 (TripleTOF 5600), respectively. An iBAQ-based classifier is at best only marginally better than a coverage-based classifier (supplemental Fig. S3, supplemental Table S6). We therefore decided to continue to focus on the sequence coverage-based classifier to account for the fact that sequence coverage information is normally readily available for proteomics data sets and does not require additional data analysis and/or data normalization.
Given the initial promise of this tool, we named it “Peptide Prediction with Abundance” (PPA) and next sought to validate PPA on other data sets.
PPA Validation
Next, we validated our ability to predict whether a specific peptide of interest is detected given a known protein abundance and whether we can predict the change in peptide detectability with changing protein abundance. This is particularly important as we envision the utility of our tool being its ability to predict the enrichment needed to detect a specific peptide of interest. We computed, over a broad range of protein abundances, the PPA score of each peptide and found significant differences even when the peptides are from the same protein (Fig. 3A, supplemental Fig. S4A). We then compared the computation to an experimental dilution series of 31 different proteins at 2–4 different concentrations (DTX3 in Fig. 3B, other 30 proteins in supplemental Fig. S4B–S4C) for a total of 110 predictions. Analyzing the data from all 31 dilution series, 96% of the peptides with PPA scores <0.15 were not detected, whereas 83% of the peptides with PPA scores >0.75 were detected. In summary, we were able to correctly predict peptide detectability 78% of the time (Fig. 3B and supplemental Fig. S4B–S4C). To the best of our knowledge there is no other peptide prediction software is able to predict changes in peptide detectability over a wide range of protein concentration.
A second way to evaluate the ability of our PPA classifier to predict abundance-based detectability is to compare predicted sequence coverage to actual sequence coverage at several different concentrations. PPA scores were again calculated over a broad range of concentrations, but here these PPA scores were converted into a single metric – sequence coverage. Predicted sequence coverage was calculated by assuming that a peptide will be detected if the PPA score was above 0.5. This value is a based on the PPA score that was equally likely to predict a peptide would be detected as not detected (Fig. 2B). Fig. 3B–3D compares our predicted sequence coverage to the actual sequence coverage from at least three dilutions of 31 proteins (supplemental Fig. S4B–S4C); the observed median difference between predicted sequence coverage and actual sequence coverage (ΔSC in Fig. 3C) is −0.6% with a standard deviation of 15%. The median of the absolute value of ΔSC is 9%. Additionally, we estimated the accuracy of our method by dividing the experimental protein abundance by the predicted protein abundance required to obtain the same sequence coverage as experimentally obtained sequence coverage (ΔPA in Fig. 3D). The median ΔPA is 1.4-fold with a standard deviation of sixfold. The median of the absolute value of ΔPA was fivefold (see also supplemental Fig. S4D). This shows again, that our PPA classifier is capable of predicting which peptides are either detected or not detected as a function of abundance, thereby underscoring PPA's utility as a tool for determining how much enrichment would be required to detect a peptide of interest.
After confirming that the PPA tool works for single protein digests and enrichment prediction, we extended the testing of PPA to complex samples. Specifically, we tested the performance of PPA on data from samples containing: (1) mixtures of eight recombinantly expressed proteins (Fig. 4A), (2) human K562 cell lysates (Fig. 4B), and (3) yeast lysates (Fig. 4C). LC-MS/MS data for all samples were collected on a Q-Exactive mass spectrometer (supplemental Tables S9–S10). For the calculation of the PPA, we used protein abundance estimates from one of two sources: (1) external sequence coverage (eSC), that is, protein abundance information from other sources than the initial data set, or 2) internal sequence information (iSC), i.e. sequence coverage from the same data set analyzed.
Fig. 4.

Validation of PPA Classification Model on Different Data sets. A–C, ROC-based evaluation of different PPA models and the ESP predictor (15) using various data sets acquired on a Q-Exactive and across instrument platforms: A, data from a pool of eight individual protein digests, B, data from an unfractionated K562 cell lysate, and C, data from an unfractionated yeast lysate. D, publicly available Orbitrap Velos data from highly fractionated colon tissue lysates (27), E, TripleTOF 5600 data from an unfractionated yeast lysate, and F, publicly available Orbitrap Fusion data from an unfractionated yeast lysate (2). ROC curves for the ESP predictor (green, “ESP”), our PPA using only the 15 physicochemical properties (red, “15”), our PPA predictor using sequence coverage information from previously published resources as (external) abundance feature (blue, “eSC”), and our PPA score using the data set-derived sequence coverage as (internal) abundance feature (orange, “iSC”). The statistical significance for the difference between “15” and “eSC,” “15” and “iSC,” and “ESP” and “iSC” was determined by modified z-test (29); p values are listed.
The following eSCs were used for the different data sets: 1) sequence coverage from our human K562 cell lysate data to calculate the eSC for the recombinant protein mixture, 2) sequence coverage information from de Godoy et al. (28) for the yeast lysate, and 3) sequence coverage information published by Wisniewski et al. (27) from a human colon tissue proteomics study for the human K562 cell lysate. Fig. 4A–4C compares PPA using eSC and iSC to the ESP predictor and our initial 15-feature classifier for these three data sets. Even though the protein abundance values used for eSCs are very crude estimates, as they are likely to vary widely based on sample type and/or condition, PPA with eSC outperformed our initial 15-feature classifier and the ESP predictor under all (except the recombinant protein mixture) tested conditions (Fig. 4), with p values of 1e-5 (modified z-test (29)) or better. As expected, the protein mixtures comprising eight recombinantly expressed proteins were the only exception as there is no reason to expect any correlation between expression in a human cell type and in vitro expression in a wheat germ extract (supplemental Tables S6–S7).
Although the PPA calculated with eSC (PPAeSC) improved the AUROC, we reasoned that an abundance measurement directly from the sample of interest should be more accurate. Protein abundance was therefore calculated directly from the sequence coverage of the measured samples to determine the PPA score (PPAiSC). PPAiSC had the highest AUROC across all samples (p values less than 1e-6 by modified z-test (29)) (Fig. 4, supplemental Tables S6–S7). Using PPA with internal sequence information from the sample itself may seem circular. However, the rationale for using this internal sequence information is that the most frequent application envisioned for PPA will be the detailed characterization of one protein or a set of proteins. Such characterization normally entails pilot experiments, that is preliminary LC-MS/MS-based analyses of some test samples whose results can be used by the researchers to estimate the amount and/or enrichment necessary to detect the peptides of interests.
To validate PPA across MS platforms we compared ESP predictor, our initial 15-feature model, PPAeSC, and PPAiSC on three different samples: (1) human colon tissue data acquired on an Orbitrap Velos (27) (Fig. 4D), (2) yeast samples analyzed on a TripleTOF 5600 (Fig. 4E, supplemental Table S11), and (3) yeast data acquired on an Orbitrap Fusion (2) (Fig. 4F). Sequence coverage from our human K562 cell lysate data was used to calculate eSC for the human colon tissue data set and sequence coverage information from de Godoy et al. (28) was used as eSC for the two yeast lysate data sets. PPAeSC improves on the AUROC over the 15-features and ESP metrics (p value for ESP versus PPAeSC <1e-6 by modified z-test (29); Supplemental Table S7). Further improvement is achieved when iSC is used for the PPA as determined by AUROC (p value ESP versus PPAiSC <1e-6 by modified z-test (29)).
We noticed that the 15-feature model, PPAeSC and PPAiSC prediction performance improvements relative to the ESP were less significant in the case of the Orbitrap Velos and Orbitrap Fusion data sets in comparison to the Q-Exactive data sets. One obvious difference between the data sets is the application of two different types of collision-induced dissociation, namely ion trap CID in the case of the Orbitrap Velos and Fusion and HCD in the case of the Q-Exactive. Therefore, to explore the effect of the fragmentation method on the prediction performance, we trained the 15-feature model using a CID-based data set acquired on an Orbitrap Velos data set (27). For performance evaluation, we applied two different models, one trained with the ion trap CID data and one with the HCD data, to a CID-based data set (Orbitrap Fusion; see Ref (2) and to a HCD-based data set (Q-Exactive; see supplementary Table S10). The ROC analysis of the results showed that the prediction performance is independent of the type of collision-induced dissociation (supplemental Fig. S5), that is, the AUROCs were almost identical irrespective of the PPA model used (supplemental Table S6). Although the improvement in AUROC for the PPAeSC and PPAiSC metric are smaller when compared with the ESP metric for our two data sets from the Orbitrap Velos and Orbitrap Fusion, collision method does not explain this difference. Instead, some other variable must exist that explains this difference perhaps differences in analytical depth in the different data sets; determining the source of this difference is out of the scope of this work.
In summary, PPA works on samples from multiple organisms and across several mass spectrometry platforms to successfully predict when a peptide is detected. The thorough testing and validation of the PPA with data sets acquired on different instrument types (Orbitrap, ion linear ion trap/orbitrap, and/or quadrupole-TOF) ensures that the PPA is very robust and is valid for use with the vast majority of the LC/MS-based proteomics data sets. That is, a single PPA model can be used: (1) irrespective of the type collision-induced dissociation (i.e. ion trap CID versus HCD/linear quadrupole CID; see above and supplemental Fig. S5), (2) independent of the ion sources from different instrument manufacturers (various generations of Thermo ion sources versus ion source from AB Sciex), and (3) independent of the mass analyzer used for the acquisition of the product ion spectra (low resolution and accuracy ion trap versus high resolution and accuracy TOF and/or Orbitrap).
Given this robustness in our PPA model, scenarios for generating new neural network parameters for a customized PPA are limited to drastic changes in the analytical strategy such as alternative fragmentation mechanisms such as electron transfer dissociation (ETD), or changes in the upfront liquid chromatography, which significantly alters the contribution from very hydrophilic and/or hydrophobic peptides. Similarly, users should consider testing the appropriate set of physicochemical properties when selected amino acid side chains are derivatized that cause significant changes in their properties. One example for such modification is lysine propionylation; this derivatization abolishes the basic properties of the side chain and significantly increases the hydrophobicity. To facilitate the extension of PPA beyond the tested conditions, we provide the source code, which will allow users to develop classifiers that are more specific to their respective instrument and data set.
CONCLUSIONS
Earlier work has shown that physicochemical properties of peptides can predict detectability by MS. However, these previous prediction algorithms focused on identifying the most detectable, that is, the “proteotypic” peptides to facilitate protein quantification. For protein quantification the choice of peptides is based on their detectabilities and researchers are able to select the most detectable peptides of the protein of interest. In contrast, in the analysis of post-translational modifications, one does not have the luxury of selecting the peptides of choice. Instead, the modification defines the peptide of interest, irrespective of its detectability. Therefore, we were interested in an algorithm to estimate the likelihood of detecting any peptide and the amount of enrichment needed to detect a particular peptide that is not initially detected. Based on a neural network approach, we identified 15 physicochemical properties that combined with a measure of protein abundance (Peptide Prediction with Abundance - PPA) allows for better detectability predictions than other published programs and algorithms. PPA was validated using data sets from various samples and instrument platforms from our own as well as other labs. Quantitative information from literature or protein sequence coverage from pilot experiments can be used as an abundance input, thereby making PPA a very useful tool to guide targeted experiments aimed at characterizing proteins and/or monitoring selected peptides.
PPA is available for download or can be used through our web interface at http://software.steenlab.org/rc4/PPA.php. Using the web-based tool, the user enters one or many peptides or protein sequences and optionally sequence coverage information (either from pilot experiments or from published studies) for the different protein sequences. Optionally, the user can also provide a protein amount in fmol, in case the protein has been precisely quantified in previous experiments. The web-based tool then performs an in silico tryptic digestion and predicts on a peptide-by-peptide basis the probability of detecting each peptide as well as the enrichment needed for a desired probability of detection, that is, PPA score.
Author Contributions
JM carried out all LC-MS/MS experiments and initial data analysis. SAB carried out the cloning, in vitro protein expression, processed the samples and helped with writing the manuscript. ST analyzed the data, generated the algorithm and contributed in writing of the manuscript. SAB was involved in the sample preparation and data acquisition. IL helped with the algorithm generation. GF helped with the library generation. HS and MS oversaw the experiments and supervised the work. JM, HS, and MS wrote the manuscript. All authors critically read and revised the manuscript.
Supplementary Material
Acknowledgments
We thank Waltraud Mair, Peter Warren, and Judith Steen for critically reading of the manuscript and for fruitful discussions.
Footnotes
Author contributions: H.S. and M.S. designed research; J.M., S.A.B., S.T., and S.A. performed research; J.M., S.T., H.S., and M.S. analyzed data; J.M., S.A.B., H.S., and M.S. wrote the paper; I.W. helped in algorithm generation; G.F. helped with library generation.
* This work is funded by US National Institutes of Health grants GM096319 (to M.S. and H.S.). The TripleTOF 5600 used for some of the experiments was funded by a US National Institutes of Health shared instrumentation grant (S10OD010706 to H.S.).
 This article contains supplemental Figs. S1 to S6 and Tables S1 to S11.
This article contains supplemental Figs. S1 to S6 and Tables S1 to S11.
1 The abbreviations used are:
- PTM
- post-translational modification
- eSC
- external sequence coverage
- ESP
- enhanced signature peptide
- FDR
- false discovery rate
- FLEXIQuant
- full-length expressed stable isotope-labeled proteins for quantification
- iBAQ
- intensity based absolute quantification
- iSC
- internal sequence coverage
- PA
- protein abundance
- PPA
- peptide prediction with abundance
- SRM
- single reaction monitoring
- SVM
- support vector machine.
REFERENCES
- 1. Mann M., Kulak N. A., Nagaraj N., Cox J. (2013) The coming age of complete, accurate, and ubiquitous proteomes. Mol. Cell 49, 583–590 [DOI] [PubMed] [Google Scholar]
- 2. Hebert A. S., Richards A. L., Bailey D. J., Ulbrich A., Coughlin E. E., Westphall M. S., Coon J. J. (2014) The one hour yeast proteome. Mol. Cell. Proteomics 13, 339–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Beck M., Schmidt A., Malmstroem J., Claassen M., Ori A., Szymborska A., Herzog F., Rinner O., Ellenberg J., Aebersold R. (2011) The quantitative proteome of a human cell line. Mol. Sys. Biol. 7, 549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nagaraj N., Wisniewski J. R., Geiger T., Cox J., Kircher M., Kelso J., Paabo S., Mann M. (2011) Deep proteome and transcriptome mapping of a human cancer cell line. Mol. Sys. Biol. 7, 548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Munoz J., Low T. Y., Kok Y. J., Chin A., Frese C. K., Ding V., Choo A., Heck A. J. (2011) The quantitative proteomes of human-induced pluripotent stem cells and embryonic stem cells. Mol. Sys. Biol. 7, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Geiger T., Wehner A., Schaab C., Cox J., Mann M. (2012) Comparative proteomic analysis of eleven common cell lines reveals ubiquitous but varying expression of most proteins. Mol. Cell. Proteomics 11, M111 014050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Grimsrud P. A., Swaney D. L., Wenger C. D., Beauchene N. A., Coon J. J. (2010) Phosphoproteomics for the masses. ACS Chem. Biol. 5, 105–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Melo-Braga M. N., Schulz M., Liu Q., Swistowski A., Palmisano G., Engholm-Keller K., Jakobsen L., Zeng X., Larsen M. R. (2014) Comprehensive quantitative comparison of the membrane proteome, phosphoproteome, and sialiome of human embryonic and neural stem cells. Mol. Cell. Proteomics 13, 311–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hornbeck P. V., Chabra I., Kornhauser J. M., Skrzypek E., Zhang B. (2004) PhosphoSite: a bioinformatics resource dedicated to physiological protein phosphorylation. Proteomics 4, 1551–1561 [DOI] [PubMed] [Google Scholar]
- 10. Kawashima S., Kanehisa M. (2000) AAindex: amino acid index database. Nucleic Acids Res. 28, 374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. (2013) Method of the Year 2012. Nature Methods 10, 1–1 [DOI] [PubMed] [Google Scholar]
- 12. Sanders W. S., Bridges S. M., McCarthy F. M., Nanduri B., Burgess S. C. (2007) Prediction of peptides observable by mass spectrometry applied at the experimental set level. BMC Bioinformatics 7, S23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Webb-Robertson B. J., Cannon W. R., Oehmen C. S., Shah A. R., Gurumoorthi V., Lipton M. S., Waters K. M. (2008) A support vector machine model for the prediction of proteotypic peptides for accurate mass and time proteomics. Bioinformatics 24, 1503–1509 [DOI] [PubMed] [Google Scholar]
- 14. Mallick P., Schirle M., Chen S. S., Flory M. R., Lee H., Martin D., Ranish J., Raught B., Schmitt R., Werner T., Kuster B., Aebersold R. (2007) Computational prediction of proteotypic peptides for quantitative proteomics. Nat. Biotechnol. 25, 125–131 [DOI] [PubMed] [Google Scholar]
- 15. Fusaro V. A., Mani D. R., Mesirov J. P., Carr S. A. (2009) Prediction of high-responding peptides for targeted protein assays by mass spectrometry. Nat. Biotechnol. 27, 190–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ishihama Y., Oda Y., Tabata T., Sato T., Nagasu T., Rappsilber J., Mann M. (2005) Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol. Cell. Proteomics 4, 1265–1272 [DOI] [PubMed] [Google Scholar]
- 17. Lu P., Vogel C., Wang R., Yao X., Marcotte E. M. (2007) Absolute protein expression profiling estimates the relative contributions of transcriptional and translational regulation. Nat. Biotechnol. 25, 117–124 [DOI] [PubMed] [Google Scholar]
- 18. Singh S., Springer M., Steen J., Kirschner M. W., Steen H. (2009) FLEXIQuant: a novel tool for the absolute quantification of proteins, and the simultaneous identification and quantification of potentially modified peptides. J. Proteome Res. 8, 2201–2210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kessner D., Chambers M., Burke R., Agus D., Mallick P. (2008) ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics 24, 2534–2536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cox J., Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 21. Schwanhausser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., Selbach M. (2011) Global quantification of mammalian gene expression control. Nature 473, 337–342 [DOI] [PubMed] [Google Scholar]
- 22. Schilling B., Rardin M. J., MacLean B. X., Zawadzka A. M., Frewen B. E., Cusack M. P., Sorensen D. J., Bereman M. S., Jing E., Wu C. C., Verdin E., Kahn C. R., Maccoss M. J., Gibson B. W. (2012) Platform-independent and label-free quantitation of proteomic data using MS1 extracted ion chromatograms in skyline: application to protein acetylation and phosphorylation. Mol. Cell. Proteomics 11, 202–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Desiere F., Deutsch E. W., King N. L., Nesvizhskii A. I., Mallick P., Eng J., Chen S., Eddes J., Loevenich S. N., Aebersold R. (2006) The PeptideAtlas project. Nucleic acids Res. 34, D655–D658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mitchell T. M. (1997) Machine Learning, McGraw-Hill, New York [Google Scholar]
- 25. Duda R. O., Hart P. E., Stork D. G. (2001) Pattern classification, 2nd Ed., Wiley, New York [Google Scholar]
- 26. Silva J. C., Gorenstein M. V., Li G. Z., Vissers J. P., Geromanos S. J. (2006) Absolute quantification of proteins by LCMSE: a virtue of parallel MS acquisition. Mol. Cell. Proteomics 5, 144–156 [DOI] [PubMed] [Google Scholar]
- 27. Wisniewski J. R., Ostasiewicz P., Dus K., Zielinska D. F., Gnad F., Mann M. (2012) Extensive quantitative remodeling of the proteome between normal colon tissue and adenocarcinoma. Mol. Sys. Biol. 8, 611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. de Godoy L. M., Olsen J. V., Cox J., Nielsen M. L., Hubner N. C., Frohlich F., Walther T. C., Mann M. (2008) Comprehensive mass-spectrometry-based proteome quantification of haploid versus diploid yeast. Nature 455, 1251–1254 [DOI] [PubMed] [Google Scholar]
- 29. Hanley J. A., McNeil B. J. (1982) The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology 143, 29–36 [DOI] [PubMed] [Google Scholar]
- 30. Singh S., Kirchner M., Steen J. A., Steen H. (2012) A practical guide to the FLEXIQuant method. Methods Mol. Biol. 893, 295–319 [DOI] [PubMed] [Google Scholar]
- 31. Vapnik V. N. (2000) The nature of statistical learning theory, 2nd Ed., Springer, New York [Google Scholar]
- 32. Breiman L., (2001) Random Forest, In: 45, ed Machine Learning, pp. 5–32 [Google Scholar]
- 33. Ahrne E., Molzahn L., Glatter T., Schmidt A. (2013) Critical assessment of proteome-wide label-free absolute abundance estimation strategies. Proteomics 13, 2567–2578 [DOI] [PubMed] [Google Scholar]
- 34. Grossmann J., Roschitzki B., Panse C., Fortes C., Barkow-Oesterreicher S., Rutishauser D., Schlapbach R. (2010) Implementation and evaluation of relative and absolute quantification in shotgun proteomics with label-free methods. J. Proteomics 73, 1740–1746 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.