

Abstract
In order to identify targets whose inhibition may enhance the efficacy of chemoradiation in pancreatic cancer, we previously conducted an RNAi library screen of 8,800 genes. We identified Mcl-1 (myeloid cell leukemia-1), an anti-apoptotic member of the Bcl-2 family, as a target for sensitizing pancreatic cancer cells to chemoradiation. In the present study we investigated Mcl-1 inhibition by either genetic or pharmacological approaches as a radiosensitizing strategy in pancreatic cancer cells. Mcl-1 depletion by siRNA produced significant radiosensitization in BxPC-3 and Panc-1 cells in association with Caspase-3 activation and PARP cleavage, but only minimal radiosensitization in MiaPaCa-2 cells. We next tested the ability of the recently identified, selective, small molecule inhibitor of Mcl-1, UMI77, to radiosensitize in pancreatic cancer cells. UMI77 caused dissociation of Mcl-1 from the pro-apoptotic protein Bak and produced significant radiosensitization in BxPC-3 and Panc-1 cells, but minimal radiosensitization in MiaPaCa-2 cells. Radiosensitization by UMI77 was associated with Caspase-3 activation and PARP cleavage. Importantly, UMI77 did not radiosensitize normal small intestinal cells. In contrast, ABT-737, an established inhibitor of Bcl-2, Bcl-XL, and Bcl-w, failed to radiosensitize pancreatic cancer cells suggesting the unique importance of Mcl-1 relative to other Bcl-2 family members to radiation survival in pancreatic cancer cells. Taken together, these results validate Mcl-1 as a target for radiosensitization of pancreatic cancer cells and demonstrate the ability of small molecules which bind the canonical BH3 groove of Mcl-1, causing displacement of Mcl-1 from Bak, to selectively radiosensitize pancreatic cancer cells.
Introduction
Pancreatic cancer is a highly aggressive disease that is associated with both local and systemic disease progression. In the metastatic disease setting, new chemotherapy regimens have produced modest survival improvements [1], [2] while in the locally advanced disease setting antimetabolite-based chemoradiation has remained the standard of care [3]. Although the combination of radiation with gemcitabine improves survival over gemcitabine alone, recent clinical studies suggest that intensification of radiation therapy in combination with concurrent gemcitabine further improves survival [4]. Despite the improvements afforded by concurrent gemcitabine and radiation therapy, however, survival for locally advanced pancreatic cancer is between 11 and 15 months [3], [4].
In order to further improve therapy for locally advanced pancreatic cancer we have previously investigated a number of strategies which build upon standard gemcitabine-based chemoradiation therapy. Incorporation of additional chemotherapeutic agents, such as cisplatin and oxaliplatin, with gemcitabine-radiation does not appear to improve survival but does increase toxicity [5], [6]. Thus, targeted therapies which are generally less toxic than standard chemotherapy have been combined with gemcitabine-radiation. Preclinically, small molecule inhibitors of Chk1 and Wee1 sensitize pancreatic tumors to gemcitabine-radiation [7], [8], [9]. In order to identify additional targets for sensitization of pancreatic cancers to gemcitabine-radiation using an unbiased approach, we previously screened an RNAi library designed to target 8,800 genes from the druggable genome [10]. In addition to previously established targets for sensitization, such as Chk1 and ATR, we identified Mcl-1 as a leading target for sensitization of pancreatic cancer cells to gemcitabine-radiation.
Mcl-1 is an anti-apoptotic member of the Bcl-2 protein family that negatively regulates the intrinsic apoptotic pathway [11]. By sequestering the pro-apoptotic proteins Bax and Bak, Mcl-1 inhibits permeabilization of the mitochondrial membrane, preventing cyctochrome C release, Caspase-9 activation, and ultimately apoptosis. Mcl-1 is subject to negative regulation by Noxa as well as other members of the BH3-only protein family (e.g. Bim, Puma, tBid), which specifically bind to the BH3 binding groove, formed by BH1-3 domains of Mcl-1, displacing Mcl-1 from Bax/Bak and thus promoting apoptosis. Overexpression of Mcl-1 has been shown to be a mechanism of resistance to various cancer therapies including radiation [12], chemotherapy [13], [14], [15], and agents targeting the Bcl-2 family of proteins [16], [17]. In contrast, genetic silencing of Mcl-1 sensitizes a spectrum of cancers including melanoma, non-small cell lung, and hepatocellular cancers to radiation [18], chemotherapy [19], [20], and Bcl-2 family inhibitors [21], [22], [23]. In pancreatic cancers, Mcl-1 protein is expressed at high levels relative to normal pancreatic tissue and is associated with advanced disease [24], and genetic silencing of Mcl-1 sensitizes pancreatic cancers to either gemcitabine or radiation [25], [26].
Given the roles of Mcl-1 in promoting resistance to radiation, chemotherapy, and Bcl-2 family inhibitors, the development of Mcl-1 selective inhibitors is an active area of investigation. While there are several established small molecule inhibitors of other Bcl-2 family members, such as ABT737 and its clinical derivative ABT263 (navitoclax), as well as ABT199 and GX15-070 (obotaclax) [27], selective Mcl-1 inhibitors are in earlier stages of development. Selective targeting of Mcl-1 has proven to be challenging relative to other Bcl-2 family members and may be attributable to the structure of Mcl-1 [28]. A few selective Mcl-1 inhibitors are in preclinical development [29], such as the stapled peptide SAHB [30], the small molecules MIM1 [31], EU-5148 [19], and the compound 53 [32], as well as a novel class of hydroxynapthalen aryl sulfonamides [33]. More recently, a small molecule inhibitor of Mcl-1, designated UMI77, that engages Mcl-1 at its canonical BH3-binding groove, was shown to have single agent activity in human pancreatic cancer models both in vitro and in vivo [34].
Based on our identification of Mcl-1 as a target for enhancing chemoradiosensitization in pancreatic cancer, in this study we sought to validate Mcl-1 as a target for radiosensitization and to investigate a novel, pharmacologic approach for Mcl-1 inhibition. Following confirmation of Mcl-1 as a radiosensitizing target in pancreatic cancer cells using siRNA, we tested the ability of the small molecule inhibitor of Mcl-1, UMI77, to radiosensitize in pancreatic cancer cells and normal small intestinal cells. To begin to understand the mechanisms of radiosensitization by Mcl-1 inhibition, we investigated the ability of UMI77 to prevent Mcl-1 and Bak complex formation and induce apoptosis assessed by Caspase-3 activity and PARP-1 cleavage, as well as sub-G1 DNA content.
Materials and Methods
Cell Culture, Drug Solutions, and RNAi
All cells were obtained from the American Type Culture Collection. Cells were grown in DMEM (Panc-1, MiaPaCa-2), RPMI1640 (BxPC-3) or HybriCare (ATCC) with 30ng/ml epidermal growth factor (CCL-241, normal human small intestine epithelial cells) media supplemented with and 10% fetal bovine serum (Invitrogen), 2 mmol/L l-glutamine (Sigma) and penicillin/streptomycin (Sigma). Experiments were conducted on exponentially growing cells. Cells were tested for Mycoplasma once every 3 months. UMI77 was dissolved in DMSO and stored at − 20°C. Non-specific and Mcl-1 siRNAs were purchased from Dharmacon and used as previously described [10]. Cells were transfected with siRNA using Oligofectamine (Invitrogen) or X-treme gene (Roche) transfection reagents.
Irradiation
Irradiations were performed using a Philips RT250 (Kimtron Medical) at a dose rate of ∼ 2 Gy/min in the University of Michigan Comprehensive Cancer Center Experimental Irradiation Core. Dosimetry was carried out using an ionization chamber connected to an electrometer system that is directly traceable to a National Institute of Standards and Technology calibration.
Clonogenic Survival Assay
Clonogenic assays were carried out using standard techniques as described previously [8], [10]. For siRNA experiments, cells were transfected at 30-50% confluency (as described above) and at 48 hours post-transfection plated at clonal densities and then irradiated. For drug experiments, cells were plated at clonal densities and then treated with UMI77 and radiation. Cells were grown at 37°C for 7–12 days for colony formation. The cytotoxicity of UMI77 or Mcl-1 siRNA in the absence of radiation treatment was calculated as the ratio of surviving treated cells relative to surviving vehicle or non-specific siRNA -treated cells, respectively. Radiation survival data from treated cells were corrected for plating efficiency by normalizing to un-irradiated control cells. Cell survival curves were fitted using the linear-quadratic equation, and the mean inactivation dose was calculated and used to determine the radiation enhancement ratio [35]. Radiosensitization is indicated by a radiation enhancement ratio of significantly greater than 1.
Immunoblotting
Cells were washed with PBS (phosphate buffered saline) and lysed in RIPA buffer (50mM Tris, pH 8.0, 150mM sodium chloride, 1.0% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) containing protease and phosphotase inhibitor cocktails (Roche). Proteins were separated by SDS-PAGE, transferred to nitrocellulose membranes (Invitrogen) and probed with primary antibodies including those recognizing Mcl-1 (S-19; Santa Cruz Biotechnology), PARP-1 (Cell Signaling), Bak (Calbiochem) and β-actin (Abcam). HRP-conjugated secondary antibodies (Santa Cruz) and chemiluminescence (ECL; Amersham Biosciences) were used to detect specific protein signals.
Immunoprecipitation
Exponentially growing pancreatic cancer cells were incubated with UMI77 for 48 hours. Cell lysates were prepared with a solution of cold NP-40 lysis buffer (50mM Tris, pH 7.4, 150mM NaCl, 1mM EDTA, 1mM DTT, 0.5% (v/v) NP-40, and Complete protease inhibitor (Roche)). Cellular debris was pelleted at 14,000g for 15 min at 4°C, and the supernatant was then collected and exposed to pre-equilibrated protein A/G Sepharose beads (Santa Cruz Biotechnology). The pre-cleared supernatant was incubated with either Mcl-1 antibody (S-19) or an isotype control (Rabbit IgG; Santa Cruz Biotechnology), overnight at 4°C, followed by the addition of protein A/G Sepharose beads for 2 hours. The beads were pelleted and washed with cold NP-40 lysis buffer followed by elution of the sample from the beads by heating at 95°C for 10 min in SDS loading buffer (60mM Tris pH 6.8, 2% SDS, 10% glycerol, and 5% 2-mercaptoethanol). The immunoprecipitates were subjected to electrophoresis and western analysis using an anti-BAK antibody (CalBiochem).
Flow Cytometry
Cells were washed with PBS and resuspended in 70% ice-cold ethanol. The cells were incubated overnight at 4°C and then washed in PBS. The cells were then resuspended in prodium iodide (PI) solution (50μg/ml PI, 200μg/ml DNase-free RNaseA in PBS) and then analyzed by flow cytometry (FACSCanto, BD Biosciences). Cell apoptosis after treatment was analyzed by measuring sub-G1 peaks using FloJo software (TreeStar).
Caspase-3 Activity Assay
The immunoabsorbent Caspase-3 activity assay kit from Roche was used to detect Caspase-3 activity after treatment with UMI77 or Mcl-1 siRNA and radiation. Briefly, after treatment, lysates from 2 × 107 cells were prepared and Caspase-3 was captured from the lysate in microplates coated with anti-Caspase-3 monoclonal antibody. Plates were washed, Ac-DEVD-AFC was added and the cleaved AFC was determined using a spectrophotometer at 405nm. Samples were calibrated to a standard curve generated from AFC stock solution.
Statistical Analysis
For radiosensitization experiments, statistical significance was determined by a Student’s T-test using GraphPad Prism (GraphPad Software Inc.).
Results
Given the identification of Mcl-1 in an RNAi library screen as a target for sensitization of pancreatic cancer cells to chemoradiation (Suppl. Fig. 1) [10], we initiated the present study by investigating the ability of Mcl-1 depletion to radiosensitize pancreatic cancer cells. Mcl-1 was depleted from BxPC-3 and Panc-1 cells via RNAi and radiation sensitivity was determined with a clonogenic survival assay in both cell lines. We found that treatment with Mcl-1 siRNA led to Mcl-1 protein depletion (Figure 1A, B, insets) and radiosensitization (Figure 1) compared to treatment with non-specific siRNA. Radiosensitization by Mcl-1 depletion was evidenced by radiation enhancement ratios between 1.3±0.04 and 1.5±0.06 in BxPC-3 and Panc-1 cells, respectively, (Figure 1C) which were significant in comparison to their respective controls. In addition, we found that MiaPaCa-2 cells were radiosensitized by Mcl-1 depletion (Suppl. Fig. 2A), although to a lesser extent than Panc-1 or BxPC-3 cells. Mcl-1 expression is relatively low in MiaPaCa-2 cells compared to Panc-1 or BxPC-3 cells [34] and may account for the relative resistance of MiaPaCa-2 cells to UMI77. Overall, these results validate Mcl-1 as a target for radiosensitization in pancreatic cancer cells.
Figure 1.
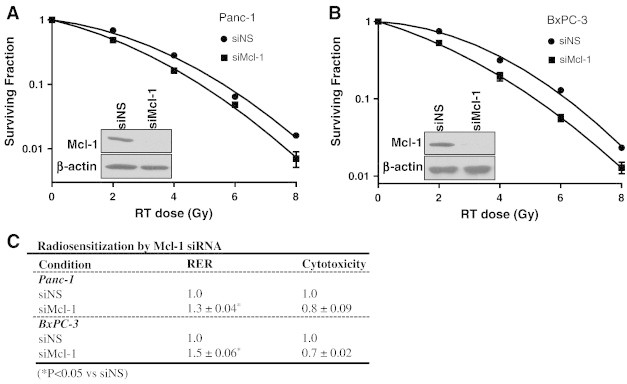
Mcl-1 depletion radiosensitizes pancreatic cancer cells. Panc-1 or BxPC-3 cells were treated with either non-specific (siNS) or Mcl-1 siRNA (siMcl-1). Seventy-two hours post-transfection cells were treated with radiation (0-8Gy). Radiosensitization was assessed by clonogenic survival and is indicated by an RER (radiation enhancement ratio) of greater than 1. Mcl-1 protein depletion by siRNA is shown (inset). Data are from a single representative experiment (A, B) or are the mean ± standard error of 3 independent experiments (C). Statistical significance versus siNS is indicated (*P<0.05). Cytotoxicity is the surviving fraction of siMcl-1-treated cells relative to siNS, in the absence of radiation.
Based on the established functions of Mcl-1 in the intrinsic apoptotic pathway, we hypothesized that Mcl-1 plays a critical role in regulating apoptosis after radiation. In order to begin to determine whether radiosensitization via Mcl-1 depletion is mediated by induction of apoptosis, we assessed both Caspase-3 activity and cleaved-PARP levels in BxPC-3 and Panc-1 cells following treatment with Mcl-1 siRNA and radiation. In Panc-1 cells, we found that depletion of Mcl-1 in the absence of radiation caused little to no increase in Caspase-3 activity or PARP cleavage or (Figure 2A, C). In combination with radiation however, Mcl-1 depletion led to both increased Caspase-3 activity and increased PARP cleavage. Likewise, depletion of Mcl-1 from BxPC-3 cells had no effect on either Caspase-3 activity or cleaved PARP, while in combination with radiation Mcl-1 depletion caused an increase in Caspase-3 activity and PARP cleavage (Figure 2B, D). Taken together, these results confirm that Mcl-1 is a target for sensitizing pancreatic cancer cells to radiation, via induction of apoptosis.
Figure 2.
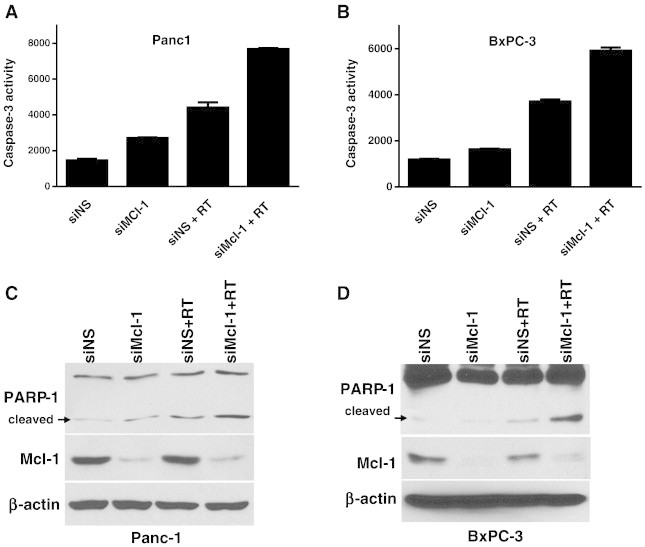
Mcl-1 depletion in combination with radiation causes Caspase-3 activation and PARP-1 cleavage. Panc-1 cells were treated with siRNA and radiation (7.5Gy) as described in Figure 1. At 48 hours post-radiation, cells were prepared for Caspase-3 activity assays (A, B) or immunoblotting (C, D). Data are the mean ± standard deviation of 2 independent experiments with each test performed in duplicate (A, B) or are from a single representative experiment (C, D).
In order to begin to develop a pharmacological strategy for radiosensitization by Mcl-1 inhibition, we tested the ability of UMI77, a recently characterized small molecule inhibitor of Mcl-1 [34], to radiosensitize pancreatic cancer cells. We began by determining the effects of minimally cytotoxic concentrations of UMI77 on radiosensitization of Panc-1 and BxPC-3 cells according to the schedule illustrated (Figure 3A). Treatment of Panc-1 cells with UMI77 resulted in a concentration-dependent increase in radiosensitization evident by a decrease in survival in response to radiation (Figure 3B) and statistically significant radiation enhancement ratios of 1.3±0.08 to 1.5±0.07 (Figure 3D). BxPC-3 cells were also significantly radiosensitized by UMI77 with a radiation enhancement ratio of 1.3±0.04 (Figure 3C, D). Since high Mcl-1 expression levels are associated with sensitivity to Mcl-1 inhibition, we also examined the ability of UMI77 to radiosensitize MiaPaCa-2 cells which express low levels of Mcl-1 relative to Panc-1 or BxPC-3 cells [34]. We found that MiaPaCa-2 cells were less radiosensitized by UMI77 (radiation enhancement ratio: 1.2; Suppl. Fig. 2B) than BxPC-3 or Panc-1 cells, a finding which is consistent with the relatively low Mcl-1 expression [34] and minimal radiosensitization by Mcl-1 siRNA (Suppl. Fig. 2A) in MiaPaCa-2 cells. Given the availability of other Bcl-2 family inhibitors, we next sought to compare UMI77 to other well-established Bcl-2 family inhibitors in the context of radiosensitization. In contrast to Mcl-1 inhibition by UMI77, we found that ABT737, an inhibitor of Bcl-2, Bcl-XL, and Bcl-w, failed to radiosensitize Panc-1 cells (Suppl. Fig. 3). This result is consistent with the lack of identification of Bcl-2, Bcl-XL, or Bcl-w in our initial RNAi library screen (Suppl. Fig. 1) [10]. Taken together, these results demonstrate that pharmacological inhibition of Mcl-1 is an effective radiosensitizing strategy in pancreatic cancer cells and furthermore, suggest that inhibition of Mcl-1 is uniquely effective relative to inhibition of other Bcl-2 family members for radiosensitizing pancreatic cancers with high Mcl-1 expression levels.
Figure 3.
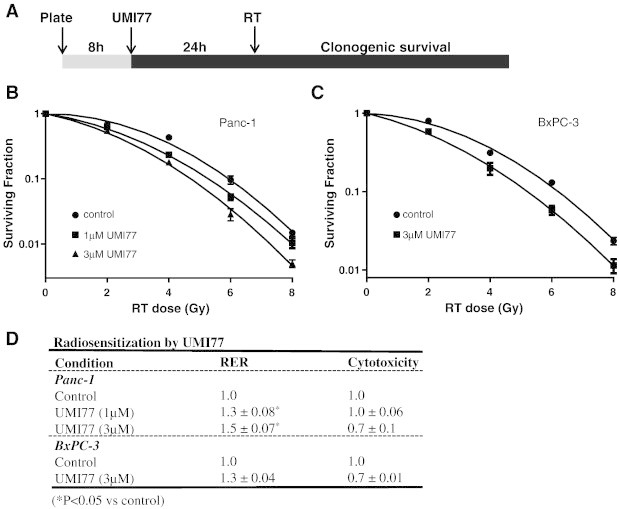
Inhibition of Mcl-1 by UMI77 radiosensitizes pancreatic cancer cells. Cells were treated with UMI77 (1-3μM) as illustrated (A) and radiosensitization was assessed by clonogenic survival. Data are from a single representative experiment (B, C) or are the mean ± standard deviation/error of 2–4 independent experiments (D). Statistical significance versus control is indicated (*P<0.05). Cytotoxicity is the surviving fraction of UMI77-treated cells relative to control cells in the absence of radiation.
Since Mcl-1 prevents apoptosis by sequestration of Bak and Bax [36], [37], we next examined the ability of UMI77 to inhibit protein-protein interactions between Mcl-1 and Bak. To this end, Panc-1 and BxPC-3 cells were treated with UMI77, followed by immunoprecipitation of Mcl-1 and detection of Bak protein levels. Consistent with a previous study [34], we found in both Panc-1 and BxPC-3 cells that UMI77 caused a concentration dependent decrease in Mcl-1-Bak interactions (Figure 4), a finding which is in agreement with the concentration dependent radiosensitization observed in response to UMI77 (Figure 3). Similarly, UMI77 inhibited Mcl-1-Bak interactions in MiaPaCa-2 cells (Suppl. Fig. 2C). These data demonstrate that UMI77 displaces Bak from Mcl-1 in pancreatic cancer cells and suggest that dissociation of Bak from Mcl-1 is a mechanism of radiosensitization by UMI77.
Figure 4.
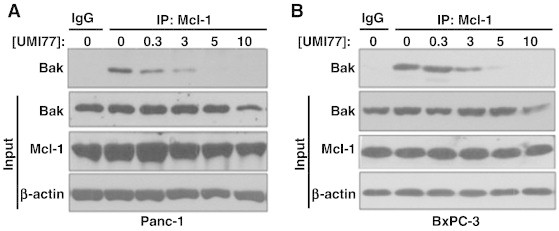
UMI77 disrupts the Mcl-1/Bak complex. Panc-1 (A) and BxPC-3 (B) cells were treated with the indicated concentrations of UMI77 for 48 hours and then prepared for immunoprecipitation and immunoblotting. Mcl-1 was immunoprecipitated from cell lysates and the levels of Bak interacting with Mcl-1 were detected by immunoblotting for Bak. The levels of Bak and Mcl-1 in total cell lysates are shown (input). Data are representative of 2–3 independent experiments.
To further examine the mechanism of radiosensitization by UMI77, we next assessed the ability of UMI77 to induce Caspase-3 activation and PARP cleavage in response to radiation in pancreatic cancer cells. In Panc-1 cells, treatment with either radiation or UMI77 alone caused a concentration-dependent increase in Caspase-3 activity and PARP cleavage (Figure 5A, C). In combination with radiation, however, UMI77 caused a higher induction of Caspase-3 activity and PARP cleavage compared to treatment with either single agent. Consistent with activation of the apoptotic pathways, UMI77 and radiation caused an increase in the number of cells with sub-G1 DNA content (Suppl. Fig. 4). Total Mcl-1 protein levels were minimally affected under these treatment conditions, suggesting UMI77 does not cause degradation of Mcl-1. Likewise, in BxPC-3 cells, UMI77 also caused Caspase-3 activation and PARP cleavage which was greater in response to the combination of UMI77 with radiation (Figure 5B, D). Taken together, these findings demonstrate that the mechanism for radiosensitization of pancreatic cancer cells by UMI77 involves displacement of Mcl-1 from the pro-apoptotic protein Bak and subsequent induction of apoptosis.
Figure 5.
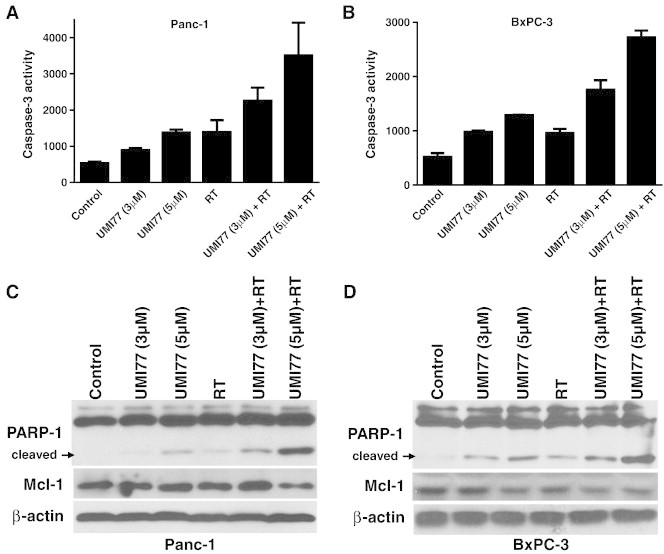
UMI77 in combination with radiation causes Caspase-3 activation and PARP-1 cleavage. Panc-1 (A, C) and BxPC-3 (B, D) cells were treated with UMI77 for 24h pre- and 48h post-radiation (7.5Gy). At the end of treatment cells were prepared for Caspase-3 activity assays (A, B) or immunoblotting (C, D). Data are the mean ± standard deviation/error of 2–3 independent experiments with each test performed in duplicate (A, B) or are from a single experiment representative of 3 independent experiments (C, D).
To begin to determine whether radiosensitization by Mcl-1 inhibition is tumor cell selective, we assessed the ability of UMI77 to radiosensitize normal cells. Given that the dose limiting toxicity for radiation treatment of pancreatic cancer is duodenum, normal human small intestinal epithelial cells were chosen for this study. Treatment of CCL-241 cells with UMI77 under identical conditions to those used for pancreatic cancer cells did not result in significant radiosensitization or cytotoxicity (Figure 6). These results suggest that UMI77 confers selective radiosensitization of pancreatic cancer cells while sparing normal intestine epithelial cells.
Figure 6.
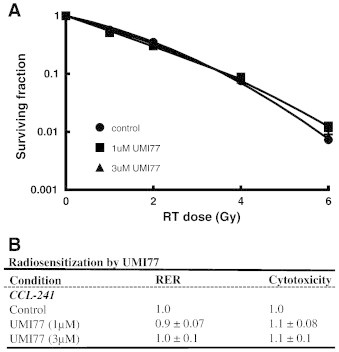
Inhibition of Mcl-1 by UMI77 does not radiosensitize normal cells. CCL-241 normal small intestinal cells were treated with UMI77 (1-3μM) as illustrated (Figure 3A) and radiosensitization was assessed by clonogenic survival. Data are from a single representative experiment (A) or are the mean ± standard error of 3 independent experiments (B). Cytotoxicity is the surviving fraction of UMI77-treated cells relative to control cells in the absence of radiation.
Discussion
Our initial identification of Mcl-1 as a sensitizing target in pancreatic cancer among a library of 8,800 other potential targets suggests the importance of Mcl-1 to the sensitivity of pancreatic cancers to chemoradiation therapy. In this study we investigated the small molecule Mcl-1 inhibitor, UMI77, as a novel radiosensitizer in pancreatic cancer. We found that UMI77 disrupts Mcl-1-Bak interactions, induces apoptosis in response to radiation, and ultimately causes radiosensitization. Further supporting the exclusive role of Mcl-1 in survival following radiation, we found that inhibition of other anti-apoptotic Bcl-2 family members (Bcl-2, Bcl-XL, and Bcl-w) failed to radiosensitize pancreatic cancer cells. In addition, we observed varying degrees of radiosensitization by Mcl-1 inhibition across a panel of pancreatic cancer cells which may be reflective of their varying Mcl-1 protein levels, with cells containing high levels of Mcl-1 being the most sensitized by Mcl-1 inhibition [34]. Importantly, we found that UMI77 was selective for pancreatic cancer cells as normal intestinal cells were not radiosensitized by UMI77. The results of this study illustrate the unique importance of Mcl-1 relative to other Bcl-2 family members for survival following radiation and suggest that pancreatic cancer cells with high Mcl-1 expression may be selectively radiosensitized by Mcl-1 inhibition.
Since Mcl-1 has an established role in regulating apoptosis, we investigated the effects of Mcl-1 siRNA or UMI77 on the apoptosis pathway. Indeed, we found that either depletion or inhibition of Mcl-1 in combination with radiation caused an increase in Caspase-3 activity and cleaved-PARP levels. These results suggest that apoptosis is a mechanism of radiosensitization by Mcl-1 inhibition. Given that Mcl-1 also has non-apoptotic functions such as regulation of the DNA damage response [38], [39], autophagy [40], mitochondrial respiration [41], and cellular senescence [42], it is possible that one of these mechanisms may contribute to radiosensitization as well. It is not clear at this time whether small molecules such as UMI77 which bind the canonical BH3 binding groove on Mcl-1, and block Mcl-1 interactions with pro-apoptotic proteins such as Bak and Bax, would affect these non-apoptotic Mcl-1 functions.
Mcl-1 is negatively regulated not only by BH3 proteins (e.g. Noxa, Puma) which displace Mcl-1 from the pro-apoptotic proteins Bak and Bax, but also by proteasomal degradation which has previously been shown to be induced by Noxa [36], [43]. Given that many of the Mcl-1 inhibitors currently under development are ‘Noxa-like’ (e.g. MIM1 and UMI77) and selectively displace Bak/Bax from Mcl-1 [27], it is of interest to know whether these Noxa-like Mcl-1 inhibitors also induce Mcl-1 degradation. Like Noxa, the putative Mcl-1 inhibitor maritoclax was reported to induce Mcl-1 degradation [44]. However, the bona fide BH3 mimetic, Mcl-1 inhibitor BIMS2A does not cause Mcl-1 degradation [45]. Consistent with the lack of effect of BIMS2A on Mcl-1 stability, we observed only minimal effects of UMI77 on Mcl-1 protein levels (Figure 5C, D). While more detailed studies of the effects of UMI77 on Mcl-1 degradation are required in the future, these results suggest that the activity of UMI77 does not require Mcl-1 degradation.
Although we did not investigate the activity of UMI77 in animal tumor models, previous studies by Abulwerdi and colleagues [34] have shown that UMI77 is well-tolerated and inhibits the growth of pancreatic tumor xenografts with no apparent toxicity in normal mouse tissues. Mechanistic studies revealed the presence of TUNNEL-positive apoptotic cells in tumors collected from UMI77-treated animals, further supporting our observations that induction of apoptosis is a mechanism of action of UMI77. Since the in vitro data in the present study demonstrated the efficacy of UMI77 as a radiosensitizing agent in pancreatic cancers, it will be important in future studies to determine the anti-tumor effects as well as toxicity of the combination of UMI77 with radiation in animal models.
Standard treatment for locally advanced pancreatic cancer is concurrent antimetabolite-based chemoradiation. In particular, the combination of gemcitabine with radiation is superior to gemcitabine alone [3], [4]. Thus, in the development of targeted agents for the treatment of pancreatic cancer, interactions with gemcitabine are an important consideration. Given that our initial RNAi screen identified Mcl-1 as a target for sensitization to gemcitabine-radiation, it is likely that small molecule Mcl-1 inhibitors will also sensitize to gemcitabine-radiation. Determining the efficacy, toxicity, and optimal schedule of administration of Mcl-1 inhibitors such as UMI77 in combination with gemcitabine and radiation are the critical next steps required in order to begin to advance Mcl-1 inhibitors to the clinic for the treatment of locally advanced pancreatic cancer. Taken together, the findings of this study support the continued development of selective inhibitors of Mcl-1 for sensitizing pancreatic cancers to chemoradiation therapy.
Footnotes
Grant support: This work was funded by NIH Grants R01CA163895 (MAM) and R01CA149442 (ZN-C), and an Alfred B. Taubman Scholarship (TSL).
Disclosure of potential conflicts of interest: None
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.tranon.2014.12.004.
Appendix A. Supplementary data
Supplementary Figures
References
- 1.Conroy T., Desseigne F., Ychou M., Bouche O., Guimbaud R., Becouarn Y. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364:1817–1825. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 2.Von Hoff D.D., Ramanathan R.K., Borad M.J., Laheru D.A., Smith L.S., Wood T.E. Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: a phase I/II trial. J Clin Oncol. 2011;29:4548–4554. doi: 10.1200/JCO.2011.36.5742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Loehrer P.J., Sr., Feng Y., Cardenes H., Wagner L., Brell J.M., Cella D. Gemcitabine alone versus gemcitabine plus radiotherapy in patients with locally advanced pancreatic cancer: an eastern cooperative oncology group trial. J Clin Oncol. 2011;29:4105–4112. doi: 10.1200/JCO.2011.34.8904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ben-Josef E., Schipper M., Francis I.R., Hadley S., Ten-Haken R., Lawrence T. A Phase I/II Trial of Intensity Modulated Radiation (IMRT) Dose Escalation With Concurrent Fixed-dose Rate Gemcitabine (FDR-G) in Patients With Unresectable Pancreatic Cancer. Int J Radiat Oncol Biol Phys. 2012;84:1166–1171. doi: 10.1016/j.ijrobp.2012.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muler J.H., McGinn C.J., Normolle D., Lawrence T., Brown D., Hejna G. Phase I trial using a time-to-event continual reassessment strategy for dose escalation of cisplatin combined with gemcitabine and radiation therapy in pancreatic cancer. J Clin Oncol. 2004;22:238–243. doi: 10.1200/JCO.2004.03.129. [DOI] [PubMed] [Google Scholar]
- 6.Hunter K.U., Feng F.Y., Griffith K.A., Francis I.R., Lawrence T.S., Desai S. Radiation therapy with full-dose gemcitabine and oxaliplatin for unresectable pancreatic cancer. Int J Radiat Oncol Biol Phys. 2012;83:921–926. doi: 10.1016/j.ijrobp.2011.08.022. [DOI] [PubMed] [Google Scholar]
- 7.Morgan M.A., Parsels L.A., Zhao L., Parsels J.D., Davis M.A., Hassan M.C. Mechanism of radiosensitization by the Chk1/2 inhibitor AZD7762 involves abrogation of the G2 checkpoint and inhibition of homologous recombinational DNA repair. Cancer Res. 2010;70:4972–4981. doi: 10.1158/0008-5472.CAN-09-3573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Engelke C.G., Parsels L.A., Qian Y., Zhang Q., Karnak D., Robertson J.R. Sensitization of pancreatic cancer to chemoradiation by the Chk1 inhibitor MK8776. Clin Cancer Res. 2013;19:4412–4421. doi: 10.1158/1078-0432.CCR-12-3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kausar T., Parsels L.A., Parsels J.D., Karnak D., Davis M.A., Maybaum J. Proceedings of the 105th Annual Meeting of the American Association for Cancer Research. 2014. Sensitization of pancreatic cancer to chemoradiation by the Wee1 inhibitor AZD1775. [Abstract No. 862] [Google Scholar]
- 10.Wei D., Parsels L.A., Karnak D., Davis M.A., Parsels J.D., Marsh A.C. Inhibition of Protein Phosphatase 2A Radiosensitizes Pancreatic Cancers by Modulating CDC25C/CDK1 and Homologous Recombination Repair. Clin Cancer Res. 2013;19:4422–4432. doi: 10.1158/1078-0432.CCR-13-0788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Akgul C. Mcl-1 is a potential therapeutic target in multiple types of cancer. Cell Mol Life Sci. 2009;66:1326–1336. doi: 10.1007/s00018-008-8637-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trivigno D., Essmann F., Huber S.M., Rudner J. Deubiquitinase USP9x confers radioresistance through stabilization of Mcl-1. Neoplasia. 2012;14:893–904. doi: 10.1593/neo.12598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nencioni A., Hua F., Dillon C.P., Yokoo R., Scheiermann C., Cardone M.H. Evidence for a protective role of Mcl-1 in proteasome inhibitor-induced apoptosis. Blood. 2005;105:3255–3262. doi: 10.1182/blood-2004-10-3984. [DOI] [PubMed] [Google Scholar]
- 14.Michels J., Obrist F., Vitale I., Lissa D., Garcia P., Behnam-Motlagh P. MCL-1 dependency of cisplatin-resistant cancer cells. Biochem Pharmacol. 2014;92:55–61. doi: 10.1016/j.bcp.2014.07.029. [DOI] [PubMed] [Google Scholar]
- 15.Zhou P., Qian L., Kozopas K.M., Craig R.W. Mcl-1, a Bcl-2 family member, delays the death of hematopoietic cells under a variety of apoptosis-inducing conditions. Blood. 1997;89:630–643. [PubMed] [Google Scholar]
- 16.Mazumder S., Choudhary G.S., Al-Harbi S., Almasan A. Mcl-1 Phosphorylation defines ABT-737 resistance that can be overcome by increased NOXA expression in leukemic B cells. Cancer Res. 2012;72:3069–3079. doi: 10.1158/0008-5472.CAN-11-4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boiani M., Daniel C., Liu X., Hogarty M.D., Marnett L.J. The stress protein BAG3 stabilizes Mcl-1 protein and promotes survival of cancer cells and resistance to antagonist ABT-737. J Biol Chem. 2013;288:6980–6990. doi: 10.1074/jbc.M112.414177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Skvara H., Thallinger C., Wacheck V., Monia B.P., Pehamberger H., Jansen B. Mcl-1 blocks radiation-induced apoptosis and inhibits clonogenic cell death. Anticancer Res. 2005;25:2697–2703. [PubMed] [Google Scholar]
- 19.Whitsett T.G., Mathews I.T., Cardone M.H., Lena R.J., Pierceall W.E., Bittner M. Mcl-1 mediates TWEAK/Fn14-induced non-small cell lung cancer survival and therapeutic response. Mol Cancer Res. 2014;12:550–559. doi: 10.1158/1541-7786.MCR-13-0458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schulze-Bergkamen H., Fleischer B., Schuchmann M., Weber A., Weinmann A., Krammer P.H. Suppression of Mcl-1 via RNA interference sensitizes human hepatocellular carcinoma cells towards apoptosis induction. BMC Cancer. 2006;6:232. doi: 10.1186/1471-2407-6-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lucas K.M., Mohana-Kumaran N., Lau D., Zhang X.D., Hersey P., Huang D.C. Modulation of NOXA and MCL-1 as a strategy for sensitizing melanoma cells to the BH3-mimetic ABT-737. Clin Cancer Res. 2012;18:783–795. doi: 10.1158/1078-0432.CCR-11-1166. [DOI] [PubMed] [Google Scholar]
- 22.Chen S., Dai Y., Harada H., Dent P., Grant S. Mcl-1 down-regulation potentiates ABT-737 lethality by cooperatively inducing Bak activation and Bax translocation. Cancer Res. 2007;67:782–791. doi: 10.1158/0008-5472.CAN-06-3964. [DOI] [PubMed] [Google Scholar]
- 23.van Delft M.F., Wei A.H., Mason K.D., Vandenberg C.J., Chen L., Czabotar P.E. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006;10:389–399. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen Z., Sangwan V., Banerjee S., Mackenzie T., Dudeja V., Li X. miR-204 mediated loss of Myeloid cell leukemia-1 results in pancreatic cancer cell death. Mol Cancer. 2013;12:105. doi: 10.1186/1476-4598-12-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wei S.H., Dong K., Lin F., Wang X., Li B., Shen J.J. Inducing apoptosis and enhancing chemosensitivity to gemcitabine via RNA interference targeting Mcl-1 gene in pancreatic carcinoma cell. Cancer Chemother Pharmacol. 2008;62:1055–1064. doi: 10.1007/s00280-008-0697-7. [DOI] [PubMed] [Google Scholar]
- 26.Guoan X., Hanning W., Kaiyun C., Hao L. Adenovirus-mediated siRNA targeting Mcl-1 gene increases radiosensitivity of pancreatic carcinoma cells in vitro and in vivo. Surgery. 2010;147:553–561. doi: 10.1016/j.surg.2009.10.033. [DOI] [PubMed] [Google Scholar]
- 27.Billard C. BH3 mimetics: status of the field and new developments. Mol Cancer Ther. 2013;12:1691–1700. doi: 10.1158/1535-7163.MCT-13-0058. [DOI] [PubMed] [Google Scholar]
- 28.Czabotar P.E., Lessene G., Strasser A., Adams J.M. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15:49–63. doi: 10.1038/nrm3722. [DOI] [PubMed] [Google Scholar]
- 29.Belmar J., Fesik S.W. Small molecule Mcl-1 inhibitors for the treatment of cancer. Pharmacol Ther. 2015;145:76–84. doi: 10.1016/j.pharmthera.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stewart M.L., Fire E., Keating A.E., Walensky L.D. The MCL-1 BH3 helix is an exclusive MCL-1 inhibitor and apoptosis sensitizer. Nat Chem Biol. 2010;6:595–601. doi: 10.1038/nchembio.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cohen N.A., Stewart M.L., Gavathiotis E., Tepper J.L., Bruekner S.R., Koss B. A competitive stapled peptide screen identifies a selective small molecule that overcomes MCL-1-dependent leukemia cell survival. Chem Biol. 2012;19:1175–1186. doi: 10.1016/j.chembiol.2012.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Friberg A., Vigil D., Zhao B., Daniels R.N., Burke J.P., Garcia-Barrantes P.M. Discovery of potent myeloid cell leukemia 1 (Mcl-1) inhibitors using fragment-based methods and structure-based design. J Med Chem. 2013;56:15–30. doi: 10.1021/jm301448p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abulwerdi F.A., Liao C., Mady A.S., Gavin J., Shen C., Cierpicki T. 3-Substituted-N-(4-hydroxynaphthalen-1-yl)arylsulfonamides as a novel class of selective Mcl-1 inhibitors: structure-based design, synthesis, SAR, and biological evaluation. J Med Chem. 2014;57:4111–4133. doi: 10.1021/jm500010b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abulwerdi F., Liao C., Liu M., Azmi A.S., Aboukameel A., Mady A.S. A novel small-molecule inhibitor of mcl-1 blocks pancreatic cancer growth in vitro and in vivo. Mol Cancer Ther. 2014;13:565–575. doi: 10.1158/1535-7163.MCT-12-0767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fertil B., Dertinger H., Courdi A., Malaise E.P. Mean inactivation dose: a useful concept for intercomparison of human cell survival curves. Radiat Res. 1984;99:73–84. [PubMed] [Google Scholar]
- 36.Willis S.N., Chen L., Dewson G., Wei A., Naik E., Fletcher J.I. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005;19:1294–1305. doi: 10.1101/gad.1304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Willis S.N., Fletcher J.I., Kaufmann T., van Delft M.F., Chen L., Czabotar P.E. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science. 2007;315:856–859. doi: 10.1126/science.1133289. [DOI] [PubMed] [Google Scholar]
- 38.Jamil S., Mojtabavi S., Hojabrpour P., Cheah S., Duronio V. An essential role for MCL-1 in ATR-mediated CHK1 phosphorylation. Mol Biol Cell. 2008;19:3212–3220. doi: 10.1091/mbc.E07-11-1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jamil S., Stoica C., Hackett T.L., Duronio V. MCL-1 localizes to sites of DNA damage and regulates DNA damage response. Cell Cycle. 2010;9:2843–2855. doi: 10.4161/cc.9.14.12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lindqvist L.M., Heinlein M., Huang D.C., Vaux D.L. Prosurvival Bcl-2 family members affect autophagy only indirectly, by inhibiting Bax and Bak. Proc Natl Acad Sci U S A. 2014;111:8512–8517. doi: 10.1073/pnas.1406425111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Perciavalle R.M., Stewart D.P., Koss B., Lynch J., Milasta S., Bathina M. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat Cell Biol. 2012;14:575–583. doi: 10.1038/ncb2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bolesta E., Pfannenstiel L.W., Demelash A., Lesniewski M.L., Tobin M., Schlanger S.E. Inhibition of Mcl-1 promotes senescence in cancer cells: implications for preventing tumor growth and chemotherapy resistance. Mol Cell Biol. 2012;32:1879–1892. doi: 10.1128/MCB.06214-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Czabotar P.E., Lee E.F., van Delft M.F., Day C.L., Smith B.J., Huang D.C. Structural insights into the degradation of Mcl-1 induced by BH3 domains. Proc Natl Acad Sci U S A. 2007;104:6217–6222. doi: 10.1073/pnas.0701297104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Doi K., Li R., Sung S.S., Wu H., Liu Y., Manieri W. Discovery of marinopyrrole A (maritoclax) as a selective Mcl-1 antagonist that overcomes ABT-737 resistance by binding to and targeting Mcl-1 for proteasomal degradation. J Biol Chem. 2012;287:10224–10235. doi: 10.1074/jbc.M111.334532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee E.F., Czabotar P.E., van Delft M.F., Michalak E.M., Boyle M.J., Willis S.N. A novel BH3 ligand that selectively targets Mcl-1 reveals that apoptosis can proceed without Mcl-1 degradation. J Cell Biol. 2008;180:341–355. doi: 10.1083/jcb.200708096. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures