

Abstract
Background
Accurate evaluation of unclassified sequence variants in cancer predisposition genes is essential for clinical management and depends on a multifactorial analysis of clinical, genetic, pathologic, and bioinformatic variables and assays of transcript length and abundance. The integrity of assay data in turn relies on appropriate assay design, interpretation, and reporting.
Methods
We conducted a multicenter investigation to compare mRNA splicing assay protocols used by members of the ENIGMA (Evidence-Based Network for the Interpretation of Germline Mutant Alleles) consortium. We compared similarities and differences in results derived from analysis of a panel of breast cancer 1, early onset (BRCA1) and breast cancer 2, early onset (BRCA2) gene variants known to alter splicing (BRCA1: c.135-1G>T, c.591C>T, c.594-2A>C, c.671-2A>G, and c.5467+5G>C and BRCA2: c.426-12_8delGTTTT, c.7988A>T, c.8632+1G>A, and c.9501+3A>T). Differences in protocols were then assessed to determine which elements were critical in reliable assay design.
Results
PCR primer design strategies, PCR conditions, and product detection methods, combined with a prior knowledge of expected alternative transcripts, were the key factors for accurate splicing assay results. For example, because of the position of primers and PCR extension times, several isoforms associated with BRCA1, c.594-2A>C and c.671-2A>G, were not detected by many sites. Variation was most evident for the detection of low-abundance transcripts (e.g., BRCA2 c.8632+1G>A Δ19,20 and BRCA1 c.135-1g>t Δ5q and Δ3). Detection of low-abundance transcripts was sometimes addressed by using more analytically sensitive detection methods (e.g., BRCA2 c.426-12_8delGTTTT ins18bp).
Conclusions
We provide recommendations for best practice and raise key issues to consider when designing mRNA assays for evaluation of unclassified sequence variants.
Germline mutations in the breast cancer susceptibility genes breast cancer 1, early onset (BRCA1)26 and breast cancer 2, early onset (BRCA2) (OMIM #113705 and #600185, respectively) are associated with a significantly increased risk of breast and other cancers (1). Although many thousands of disease-associated mutations have been identified in these genes, many DNA sequence changes found during genetic screening fall into the category of unclassified variants because their functional and clinical significance is not immediately clear. Such unclassified variants pose a challenge for clinical management of variant carriers.
Unclassified variants have the potential to alter protein function by changing the coding sequence of a transcript, or the level or structure of the gene transcript, and by disrupting regulatory regions in promoters, untranslated regions, exons, or introns (2-5). Such regulatory variants include those affecting normal splicing of BRCA1 and BRCA2, many of which have been shown to be clinically significant by use of cDNA studies and multifactorial likelihood analysis methods that combine bioinformatic, pathologic, and clinical information (6-8). These variants include those that affect splicing by disrupting or weakening the motifs at intron-exon boundaries, introducing de novo splice acceptor or donor sites, activating cryptic splice sites, or disrupting enhancer and silencer sequences. Several studies have shown that bioinformatic prediction tools can be used to prioritize variants for splicing assays (9-14).
To date, a total of 82 studies have reported findings related to splicing in BRCA1 or BRCA2 (15). The majority of these used reverse transcriptase PCR (RT-PCR)27 analysis of RNA extracted from blood of variant carriers or alternatively, minigene constructs containing the variant and assayed in non-patient-derived cell lines. The interpretation of splicing results for variant carriers can be complicated by the detection of normal alternatively spliced transcripts that occur in healthy individuals—an issue that has yet to be extensively addressed in the literature. The effect of the range of variables found in protocols used in research and clinical testing laboratories, including the PCR assay design, reagents used, and tools for visualizing and characterizing transcripts identified by PCR on assay result interpretation, is also unclear.
There are 4 instances of inconsistent or conflicting splicing results (6, 8, 14, 16-19). These include BRCA1 c.212+3A>G, c.670+8C>T, and c.736T>G and BRCA2 c.517-19C>T (4, 19-25). Reports of splicing results from a further 7 variants differed in the number of aberrant bands found in each study. The potential clinical implications of such inconsistencies highlight the need to establish the advantages and limitations of the various techniques in practice.
Guidelines for clinical interpretation and reporting of unclassified variants analyzed using splicing assays are available in the UK and Netherlands via the UK Clinical Molecular Genetics Society (http://www.cmgs.org/BPGs/Best_Practice_Guidelines.htm) and Dutch Society of Clinical Genetic Laboratory Specialists (http://www.vkgl.nl/). In addition, a range of in silico approaches have been compared with one another, and with transcript analysis, by the splice network of the French BRCA diagnostic testing laboratories, recently reported by Houdayer et al. (11). In this study (11), Houdayer et al. investigated the value of combining Splice-site Finder and MaxEntScan prediction tools and showed that major splice defects were consistently identified across a number of different laboratories. The authors did find some discrepancies with results previously reported in the literature and recommended a large cross-validation study as a future priority.
The Evidence-Based Network for the Interpretation of Germline Mutant Alleles (ENIGMA) consortium was established in 2009 with the purpose of sharing data, methods, and resources to facilitate classification of unclassified variants (21). To date, a total of 3286 unique BRCA1 and BRCA2 variants considered to be of uncertain clinical significance have been submitted to ENIGMA from more than 43 sites in 19 countries. The consortium has established several working groups, including one dedicated to examining variants that potentially alter RNA splicing.
Here we describe the outcome of an ENIGMA Splicing Working Group study to assess the importance of various mRNA assay components on consistency of results. We identified a variety of differences in protocols from 23 laboratories, the majority of which conduct routine clinical assays (see Table 1 in the Data Supplement that accompanies the online version of this article at http://www.clinchem.org/content/vol60/issue2). We report the critical elements on assay design that should be considered in the analysis of variants that may impact RNA splicing.
Table 1.
Phase 1 results submitted by 17 sites.a
| Variant | Transcript | Site (n = 17) | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6b | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | ||||
| Unequivocal | ||||||||||||||||||||
| BRCA2 c.9501+3A>T | Full length | + | + | + | + | + | + | NAc | + | + | + | + | + | + | + | + | + | + | ||
| Δ25d | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | ||||
| BRCA2 c.8632+1G>A | Full length | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | ||
| Δ20d | + | + | + | - | + | + | + | + | + | + | + | + | + | - | + | + | - | |||
| Δ19&20d | - | - | - | - | + | + | - | - | - | - | - | - | - | - | - | + | - | |||
| ins i20 | + | + | - | + | + | + | - | - | - | + | - | - | - | - | - | - | - | |||
| BRCA1 c.135–1G>T | Full length | + | + | + | + | + | + | NA | + | + | + | + | + | + | + | + | + | + | ||
| Δ5 | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | ||||
| Δ5q | - | + | - | - | - | + | - | - | + | - | - | - | - | - | - | - | ||||
| Δ3 | - | + | - | - | - | - | - | - | - | - | - | - | - | - | - | - | ||||
| BRCA1 c.5467+5G>C | Full length | + | + | + | + | + | + | NA | + | + | + | + | + | + | + | + | + | + | ||
| Δ23d | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | - | ||||
| Equivocal | ||||||||||||||||||||
| BRCA2 c.7988A>T | Full length | + | + | + | + | + | + | NA | + | + | + | + | + | + | + | + | + | + | ||
| Δ18 | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | - | ||||
| Δ17&18 | + | + | - | - | - | + | + | + | + | - | + | + | - | - | + | + | ||||
| BRCA2 c.426-12_8delGTTTT | Full length | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | ||
| Δ6q,7 | + | + | - | - | + | - | - | - | - | - | - | - | - | - | - | - | - | |||
| Δ3,5d | - | + | - | - | - | + | - | - | - | - | - | - | - | - | - | - | - | |||
| Δ3,4,5d | - | + | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | |||
| ins 18bpd | - | - | - | - | - | - | - | - | - | - | - | - | - | + | - | - | - | |||
| Δ4,5,6,7d | - | - | - | - | - | - | + | - | - | - | - | - | - | - | - | - | - | |||
| Δ5 | + | + | + | + | + | + | - | + | + | + | + | + | + | + | + | + | - | |||
| Δ5,6,7d | - | - | - | - | + | - | - | - | - | - | - | - | - | - | - | - | - | |||
| Δ6,7d | - | - | - | - | - | - | - | - | - | - | - | - | + | - | - | - | - | |||
| BRCA1 c.591C>T | Full length | + | + | + | + | + | + | NA | + | + | + | + | + | NA | + | + | + | + | ||
| Δ8&9 | - | - | - | - | + | - | - | - | - | - | - | - | - | - | - | |||||
| Δ8,9,10 | - | - | - | - | + | - | - | - | - | - | - | - | - | - | + | |||||
| Δ9 | - | + | + | + | - | + | + | - | + | + | - | + | - | + | - | |||||
| Δ9,10 | + | + | + | - | - | + | + | + | + | - | + | - | + | + | + | |||||
| Δ9,10,11q | + | + | - | - | - | - | - | - | + | - | - | - | - | + | - | |||||
| Δ11q | + | - | - | - | - | - | - | - | + | - | - | - | - | + | - | |||||
| Δ9,11qd | + | + | - | - | - | - | - | - | + | - | - | - | - | + | - | |||||
| Δ10d | - | - | - | - | - | - | + | - | - | - | - | - | - | - | - | |||||
| BRCA1 c.594-2A>C | Full length | + | + | + | + | + | + | - | + | + | + | + | + | + | + | + | + | NA | ||
| Δ9 | - | + | - | - | - | + | - | - | - | + | - | - | - | - | + | - | ||||
| Δ9,10 | + | + | + | + | + | + | + | + | + | - | - | - | + | - | + | |||||
| Δ9,10,11q | + | + | - | - | - | - | - | - | - | + | - | - | - | - | - | + | ||||
| Δ9,10,11 | + | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | ||||
| Δ11q | + | + | - | - | - | - | - | - | - | + | - | - | - | - | - | + | ||||
| Ins 21bpd Intron 9d | + | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | ||||
| Δ9,11qd | - | + | - | - | - | - | - | - | - | + | - | - | - | - | - | + | ||||
| Δ10,11qd | - | + | - | - | - | - | - | - | - | + | - | - | - | - | - | + | ||||
| Δ10d | + | + | + | + | + | + | + | + | + | + | + | + | + | - | + | + | ||||
| BRCA1 .671-2A>G | Full length | + | + | + | - | - | + | - | + | NA | + | - | - | + | + | - | + | NA | ||
| Δ9,11d | - | + | - | - | - | - | - | - | - | - | - | - | - | - | - | |||||
| Δ10,11d | + | + | + | - | - | - | - | + | + | + | + | - | - | + | + | |||||
| Δ11&12d | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | |||||
| Δ∼3.2 kb exon 11d | + | - | + | - | - | - | - | + | - | + | - | - | - | - | - | |||||
| Δ9,10,11,12d | - | - | - | - | - | - | - | - | - | - | - | - | - | - | + | |||||
| Δ9 | - | + | - | - | + | - | - | - | + | - | - | - | - | - | - | |||||
| Δ9,10 | + | + | - | - | - | - | - | + | + | - | - | - | + | - | + | |||||
| Δ9,10,11 | + | + | - | - | + | - | - | + | + | - | - | - | - | + | + | |||||
| Δ9,10,11q | + | + | - | - | - | - | + | + | + | - | - | - | - | - | - | |||||
| Δ11 | + | + | + | + | + | + | - | + | + | + | - | + | - | + | + | |||||
| Δ11q | - | + | - | + | + | + | - | + | + | + | - | + | - | - | - | |||||
+, Detection of the transcript; –, no detection.
Site 6 reported Δ1q and Δ5,1q (BRCA1 c.135-1G>T) and Δ3 (BRCA2 c.426-12_8delGTTTT) following a review of the data after results were initially compiled. HGVS (Human Genome Variation Society) nomenclature descriptions are available in online Supplemental Table 3. The Δ6,7 transcript identified in the analysis of BRCA2 c.426-12_8delGTTTT is not sequence confirmed, and it is possible that this transcript is actually Δ6q,7, which differs by only 2 bp. Additional minor and uncharacterized peaks were detected by some sites but have not been included in the table. For example, site 17 also reported Δ8p and Δ14p splice slipping events.
NA, not assessed.
Transcripts detected in the variant carrier only. Other transcripts were identified in at least 1 control.
Materials and Methods
Each participating laboratory submitted information about the mRNA splicing protocol in use at their site. These protocols were then compared on the basis of the source of biological material; the use of a nonsense mediated decay (NMD) inhibitor, RNA extraction, or removal of contaminating genomic DNA; the choice of cDNA synthesis primer, reverse transcriptase, and DNA Taq polymerase; the method of PCR product detection; and whether products were isolated, subcloned or sequenced (see online Supplemental Table 1).
To compare the assays used by laboratories within the ENIGMA consortium, 23 sites were sent aliquots of samples from the same lymphoblastoid cell lines (LCLs) that had been generated by the Kathleen Cuningham Consortium for Research into Familial Breast Cancer (kConFab) from 9 carriers of BRCA1 or BRCA2 variants known to be associated with splicing defects (Fig. 1) and from 11 controls. Four LCLs carried variants that produce unequivocal splicing aberrations resulting in a clear exon-skipping event. Five LCLs carried variants considered to produce equivocal splicing aberrations, based on the observation that they confer more subtle and variable effects, such as altering the availability of naturally occurring isoforms to avariable extent and/or producing a large and variable number of uncharacterized splicing products.
Fig. 1. BRCA1 and BRCA2 exons showing the positions of the variants studied.

BRCA1 c.135-1 G>T, BRCA1 c.5467+5 G>C and BRCA2 c.9501 + 3A>T and c.8632 + 1G>A were considered to produce unequivocal splicing aberrations. BRCA1 c.591 C>T, c.594–2 A>C and c.671–2 A>G and BRCA2 c.426-12_8delGTTTT and c.7988 A>T were considered to produce equivocal splicing aberrations.
The project was conducted in 2 phases. In the initial phase (phase 1), 16 sites used an mRNA splicing protocol they routinely use in their laboratory (see online Supplemental Table 1), summarized their results, and submitted these to PJW and MAB. for collation. Following an analysis of phase 1 results, phase 2, informed by the phase 1 findings, was initiated, during which some sites repeated each assay using a standard set of PCR primers and cycling conditions (see online Supplemental Table 2). All other components of the protocol were per phase 1, apart from site 8, which used a Bioanalyzer in phase 1 and capillary electrophoresis (CE) in phase 2. Seven sites that participated in phase 1 repeated the assays under the controlled conditions of phase 2. An additional 3 sites joined the study to assay all variants for phase 2. A further 3 sites joined phase 2 to specifically assay BRCA1 c.671-2A>G, following the finding that this equivocal variant gave rise to the greatest range of alternatively spliced transcripts.
Results
The initial comparison of protocols used by participating laboratories revealed that cycloheximide or puromycin was sometimes used for NMD treatment, with incubation times between 4 and 8 h and concentrations between 100 and 250 μg/mL, the use of 8 cDNA synthesis kits, 12 different DNA polymerases, and transcript isolation strategies that included band excision, subcloning, and sequencing. The majority of laboratories used agarose gel electrophoresis for visualizing transcripts, but several used digital visualization strategies.
In phase 1 of the study, all sites detected the fulllength transcript for each of the 4 unequivocal variants (Table 1). All sites also detected the most prominent single-exon skipping events not seen in controls for each of the unequivocal variants, apart from site 4 and 14, which did not detect the Δ20 transcript for the unequivocal variant BRCA2 c.8632+1G>A. Not all sites detected all of the less abundant transcripts from this variant, however, with only 3/16 sites detecting the Δ19&20 transcript and only 6/16 sites detecting the ins i20 transcript. For the unequivocal variant BRCA1 c.135-1g>t, which has been associated with multiple splice isoforms (22), only 3/16 detected the Δ5q transcript, and only 1 site detected the Δ3 transcript.
Detailed analysis of each of the protocols and resulting data revealed that the range of PCR design strategies contributed to the variation in detection of transcripts, in particular PCR primer design and PCR cycling conditions. For example, 11 out of 16 sites that analyzed equivocal BRCA1 c.671-2A>G were unable to detect all of the transcripts because primer position did not allow some, clearly unanticipated, fragments to be amplified (Table 1). Forward primers positioned in exon 9 or 10 were unable to amplify isoforms lacking those exons, including Δ9/10, Δ9/10/11, or Δ9/10/11q isoforms seen in controls, or the Δ9/11 or Δ10/11 variant-associated isoforms detected by other sites.
The length of extension time during PCR amplification was also found to be a contributing factor, with several protocols using times that were likely to be too short to detect the longer PCR products amplified from some splice isoforms. For example, 5 sites used elongation times of 3 min or less and were unable to amplify full-length transcripts or transcripts containing exon 11 (Δ9 or Δ9/10) for BRCA1 c.671-2A>G, which are longer than 3 kb. As for the results observed for unequivocal variants, an additional explanation for variation in detection of transcripts was the low abundance of some transcripts, including those identified in the variant carrier only (e.g., Δ9/11, Δ10/11, and Δ>3kb exon 11 transcripts), which is known to lead to variable PCR amplification. PCR cycle number was also important, with site 23 detecting only a limited number of transcripts (Table 1), likely reflecting the use of only 25 cycles (see online Supplemental Table 1).
Given that phase 1 showed that many transcripts were not observable due to the positioning of primers or elongation time, phase 2 of the study was initiated. Phase 2 included assays conducted by 10-12 sites (depending on the variant analyzed) using a standard set of primers and elongation times appropriate for the expected lengths of the transcripts (see online Supplemental Table 2). The outcome was a much greater analytical sensitivity and consistency of results (Table 2). For example, all sites were now able to detect relatively high-abundance isoforms or variant-associated transcripts reported in previous studies, but not consistently reported in phase 1 [Δ17,18 for BRCA2 c.7988A>T, Δ20 for BRCA2 c.8632+1G>A, Δ5 for BRCA2 c.426-12_8delGTTTT, and Δ10 for BRCA1 c.594-2A>C (5, 7, 8)]. Importantly, unlike phase 1, in phase 2 all study sites were able to detect at least 1 aberrant band (cf. controls) and thus may have been able to better classify the variant using the IARC (International Agency for Research on Cancer) 5-tier classification scheme.
Table 2.
Summary of results from phase 2 of the study, in which PCR primers and conditions were controlled.a,b
| Variant | Transcripts | Phase 2 results (10 sites)c | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1* | 2^ | 3* | 4* | 8^ | 9^ | 16* | 17* | 18* | 19* | ||||
| Unequivocal | |||||||||||||
| BRCA2 c.9501+3A>T | Full length | + | + | + | + | + | + | + | + | + | + | ||
| Δ25d | + | + | + | + | + | + | + | + | + | + | |||
| ins i23d | - | + | - | - | - | - | - | - | - | - | |||
| BRCA2 c.8632+1G>A | Full length | + | + | + | + | + | + | NA | + | + | + | ||
| Δ19d | - | - | - | - | + | - | - | - | - | ||||
| Δ20d | + | + | + | + | + | + | + | + | + | ||||
| Δ19&20d | + | + | - | - | + | + | + | - | - | ||||
| ins i20d | - | + | - | + | - | - | - | - | - | ||||
| Extra peaksd | - | + | - | - | - | - | - | - | - | ||||
| BRCA1 c.135-1G>T | Full length | + | + | + | + | + | + | NA | + | + | + | ||
| Δ5qd | + | + | - | - | + | - | + | - | - | ||||
| Δ5d | + | + | + | + | + | + | + | + | + | ||||
| Δ3d | - | + | - | - | + | - | - | - | - | ||||
| Δ3,5d | - | + | - | - | + | - | - | - | - | ||||
| Δ5,6d | - | + | - | - | - | - | - | - | - | ||||
| ins i3d | - | + | - | - | + | - | - | - | - | ||||
| ins i3+Δ5d | - | + | - | - | - | - | - | - | - | ||||
| BRCA1 c.5467+5G>C | Full length | + | + | + | + | + | + | + | + | + | + | ||
| Δ21d | - | - | - | - | + | - | - | - | - | - | |||
| Δ21,23d | - | - | - | - | + | - | - | - | - | - | |||
| Δ22,23d | - | + | - | - | + | - | - | + | - | - | |||
| Δ23d | + | + | + | + | + | + | + | + | + | + | |||
| Δ22d | - | + | - | + | + | - | - | - | - | - | |||
| Equivocal | |||||||||||||
| BRCA2 c.7988A>T | Full length | + | + | + | + | + | + | + | + | + | + | ||
| Δ18 | + | + | + | + | + | + | + | + | + | + | |||
| Δ17&18 | + | + | + | + | + | + | + | + | + | + | |||
| BRCA2 c.426-12_8delGTTTT* | FL | + | + | + | + | + | + | NA | + | + | + | ||
| Δ6q,7 | + | + | - | + | + | - | + | - | - | ||||
| ins 18bpd | - | - | - | - | - | + | - | - | - | ||||
| Δ5d | + | + | + | + | + | + | + | + | + | ||||
| Δ5,6,7d | - | + | - | - | + | - | + | - | - | ||||
| Δ6,7d | - | - | - | - | - | + | - | - | + | ||||
| BRCA1 c.591C>T | Full length | + | + | + | + | + | + | + | + | + | NA | ||
| Δ9 | - | + | + | + | + | - | + | + | - | ||||
| Δ9,10 | + | + | + | + | + | + | + | + | + | ||||
| Δ9,10,11q | + | + | + | - | + | + | + | + | - | ||||
| Δ9,10,11 | - | + | - | + | + | - | - | - | - | ||||
| Δ11q | + | + | + | - | + | + | + | + | - | ||||
| Δ9,11qd | + | + | - | - | + | - | + | - | - | ||||
| Δ10,11qd | - | - | - | - | + | - | - | - | - | ||||
| Δ10 | - | + | - | - | + | - | - | - | - | ||||
| Δ11q+insi13d | - | + | - | - | + | - | - | - | - | ||||
| ins i21 intron 9d | - | - | - | - | + | - | - | - | - | ||||
| Extra peaksd | - | + | - | - | - | - | - | - | - | ||||
| BRCA1 c.594-2A>C | Full length | + | + | + | + | + | + | + | + | + | NA | ||
| Δ9 | - | + | - | - | + | + | - | - | - | ||||
| Δ9,10 | + | + | + | + | + | + | + | + | + | ||||
| Δ9,10,11q | + | + | + | - | + | + | + | + | - | ||||
| Δ9,10,11 | + | + | - | - | - | + | - | - | - | ||||
| Δ10d | + | + | + | + | + | + | + | + | + | ||||
| Δ11q | + | + | + | + | + | + | + | + | - | ||||
| ins i21d | - | + | - | - | + | - | - | - | - | ||||
| Δ9,11qd | - | + | - | - | + | + | + | - | - | ||||
| Δ10,11qd | - | + | + | - | + | + | + | + | - | ||||
| Δ11q+insi13d | - | + | - | - | + | - | - | - | - | ||||
| Phase 2 BRCA1 c.67-2A>G results (12 sites)c | |||||||||||||
| 1* | 2^ | 3* | 4* | 8^ | 9^ | 16* | 17* | 18* | 20^ | 21* | 22^ | ||
| BRCA1 c.671-2A>G | Full length | + | + | + | + | + | + | + | + | + | + | + | + |
| Δ9 | - | + | + | - | + | - | - | - | - | + | - | - | |
| Δ9,10 | + | + | + | + | + | + | + | + | + | + | + | - | |
| Δ9,10,11 | + | + | + | + | + | + | + | + | - | + | + | - | |
| Δ9,10,11q | + | + | - | - | + | + | - | - | + | - | - | - | |
| Δ10d | - | + | - | - | + | - | - | - | - | - | - | - | |
| Δ11 | + | + | + | + | - | + | + | + | + | + | - | + | |
| Δ11q | - | + | - | + | + | + | - | - | + | + | - | + | |
| Δ9,10,11,12d | - | - | - | - | - | - | + | - | - | - | - | - | |
| Δ9,11d | - | + | - | - | - | - | - | - | - | - | - | - | |
| Δ10,11d | + | + | + | + | + | + | + | + | - | - | - | + | |
| Δ10,11qd | - | - | - | - | + | - | - | - | - | - | - | - | |
| Δ11&12d | - | + | - | - | - | - | - | - | - | + | - | - | |
| Δ∼3.2kb exon 11d | + | - | - | - | - | - | - | - | - | - | - | + | |
| Δ9,11qd | - | + | - | - | + | - | - | - | - | - | - | - | |
| ins i21d | - | - | - | - | + | - | + | - | - | - | - | - | |
| Δ11q+insi13d | - | + | - | - | + | - | - | - | - | - | - | - | |
+, Detection of the transcript; –, no detection.
Transcripts were identified in at least one control. The Δ6,7 transcript identified in the analysis of BRCA2:c.426-12_8delGTTTT is not sequence confirmed and it is possible that this transcript is actually Δ6q,7, which differs by only 2 bp. Splice slipping events were also reported by several sites, for example 442_444del3 from BRCA1c.591C>T by site 11. Δ8pand Δ14p splice slipping events were also reported by site 11 and 17 following a review of the data after results were initially compiled. Additional minor and uncharacterized peaks were detected by some sites but have not been included in the table. HGVS (Human Genome Variation Society) nomenclature descriptions are available in online Supplemental Table 3.
Agarose gel detection;
Agarose gel detection;
other detection; NA, not assessed. Sites 2 and 8 used capillary EP, 9 used Qiaxcel, 20 used MultiNA, 22 used Labchip GX.
Transcripts detected in the variant carrier only. Other transcripts were identified in at least 1 control.
There remained some inconsistencies in the phase 2 data. Further comparison of protocols suggested that the method of PCR product detection was likely to be a contributing factor. Sites 2 and 8 in phase 2 were the only sites to use CE exclusively for detection of transcripts. Site 2 had higher overall detection compared to the other sites. Indeed, 10 of the 23 transcripts (43.5%) identified across all sites in the phase I analysis of unequivocal variants were detected only by CE, demonstrating it to be a comparatively more analytically sensitive detection method. This trend continued for equivocal variants analyzed in phase 2, with 12 of the 49 (24.5%) transcripts detected only by capillary CE. The sites employing a Qiaxcel visualization, Bioanalyzer, or MultiNA systems demonstrated that these systems were often more analytically sensitive than gel electrophoresis. For example, sequencing of transcripts identified by Qiaxcel analysis of BRCA2 c.426-12_8delGTTTT (site 9, phase 2) showed that it was the only system to discriminate the small insertion of 18 nucleotides from the full-length transcript (Table 2; also see online Supplemental Fig. 1).
Analysis of BRCA1 c.594-2A>C in phase 2 identified 11 different transcripts. Excising bands from agarose gel or sequencing PCR products directly enabled detection of 3-6 transcripts (sites 3, 4, 17, and 18). Cloning PCR products followed by sequencing detected 6-7 transcripts (sites 1 and 16), and CE detected 10-11 transcripts (sites 2 and 8). This showed that cloning PCR products improved analytical sensitivity, and visualization by the Qiaxcel system or capillary CE together with sequence analysis is optimal to identify and characterize transcripts. The number of clones sequenced also appeared to improve analytical sensitivity; screening 40 clones (site 16) in comparison to 24 clones (site 1) enabled the detection of 1 additional transcript.
Finally, we examined the effect of using different reverse transcriptase enzymes with the same RNA, cDNA synthesis primers, and PCR primers, enzymes, and conditions. As shown in Fig. 2, the amplification of the longest transcripts was not possible with GoScript; with M-MuLV we missed in the patient with the c.671-2A>G variant the wild-type transcript; only Superscript II allowed amplification of the longest transcript in both controls and variant carriers.
Fig. 2. Comparison of cDNA synthesis enzymes for the detection of different isoforms arising from the variant BRCA1: c.671-2A>G variant.
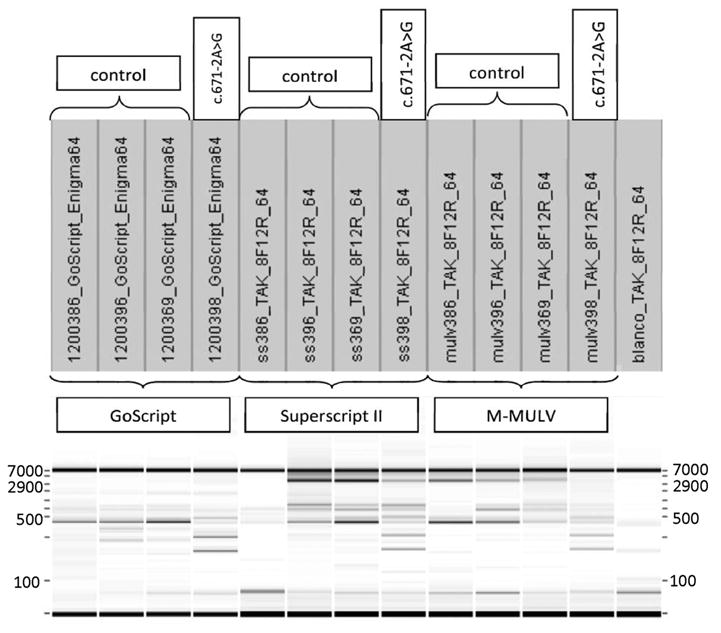
RT-PCR results (obtained by capillary electrophoresis on the Labchip, Caliper) obtained by using the same RNA, Taq polymerase (Takara), and PCR program. cDNA was synthesized with 3 different kits [GoScript (Promega), M-MuLV (New England BioLabs), SuperscriptII (Invitrogen)].
It is important to note that all transcripts shown in Tables 1 and 2 were the outcome of results by scorers who were blind to the transcripts identified by other participants, to avoid biasing the interpretation and thus the value of each approach. Once the full range of transcripts was known, however, it was possible to find some missing transcripts, demonstrating the importance of prior knowledge in both the design of the assays and the interpretation of results.
There was no clear evidence of any differences as a result of using (a) cycloheximide vs puromycin treatment for NMD inhibition; (b) differing RNA extraction methods; (c) oligo d(T) and random hexamers vs gene specific primers; (e) various methods of DNase treatment; and (f) a particular type or brand of Taq polymerase.
A summary of the recommendations arising from this study is provided in Table 3.
Table 3.
Protocol recommendations.
| Protocol | Recommendation |
|---|---|
| NMD inhibitor | Need for this depends on sensitivity of detection method. |
| For agarose gel detection, either CHX (cycloheximide) or puromycin are recommended. | |
| RNA extraction | RNA extraction protocols were indistinguishable. |
| Dnase treatment | Recommended to avoid genomic DNA contamination. |
| cDNA synthesis primer(s) | Gene-specific or oligodT+random hexamers are recommended. |
| cDNA synthesis | SuperscriptII reverse transcriptase is better for longer transcripts. |
| PCR primers | Forward and reverse primers must be at least 1 whole exon 5′ or 3′ of variant, respectively. |
| PCR polymerase | Different PCR polymerases were indistinguishable. |
| PCR conditions | Extension time long enough to copy amplicon (see enzyme manufacturer's instructions). At least 30 cycles. |
| Detection | CE was the most sensitive, followed by Qiaxcel, and then sequencing and agarose gel electrophoresis. |
| Sequencing | Cloning and sequencing is more sensitive than direct sequencing, but need to sequence at least 40 clones. |
Discussion
RNA splicing assays are commonly used in diagnostic and research settings to assess the potential effects of unclassified variants in multiple genes, including BRCA1 and BRCA2. There are a multitude of differing protocols used in clinical and research laboratories, including those within the ENIGMA consortium, and this prompted a study aimed at establishing assay guidelines.
This study shows that prior knowledge of the expected transcripts, including naturally occurring isoforms and aberrant transcripts predicted to occur in variant-carrying samples, is important for assay design. Phase 1, followed by phase 2, demonstrated that the selection of primers used to amplify exons and the design of cycling conditions appropriate for that primer design explain the vast majority of the differential success of detecting some isoforms. In phase 2 of the project, during which primer design and extension time were controlled, all sites detected the fulllength transcript and the predominant alternative transcripts, suggesting that high-abundance aberrant transcripts will be detectable regardless of assay protocol, which is consistent with the conclusions of Houdayer et al. (12).
Variability in overall detection increased as the apparent abundance of individual transcripts in a sample decreased, and thus detection became more dependent on the sensitivity of the method of analysis. This variability is also likely to occur between replicates done in a single laboratory, in addition to that between different laboratories. A controlled comparison of different reverse transcriptases showed that Superscript is much better able to copy longer transcripts (Fig. 2). It is also possible that the maximum span length of some PCR polymerases contributed to the ability of some groups to detect longer transcripts. Furthermore, primer pairs that selectively amplify disease-associated isoforms rather than naturally occurring isoforms could increase assay sensitivity.
Sites that used gel electrophoresis visualization alone were unable to detect some bands because of the inherent insensitivity of this technique, combined with the stochastic nature of PCR when analyzing low levels of target (26). An example of this is site 1, which when analyzing the equivocal variant BRCA2 c.8632+1G>A detected the ins i21bp intron 9 in phase 1 but not in phase 2, despite using the same primers and PCR conditions.
Some sites sequenced PCR products. Sites that directly sequenced the products of PCR reactions experienced some challenges in determining the sequence of low-level transcripts. An accurate assessment of transcript sequence was also confounded by the presence of multiple (3 or more) PCR products of similar lengths. In these instances, adjustments to the concentration of agarose and running times of electrophoresis may improve analytical sensitivity. However, it appears that this may be less relevant if CE systems are adopted (see below). Cloning single PCR products into a vector system is a useful alternative for isolating transcripts and appears to improve sensitivity over band excision and sequencing alone. Furthermore, by increasing the number of clones screened it is possible to marginally increase the number of transcripts detected. However, to identify low-abundance transcripts, analysis of very large numbers of clones (100s or 1000s) or next generation sequencing would be necessary.
Of all the detection methods used, CE was shown to be the most analytically sensitive. For example, site 8 showed an increase in sensitivity from phase 1 to phase 2 after switching from using a Bioanalyzer to using CE. In addition to analytical sensitivity, the CE system has the added advantage of a greater resolution (1–2 bp) compared to Qiaxcel (3–5 bp). However, the limitation with both the Bioanalyzer and CE is the inability to harvest and thus perform sequence analysis of the PCR product. Also, CE relies on a prediction of the splicing event based on the length of the product observed, which can be limited by the inaccuracy of size standards, so a secondary set of primers may be required. It is also worth noting that very long full-length (or alternative) transcripts (like those involving BRCA1 exon11 and BRCA2 exons 10 and 11) cannot be analyzed by CE.
The results presented here represent each laboratory's initial assessment of each variant. Each site had the opportunity to reassess their results after the data from all sites were released to the group and several sites reported that they detected additional transcripts in addition to (and thus not shown) the initial conclusions reported in Tables 1 and 2. This finding suggests that a prior knowledge of all potential splice transcripts related to variant carriers, from studies such as these, as well as those that occur as naturally occurring isoforms in healthy controls, is essential not only to design detection strategies (see above) but to interpret results.
The use of analytically sensitive PCR product detection (CE and Qiaxcel in phases 1 and 2, Bioanalyzer in phase 1) enabled the identification of several novel low-abundance transcripts, in both normal controls and variant carriers. This raises the question of which detectable transcripts are functional and thus relevant for determining the pathogenicity of clinically identified unclassified variants, and whether or not low abundance transcripts are of biological or pathological significance in vivo. It is generally accepted that variants resulting in single major transcripts that lack an open reading frame will be deleterious (27). However, it is much less clear whether changes in the levels of low-abundance alternative splicing events will have an impact either directly or through altering the function or levels of endogenous transcripts including fulllength mRNA.
It is possible, for example, that a reduction in the full-length expression will have a deleterious effect on known BRCA1 functions (DNA repair, cell cycle control) (28). A quantitative analysis of the range of naturally occurring isoforms relative to full-length expression and relative to other BRCA1 or BRCA2 isoforms is required, as is a comprehensive analysis of the functional role of each of these isoforms in both the healthy functioning of BRCA genes and the consequences of sequence variation on this process (29). It will also be important to extend this investigation to breast and ovarian tissue, to gain a broader understanding of the tissue-specific nature of splice-isoform regulation. Importantly, this information will be essential to determine whether knowing the full complement of transcripts has the potential to have an impact on the final classification of the variant as pathogenic or otherwise. For example, does the expression profile of the 16 alternately spliced transcripts detected in BRCA1 c.671-2A>G carriers change at different tissue sites, and will this new information influence the classification of the variant?
In summary, we have shown that primer design, PCR conditions, and PCR product detection methodology, together with prior knowledge of potential transcripts, are important contributors to the analytical sensitivity of PCR-based assays for detecting alternatively spliced RNA transcripts from variant carriers and from wild-type sequences. These factors must be considered when designing assays, particularly when they form the basis of clinical decision-making. Furthermore, the formulation of standard assay design and detection methods is indicated for all variants, but particularly for those that may impact on isoform expression.
Supplementary Material
Acknowledgments
Research Funding: The Clinical Follow Up Study was funded 2001–2009 by NHMRC and currently by the National Breast Cancer Foundation and Cancer Australia #628333. kConFab is supported by grants from the National Breast Cancer Foundation, the National Health and Medical Research Council (NHMRC) and by the Queensland Cancer Fund, the Cancer Councils of New South Wales, Victoria, Tasmania and South Australia, and the Cancer Foundation of Western Australia. The Beckman Research Institute of City of Hope (BRICOH) study was supported by Grant Number P30 CA033572 from the National Cancer Institute. R. Brandao, Fundação para a Ciência e Tecnologia; A.B. Spurdle, Australian NHMRC Project grant ID #1010719, The University of Queensland, NHMRC Senior Research Fellowship; M.A. Brown, Australian NHMRC Project grant ID #1010719, The University of Queensland; C. Lazaro, the Spanish Health Research Fund, Carlos III Health Institute, Catalan Health Institute, Government of Catalonia and the Spanish Association Against Cancer (Contract grant numbers: ISCIIIRETIC: RD06/0020/1051, 2009SGR290, PI10/01422); C. Houdayer, INCA DHOS 2010 Recherche Translationnelle sur le Cancer, FASDEC grant; L.C. Walker, Health Research Council NZ Sir Charles Hercus Health Research Fellowship; D. Baralle, CRUK Project grant; M. Colombo, the Italian Association for Cancer Research (AIRC; grant number 11897); G. De Vecchi, the Italian Association for Cancer Research (AIRC; grant number 11897); P. Radice, the Italian Association for Cancer Research (AIRC; grant number 11897); M. de la Hoya, the NEYE Foundation, the Xunta de Galicia (10PXIB 9101297PR) and FMM Foundation and a Fondo de Investigación Sanitaria (FIS) research grant PI 12/00539, an initiative of the Insituto de Salud Carlos III (Spain), partially supported by European Regional; S. Gutiérrez-Enríquez, Miguel Servet contract from the Spanish Carlos III Health Institute - Miguel Servet Project grant CP10/00617; A. Tenes, Miguel Servet contract from the Spanish Carlos III Health Institute; A. Vega, Xunta de Galicia, FMM Foundation; O. Diez, Fondo de Investi-gación Sanitaria (FIS) research grant PI 12/02585 from the Spanish Carlos III Health Institute.
We thank all members of the ENIGMA consortium Splicing Working Group for useful suggestions relating to study execution and interpretation. We also wish to thank Heather Thorne, Eveline Niedermayr, all the kConFab research nurses and staff, the heads and staff of the Family Cancer Clinics, and the Clinical Follow-Up Study for their contributions to this resource, and the many families who contribute to kConFab. K. Claes and K. De Leeneer would like to thank Anneke Grunewald for her contributions to the project.
Role of Sponsor: The funding organizations played no role in the design of study, choice of enrolled patients, review and interpretation of data, or preparation or approval of manuscript.
Footnotes
Disclaimer: The contents of this paper are solely the responsibility of the authors and do not necessarily represent the official views of the National Cancer Institute or NIH.
Human genes: BRCA1, breast cancer 1, early onset; BRCA2, breast cancer 2, early onset
Nonstandard abbreviations: RT-PCR, reverse transcriptase PCR; ENIGMA, Evidence-Based Network for the Interpretation of Germline Mutant Alleles; NMD, nonsense-mediated decay; LCL, lymphoblastoid cell lines; kConFab, Kathleen Cuningham Consortium for Research into Familial Breast Cancer; CE, capillary electrophoresis.
Author Contributions: All authors confirmed they have contributed to the intellectual content of this paper and have met the following 3 requirements: (a) significant contributions to the conception and design, acquisition of data, or analysis and interpretation of data; (b) drafting or revising the article for intellectual content; and (c) final approval of the published article.
Authors' Disclosures or Potential Conflicts of Interest: Upon manuscript submission, all authors completed the author disclosure form. Disclosures and/or potential conflicts of interest:
Employment or Leadership: S. Neuhausen, the Morris and Horowitz Families Endowed Professor, The Beckman Research Institute of City of Hope.
Consultant or Advisory Role: None declared.
Stock Ownership: None declared.
Honoraria: None declared.
Expert Testimony: None declared.
Patents: None declared.
References
- 1.Antoniou AC, Pharoah PD, McMullan G, Day NE, Stratton MR, Peto J, et al. A comprehensive model for familial breast cancer incorporating BRCA1, BRCA2 and other genes. Br J Cancer. 2002;86:76–83. doi: 10.1038/sj.bjc.6600008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chatterjee S, Pal JK. Role of 5′- and 3′- untranslated regions of mRNAs in human diseases. Biol Cell. 2009;101:251–62. doi: 10.1042/BC20080104. [DOI] [PubMed] [Google Scholar]
- 3.Brewster BL, Rossiello F, French JD, Edwards SL, Wong M, Wronski A, et al. Identification of fifteen novel germline variants in the BRCA1 3′UTR reveals a variant in a breast cancer case that introduces a functional miR-103 target site. Hum Mutat. 2012;33:1665–75. doi: 10.1002/humu.22159. [DOI] [PubMed] [Google Scholar]
- 4.Chen X, Truong TTN, Weaver J, Bove BA, Cattie K, Armstrong BA, et al. Intronic alterations in BRCA1 and BRCA2: effect on mRNA splicing fidelity and expression. Hum Mutat. 2006;27:427–35. doi: 10.1002/humu.20319. [DOI] [PubMed] [Google Scholar]
- 5.Lovelock PK, Healey S, Au W, Sum EY, Tesoriero A, Wong EM, et al. Genetic, functional, and histopathological evaluation of two C-terminal BRCA1 missense variants. J Med Genet. 2006;43:74–83. doi: 10.1136/jmg.2005.033258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Farrugia DJ, Agarwal MK, Pankratz VS, Deffenbaugh AM, Pruss D, Frye C, et al. Functional assays for classification of BRCA2 variants of uncertain significance. Cancer Res. 2008;68:3523–31. doi: 10.1158/0008-5472.CAN-07-1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thomassen M, Blanco A, Montagna M, Hansen TV, Pedersen IS, Gutierrez-Enriquez S, et al. Characterization of BRCA1 and BRCA2 splicing variants: a collaborative report by ENIGMA consortium members. Breast Cancer Res Treat. 2012;132:1009–23. doi: 10.1007/s10549-011-1674-0. [DOI] [PubMed] [Google Scholar]
- 8.Walker LC, Whiley PJ, Couch FJ, Farrugia DJ, Healey S, Eccles DM, et al. Detection of splicing aberrations caused by BRCA1 and BRCA2 sequence variants encoding missense substitutions: implications for prediction of pathogenicity. Hum Mutat. 2010;31:E1484–505. doi: 10.1002/humu.21267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vreeswijk MP, Kraan JN, van der Klift HM, Vink GR, Cornelisse CJ, Wijnen JT, et al. Intronic variants in BRCA1 and BRCA2 that affect RNA splicing can be reliably selected by splice-site prediction programs. Hum Mutat. 2009;30:107–14. doi: 10.1002/humu.20811. [DOI] [PubMed] [Google Scholar]
- 10.Mucaki EJ, Ainsworth P, Rogan PK. Comprehensive prediction of mRNA splicing effects of BRCA1 and BRCA2 variants. Hum Mutat. 2011;32:735–42. doi: 10.1002/humu.21513. [DOI] [PubMed] [Google Scholar]
- 11.Houdayer C, Caux-Moncoutier V, Krieger S, Barrois M, Bonnet F, Bourdon V, et al. Guidelines for splicing analysis in molecular diagnosis derived from a set of 327 combined in silico/in vitro studies on BRCA1 and BRCA2 variants. Hum Mutat. 2012;33:1228–38. doi: 10.1002/humu.22101. [DOI] [PubMed] [Google Scholar]
- 12.Houdayer C, Dehainault C, Mattler C, Michaux D, Caux-Moncoutier V, Pages-Berhouet S, et al. Evaluation of in silico splice tools for decision-making in molecular diagnosis. Hum Mutat. 2008;29:975–82. doi: 10.1002/humu.20765. [DOI] [PubMed] [Google Scholar]
- 13.Radice P, De Summa S, Caleca L, Tommasi S. Unclassified variants in BRCA genes: guidelines for interpretation. Ann Oncol. 2011;22(Suppl 1):i18–23. doi: 10.1093/annonc/mdq661. [DOI] [PubMed] [Google Scholar]
- 14.Sanz DJ, Acedo A, Infante M, Duran M, Perez-Cabornero L, Esteban-Cardenosa E, et al. A high proportion of DNA variants of BRCA1 and BRCA2 is associated with aberrant splicing in breast/ovarian cancer patients. Clin Cancer Res. 2010;16:1957–67. doi: 10.1158/1078-0432.CCR-09-2564. [DOI] [PubMed] [Google Scholar]
- 15.Walker LC, Whiley PJ, Houdayer C, Hansen TO, Vega A, Santamarina M, et al. Evaluation of a 5-tier scheme proposed for classification of sequence variants using bioinformatic and splicing assay data: inter-reviewer variability and promotion of minimum reporting guidelines. Hum Mutat. 2013;34:1424–31. doi: 10.1002/humu.22388. [DOI] [PubMed] [Google Scholar]
- 16.Claes K, Poppe B, Machackova E, Coene I, Foretova L, De Paepe A, Messiaen L. Differentiating pathogenic mutations from polymorphic alterations in the splice sites of BRCA1 and BRCA2 Genes. Chrom Cancer. 2003;37:314–20. doi: 10.1002/gcc.10221. [DOI] [PubMed] [Google Scholar]
- 17.Gayther SA, Warren W, Mazoyer S, Russell PA, Harrington PA, Chiano M, et al. Germline mutations of the BRCA1 gene in breast and ovarian cancer families provide evidence for a genotype-phenotype correlation. Nat Genet. 1995;11:428–33. doi: 10.1038/ng1295-428. [DOI] [PubMed] [Google Scholar]
- 18.Machackova E, Foretova L, Lukesova M, Vasickova P, Navratilova M, Coene I, et al. Spectrum and characterisation of BRCA1 and BRCA2 deleterious mutations in high-risk Czech patients with breast and/or ovarian cancer. BMC Cancer. 2008;8:140. doi: 10.1186/1471-2407-8-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Menendez M, Castellsague J, Mirete M, Pros E, Feliubadalo L, Osorio A, et al. Assessing the RNA effect of 26 DNA variants in the BRCA1 and BRCA2 genes. Breast Cancer Res Treat. 2012;132:979–92. doi: 10.1007/s10549-011-1661-5. [DOI] [PubMed] [Google Scholar]
- 20.Vega A, Campos B, Bressac-De-Paillerets B, Bond PM, Janin N, Douglas FS, et al. The R71G BRCA1 is a founder Spanish mutation and leads to aberrant splicing of the transcript. Hum Mutat. 2001;17:520–1. doi: 10.1002/humu.1136. [DOI] [PubMed] [Google Scholar]
- 21.Spurdle AB, Healey S, Devereau A, Hogervorst FB, Monteiro AN, Nathanson KL, et al. ENIGMA-evidence-based network for the interpretation of germline mutant alleles: an international initiative to evaluate risk and clinical significance associated with sequence variation in BRCA1 and BRCA2 genes. Hum Mutat. 2012;33:2–7. doi: 10.1002/humu.21628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tesoriero AA, Wong EM, Jenkins MA, Hopper JL, Brown MA, Chenevix-Trench G, et al. Molecular characterization and cancer risk associated with BRCA1 and BRCA2 splice site variants identified in multiple-case breast cancer families. Hum Mutat. 2005;26:495. doi: 10.1002/humu.9379. [DOI] [PubMed] [Google Scholar]
- 23.Bonnet C, Krieger S, Vezain M, Rousselin A, Tournier I, Martins A, Berthet P, et al. Screening BRCA1 and BRCA2 unclassified variants for splicing mutations using reverse transcription PCR on patient RNA and an ex vivo assay based on a splicing reporter minigene. J Med Genet. 2008;45:438–46. doi: 10.1136/jmg.2007.056895. [DOI] [PubMed] [Google Scholar]
- 24.Thery JC, Krieger S, Gaildrat P, Revillion F, Buisine MP, Killian A, et al. Contribution of bioinformatics predictions and functional splicing assays to the interpretation of unclassified variants of the BRCA genes. Eur J Hum Genet. 2011;19:1052–8. doi: 10.1038/ejhg.2011.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Colombo M, De Vecchi G, Caleca L, Foglia C, Ripamonti CB, Ficarazzi F, et al. Comparative in vitro and in silico analyses of variants in splicing regions of BRCA1 and BRCA2 genes and characterization of novel pathogenic mutations. PLoS One. 2013;8:e57173. doi: 10.1371/journal.pone.0057173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taberlet P, Griffin S, Goossens B, Questiau S, Manceau V, Escaravage N, et al. Reliable genotyping of samples with very low DNA quantities using PCR. Nucleic Acids Res. 1996;24:3189–94. doi: 10.1093/nar/24.16.3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spurdle AB, Couch FJ, Hogervorst FB, Radice P, Sinilnikova OM. Prediction and assessment of splicing alterations: implications for clinical testing. Hum Mutat. 2008;29:1304–13. doi: 10.1002/humu.20901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Orban TI, Olah E. Expression profiles of BRCA1 splice variants in asynchronous and in G1/S synchronized tumor cell lines. Biochem Biophys Res Commun. 2001;280:32–8. doi: 10.1006/bbrc.2000.4068. [DOI] [PubMed] [Google Scholar]
- 29.Sevcik J, Falk M, Macurek L, Kleiblova P, Lhota F, Hojny J, et al. Expression of human BRCA1 Delta17–19 alternative splicing variant with a truncated BRCT domain in MCF-7 cells results in impaired assembly of DNA repair complexes and aberrant DNA damage response. Cell Signal. 2013;25:1186–93. doi: 10.1016/j.cellsig.2013.02.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.