

Abstract
GM2 gangliosidoses are severe neurodegenerative disorders resulting from a deficiency in β-hexosaminidase A activity and lacking effective therapies. Using a Sandhoff disease (SD) mouse model (Hexb−/−) of the GM2 gangliosidoses, we tested the potential of systemically delivered adeno-associated virus 9 (AAV9) expressing Hexb cDNA to correct the neurological phenotype. Neonatal or adult SD and normal mice were intravenously injected with AAV9-HexB or –LacZ and monitored for serum β-hexosaminidase activity, motor function, and survival. Brain GM2 ganglioside, β-hexosaminidase activity, and inflammation were assessed at experimental week 43, or an earlier humane end point. SD mice injected with AAV9-LacZ died by 17 weeks of age, whereas all neonatal AAV9-HexB–treated SD mice survived until 43 weeks (P < 0.0001) with only three exhibiting neurological dysfunction. SD mice treated as adults with AAV9-HexB died between 17 and 35 weeks. Neonatal SD-HexB–treated mice had a significant increase in brain β-hexosaminidase activity, and a reduction in GM2 ganglioside storage and neuroinflammation compared to adult SD-HexB– and SD-LacZ–treated groups. However, at 43 weeks, 8 of 10 neonatal-HexB injected control and SD mice exhibited liver or lung tumors. This study demonstrates the potential for long-term correction of SD and other GM2 gangliosidoses through early rAAV9 based systemic gene therapy.
Introduction
Sandhoff disease (SD; OMIM 268800) is an autosomal recessive, lysosomal storage disorder characterized by severe progressive neurodegeneration because of accumulating GM2 ganglioside in the brain.1 GM2 ganglioside is normally degraded to GM3 ganglioside by an activating protein (GM2A), that solubilizes GM2 in the lysosomal membrane, and β-hexosaminidase A (Hex A) which removes GM2's terminal N-acetylgalactosamine. Hex A is a heterodimer comprised of α- and β-subunits encoded by the HEXA and HEXB genes respectively. Mutations in these genes or the activator-encoding gene, GM2A, can result in GM2 gangliosidosis. Hex S (αα) or Hex B (ββ) are present in cells lacking Hex A, but these isoenzymes cannot hydrolyze GM2 ganglioside to compensate for Hex A deficiency.2
Tay-Sachs disease (TSD; OMIM 272800) results from mutations in HEXA and has a carrier frequency in Ashkenazi Jews of 1/25.3 The carrier frequency of HEXB mutations causing SD is about 1/276 in the general population and is increased in specific populations,4,5,6,7,8 whereas GM2A mutations (OMIM 272750) are rare. GM2 gangliosidoses have infantile-, juvenile- or adult-onset forms whose severity is inversely correlated with the residual Hex A activity.9 The infantile form has <2% residual activity, is evident by 6 months of age, and exhibits neurological deterioration that typically culminates in death before the age of 4 years. Small increases in activity (<10%) result in later-onset forms, but the neurodegeneration remains debilitating and often lethal.
To further understand the pathology of the GM2 gangliosidoses, TSD and SD mouse models were attempted by targeting the Hexa or Hexb gene. The disruption of Hexa resulted in a late- rather than an early-onset neurological phenotype,10 as mouse sialidase converts GM2 to its asialo form (GA2), which is degraded by Hex B, bypassing the need for Hex A.11 In contrast, the disruption of Hexb leads to extensive GM2 ganglioside accumulation and severe neurological disease resembling that of the human infantile disease.10,11,12,13 Thus, the SD mouse model is often used as a model for studying potential therapies for severe GM2 gangliosidoses.
Various therapies for the GM2 gangliosidoses have been tested. Bone marrow transplant trials had little success.14 Enzyme replacement had a therapeutic effect in one study where a modified form of HexA was injected into the brain of mice.15 Gene therapy using AAV injected into the brain corrected the disease in mice, especially in areas close to the injection site.16,17,18 Other strategies, including substrate reduction,19 inhibitors of glycosylceramide synthase,20 and chaperone-mediated stabilization of mutant enzyme subunits,19 did not correct the infantile disease, and have been successful for less severe forms only when used in combination.
Recombinant AAV vectors are effective gene delivery vehicles because they can transduce nondividing cells and confer long-term stable gene expression.21 The AAV9 serotype penetrates the blood brain barrier and transduces neurons and astrocytes, as well as liver and heart.22,23 AAV9 has been used successfully to treat different neurological conditions.24 No study investigating the systemic treatment of GM2 gangliosidoses using rAAV9 has been reported. Here, we present proof-of-concept therapeutic evaluation of AAV9 for the long-term correction of SD in a mouse model. After a single intravenous administration of rAAV9 into neonatal mice, we were able to achieve prolonged survival, increased β-hexosaminidase activity, and reduction of lysosomal storage of GM2 gangliosides.
Results
To evaluate the efficacy of a single intravenous injection of rAAV9 expressing the mouse Hexb cDNA (AAV9-HexB) in ameliorating the biochemical and neurological phenotype in SD, AAV9-HexB or AAV9-LacZ (β-galactosidase expressing control vector) was administered to postnatal day 1 or 2 neonates (2.5 × 1014 vector genomes (vg)/kg via superficial temporal vein), and 6-week-old adults (3.5 × 1013 vg/kg via tail vein). For a description of the experimental design, see Supplementary Table S1. Mice were followed until they reached 43 weeks of age, or a humane end point (see Materials and Methods).
Impact of AAV9-HexB on survival and motor activity of SD mice
Progressive neuronal damage in untreated SD mice results in tremors, muscle weakness, spasticity, seizures, and ultimately death by 15–17 weeks of age.11 In our study, we examined the survival of SD (Hexb−/−) and control (Hexb+/− or Hexb+/+) mice, injected as adults or neonates with AAV9-HexB or AAV9-LacZ, until 43 weeks. A significantly extended life span was observed for all SD mice treated with AAV9-HexB (Figure 1a). Compared to AAV9-LacZ–injected SD mice (SD-LacZ), with a median survival of 17 weeks (mean = 16 weeks), a single intravenous injection of AAV9-HexB into neonatal SD mice (nSD-HexB) was sufficient to ensure their long-term survival (mean = 43 weeks, P < 0.0001; Figure 1a); they were sacrificed at the experimental end point of 43 weeks with no evidence of paralysis or seizures although two exhibited a slight tremor and three had a tendency to clasp their limbs on a tail suspension test. The prolonged life span within the five adult SD-treated mice (aSD-HexB) that received the rAAV9-HexB intravenous treatment at 6 weeks was highly variable (Figure 1a), with a median of 19 weeks (mean = 22.4 weeks, P < 0.01, Figure 1a), demonstrating a possible beneficial outcome even when rAAV9 intravenous treatment is delayed.
Figure 1.

Survival and motor activity of rAAV9–injected mice. SD or control mice were injected as neonates or adults with AAV9-LacZ or AAV9-HexB and monitored for up to 43 weeks. (a) Survival of mice over 43 weeks. Each point represents the percentage of the mice surviving at that time. Note that AAV9-HexB–injected neonates (n = 5) were the only control animals held to 43 weeks because other controls were sacrificed as the same time as their LacZ-injected or adult-treated SD counterparts. Control (n = 5); SD-LacZ (n = 10); aSD-HexB (n = 5); neonatal (n) SD-HexB (n = 5) (b) Distance travelled over time. Mice were videotaped in an open field at 3–5 time points after administration of the rAAV9, and the distance travelled was analyzed using ANY-maze software. Values at each time point represent the mean ± SEM. The control group includes mice treated as neonates or adults with either LacZ or HexB, as no significant difference was found among these groups (Supplementary Table S2). In b, no statistical comparison of the HexB- and LacZ-injected SD mice was possible after 16 weeks because all members of the comparison groups had died; however, there was no statistically significant difference between the nHexB-treated SD group and the control group. *P < 0.5, **P < 0.01, ***P < 0.0001; ns, not significant.
To determine if administration of AAV9-HexB improved motor activity in the adult and neonatally treated SD mice, we analyzed distance travelled and number of head rotations in an open-field test beginning at 14 weeks when motor deterioration in SD mice is predictably severe, and continued to 30 weeks. At all times tested, neonatal SD mice injected with AAV9-HexB travelled further than AAV9-LacZ–injected SD mice (Figure 1b). In contrast, the SD mice injected with AAV9-HexB as adults have a similar activity level to that of the AAV9-LacZ–injected SD mice (P > 0.05; Figure 1b). However, the decline in the adult-injected SD-HexB group extended over a longer time period compared to the SD-LacZ mice, indicating a delay in disease progression, albeit not statistically significant (Figure 1b). When comparing head rotations, SD mice injected with AAV9-HexB as neonates had a significantly higher number than that observed in SD mice injected with AAV9-LacZ at each time point leading up to the end point (Supplementary Figure S1, P < 0.01). Similar to the survival curve, SD mice injected with AAV9-HexB as adults showed no statistically significant improvement in head rotations (Supplementary Figure S1). Videos at 16 weeks of representative SD mice treated with AAV9-HexB either as a neonate or adult, each compared to an AAV9-LacZ–injected SD mouse and a normal control, are provided in Supplementary Videos S1 and S2.
Effect of AAV9-HexB treatment on GM2 ganglioside storage
The increased survival and motor activity in SD mice treated neonatally with AAV9-HexB suggested that the intravenously administered viral vector had crossed the blood brain barrier (BBB) and reduced the level of GM2 ganglioside storage. To assess GM2 ganglioside in the brain, we used toluidine blue staining to visualize the extent of lysosomal vacuolization (Figure 2a,b), and high performance thin layer chromatography (HPTLC) to examine the ganglioside levels (Figure 2c,d). Extensive vacuolization was evident in the brains of SD mice treated with AAV9-LacZ (Figure 2a). The vacuolization was similar in SD mice treated as adults with AAV9-HexB, but was substantially reduced in most areas of the brain of AAV9-HexB–treated neonatal mice (Figure 2a). A semiquantitative analysis where a numerical value was applied to the extent of vacuolization showed significantly reduced vacuolization in all regions of the AAV9-HexB–injected neonatal SD mice (P < 0.0001), although the level of vacuolization was still significantly higher than that in normal control mice (Figure 2b). The level of vacuolization in SD mice injected with AAV9-HexB as adults was similar to that of LacZ-treated SD mice; a modest reduction in vacuolization was observed in only three areas of the brain (Figure 2b). Biochemical analysis using HPTLC supported these findings as GM2 ganglioside was not detectable in normal mice, and at reduced levels in SD mice injected with AAV9-HexB as neonates (Figure 2c,d). In contrast, GM2 ganglioside levels were significantly higher in SD mice injected with AAV9-LacZ or AAV9-HexB (as adults), than normal or SD mice treated with AAV9-HexB as neonates, (Figure 2c,d).
Figure 2.
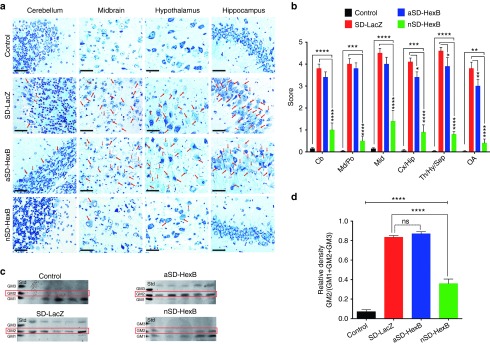
Assessment of GM2 ganglioside accumulation in AAV9-treated brains. Brains from SD and age-matched normal control mice were collected at either the humane end point (17–19 weeks) or the end of the study at 43 weeks. (a) Toluidine blue staining of brain sections. Paraffin sections (1.5 microns) were examined for vacuolization using a compound microscope. Representative images are shown for four different brain regions. The brain images in the control group were taken from LacZ-treated adult or neonatal controls. Red arrows identify cells containing vacuoles. Scale bars represent 50 µm.(b) Semiquantitative analysis of cellular vacuolization. Multiple fields (10–30) in each region of the brain, were scored based on severity of vacuolization with 0 indicating no vacuoles and 5 indicating severely vacuolated. Columns represent the mean ± SEM. The control group includes mice treated as neonates or adults with either LacZ or HexB (n = 20), as no significant difference was found among these groups (Supplementary Table S2); SD-LacZ (n = 10); aSD-HexB (n = 5); neonatal (n) SD-HexB (n = 5). (c) Analysis of ganglioside content. Gangliosides isolated from flash-frozen brains as well as ganglioside standards were separated by HPTLC and detected with resorcinol. Samples from the brains of all 5 mice in the adult (a) SD-HexB group, and nSD-HexB group are shown as well as 5 representative samples from the normal control and SD-LacZ–injected groups. The position of GM2 ganglioside is indicated by a red box. Although no standard was included on the plates for GA2, the upper band that is more abundant in SD mice injected with AAV9-LacZ is likely to be GA2. (d) Densitometry-based quantification of GM2 in the brain. HPTLC plates of brain gangliosides were analyzed by densitometry and GM2 ganglioside levels are represented as a proportion of the combined total of GM1, GM2, and GM3 ganglioside levels. The control group (n = 20) includes mice treated as neonates or adults treated with either LacZ or HexB, as no significant difference was found among these groups (Supplementary Table S2); SD-LacZ (n = 10); aSD-HexB (n = 5); nSD-HexB (n = 5). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; ns, not significant. Cb, cerebellum; Cx, cerebral cortex; Hip, hippocampus; Hy, hypothalamus; Md, medulla; OA, olfactory bulb; Po, Pons; Mid, midbrain; Sep, septum; Th, thalamus.
β-Hexosaminidase analyses
Consistent with the reduced GM2 ganglioside in the AAV9-HexB neonatally treated SD mice, we anticipated increased levels of β-hexosaminidase activity in the brains of this group. Total serum β-hexosaminidase activity was significantly higher at all tested time points in AAV9-HexB–treated mice (Figure 3a). Although there was a notable decrease in β-hexosaminidase over time in the neonatally treated group, both AAV9-HexB–treated adult and neonatal groups had significantly higher levels of activity than LacZ-treated controls at the end of the study (Figure 3a). The normal control group included only activities from LacZ-injected mice, and not mice injected with AAV9-HexB because their β-hexosaminidase activity was significantly increased above normal levels (see Supplementary Figure S2a,b). Increased β-hexosaminidase activity in the serum indicated successful and sustained transduction of noncentral nervous system tissues, especially the liver. The levels were particularly high in the adult-treated mice, but these may not reflect the rAAV9 transduction levels in the brain. Therefore, total β-hexosaminidase activity levels were also measured in lysates prepared from brains collected at the endpoint when mice were sacrificed. As expected, brain β-hexosaminidase activity in neonatal mice injected with AAV9-HexB was between 82 and 594 nm/µg/h of protein, significantly higher than the levels in AAV9-LacZ–injected SD mice (between 11 and 39 nm/µg/h, P < 0.01; Figure 3b). The levels of β-hexosaminidase activity in adult SD mice injected with AAV9-HexB did not differ significantly from those injected with AAV9-LacZ, indicating that the level of transduction in the brain was low and/or not sustained until the humane end point.
Figure 3.

Effect of AAV9-HexB injection on serum and brain β-hexosaminidase activity. Serum, or lysates prepared from a portion of the brain frozen immediately after sacrifice, was used for determination of β-hexosaminidase activity using the substrate, 4-MUG. (a) Serum enzyme activity. Columns represent the mean β-hexosaminidase activity per ml of serum ±SEM and include the values collected at all time points. (b) Brain enzyme activity. The columns represent mean brain β-hexosaminidase activity normalized to protein ± SEM. For both representation and statistical analyses of the enzyme activity in a and b, only the 10 normal animals injected as adults or neonates with AAV9-LacZ (n = 10) are shown in the control group to allow for comparison to normal enzyme levels. As expected, the β-hexosaminidase activity levels of adults and neonates injected with AAV9-HexB (n = 10) are significantly higher than normal (Supplementary Table S2), and therefore were excluded from the control group in this figure. However, the β-hexosaminidase levels for all control groups, and their statistical analyses are shown in Supplementary Figure S2b. Control (n = 10); SD-LacZ (n = 10); aSD-HexB (n = 5); nSD-HexB (n = 5). **P < 0.01, ***P < 0.0001, ****P < 0.00001.
Inflammation in the brains of AAV9-HexB–treated mice
Inflammation has been demonstrated to be an important mediator of neurodegeneration in SD mice,25 where increased numbers of activated microglia are associated with neural cell death.26 We qualitatively evaluated the level and activation of glial cells in the brains of AAV9-HexB– or -LacZ–injected SD and normal control mice using the macrophage marker F4/80. Activated glial cells, indicated by strong orange/brown staining in Figure 4 were abundant in AAV9-LacZ–injected SD mice compared to AAV-LacZ– or -HexB–injected control and AAV9-HexB–injected neonatal SD mice (Figure 4). However, in AAV9-HexB–injected adult SD mice, levels of activated glial cells were similar to that in the AAV9-LacZ–injected SD mice (Figure 4). Although neuroinflammation was still clearly evident in SD mice injected with AAV9-HexB as neonates, in general, decreased neuroinflammation was associated with decreased GM2 ganglioside storage.
Figure 4.

Activation of glial cells. Immunohistochemistry with the F4/80 antibody which detects activated microglial cells was used to examine sections from the brains of rAAV9-treated mice collected when they were sacrificed at the humane end point of 15–17 weeks (SD-LacZ), 17–35 weeks (aSD-HexB), or the experimental end point of 43 weeks for the neonatal (n) SD-HexB group. Representative images from each group of mice are presented; for the control group, as inflammation does increase with aging, representative images are from the oldest control group, mice treated neonatally with HexB (n = 5). Each panel represents a different section of the brain and the brown staining indicates activated glial cells (arrows). Original photos taken at 20×; scale bars represent 50 µm.
Copy number in the brain and liver of rAAV9-treated mice
The vg copies per diploid genome (dg) in mice treated as neonates or adults were determined in both the brain and liver by quantitative PCR. Given that the dose per kg was approximately sevenfold lower in adult-treated mice, we expected fewer cells to be transduced in adult brains. As shown in Figure 5a, the number of vg in the brain of adult-injected mice was variable, with a mean of 0.63 vg/dg, more than ninefold lower than the mean of 6 vg/dg in neonates. However, the opposite was found in the liver (Figure 5a), with a mean of 272 vg/dg in adult-injected mice, approximately tenfold higher than the mean of 22 vg/dg in neonatally injected mice. This was also reflected in the ratio of the brain to liver vg, which was ~1 in neonates, and although variable, 10- to 1,000-fold lower in most of the adults (Figure 5b). Although different doses of virus were introduced into adults and neonates, it appears that the liver is more readily transduced in adults.
Figure 5.

AAV9 vg copy number in the brains and livers of rAAV9-treated mice. PCR quantification of the AAV9 vg copy numbers were performed on DNA isolated from frozen liver or brain tissue lysates and calculated per diploid genome (dg). (a) Scatter plot of the vg copy number in liver and brain. Male (triangle) and female (circle) mice injected as adults (closed fill) and neonates (open fill) showed no significant difference in transduction efficiencies (Supplementary Table S2). All points are plotted (n = 20 for each group) and the horizontal line represents the mean, and the vertical line represents the SEM. **P < 0.01, ****P < 0.0001. (b) Scatter plot of the ratio of brain to liver vg in each animal (n = 20 per group). Symbols were used as defined in a above.
Tumor pathology in rAAV9-treated mice
At the 43 weeks experimental end point, gross examination during necropsy revealed tumors in 8 of 10 neonatally AAV9-HexB–injected SD and control mice; seven animals had liver tumors and one had multiple lung tumors. Tumors were not observed in the mice that reached a humane end point at 24 weeks or earlier, or in the single pair of adult mice that survived for 35 weeks, suggesting that these tumors had developed only in the neonatally injected mice as they aged. The pathology report based on hematoxylin and eosin stained paraffin sections from the liver tumors indicated benign hyperplasia, but did not rule out hepatocellular adenoma (Figure 6).
Figure 6.

Histological analysis of tumor tissues. Paraffin sections (five microns) prepared from tumors collected at 43 weeks when the animals were sacrificed, were stained with hematoxylin and eosin. The top panel is from normal liver and lung tissue and the bottom three panels are from liver and lung tumor tissue. (a) Images of normal liver and liver tumors from independent mice at 20× magnification. (b) Images of normal lung and lung tumor at 20× magnification. Scale bars represent 50 µm.
Tissue from the tumors, which contained numerous nodules as well as normal tissue, was also used for the analysis of insertion sites using inverse PCR and linker-ligation mediated PCR to amplify junctions between the AAV vectors and mouse cellular DNA using two unique barcodes per sample. Among the 579 unique integration sites we mapped, we found chromosome 2 to have the highest absolute number of integrations, while the Mir341 gene on chromosome 12 had the highest rate of insertion per kb of DNA (Supplementary Figure S3). Although no direct link with tumorigenesis can be made because the DNA was from a mixture of tumor and nontumor tissues, insertions in the previously identified hepatocellular carcinoma site in Rian27 were present in two mice injected neonatally with AAV9-HexB, one SD and one control. Furthermore, a unique insertion site in the growth factor receptor gene, FGFR2, a known cause of lung cancer,28 was found in the sample that exhibited lung tumors.
Discussion
Our studies show that a single intravenous dose of rAAV9-HexB to neonatal SD mice can provide enough β-hexosaminidase to the brain to reduce/prevent GM2 ganglioside storage and inflammation, correct motor function, and prolong survival. Treatment of adult mice, however, was not nearly as effective, leaving ganglioside storage similar to that of mice treated with the control vector, AAV9-LacZ, and only minimal improvement in motor function or survival.
The efficiency of rAAV9 transduction in adults and neonates cannot be directly compared in this study because the dose of vg/kg was lower in the adult mice. However, it is clear that AAV9 crossed the BBB and transduced neurons in both adults and neonates, as seen in previous studies.22,23 The increased number of vg in the brain of neonates compared to adult mice, and vice versa in the liver, suggests that the liver of adult mice is transduced much more effectively than that of neonates. However, there are several factors that need to be considered in the interpretation of these findings. First, the neonatal livers would have undergone substantial growth after AAV9 treatment, leading to a decrease in the vg/dg compared to adult-treated mice. In addition, the low levels of transduced brain cells in the adult mouse may be because the level of damage was already so severe that new enzyme activity could not rescue cell death. Nonetheless, it seems likely that the brain of neonates was more exposed to AAV9 than that of adults as the BBB is more permeable in neonates,29 and the injected volume was large. For example, it has been demonstrated previously that a single intravenous injection of a first generation AAV2 vector in neonatal animals resulted in therapeutic levels of lysosomal enzyme in the brain.30 However, because we used more rAAV9 in the treatment of neonates, it is impossible to be certain of the basis of the increased transduction.
Early treatment with AAV9-HexB appeared to be very important in correcting the storage of GM2 gangliosides and the subsequent impact on neuron death and neurological function. This is consistent with previous studies showing that GM2 ganglioside storage starts before birth. The importance of early treatment has been supported by recent studies showing that intracranial injection of rAAV is only effective when the therapy is delivered before symptoms become apparent or very early in the manifestations of the disease.18
The use of only the β-subunit for the gene therapy was very effective, and resulted in an increase in total β-hexosaminidase activity. This is consistent with a previous study where direct injection of the β-subunit into the brain of SD mice resulted in a therapeutic effect.16 In this study, the authors showed that only the β-subunit dimer HexB is formed, and thus it is likely that the therapeutic effect in our study is largely the result of Hex B which is able to degrade GA2 generated from GM2 by the sialidase bypass. Indeed, very little activity is detected in AAV9-Hexb–injected mice using the sulfated form of 4-MUG which is specific for α-subunit containing dimers (data not shown). Many gene therapy studies have used both α- and β-subunits of β-hexosaminidase together, and this is likely to result in higher levels of Hex A because the endogenous subunits may become limiting. This will be important in humans where the sialidase bypass does not function. Thus, the intravenous administration of a vector expressing both α- and β-subunits, or vectors expressing the two subunits independently, are likely needed to treat human GM2 gangliosidoses.
Inflammation is very important in the progression of neurodegeneration in all forms of GM2 gangliosidosis. The AAV9-HexB treatment clearly decreased inflammation, presumably by decreasing the levels of GM2 ganglioside storage. A recent study that introduced a deletion of tumor necrosis factor-α into SD mice, showed that decreasing inflammation greatly slowed the progression of the disease.31 This raises the possibility of combining gene therapy with agents targeting inflammation to provide therapies for patients with GM2 gangliosidoses.
The detection of tumors in the neonatally treated mice was disappointing, but not surprising as our neonatal dose of rAAV9 was high and similar findings have been reported in mice treated neonatally for β-glucuronidase32 and as 9- to 11-week-old adults for ornithine transcarbamylase deficiencies.33,34 In both cases, there was an increased occurrence of hepatocellular carcinoma in mice that survived for more than 13 months. This was attributed to insertional mutagenesis by the AAV vectors which integrated, in some of the tumors, within a 6-kb window on chromosome 12, near the Rian and Mirg genes.27,35 Studies have shown that in mice, integration of the provirus in this region leads to increased expression of surrounding genes, resulting in hepatocellular carcinoma.27 Two of our mice did show integrations in this same region. Our results are difficult to interpret however, as multiple nodules surrounded by normal tissue were used in the analysis of the insertion sites. Although higher rates of hepatocellular carcinoma or insertional activation of oncogenes have been variably detected in adult mice injected with high doses of rAAV,36,37 neonates would be the target of gene therapy for the GM2 gangliosidoses. Although we did detect multiple nodules/tumors, the histopathological report diagnosed them as nodular hyperplasia. However, the endpoint of our study was only 10 months, so it is possible that these benign nodules might have turned malignant in the future had we continued our study to a later time point. These findings emphasize the importance of continued awareness in ongoing gene therapy studies using AAV, as had been suggested previously.38
This positive outcome with just a single intravenous dose of AAV9-HexB provides hope for less invasive therapies for the GM2 gangliosidoses than direct injection in the brain. Systemic delivery will also benefit the peripheral tissues affected by lysosomal storage of GM2 ganglioside and other glycoconjugates, which become prominent when animals live longer because the neurological disease is treated. The recent development of a novel hybrid protein that combines the activities of the α- and β-subunits of β-hexosaminidase sets the stage for the construction of a single rAAV vector expressing an enzyme capable of GM2 ganglioside degradation.39 Taken together, the potential for easily delivered therapies for the human GM2 gangliosidoses is very promising.
Materials and Methods
Experimental animals. A SD mouse model on a C57BL/6:C129 background has been previously described11 and was a generous gift of Dr Roy Gravel (University of Calgary, Calgary, Alberta, Canada). This mouse model does not make β-hexosaminidase A or B because a neomycin resistance expression cassette is inserted into exon 2 of Hexb. All experimental animals were derived from heterozygous intercrosses. The Hexb genotype was determined by PCR of DNA isolated from ear clippings using the primers and experimental conditions described previously.11
Mice were euthanized when they reached a humane end point, indicated by significant paralysis or frequent seizure episodes or an experimental end point of 43 weeks. All procedures and care of the animals were in compliance with the Canadian Council on Animal Care and approved by the Animal Care Committee at the University of Manitoba.
Analysis of motor function. To assess motor function, mice were analyzed in an open field test. Motor function was assessed as distance travelled and number of head rotations by analysis of the videos using ANY-maze software. Representative videos of mice were taken for demonstration of motor activity and tail suspension.
Construction of rAAV2/9 viral vectors. The pAAV2.1 CMV-HexB vector, expressing the murine HEXB cDNA, was constructed by PCR-amplifying the HEXB cDNA from pβHex54 (ref. 40) using primers (sense 5′-gccggccgggagcagtcatgccgcag-3′; antisense 5′-cgggatcccagaatcaacatgatcatagctgg-3′) that incorporated EagI and BamHI restriction enzyme sites onto the 5′ and 3′ ends, respectively. This fragment was subcloned into pAAV2.1 CMV lacZ41 by removing the lacZ sequence with Eag1/BamHI and replacing it with the Hexb cDNA to create pAAV2.1 CMV-HexB. The sequence of the vector was confirmed by dideoxy sequencing at the Toronto Centre for Applied Genomics. AAV2/9 Viral particles expressing HEXB were constructed by triple cotransfection of HEK293 cells with pAAV2.1 CMV-HEXB, pAdΔF6, and pAAV/SP70 adenovirus derivative helper plasmid, followed by purification as described previously.41 The titers were determined by PCR amplification following previously described procedures.41 AAV2/9.CMV.LacZ.bGH was purchased from the University of Pennsylvania Vector Core Facility (Philadelphia, PA).
Intravenous injection of rAAV9. The neonatal SD (n = 10) and normal control (n = 10) mice received 2.5 × 1014 vg/kg of rAAV9 vector expressing either HexB (n = 5) or the control vector, LacZ (n = 5) at postnatal day 1 or 2 in a 100 µl volume through the superficial temporal vein. The adult group of SD (n = 10) or normal control (n = 10) mice received 3.5 × 1013 vg/kg of rAAV9 vector expressing either HexB (n = 5), or the control LacZ (n = 5) at 6 weeks of age in a 100 µl volume through the tail vein. Normal control mice were age-matched littermates that were either wild-type (Hexb+/+) or heterozygous (Hexb+/−) for the targeted Hexb gene (see experimental design in Supplementary Table S1).
Tissue and serum processing. Blood samples were collected from the saphenous vein at 10, 16, 28, and by cardiac puncture at 43 weeks, or at the humane end point if it preceded 43 weeks. Serum was collected from clotted blood samples and stored at −20 °C for future assays. Organs harvested from euthanized mice were divided in two parts—one portion was snap-frozen and stored at −20 °C, while the second was fixed overnight with 10% buffered formalin and processed for embedding. Before use, the frozen brain tissue was homogenized in phosphate-buffered saline at 10% weight per volume and stored at −20 °C to allow it to be used for multiple assays.
Determination of vg number. The vg copy number was determined from genomic DNA extracted from brain or liver tissue that was collected at death by quantitative PCR. Primers were designed to target the bGHpoly(A): BGH FW and BGH REV (5′-tctagttgccagccatctgttgt-3′ and 5′tgggagtggcaccttcca-3′, respectively). The probe was 5′-(6-FAM)tcccccgtgccttccttgacc-(BHQ1a-Q)-3′. Samples were run on a LightCycler 480 Instrument II (Roche, Mississauga, Ontario, Canada).
β-Hexosaminidase assay. Total β-hexosaminidase activity was determined in serum or brain lysates using 4-methylumbelliferyl-2-acetamido-2-deoxy-β-D-glucopyranoside (4-MUG) as a substrate, and following established procedures.42 For serum, the activity was determined per µl, while brain activities were calculated per µg of protein as determined using the Bradford assay with a kit supplied by ThermoFisher Scientific (Ottawa, Ontario, Canada).
Ganglioside analysis. Gangliosides were extracted from brain homogenates (300 mg of protein) with a chloroform-methanol (1:2) mixture and GM2 was phase partitioned using chloroform: methanol: ddH2O (1:2:1.4). The upper phase containing GM2 ganglioside was collected, dried, suspended in ddH2O and dialyzed against water using an 8,000 kDa cut-off membrane. After drying, the sample was dissolved in 100 µl of chloroform: methanol: ddH2O (60:30:4.4), and half was spotted alongside monosialoganglioside standards (Matreya LLC, Pleasant Gap, PA) on a HPTLC silica gel 60 plate with a concentrating zone (Millipore Canada, Etobicoke, Ontario, Canada), and separated using 55:45:10 chloroform: methanol: 0.2% CaCl2 as the mobile phase. The bands were visualized with resorcinol reagent and the plates were dried at 100 °C for 30 minutes. Densitometry to quantify the bands was performed with a BioRad ChemiDoc MP instrument and using Image Lab 4.1 software.
Histology and microscopy. For evaluation of cytoplasmic vacuolation, 1.5 micron paraffin-embedded brain sections were stained with toluidine blue. Hematoxylin and eosin staining was performed to evaluate overall morphology of tumor tissues. Immunohistochemical detection of microglia was performed on five micron brain sections with a 1/50 dilution of the mouse monoclonal anti-F4/80 (AbDSerotec, Cedarlane Laboratories, Burlington, Ontario, Canada). The primary antibody was detected with biotinylated goat anti-rat IgG (Invitrogen, ThermoFisher Scientific), followed by an avidin-linked horse-radish peroxidase and the 3,3′-diaminobenzidine substrate (Vector Laboratories (Canada), Burlington, Ontario, Canada). Slides were viewed with a Zeiss AxioCam A1 compound microscope equipped with a color AxioCamMRc camera, and photomicrographs were processed using AxioVision software.
Analysis of AAV9 insertion sites. The sequencing library was prepared as previously described, isolating AAV-containing fragments via linker-mediated PCR.43 The first round of PCR used the AAV ITR primer, 5′-GGAGTTGGCCACTCCCTCTCTG-3′ and the linker primer, 5′-GTAATACGACTCACTATAGGGCACGCGTG-3′ using cycle conditions of 95 °C for 2 minutes followed by 25 cycles of 95 °C for 15 seconds, 55 °C for 30 seconds and 72 °C for 1 minute. The resulting amplicons were diluted 1:50 and a second PCR was performed with AAV ITR nested primer, 5′-TCTCTGCGCGCTCGCTCG-3′ and nested linker primer, 5′-GCGTGGTCGACTGCGCAT-3′. Cycle conditions were 95 °C for 2 minutes followed by 20 cycles of 95 °C for 15 seconds, 58 °C for 30 seconds, and 72 °C for 1 minute. The sequence barcodes on the linkers were used here to both differentiate the AAV samples, and to multiplex the samples with unrelated samples. Each sample received two barcodes. Integration sites were identified by using AAV_GeIST, a modification of the GeIST program used for murine leukemia virus (MLV) detection.44 The steps that had trimmed the MLV long terminal repeat were each replaced with a step that trimmed the portion of the AAV ITR matching the ITR primer used in the second round of linker-mediated PCR, and a step that trimmed the remainder of the variable-length ITR. All versions of GeIST are designed to identify the junction at which viral DNA meets cellular genomic DNA. As such, the program requires that each amplicon contain both viral DNA and genomic DNA, thereby filtering out potential episomal contamination. AAV_GeIST used BamTools version 2.3.0, Cutadapt version 0.9.3, and Bowtie version 0.12.7, and aligned reads to assembly GRCm38.45,46,47 A minimum cutoff of 30 fragments per integration per barcode was employed as a means of filtering out spurious alignments. The 415 putative integrations that mapped within Hexb (chr13:97,176,332-97,198,357) were discarded, as these reads were more likely to represent amplification of the vector than actual integration events.48 The integrations were annotated using Ensembl genes 75, downloaded from BioMart.49,50 The DNA sequences used for these analyses have been deposited in GenBank as BioProject PRJNA257830.
Statistical analyses. All statistical analyses were performed using GraphPad V6 software. The log-rank (Mantel-Cox) test was used for the analysis of survival and a one-way ANOVA was used to analyze the increase in mice life span, motor activity and the severity of vacuolization in different regions of the brain. For all other analyses unpaired, one or two-tailed Student's t-tests were used to test significance. Significance was taken as P < 0.05.
SUPPLEMENTARY MATERIAL Figure S1. Head rotations. Figure S2. β-hexosaminidase activity levels in control groups. Figure S3. AAV9 integration sites. Table S1. Study design. Table S2. Statistical comparisons of control groups that are merged within figures. Video S1. Comparison of the activity of neonatally treated mice. Video S2. Comparison of the activity of adult-treated mice.
Acknowledgments
The authors thank Xin-Min Li for access to open-field testing equipment and ANY-maze software. This research was supported in part by the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health (SB), by the Public Health Agency of Canada (GK) and by grants from the Manitoba Institute of Child Health and The Manitoba Medical Service Foundation to JW/BTR. NA was supported by funding from the Saudi Arabian Cultural Bureau in Canada. The work was primarily done in Winnipeg, Manitoba, Canada. The authors declare no conflict of interest.
Supplementary Material
Comparison of the activity of neonatally treated mice.
Comparison of the activity of adult-treated mice.
References
- Sandhoff K, Andreae U, Jatzkewitz H. Deficient hexosaminidase activity in an exceptional case of Tay-Sachs disease with additional storage of kidney globoside in visceral organs. Pathol Eur. 1968;3:278–285. [PubMed] [Google Scholar]
- Gravel RA, Triggs-Raine BL, Mahuran DJ. Biochemistry and genetics of Tay-Sachs disease. Can J Neurol Sci. 1991;18 suppl. 3:419–423. doi: 10.1017/s0317167100032583. [DOI] [PubMed] [Google Scholar]
- Myrianthopoulos NC. Some Epidemiologic and Genetic Aspects of Tay Sachs Disease. Academic Press; New York; 1962. [Google Scholar]
- Andermann E, Scriver CR, Wolfe LS, Dansky L, Andermann F. Genetic variants of Tay-Sachs disease: Tay-Sachs disease and Sandhoff's disease in French Canadians, juvenile Tay-Sachs disease in Lebanese Canadians, and a Tay-Sachs screening program in the French-Canadian population. Prog Clin Biol Res. 1977;18:161–188. [PubMed] [Google Scholar]
- Lowden JA, Ives EJ, Keene DL, Burton AL, Skomorowski MA, Howard F. Carrier detection in Sandhoff disease. Am J Hum Genet. 1978;30:38–45. [PMC free article] [PubMed] [Google Scholar]
- Kaya N, Al-Owain M, Abudheim N, Al-Zahrani J, Colak D, Al-Sayed M.et al. (2011GM2 gangliosidosis in Saudi Arabia: multiple mutations and considerations for future carrier screening Am J Med Genet A 155A1281–1284. [DOI] [PubMed] [Google Scholar]
- Cantor RM, Roy C, Lim JS, Kaback MM. Sandhoff disease heterozygote detection: a component of population screening for Tay-Sachs disease carriers. II. Sandhoff disease gene frequencies in American Jewish and non-Jewish populations. Am J Hum Genet. 1987;41:16–26. [PMC free article] [PubMed] [Google Scholar]
- Kleiman FE, de Kremer RD, de Ramirez AO, Gravel RA, Argaraña CE. Sandhoff disease in Argentina: high frequency of a splice site mutation in the HEXB gene and correlation between enzyme and DNA-based tests for heterozygote detection. Hum Genet. 1994;94:279–282. doi: 10.1007/BF00208283. [DOI] [PubMed] [Google Scholar]
- Conzelmann E, Sandhoff K. Partial enzyme deficiencies: residual activities and the development of neurological disorders. Dev Neurosci. 1983;6:58–71. doi: 10.1159/000112332. [DOI] [PubMed] [Google Scholar]
- Miklyaeva EI, Dong W, Bureau A, Fattahie R, Xu Y, Su M.et al. (2004Late onset Tay-Sachs disease in mice with targeted disruption of the Hexa gene: behavioral changes and pathology of the central nervous system Brain Res 100137–50. [DOI] [PubMed] [Google Scholar]
- Phaneuf D, Wakamatsu N, Huang JQ, Borowski A, Peterson AC, Fortunato SR.et al. (1996Dramatically different phenotypes in mouse models of human Tay-Sachs and Sandhoff diseases Hum Mol Genet 51–14. [DOI] [PubMed] [Google Scholar]
- Sango K, Yamanaka S, Hoffmann A, Okuda Y, Grinberg A, Westphal H.et al. (1995Mouse models of Tay-Sachs and Sandhoff diseases differ in neurologic phenotype and ganglioside metabolism Nat Genet 11170–176. [DOI] [PubMed] [Google Scholar]
- Gulinello M, Chen F, Dobrenis K. Early deficits in motor coordination and cognitive dysfunction in a mouse model of the neurodegenerative lysosomal storage disorder, Sandhoff disease. Behav Brain Res. 2008;193:315–319. doi: 10.1016/j.bbr.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogerbrugge PM, Brouwer OF, Bordigoni P, Ringden O, Kapaun P, Ortega JJ.et al. (1995Allogeneic bone marrow transplantation for lysosomal storage diseases. The European Group for Bone Marrow Transplantation Lancet 3451398–1402. [DOI] [PubMed] [Google Scholar]
- Tsuji D, Akeboshi H, Matsuoka K, Yasuoka H, Miyasaki E, Kasahara Y.et al. (2011Highly phosphomannosylated enzyme replacement therapy for GM2 gangliosidosis Ann Neurol 69691–701. [DOI] [PubMed] [Google Scholar]
- Cachón-González MB, Wang SZ, Lynch A, Ziegler R, Cheng SH, Cox TM. Effective gene therapy in an authentic model of Tay-Sachs-related diseases. Proc Natl Acad Sci USA. 2006;103:10373–10378. doi: 10.1073/pnas.0603765103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cachón-González MB, Wang SZ, McNair R, Bradley J, Lunn D, Ziegler R.et al. (2012Gene transfer corrects acute GM2 gangliosidosis–potential therapeutic contribution of perivascular enzyme flow Mol Ther 201489–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cachón-González MB, Wang SZ, Ziegler R, Cheng SH, Cox TM. Reversibility of neuropathology in Tay-Sachs-related diseases. Hum Mol Genet. 2014;23:730–748. doi: 10.1093/hmg/ddt459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke JT, Mahuran DJ, Sathe S, Kolodny EH, Rigat BA, Raiman JA.et al. (2011An open-label Phase I/II clinical trial of pyrimethamine for the treatment of patients affected with chronic GM2 gangliosidosis (Tay-Sachs or Sandhoff variants) Mol Genet Metab 1026–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashe KM, Bangari D, Li L, Cabrera-Salazar MA, Bercury SD, Nietupski JB.et al. (2011Iminosugar-based inhibitors of glucosylceramide synthase increase brain glycosphingolipids and survival in a mouse model of Sandhoff disease PLoS ONE 6e21758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Büning H, Perabo L, Coutelle O, Quadt-Humme S, Hallek M. Recent developments in adeno-associated virus vector technology. J Gene Med. 2008;10:717–733. doi: 10.1002/jgm.1205. [DOI] [PubMed] [Google Scholar]
- Gray SJ, Matagne V, Bachaboina L, Yadav S, Ojeda SR, Samulski RJ. Preclinical differences of intravascular AAV9 delivery to neurons and glia: a comparative study of adult mice and nonhuman primates. Mol Ther. 2011;19:1058–1069. doi: 10.1038/mt.2011.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM, Kaspar BK. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol. 2009;27:59–65. doi: 10.1038/nbt.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzo A, Marcó S, García M, Villacampa P, Ribera A, Ayuso E.et al. (2012Correction of pathological accumulation of glycosaminoglycans in central nervous system and peripheral tissues of MPSIIIA mice through systemic AAV9 gene transfer Hum Gene Ther 231237–1246. [DOI] [PubMed] [Google Scholar]
- Wu YP, Proia RL. Deletion of macrophage-inflammatory protein 1 alpha retards neurodegeneration in Sandhoff disease mice. Proc Natl Acad Sci USA. 2004;101:8425–8430. doi: 10.1073/pnas.0400625101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JQ, Trasler JM, Igdoura S, Michaud J, Hanal N, Gravel RA. Apoptotic cell death in mouse models of GM2 gangliosidosis and observations on human Tay-Sachs and Sandhoff diseases. Hum Mol Genet. 1997;6:1879–1885. doi: 10.1093/hmg/6.11.1879. [DOI] [PubMed] [Google Scholar]
- Wang PR, Xu M, Toffanin S, Li Y, Llovet JM, Russell DW. Induction of hepatocellular carcinoma by in vivo gene targeting. Proc Natl Acad Sci USA. 2012;109:11264–11269. doi: 10.1073/pnas.1117032109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh M. Cancer genomics and genetics of FGFR2 (Review) Int J Oncol. 2008;33:233–237. [PubMed] [Google Scholar]
- Moos T, Møllgård K. Cerebrovascular permeability to azo dyes and plasma proteins in rodents of different ages. Neuropathol Appl Neurobiol. 1993;19:120–127. doi: 10.1111/j.1365-2990.1993.tb00416.x. [DOI] [PubMed] [Google Scholar]
- Daly TM, Vogler C, Levy B, Haskins ME, Sands MS. Neonatal gene transfer leads to widespread correction of pathology in a murine model of lysosomal storage disease. Proc Natl Acad Sci USA. 1999;96:2296–2300. doi: 10.1073/pnas.96.5.2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abo-Ouf H, Hooper AW, White EJ, van Rensburg HJ, Trigatti BL, Igdoura SA. Deletion of tumor necrosis factor-a ameliorates neurodegeneration in Sandhoff disease mice. Hum Mol Genet. 2013;22:3960–3975. doi: 10.1093/hmg/ddt250. [DOI] [PubMed] [Google Scholar]
- Donsante A, Vogler C, Muzyczka N, Crawford JM, Barker J, Flotte T.et al. (2001Observed incidence of tumorigenesis in long-term rodent studies of rAAV vectors Gene Ther 81343–1346. [DOI] [PubMed] [Google Scholar]
- Zhong L, Malani N, Li M, Brady T, Xie J, Bell P.et al. (2013Recombinant adeno-associated virus integration sites in murine liver after ornithine transcarbamylase gene correction Hum Gene Ther 24520–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell P, Moscioni AD, McCarter RJ, Wu D, Gao G, Hoang A.et al. (2006Analysis of tumors arising in male B6C3F1 mice with and without AAV vector delivery to liver Mol Ther 1434–44. [DOI] [PubMed] [Google Scholar]
- Donsante A, Miller DG, Li Y, Vogler C, Brunt EM, Russell DW.et al. (2007AAV vector integration sites in mouse hepatocellular carcinoma Science 317477. [DOI] [PubMed] [Google Scholar]
- Li H, Malani N, Hamilton SR, Schlachterman A, Bussadori G, Edmonson SE.et al. (2011Assessing the potential for AAV vector genotoxicity in a murine model Blood 1173311–3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao CY, Yang SJ, Tao MH, Jeng YM, Yu IS, Lin SW. Incorporation of the factor IX Padua mutation into FIX-Triple improves clotting activity in vitro and in vivo. Thromb Haemost. 2013;110:244–256. doi: 10.1160/TH13-02-0154. [DOI] [PubMed] [Google Scholar]
- Russell DW. AAV vectors, insertional mutagenesis, and cancer. Mol Ther. 2007;15:1740–1743. doi: 10.1038/sj.mt.6300299. [DOI] [PubMed] [Google Scholar]
- Sinici I, Yonekawa S, Tkachyova I, Gray SJ, Samulski RJ, Wakarchuk W.et al. (2013In cellulo examination of a beta-alpha hybrid construct of beta-hexosaminidase A subunits, reported to interact with the GM2 activator protein and hydrolyze GM2 ganglioside PLoS ONE 8e57908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bapat B, Ethier M, Neote K, Mahuran D, Gravel RA. Cloning and sequence analysis of a cDNA encoding the beta-subunit of mouse beta-hexosaminidase. FEBS Lett. 1988;237:191–195. doi: 10.1016/0014-5793(88)80199-6. [DOI] [PubMed] [Google Scholar]
- Bello A, Tran K, Chand A, Doria M, Allocca M, Hildinger M.et al. (2009Isolation and evaluation of novel adeno-associated virus sequences from porcine tissues Gene Ther 161320–1328. [DOI] [PubMed] [Google Scholar]
- Kaback MM, Shapiro LJ, Hirsch P, Roy C. Tay-Sachs disease heterozygote detection: a quality control study. Prog Clin Biol Res. 1977;18:267–279. [PubMed] [Google Scholar]
- Varshney GK, Lu J, Gildea DE, Huang H, Pei W, Yang Z.et al. (2013A large-scale zebrafish gene knockout resource for the genome-wide study of gene function Genome Res 23727–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFave MC, Varshney GK, Gildea DE, Wolfsberg TG, Baxevanis AD, Burgess SM. MLV integration site selection is driven by strong enhancers and active promoters. Nucleic Acids Res. 2014;42:4257–4269. doi: 10.1093/nar/gkt1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett DW, Garrison EK, Quinlan AR, Strömberg MP, Marth GT. BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics. 2011;27:1691–1692. doi: 10.1093/bioinformatics/btr174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10–12. [Google Scholar]
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flicek P, Amode MR, Barrell D, Beal K, Billis K, Brent S.et al. (2014Ensembl 2014 Nucleic Acids Res 42Database issueD749–D755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsella RJ, Kähäri A, Haider S, Zamora J, Proctor G, Spudich G.et al. (2011Ensembl BioMarts: a hub for data retrieval across taxonomic space Database (Oxford) 2011bar030. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Comparison of the activity of neonatally treated mice.
Comparison of the activity of adult-treated mice.