

Key Points
Treatment with alexidine dihydrochloride, a Ptpmt1 inhibitor, reprograms cellular metabolism and preserves long-term stem cells ex vivo.
Inhibition of mitochondrial metabolism by metformin also decreases differentiation and helps maintain stem cells in culture.
Abstract
The difficulty in maintaining the reconstituting capabilities of hematopoietic stem cells (HSCs) in culture outside of the bone marrow microenvironment has severely limited their utilization for clinical therapy. This hurdle is largely due to the differentiation of long-term stem cells. Emerging evidence suggests that energy metabolism plays an important role in coordinating HSC self-renewal and differentiation. Here, we show that treatment with alexidine dihydrochloride, an antibiotic and a selective inhibitor of the mitochondrial phosphatase Ptpmt1, which is crucial for the differentiation of HSCs, reprogrammed cellular metabolism from mitochondrial aerobic metabolism to glycolysis, resulting in a remarkable preservation of long-term HSCs ex vivo in part through hyperactivation of adenosine 5′-monophosphate–activated protein kinase (AMPK). In addition, inhibition of mitochondrial metabolism and activation of AMPK by metformin, a diabetes drug, also decreased differentiation and helped maintain stem cells in culture. Thus, manipulating metabolic pathways represents an effective new strategy for ex vivo maintenance of HSCs.
Introduction
Despite the success of hematopoietic stem cell (HSC) transplantation therapy in controlling hematopoietic malignancies and other blood disorders, the difficulty in maintaining functional long-term stem cells in culture outside of the bone marrow (BM) microenvironment has impeded our ability to safely and effectively transplant HSCs in certain clinical contexts. As differentiation is favored over expansion under most culture conditions, approaches that can maintain limited functional stem cells and block differentiation are of key importance for stem cell–based therapy. Insight into the coordination of energy metabolism with HSC self-renewal and differentiation has emerged.1-3 Distinct from differentiated progenitors and mature blood cells, HSCs use glycolysis instead of mitochondrial oxidative phosphorylation for energy production.1,4,5 However, they need to switch to mitochondrial metabolism to meet rapidly increasing energy demands for differentiation.6,7 This metabolic requirement provides the possibility that forcing HSCs to use glycolysis or preventing the differentiation-associated switch to mitochondrial metabolism could block differentiation, thereby facilitating HSC maintenance and expansion. We have recently shown that Ptpmt1, a mitochondrial Pten-like phosphatase,8 plays a crucial role in embryonic stem (ES) cells9 and HSCs.7 Ptpmt1 depletion blocks differentiation in ES cells and HSCs without affecting cell survival.7,9 Inspired by these findings and given that a known antibiotic, alexidine dihydrochloride (AD), has been identified as a selective and potent Ptpmt1 inhibitor,10 we investigated whether HSCs could be better maintained/expanded ex vivo by pharmacologic inhibition of Ptpmt1.
Study design
Competitive repopulation assay
Lin−Sca-1+c-Kit+ (LSK) cells (CD45.2+) cultured in the presence of AD or vehicle for 7 days were harvested (5 × 104), mixed with freshly isolated CD45.1+ BM cells (1 × 105), and then transplanted into lethally irradiated (11 Gy) BoyJ (CD45.1+) recipients. Donor cell reconstitution (CD45.2+) was determined at 4, 8, 12, 16, and 20 weeks after transplant. For secondary transplant, BM cells harvested (1 × 106) from primary recipients 20 weeks after primary transplant were transplanted into secondary recipients. These animals were euthanized 16 weeks after transplant, and reconstitution of donor cells was analyzed.
Oxygen consumption and extracellular flux measurement
Oxygen consumption rate and extracellular acidification rates were measured using a metabolic flux analyzer (Seahorse Bioscience, North Billerica, MA) under basal conditions and in the presence of the mitochondrial inhibitor oligomycin (1 μM), the uncoupling compound carbonylcyanide-4-(trifluoromethoxy)phenylhydrazone (1 μM), and the respiratory chain inhibitor rotenone (1 μM).
Results and discussion
We first determined the specificity of AD, a reported inhibitor of Ptpmt1.10 Treatment with this compound decreased proliferation and differentiation in wild-type ES cells (supplemental Figure 1, available on the Blood Web site), recapitulating the phenotypes of Ptpmt1 knockout ES cells.9 However, these effects of the compound were barely detectable in Ptpmt1-deleted cells, verifying the specificity of this inhibitor. To determine whether HSCs could be better maintained ex vivo by pharmacologic inhibition of Ptpmt1, mouse lineage negative (Lin−) cells containing HSCs were cultured for 5 days in serum-free StemSpan medium supplemented with Tpo, Flt3L, and stem cell factor, which is helpful for maintaining stem cells, with or without AD, and then transplanted into lethally irradiated mice (3 × 104 cells per mouse). Ninety percent of the mice receiving vehicle-treated cells died due to hematopoietic failure. In contrast, all of the animals transplanted with AD-treated cells survived (Figure 1A). Furthermore, the frequency of competitive repopulating units (CRUs) in AD-treated cultures determined by limiting dilution and competitive repopulation assays was increased by approximately threefold compared to that of fresh Lin− cells (Figure 1B), suggesting a moderate expansion of functional stem cells.
Figure 1.
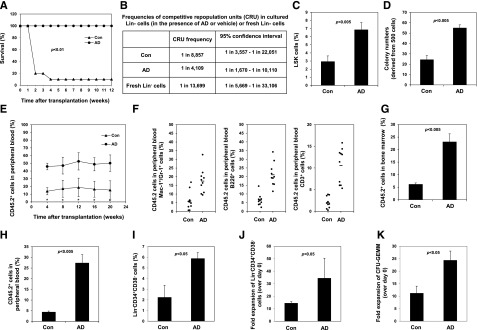
Inhibition of Ptpmt1 maintains long-term HSCs. (A) Lin− cells were cultured for 5 days in StemSpan medium supplemented with Tpo (50 ng/mL), Flt3L (100 ng/mL), and stem cell factor (100 ng/mL) in the presence of AD (200 nM) or vehicle (Con). Cells were collected and transplanted into lethally irradiated isogenic mice (3 × 104 cells per mouse). Survival curves of recipient animals were determined (n = 10 mice/group). (B) Lin− cells (CD45.2+) as cultured in panel A or freshly isolated Lin− cells (2 × 103, 1 × 104, and 5 × 104) were mixed with 1 × 106 fresh BM cells (CD45.1+) and transplanted into CD45.1+ recipients (4-5 mice per group). CRUs in test cell populations were determined 16 weeks after transplant. Recipient mice that had more than 1% contribution from test donor cells in peripheral blood were considered to have test cell reconstitution. (C-H) Sorted LSK cells were cultured as in panel A. The percentage of LSK cells was analyzed by FACS 7 days later (C), and cultured cells were harvested, washed with phosphate-buffered saline, and assessed by the colony-forming unit (CFU) assay (D); data are presented as mean ± standard error of the mean from 3 independent experiments. (E) Overall reconstituting capabilities of the cultured cells were determined by the competitive repopulation assay as described in “Methods” (n = 10 mice per group); *P < .05. CD45.2+ donor–derived myeloid (Mac-1+/Gr-1+), B-lymphoid (B220+), and T-lymphoid (CD3+) populations in the peripheral blood (F) and CD45.2+ donor–derived cells in the BM (G) of recipient animals were assessed by FACS 20 weeks after transplant. (H) BM cells harvested from primary transplants 20 weeks after transplant were transplanted into lethally irradiated secondary recipients (n = 5 mice per group). CD45.2+ donor cell reconstitution in peripheral blood was determined by FACS. (I-K) Human apheresis cells were cultured in the presence of AD (250 nM) or vehicle as described in panel A (except that human stem cell factor was used). The percentage (I) and absolute number (J) of Lin−CD34+CD38− cells were quantified by FACS. (K) CFUs of granulocytes, erythrocytes, macrophages, and megakaryocytes (CFU-GEMM) were determined by the CFU assay. Con, control vehicle; FACS, fluorescence-activated cell sorter.
We then cultured LSK cells (enriched for HSCs) with or without AD. Compared with controls, Lin− progenitors that were maintained during 7 days of culture in the presence of AD were markedly increased (supplemental Figure 2). Phenotypic (Figure 1C) and functional (Figure 1D) assays showed that LSK cells and colony forming units in AD-treated cultures were increased by two- to threefold relative to controls. Cultured cells (5 × 104) (CD45.2+) were also mixed with 1 × 105 BM cells (CD45.1+) and transplanted into CD45.1+ recipients. The overall and lineage reconstitution from AD-treated cultures was substantially increased compared to that of control cells in both primary (Figure 1E-G) and secondary (Figure 1H) transplants, strongly suggesting that pharmacologic inhibition of Ptpmt1 preserves functional long-term mouse stem cells. Remarkably, AD treatment of ex vivo cultured human apheresis cells also significantly increased the percentage of phenotypic stem cells (Lin−CD34+CD38−) (Figure 1I). Moreover, ex vivo expansion efficiencies of phenotypic stem cells (Figure 1J) and functional multipotential progenitors (colony forming units of granulocyte, erythrocyte, macrophage, and megakaryocyte) (Figure 1K) under these culture conditions were increased to nearly 35- and 25-fold, respectively, by additional AD treatment.
To further validate the effect of the Ptpmt1 inhibitor, we treated wild-type mice with AD or vehicle for 2 weeks. Both the percentage and absolute number of BM phenotypic HSCs (Lin−Sca-1+c-Kit+ CD150+CD41−CD48-)11 were increased compared to controls (supplemental Figure 3A-B), although the treatment lasted only 2 weeks and the achieved plasma levels of the drug were unknown. Competitive repopulation assays demonstrated that the repopulation capabilities of BM cells from AD-treated mice were greatly increased (supplemental Figure 3C-D). CRU assessment showed that the frequency of CRUs in AD-treated mice was nearly doubled, confirming that functional stem cells were expanded in these animals (supplemental Figure 3E).
To understand the mechanisms of the stem cell protection provided by AD, we analyzed the cell cycle. The percentage of AD-treated LSK cells at the G1 phase was decreased, whereas the percentages of S/G2/M cells were not changed (data not shown). More importantly, the percentage of AD-treated cells at the G0 phase was increased by more than threefold (Figure 2A), indicating that inhibition of Ptpmt1 helps maintain stem/progenitor cells in quiescence in culture, which is known to be critical for protecting stem cells from exhaustion under various stresses.12,13 In agreement with the cell cycle changes, p21 and p57 (cyclin-dependent kinase inhibitors controlling HSC cycling)14-16 were upregulated in the presence of AD (Figure 2B). We next sought to define the mechanisms underlying the cell cycle changes. AD-treated cells had much lower basal oxygen consumption and maximal oxidative capacity compared to control cells (Figure 2C). Measurement of extracellular proton flux revealed greatly increased extracellular acidification rates in AD-treated cells (Figure 2D), indicative of enhanced glycolysis. Intracellular levels of pyruvate, the key metabolite at the intersection of cytosolic glycolysis and mitochondrial aerobic metabolism, were elevated in AD-treated cells (Figure 2E), implying that mitochondrial utilization of this major substrate is decreased. The activity of lactate dehydrogenase, the enzyme responsible for converting pyruvate to lactate in the cytosol, was also increased (Figure 2F). Adenosine 5′-monophosphate–activated protein kinase (AMPK), an intracellular energy sensor linking metabolic stress to changes in the cell cycle,17-19 was highly activated in AD-treated LSK cells (Figure 2G). Acetyl-CoA carboxylase, one of the targets of AMPK and a negative regulator of fatty acid oxidation20 was inhibited, as evidenced by the increase in the inhibitory phosphorylation of this enzyme (Figure 2G), indicating that fatty acid oxidation is enhanced in AD-treated cells. Because glycolysis and fatty acid metabolism are required for the quiescent state of HSCs,1,3 the Ptpmt1 inhibitor appears to maintain stem cell quiescence and prevent them from differentiation by reprograming cellular metabolism.
Figure 2.

Inhibition of Ptpmt1 maintains stem cell quiescence by reprogramming cellular metabolism and activation of AMPK. (A) Sorted LSK cells were cultured as described in Figure 1C. After 7 days, cells were collected, fixed, and stained with fluorescein isothiocyanate –labeled anti-Ki67 antibody and propidium iodide (PI). Percentage of Ki67-negative cells (quiescent cells) was quantified by FACS. (B) Messenger (m)RNA levels of p21, p57, and p53 in cultured LSK cells were determined by quantitative reverse-transcription polymerase chain reaction. (C-D) Sorted LSK cells were treated with AD or vehicle as in panel A. Oxygen consumption rate (OCR) (C) and extracellular acidification rates (ECAR) (D) were measured. Experiments were repeated 3 times, and similar results were obtained in each. Representative results from 1 experiment are shown; *P < .05. (E) Lin− cells were cultured in the presence of AD (250 nM) or vehicle for 24 hours. Intracellular pyruvate concentration was determined using the pyruvate assay kit (Cayman Chemical Company, Ann Arbor, MI). (F) Intracellular lactate dehydrogenase (LDH) activity was determined using the LDH assay kit (Cayman Chemical Company). (G) Sorted LSK cells were treated with AD or vehicle as in panel A. Whole-cell lysates were prepared and examined by western blot analysis with anti-phospho-AMPK (Thr172) and anti-phospho-ACC (Ser79) antibodies. Blots were stripped and reprobed with anti-AMPK and anti-ACC antibodies. Relative phospho- (p-)AMPK and p-ACC levels normalized against pan protein levels are shown. Representative results from 3 independent experiments are shown. ACC, acetyl-CoA carboxylase.
To further verify that hyperactivation of AMPK is important for the function of AD, we treated LSK cells with compound C, an inhibitor of AMPK,21 in combination with AD. The stem cell protective effect of AD was abolished by compound C (supplemental Figure 4). Conversely, treatment with the mitochondrial respiratory chain (complex I) inhibitor metformin, which activates AMPK,22 better preserved LSK cells in culture than the vehicle (supplemental Figure 5A-B), providing additional support for the role of hyperactivated AMPK in maintaining stem cells ex vivo, although depletion of this kinase has minimal effects on HSCs in mice.23-25 Assessment of cellular metabolism showed that metformin indeed significantly decreased mitochondrial aerobic metabolism and caused a marked increase in glycolysis (supplemental Figure 5C-D). Taken together, this study provides proof-of-concept for a novel strategy; that is, reprograming cellular metabolism to preserve functional HSCs ex vivo, and suggests that AD and metformin may be used in combination with other agents to achieve better maintenance and expansion of stem cells in ex vivo procedures.
Acknowledgments
This work was supported by the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases grant DK092722 and National Cancer Institute grant CA181754 (C.-K.Q.).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: X.L., H.Z., and W.-M.Y. conducted the research and summarized the data; T.M.C. and K.D.B. discussed the work and edited the manuscript; C.-K.Q. designed the experiments and provided technical training to the first 3 authors; and X.L. and C.-K.Q. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Cheng-Kui Qu, Division of Hematology/Oncology/Bone Marrow Transplant, Department of Pediatrics, Aflac Cancer and Blood Disorders Center, Emory University School of Medicine, 1760 Haygood Dr NE, HSRB E302, Atlanta, GA 30322; e-mail: cheng-kui.qu@emory.edu.
References
- 1.Takubo K, Nagamatsu G, Kobayashi CI, et al. Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell. 2013;12(1):49–61. doi: 10.1016/j.stem.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kocabas F, Zheng J, Thet S, et al. Meis1 regulates the metabolic phenotype and oxidant defense of hematopoietic stem cells. Blood. 2012;120(25):4963–4972. doi: 10.1182/blood-2012-05-432260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ito K, Carracedo A, Weiss D, et al. A PML–PPAR-δ pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat Med. 2012;18(9):1350–1358. doi: 10.1038/nm.2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Simsek T, Kocabas F, Zheng J, et al. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell. 2010;7(3):380–390. doi: 10.1016/j.stem.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Suda T, Takubo K, Semenza GL. Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell. 2011;9(4):298–310. doi: 10.1016/j.stem.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 6.Baumann K. Stem cells: A metabolic switch. Nat Rev Mol Cell Biol. 2013;14(2):64–65. doi: 10.1038/nrm3515. [DOI] [PubMed] [Google Scholar]
- 7.Yu WM, Liu X, Shen J, et al. Metabolic regulation by the mitochondrial phosphatase PTPMT1 is required for hematopoietic stem cell differentiation. Cell Stem Cell. 2013;12(1):62–74. doi: 10.1016/j.stem.2012.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pagliarini DJ, Wiley SE, Kimple ME, et al. Involvement of a mitochondrial phosphatase in the regulation of ATP production and insulin secretion in pancreatic beta cells. Mol Cell. 2005;19(2):197–207. doi: 10.1016/j.molcel.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 9.Shen J, Liu X, Yu WM, et al. A critical role of mitochondrial phosphatase Ptpmt1 in embryogenesis reveals a mitochondrial metabolic stress-induced differentiation checkpoint in embryonic stem cells. Mol Cell Biol. 2011;31(24):4902–4916. doi: 10.1128/MCB.05629-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Doughty-Shenton D, Joseph JD, Zhang J, et al. Pharmacological targeting of the mitochondrial phosphatase PTPMT1. J Pharmacol Exp Ther. 2010;333(2):584–592. doi: 10.1124/jpet.109.163329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121(7):1109–1121. doi: 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 12.Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature. 2014;505(7483):327–334. doi: 10.1038/nature12984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ito K, Suda T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat Rev Mol Cell Biol. 2014;15(4):243–256. doi: 10.1038/nrm3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng T, Rodrigues N, Shen H, et al. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science. 2000;287(5459):1804–1808. doi: 10.1126/science.287.5459.1804. [DOI] [PubMed] [Google Scholar]
- 15.Matsumoto A, Takeishi S, Kanie T, et al. p57 is required for quiescence and maintenance of adult hematopoietic stem cells. Cell Stem Cell. 2011;9(3):262–271. doi: 10.1016/j.stem.2011.06.014. [DOI] [PubMed] [Google Scholar]
- 16.Zou P, Yoshihara H, Hosokawa K, et al. p57(Kip2) and p27(Kip1) cooperate to maintain hematopoietic stem cell quiescence through interactions with Hsc70. Cell Stem Cell. 2011;9(3):247–261. doi: 10.1016/j.stem.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 17.Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13(4):251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Imamura K, Ogura T, Kishimoto A, Kaminishi M, Esumi H. Cell cycle regulation via p53 phosphorylation by a 5′-AMP activated protein kinase activator, 5-aminoimidazole- 4-carboxamide-1-beta-D-ribofuranoside, in a human hepatocellular carcinoma cell line. Biochem Biophys Res Commun. 2001;287(2):562–567. doi: 10.1006/bbrc.2001.5627. [DOI] [PubMed] [Google Scholar]
- 19.Jones RG, Plas DR, Kubek S, et al. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005;18(3):283–293. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 20.Viollet B, Guigas B, Leclerc J, et al. AMP-activated protein kinase in the regulation of hepatic energy metabolism: from physiology to therapeutic perspectives. Acta Physiol (Oxf) 2009;196(1):81–98. doi: 10.1111/j.1748-1716.2009.01970.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bain J, Plater L, Elliott M, et al. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408(3):297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stephenne X, Foretz M, Taleux N, et al. Metformin activates AMP-activated protein kinase in primary human hepatocytes by decreasing cellular energy status. Diabetologia. 2011;54(12):3101–3110. doi: 10.1007/s00125-011-2311-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakada D, Saunders TL, Morrison SJ. Lkb1 regulates cell cycle and energy metabolism in haematopoietic stem cells. Nature. 2010;468(7324):653–658. doi: 10.1038/nature09571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gan B, Hu J, Jiang S, et al. Lkb1 regulates quiescence and metabolic homeostasis of haematopoietic stem cells. Nature. 2010;468(7324):701–704. doi: 10.1038/nature09595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gurumurthy S, Xie SZ, Alagesan B, et al. The Lkb1 metabolic sensor maintains haematopoietic stem cell survival. Nature. 2010;468(7324):659–663. doi: 10.1038/nature09572. [DOI] [PMC free article] [PubMed] [Google Scholar]