

Abstract
Objective:
To delineate the phenotype of early childhood epileptic encephalopathy due to de novo mutations of CHD2, which encodes the chromodomain helicase DNA binding protein 2.
Methods:
We analyzed the medical history, MRI, and video-EEG recordings of 9 individuals with de novo CHD2 mutations and one with a de novo 15q26 deletion encompassing CHD2.
Results:
Seizures began at a mean of 26 months (12–42) with myoclonic seizures in all 10 cases. Seven exhibited exquisite clinical photosensitivity; 6 self-induced with the television. Absence seizures occurred in 9 patients including typical (4), atypical (2), and absence seizures with eyelid myoclonias (4). Generalized tonic-clonic seizures occurred in 9 of 10 cases with a mean onset of 5.8 years. Convulsive and nonconvulsive status epilepticus were later features (6/10, mean onset 9 years). Tonic (40%) and atonic (30%) seizures also occurred. In 3 cases, an unusual seizure type, the atonic-myoclonic-absence was captured on video. A phenotypic spectrum was identified with 7 cases having moderate to severe intellectual disability and refractory seizures including tonic attacks. Their mean age at onset was 23 months. Three cases had a later age at onset (34 months) with relative preservation of intellect and an initial response to antiepileptic medication.
Conclusion:
The phenotypic spectrum of CHD2 encephalopathy has distinctive features of myoclonic epilepsy, marked clinical photosensitivity, atonic-myoclonic-absence, and intellectual disability ranging from mild to severe. Recognition of this genetic entity will permit earlier diagnosis and enable the development of targeted therapies.
The epileptic encephalopathies are severe epilepsy syndromes characterized by multiple seizure types and developmental slowing, often associated with regression; many begin in infancy or childhood. The etiology of these disorders is increasingly being recognized as due to de novo genetic mutations with a recent explosion in the number of causative genes identified.1,2
In a cohort of 500 patients, targeted massively parallel sequencing identified de novo CHD2 mutations as the cause of 6 (1.2%) cases of epileptic encephalopathy and was the fourth most frequently mutated gene after SCN1A, CDKL5, and STXBP1.1 Two of the 6 patients had been diagnosed with epilepsy with myoclonic-atonic seizures (MAE) but their detailed phenotype was not reported; one had Lennox-Gastaut syndrome with prominent photosensitivity and 3 had nonspecific epileptic encephalopathies. In a later report,3 3 patients with de novo CHD2 mutations had an epileptic encephalopathy with fever sensitivity. CHD2 encodes the chromodomain helicase DNA binding protein 2, which likely alters gene expression via chromatin modification. We present the phenotype of CHD2 encephalopathy in 10 patients including 3 novel cases, further analysis of our 6 published cases, and a case with a 15q26 deletion encompassing several genes including CHD2.4
METHODS
Nine cases were recruited to the Epilepsy Genetics Research Program at the University of Melbourne. They formed part of a large study of patients with epileptic encephalopathies and were identified through genetic testing.1 Eight had CHD2 mutations identified via targeted next-generation sequencing of candidate genes for epileptic encephalopathies.1 Our ninth case harbored a de novo 15q26 deletion including CHD2 (chr15: 91,027,533–93,477,874 [hg19]; 2.4 Mb).4 This case was included because the phenotype closely matched our other cases with CHD2 encephalopathy. The tenth case was ascertained via whole-exome sequencing of the proband and his parents by the EuroEPINOMICS-RES Consortium. De novo status was confirmed through parental sequencing of CHD2 in all cases. We reviewed the medical history, video, and EEG recordings of 10 individuals with CHD2 encephalopathy. Where possible, we reviewed their inpatient video-EEG monitoring records.
Estimates of intellectual disability were made from psychometric testing and educational assessments when available. Autism spectrum disorder (ASD) was diagnosed by a developmental pediatrician. EEG with photic stimulation was performed using the Grass PS33 plus stroboscope. Studies typically included a superimposed pattern protocol together with a protocol in which photic stimulation frequencies were repeated.
Standard protocol approvals, registrations, and patient consents.
The participants' parents provided informed consent for participation in the research study. The Austin Health Human Research Ethics Committee approved the study.
RESULTS
Seizures.
The 10 patients had a mean age of 17.9 years (range 6–36 years) with a mean age at seizure onset of 26 months (10–42 months); 6 were boys (table e-1 on the Neurology® Web site at Neurology.org). Seven cases presented with daily myoclonic seizures, 4 of whom also had absence seizures at onset. Seizure onset was explosive in 6 individuals with multiple daily myoclonic and absence seizures.
All patients developed multiple seizure types. Myoclonic seizures occurred in all and were symmetric, involving the upper limbs. Myoclonus was both spontaneous and triggered by environmental photic stimuli in 6. A seventh case self-induced generalized tonic-clonic seizures with the television; other reflex seizures were not seen. Absence seizures occurred in 9 cases, including typical (4), atypical absence (2), and eyelid myoclonia with absence (4). Prolonged absence seizures and focal dyscognitive seizures were not observed.
The pattern evolved with the development of tonic seizures in 4 patients at a mean of 3.6 years, typically nocturnal, and atonic seizures in 3 at a mean of 4.5 years. Generalized tonic-clonic seizures were a later feature, present in 9 patients with a mean onset of 5.8 years. The pattern of generalized tonic-clonic seizures and tonic seizures changed from seizures at any time to predominantly from sleep after 8 years.
Convulsive status epilepticus began at a mean of 9.7 years in 3, and nonconvulsive status in 5 at mean 9.2 years. Nonconvulsive status epilepticus often included prominent myoclonic components. Fever was not a prominent trigger; only one boy had febrile seizures at 2 years.
The waking interictal EEG showed mild diffuse background slowing at seizure onset in 7 patients. The degree of slowing did not correlate with the degree of intellectual disability.
Bursts of generalized spike wave (GSW) and generalized polyspike wave were frequently seen awake and often markedly increased in sleep. GSW was often slow at a frequency of 2 to 2.5 Hz (range 1–5 Hz) with a frontal predominance. The GSW was brought out by eye closure in 3 patients.
Cases 1, 3, and 4 (at 20, 13, and 6 years, respectively) progressed to abundant, almost continuous slow spike wave that activated in sleep. Cases 1, 2, and 8 exhibited interictal multifocal epileptiform activity (table e-1).
Atonic-myoclonic-absence seizure.
An unusual seizure type was observed in 3 patients (cases 3, 4, 5) on home videos and had a mean onset of 22 months (table e-1, figure 1, videos 1–5). These seizures commenced with an atonic component causing an abrupt head nod or atonic fall with simultaneous eye elevation. The patients sometimes sustained injuries with the sudden loss of tone. The seizure progressed to a myoclonic absence phase characterized by ratchet-like tonic abduction of the upper limbs; myoclonic activity began while truncal atonia was still present. Seizures were brief, lasting 2 to 8 seconds, with rapid return of awareness. We termed this distinctive seizure type as an “atonic-myoclonic-absence seizure.”
Figure 1. EEG of atonic-myoclonic-absence seizure.

(A) Case 4, aged 14 years. Bipolar montage; (B) Case 5, aged 5.5 years. Bipolar montage. Ictal recordings show a paroxysm of 3 Hz generalized polyspike wave activity. The seizure begins with the head dropping forward or backward followed by bilateral myoclonic ratcheting abduction of the upper limbs. The jerks correlate with the spike of the spike wave complex.
The ictal EEG showed paroxysms of 3- to 4-Hz GSW and generalized polyspike wave activity (figure 1). The atonic component of the atonic-myoclonic-absence seizure correlated with the aftergoing slow wave of the initial GSW discharge, and each myoclonic jerk corresponded with the spike of the spike wave complex (figure 1).
This seizure type was noted only in children aged 2 to 7 years and captured on home video. Patient 5 also had seizures recorded at 14 and 18 years that resembled typical myoclonic absence seizures without the preceding atonic component (videos 6 and 7).
Photosensitivity.
Seven of 10 patients exhibited extreme clinical photosensitivity with atonic myoclonic absence or absence seizures with eyelid myoclonia (table e-1). Six patients were compelled to self-induce these seizures by placing their face adjacent to the television screen and had to be restrained from this activity (video 5). In addition, case 1 had seizures induced by the repetitive flash of a digital camera as an infant. In contrast to the striking clinical history of photosensitivity, only one case (8) had a recorded photoparoxysmal response on EEG. This comprised a grade 4 response showing GSW across all frequencies. No cases demonstrated head turning toward a light stimulus or “hand-flapping” in sunlight to induce seizures.
Developmental course and behavioral features.
Development was normal in the first year of life in all 10 patients. All patients walked by 18 months. Speech delay became evident between 1 and 2 years and preceded seizure onset (table 1). Seven had moderate to severe intellectual disability and 3 had mild impairment (table e-1, figure 2). Six of the 7 more-severe cases had a history of regression that correlated temporally with the explosive onset of seizures. In general, regression tended to occur during periods of seizure exacerbation. At the milder end, case 8 was succeeding in mainstream schooling until he first regressed at age 9 years with seizure recurrence; multiple seizures occurred and proved refractory to treatment. Case 9 had speech regression at 24 months.
Table 1.
Comparison of electroclinical features of CHD2 encephalopathy with Dravet syndrome, epilepsy with myoclonic-atonic seizures, and Lennox-Gastaut syndrome
Figure 2. CHD2 encephalopathy spectrum.

CHD2 encephalopathy presents with a spectrum of severity including age of seizure onset, intellectual outcome, and seizure types. Cases are ranked by their level of intellectual disability and age at onset. All cases had myoclonic seizures and 7 had photic-induced seizures. Cases with a milder intellectual disability had fewer seizure types and were less likely to have atonic seizures, tonic seizures, and convulsive status epilepticus. Photic seizures were equally likely across the spectrum. *A-M-A is likely to be underrecognized. AA = atypical absence; Abs = all absence seizure types; A-M-A = atonic-myoclonic-absence seizure; CSE = convulsive status epilepticus; EMA = absence with eyelid myoclonia; GTCS = generalized tonic-clonic seizures; MS = myoclonic seizures; NCSE = nonconvulsive status epilepticus; TA = typical absence.
Four patients had a formal diagnosis of ASD in addition to their intellectual disability. One also had attention deficit hyperactivity disorder as did 2 without ASD. Challenging behavior was reported in 8 patients and was independent of the degree of intellectual disability. It primarily involved aggressive behavior, and required risperidone in 3 cases. In addition, patient 10 developed a prominent psychotic illness at 19 years requiring hospitalization and quetiapine.
Five individuals have reached adulthood. They showed a broad spectrum of disability, with case 1 (31 years) at the severe end of the spectrum being fully dependent on others for support, whereas case 9 (35 years) had the second mildest outcome in our series. She lived at home with her family and never worked but sang in a choir for “disadvantaged” people.
Four individuals had short stature (less than third centile) and the 2 most severe cases had head circumferences less than third centile (table e-1) but there were no consistent dysmorphic features. Neurologic examination was normal. Five patients had a crouch gait and 4 had ataxia. At 5 years, case 3 exhibited chorea with facial dyskinesia and ataxia that improved after the withdrawal of sodium valproate. While case 5 at 9 years had ataxia and vomiting associated with valproate, ataxia in case 8 from age 13 years was independent of the withdrawal and reintroduction of sodium valproate.
Seizures remained refractory to treatment in 7 of 10 cases; 3 cases, before the age of 10 years, had a period of seizure control of more than a year's duration. Three individuals trialled the ketogenic diet, which did not produce significant benefit.
Neuroimaging.
MRI brain studies were reviewed in 7 cases and showed progressive atrophy in 3 of 4 individuals with sequential studies (figure 3, table e-1). Parenchymal atrophy was most marked posteriorly and associated with atrophy of the splenium of the corpus callosum and mild diffuse cerebellar atrophy in 3 cases (figure 3). Case 8 had a combination of callosal truncation (presumably developmental) and progressive atrophy. Two of the 4 cases with ataxia had cerebellar atrophy on MRI. Generalized atrophy correlated to some extent with the degree of intellectual disability as the 2 mildest cases (9 and 10) had normal MRI scans in adult life. White matter maturation was age-appropriate and hippocampal sclerosis was not seen.
Figure 3. MRI of CHD2 encephalopathy.
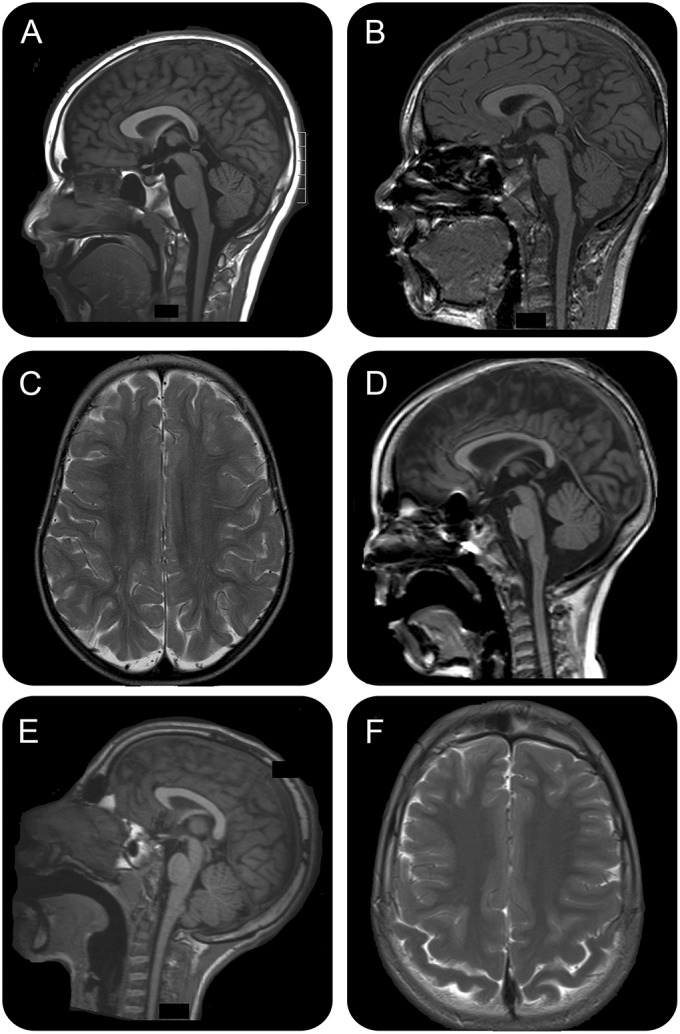
MRIs demonstrating progressive posterior atrophy. (A, B) Case 8 sagittal T1 MRIs at 10 years (A) and sagittal T1 at 11 years (B) demonstrate a truncated, hypoplastic corpus callosum with progressive atrophy and an atrophic cerebellar vermis. (C, D) Case 3 axial T2 at 2 years (C) and T1 sagittal at 7 years (D) demonstrate generalized atrophy more marked posteriorly and atrophic cerebellar vermis. The callosal atrophy is also more pronounced posteriorly. (E, F) Case 6 sagittal T1 (E) and axial T2 (F) at 12 years demonstrate generalized atrophy more prominent posteriorly.
Molecular results.
Our cohort included 3 new cases with novel mutations, 6 from our previous publication identifying the role of CHD2 in epileptic encephalopathies,1 and one from our study of copy number variants.4 All cases have de novo mutations. Seven had mutations resulting in premature truncation, 2 had missense mutations, and case 9 had a 2.4-Mb deletion that included the 5′ end of CHD2 resulting in a partial gene deletion.3 The phenotype of this patient was consistent with CHD2 encephalopathy. The location of the mutations is shown in figure 4.
Figure 4. Location of mutations in CHD2.
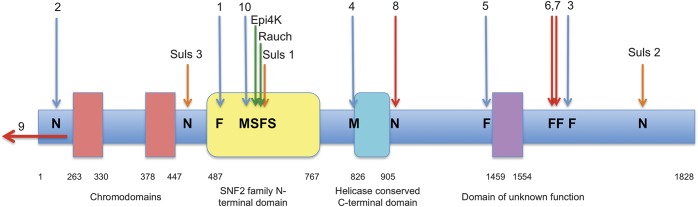
The location of all reported de novo CHD2 mutations associated with epilepsy is shown. The red arrows denote the 3 novel mutations reported here, blue arrows our 6 cases in Carvill et al.,1 orange arrows the 3 Suls et al.3 cases, and green arrows the single cases of Epi4k2 and Rauch et al.12 Suls 1 is an approximated position based on where the splice site mutation is predicted to truncate the protein.3 The arrow (case 9) represents the partial gene deletion. F = frameshift; M = missense mutation; N = nonsense mutation; S = splice site mutation.
DISCUSSION
With the recent explosion in gene discovery, an increasing number of novel genetic epileptic encephalopathies are being recognized. In contrast to Dravet syndrome in which the electroclinical phenotype was described well before the genetic finding of SCN1A mutations, we are now in a position to delineate the electroclinical phenotype of patients who share the same genetic cause. The power of massively parallel sequencing to rapidly and inexpensively screen panels of known genes1 makes possible the recognition of novel electroclinical syndromes through deep phenotypic analysis following gene identification.
CHD2 encephalopathy begins in the second year of life and is characterized by myoclonic, absence, and generalized tonic-clonic seizures and clinical photosensitivity. We distinguished a spectrum of severity in terms of age at onset, epilepsy severity, and cognitive outcome (figure 2). Microcephaly occurred in only the 2 most severe cases. Mean seizure onset in our 10 cases was 26 months with the more severe end of the spectrum (7 cases) having a mean onset of 23 months vs 34 months at the milder end (3 cases) (figure 2). CHD2 encephalopathy is associated with refractory epilepsy but, in the milder cases, a period of seizure freedom of more than a year may occur. The hallmark of CHD2 encephalopathy, self-induced photic seizures with the television, was common across the spectrum.
The severe phenotype was characterized by moderate to severe intellectual disability and tonic seizures. A similar pattern is described in other epileptic encephalopathies such as MAE in which the severe cases develop tonic seizures.5
We identified an unusual seizure type beginning with atonia and followed by myoclonic absence (videos 1–5, figure 1), which we coined an atonic-myoclonic-absence seizure. It is unclear whether this seizure type occurs in all cases as we first recognized it through home videotapes in a few cases. It may be an age-dependent seizure type and therefore only recordable for a limited time. It is of interest that these appear to have the inverse sequence of classic myoclonic-atonic seizures whereby a single myoclonic jerk heralds the onset of the seizure followed by atonia.6 Patient 8 had a history of seizures that was suspicious of the atonic-myoclonic-absence seizure type at age 2.5 years, but video was not captured. Confirmation of this seizure type using video-EEG with EMG is required to determine whether it is sensitive and specific for CHD2 encephalopathy.
How were these patients previously conceptualized regarding their epilepsy syndrome? While 4 cases were thought to have severe genetic (formerly idiopathic) generalized epilepsies, 3 had been previously diagnosed with MAE,1 described by Doose et al.7 This was partly because of their prominent drop attacks and the explosive onset of multiple seizure types associated with GSW. While these epilepsies share many features, there are elements that distinguish them (table 1). First and foremost, the CHD2 encephalopathy cases do not have the archetypal seizure that defines the syndrome of MAE, the myoclonic-atonic seizure. In contrast to MAE, CHD2 encephalopathy has the following features: (1) development is usually delayed before seizure onset; (2) seizure remission may occur in milder cases; and (3) clinical photosensitivity is seen in the majority of cases compared with less than a third of patients with MAE.8 It was surprising that only one patient showed electrical photosensitivity despite the marked clinical photosensitivity in 7 cases and self-induction with television screens seen in 6 patients.
CHD2 encephalopathy can be readily distinguished from Dravet syndrome by its later mean seizure onset of 26 months compared with 6 months, its high frequency of clinical photosensitivity compared with <50%, and the archetypal feature of self-induced seizures, which is much rarer in Dravet syndrome (table 1).9,10 The paucity of febrile seizures and abnormal development before seizure onset are also key features that separate CHD2 encephalopathy from Dravet syndrome.
Three patients have been recently described with de novo mutations who had mild intellectual disability, epileptic encephalopathy, and a predilection to seizures with fever (table 1, figure 4).3 Clinical photosensitivity was not noted. One case had ASD and 2 had ataxia, one of whom showed atrophy on MRI in keeping with our findings. We observed a more posterior pattern of atrophy involving the parenchyma and corpus callosum with additional cerebellar involvement in those cases with ataxia.
Our findings are consistent with the previous case reports in the literature of copy number variants incorporating CHD2 and intragenic CHD2 mutations (figure 4). The most similar phenotype of photosensitive myoclonic encephalopathy was reported in a girl with a large 5-Mb deletion that included 56 genes.11 Among her seizure types was “massive myoclonias with head drop associated with irregular spike and slow waves,”11 reminiscent of the atonic-myoclonic-absence seizure. MRI demonstrated cerebellar hypoplasia with thinning of the posterior body of the corpus callosum. She had multiple dysmorphic features that were not evident in our cases and may relate to deletion of other genes. An earlier onset of seizures at 6 months was noted in the single case with a de novo splice site mutation from the EPI4K study.11 A de novo CHD2 mutation has also been reported in a child with intellectual disability and absence epilepsy (T604Lfs*19)12 and another case with ASD alone (D856G).13
Case 9 had a chromosome 15q26 microdeletion that results in partial deletion of CHD2. Her phenotype was consistent with CHD2 encephalopathy but, remarkably, given the number of contiguous genes deleted, was at the mild end of the CHD2 phenotypic spectrum. Of the 5 published cases with deletions of varying size that encompass CHD2, all had seizures and 4 had delayed development before seizure onset at 6 to 48 months. Intellectual disability was mild to severe.11,14–17
The missense, nonsense, frameshift, and splice site mutations in CHD2 encephalopathy do not cluster in any definite pattern within the gene (figure 4), and the location or type of mutation does not correlate with disease severity. CHD2 is among the genes that are most intolerant to functional variation (ranked in top 2.5%).18 In keeping with this, all functional variation in the exome variant server are missense changes, and only 2 missense changes in 6,500 individuals are in the SNF2 (sucrose non-fermentable 2) domain (figure 4). Notably, 8 of 10 mutations reported here cause premature truncation; only 2 cases carry missense mutations and both lie within highly conserved helicase domains. Therefore, the phenotype in patients with CHD2 encephalopathy is likely due to haploinsufficiency of the CHD2 protein.
It is thought that CHD2 belongs to the SNF2-like family of ATPase (adenosine triphosphatase) proteins that have a role in chromatin remodeling and therefore may affect transcription of many genes. However, our knowledge of the function of CHD2, particularly in the brain, is incomplete and warrants further investigation to delineate its biological role. Sodium valproate, one of the key antiepileptic drugs for myoclonic and generalized epilepsies, inhibits histone deacetylase activity and alters chromatin structure.19 Valproate was generally a favorable drug for patients with CHD2 encephalopathy.
Knocked down chd2 zebrafish exhibit a seizure-like phenotype as well as structural abnormalities of microcephaly, body curvature, absent swim bladder, and stunted growth.3 A murine Chd2 model confirmed that the CHD2 protein is widely expressed during development in many tissues, although no aberrations were noted in the brain.20
CHD2 mutations produce a distinctive myoclonic epileptic encephalopathy with prominent clinical photosensitivity in the majority of cases. The atonic-myoclonic-absence seizures may prompt investigation for a CHD2 mutation. Recognition of CHD2 encephalopathy will lead to improved diagnosis for patients and families. Understanding the neurobiology of this disorder will form the basis for the development of genetically targeted therapies to improve outcome of this severe epileptic encephalopathy.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the patients and their families for participation in the study. The authors are grateful to Prof. Eliane Roulet-Perez for her thoughtful review of the manuscript.
GLOSSARY
- ASD
autism spectrum disorder
- CHD2
chromodomain helicase DNA binding protein 2
- GSW
generalized spike wave
- MAE
epilepsy with myoclonic-atonic seizures
Footnotes
Supplemental data at Neurology.org
Contributor Information
Collaborators: Aarno Paalotie, Anna-Elina Lehesjoki, Bobby Koeleman, Carla Marini, Christel Depienne, Dana Craiu, Deb Pal, Dorota Hoffman-Zacharska, Eric Leguern, Federico Zara, Felix Rosenow, Hande Caglayan, Helle Hjalgrim, Hiltrud Muhle, Holger Lerche, Ingo Helbig, Johanna Jähn, Johannes Lemke, Jose M Serratosa, Kaja Selmer, Karl Martin Klein, Katalin Sterbova, Manuela Pendziwiat, Nina Barisic, Padhraig Gormley, Pasquale Striano, Patrick May, Peter De Jonghe, Renzo Guerrini, Rikke Møller, Roland Krause, Rudi Balling, Sanjay Sisodiya, Sarah von Spiczak, Sarah Weckhuysen, Stéphanie Baulac, Arvid Suls, Tania Djemie, Tiina Talvik, Ulrich Stephani, Vladimir Komarek, and Yvonne Weber
AUTHOR CONTRIBUTIONS
R.H. Thomas: draft/revise manuscript, data analysis, acquisition of data. L.M. Zhang: draft manuscript, data analysis, acquisition of data. G.L. Carvill: revise manuscript, data analysis, acquisition of data. J.S. Archer: revise manuscript, data analysis. S.B. Heavin: revise manuscript, acquisition of data. S.A. Mandelstam: revise manuscript, data analysis. D. Craiu: revise manuscript, acquisition of data. EuroEPINOMICS-RES Consortium: acquisition of data. S.F. Berkovic: revise manuscript, acquisition of data. D.S. Gill: revise manuscript, acquisition of data. H.C. Mefford: revise manuscript, study design, data analysis, acquisition of data, obtain funding. I.E. Scheffer: draft/revise manuscript, study design, data analysis, acquisition of data, obtain funding.
STUDY FUNDING
Supported by the National Health and Medical Research Council of Australia.
DISCLOSURE
R. Thomas is supported by the National Institute of Social Care and Health Research in the form of a WCAT Clinical Lectureship; receives/has received research support from Epilepsy Research UK, Action Medical Research, Epilepsy Action, and the Dravet Society; and is an associate editor of Practical Neurology. L. Zhang is supported by a fellowship from Department of Neurology, Children's Hospital of Fudan University, Shanghai, China. G. Carvill, J. Archer, S. Heavin, S. Mandelstam, and D. Craiu report no disclosures relevant to the manuscript. S. Berkovic has received a grant(s) from the National Health and Medical Research Council; has received honoraria from UCB; has a patent for PCDH19 testing planned; has received payment for development of educational presentations from UCB Pharma, Novartis Pharmaceuticals, Sanofi-Aventis, and Janssen-Cilag; has a patent for SCN1A testing held by Bionomics Inc. and licensed to various diagnostic companies, with no financial return; and was a consultant to Bionomics and Athena diagnostics more than 3 years ago. D. Gill reports no disclosures relevant to the manuscript. H. Mefford has received grant funding from the NIH/National Institute of Neurological Disorders and Stroke and the Burroughs Wellcome Fund and is a consultant for the Simons Foundation (SFARI Gene Advisory Board). I. Scheffer serves on the editorial boards of the Annals of Neurology, Neurology®, and Epileptic Disorders; may accrue future revenue on a pending patent re: Therapeutic compound; has received speaker honoraria from Athena Diagnostics, UCB, GSK, and Transgenomics; has received funding for travel from Athena Diagnostics, UCB, and GSK; and receives/has received research support from the National Health and Medical Research Council of Australia, Australian Research Council, Health Research Council of New Zealand, The University of Melbourne, American Epilepsy Society, the Jack Brockhoff Foundation, the Weizmann Institute, CURE, US Department of Defense, and the Perpetual Charitable Trustees. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Carvill GL, Heavin SB, Yendle SC, et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet 2013;45:825–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Epi4K Consortium; Epilepsy Phenome/Genome Project, Allen AS, et al. De novo mutations in epileptic encephalopathies. Nature 2013;501:217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Suls A, Jaehn JA, Kecskes A, et al. De novo loss-of-function mutations in CHD2 cause a fever-sensitive myoclonic epileptic encephalopathy sharing features with Dravet syndrome. Am J Hum Genet 2013;93:967–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mullen SA, Carvill GL, Bellows S, et al. Copy number variants are frequent in genetic generalized epilepsy with intellectual disability. Neurology 2013;81:1507–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guerrini R, Mari F, Dravet C. Idiopathic myoclonic epilepsies in infancy and early childhood. In: Beureau M, Genton P, Dravet C, et al., editors. Epileptic Syndromes in Infancy, Childhood and Adolescence, 5th ed Montrouge, France: John Libbey; 2012:166. [Google Scholar]
- 6.Caraballo RH, Chamorro N, Darra F, Fortini S, Arroyo H. Epilepsy with myoclonic atonic seizures: an electroclinical study of 69 patients. Pediatr Neurol 2013;48:355–362. [DOI] [PubMed] [Google Scholar]
- 7.Doose H, Gerken H, Leonhardt R, Volzke E, Volz C. Centrencephalic myoclonic-astatic petit mal: clinical and genetic investigation. Neuropadiatrie 1970;2:59–78. [DOI] [PubMed] [Google Scholar]
- 8.Doose H, Waltz S. Photosensitivity: genetics and clinical significance. Neuropediatrics 1993;24:249–255. [DOI] [PubMed] [Google Scholar]
- 9.Dravet C, Bureau M, Oguni H, Cokar O, Guerrini R. Dravet syndrome (severe myoclonic epilepsy in infancy). In: Beureau M, Genton P, Dravet C, et al., editors. Epileptic Syndromes in Infancy, Childhood and Adolescence, 5th ed Montrouge, France: John Libbey; 2012:125–156. [Google Scholar]
- 10.Guerrini R, Aicardi J. Epileptic encephalopathies with myoclonic seizures in infants and children (severe myoclonic epilepsy and myoclonic-astatic epilepsy). J Clin Neurophysiol 2003;20:449–461. [DOI] [PubMed] [Google Scholar]
- 11.Veredice C, Bianco F, Contaldo I, et al. Early onset myoclonic epilepsy and 15q26 microdeletion: observation of the first case. Epilepsia 2009;50:1810–1815. [DOI] [PubMed] [Google Scholar]
- 12.Rauch A, Wieczorek D, Graf E, et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet 2012;380:1674–1682. [DOI] [PubMed] [Google Scholar]
- 13.Neale BM, Kou Y, Liu L, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 2012;485:242–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Capelli LP, Krepischi AC, Gurgel-Giannetti J, et al. Deletion of the RMGA and CHD2 genes in a child with epilepsy and mental deficiency. Eur J Med Genet 2012;55:132–134. [DOI] [PubMed] [Google Scholar]
- 15.Li MM, Nimmakayalu MA, Mercer D, Andersson HC, Emanuel BS. Characterization of a cryptic 3.3Mb deletion in a patient with a “balanced t(15;22) translocation” using high density oligo array CGH and gene expression arrays. Am J Med Genet A 2008;146:368–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dhamija R, Patterson MC, Wirrell EC. Epilepsy in children: when should we think neurometabolic disease? J Child Neurol 2012;27:663–671. [DOI] [PubMed] [Google Scholar]
- 17.Lund C, Brodtkorb E, Røsby O, Rødningen OK, Selmer KK. Copy number variants in adult patients with Lennox-Gastaut syndrome features. Epilepsy Res 2013;105:110–117. [DOI] [PubMed] [Google Scholar]
- 18.Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet 2013;9:e1003709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marchion DC, Bicaku E, Daud AI, Sullivan DM, Munster PN. Valproic acid alters chromatin structure by regulation of chromatin modulation proteins. Cancer Res 2005;65:3815–3822. [DOI] [PubMed] [Google Scholar]
- 20.Kulkarni S, Nagarajan P, Wall J, et al. Disruption of chromodomain helicase DNA binding protein 2 (CHD2) causes scoliosis. Am J Med Genet A 2008;146A:1117–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lund C, Brodtkorb E, Oye AM, Røsby O, Selmer KK. CHD2 mutations in Lennox-Gastaut syndrome. Epilepsy Behav 2014;33:18–21. [DOI] [PubMed] [Google Scholar]
- 22.Crespel A, Gelisse P, Nikanorova M, Ferlazzo E, Genton P. Lennox-Gastaut syndrome. In: Beureau M, Genton P, Dravet C, et al., editors. Epileptic Syndromes in Infancy, Childhood and Adolescence, 5th ed Montrouge, France: John Libbey; 2012:189–216. [Google Scholar]
- 23.Scheffer IE, Berkovic SF. Generalized epilepsy with febrile seizures plus: a genetic disorder with heterogeneous clinical phenotypes. Brain 1997;120:479–490. [DOI] [PubMed] [Google Scholar]
- 24.Singh R, Scheffer IE, Crossland K, Berkovic SF. Generalized epilepsy with febrile seizures plus: a common childhood-onset genetic epilepsy syndrome. Ann Neurol 1999;45:75–81. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.