

Abstract
Objective:
To determine the independent association of the TMEM106B variants with transactive response DNA binding protein 43 (TDP-43) pathology in older persons without frontotemporal lobar degeneration (FTLD) and to explore functional pathways that link the risk variants to the pathology, including a GRN mRNA pathway.
Methods:
Data came from 544 autopsied participants without FTLD in 2 community-based studies of aging. Participants underwent uniform neuropathologic evaluations, including TDP-43 cytoplasmic inclusions. We examined the association of TMEM106B variants with a semiquantitative measure of TDP-43 pathology in a series of regression analysis. We explored potential pathways by leveraging genetic, brain DNA methylation, miRNA, and transcriptomic data collected from this same group of participants.
Results:
TDP-43 pathology was identified in 51.7% of the participants. The index single-nucleotide polymorphism (SNP), rs1990622A, was associated with more advanced TDP-43 pathology. Top hits from fine mapping of the locus were in linkage disequilibrium of the index SNP. The association remained significant after adjustment for other neuropathologies including Alzheimer disease and hippocampal sclerosis (odds ratio = 1.351, 95% confidence interval = 1.068–1.709, p = 0.012). GRN expression was upregulated in rs1990622AA/AG carriers, and was associated with more advanced TDP-43 pathology. The TMEM106B variants were associated with lower level of DNA methylation in an active enhancer in GRN.
Conclusions:
Common variants in TMEM106B serve as a distinct risk factor for TDP-43 pathology in older persons without FTLD. The role of GRN expression and epigenetic mechanisms associating TMEM106B in the accumulation of TDP-43 in older persons require further study.
Transactive response DNA binding protein 43 (TDP-43) is the major disease protein in frontotemporal lobar degeneration (FTLD) with ubiquitin-positive inclusions and amyotrophic lateral sclerosis (ALS).1 TDP-43 immunoreactivity is also present in other neurodegenerative diseases such as Alzheimer disease (AD), Lewy body disease, and hippocampal sclerosis.2–5 Recent genome-wide association studies (GWAS) suggest that common variants in the transmembrane protein 106B (TMEM106B) locus is a risk factor for susceptibility of FTLD with TDP inclusions (FTLD-TDP).6,7 However, it is unclear whether the same risk variants are associated with the much more common TDP-43 pathology found in community-based older persons without FTLD. Further, as comorbid pathologies are common in the aging brain8–10 and the TMEM106B variants are implicated in patients with pathologic AD diagnosis,11 it is important to determine whether such an association is independent of other neuropathologies.
Notably, the physiologic function of the TMEM106B locus in TDP-43 pathology remains elusive. Initial studies report that TMEM106B RNA expression is elevated in FTLD-TDP and could be cis regulated by the risk variants6,12; however, these findings are inconclusive.7 Separately, TMEM106B variants are posited to influence the risk of FTLD-TDP by potentially regulating expression of progranulin (GRN), a mendelian risk gene for FTLD13,14; yet there are also reports that GRN level is not altered by TMEM106B expression.15 In addition, epigenetic mechanisms such as micro RNA (miRNA) and DNA methylation could also be involved.16,17
In this study, we first tested the hypothesis that the TMEM106 risk variants are associated with TDP-43 pathology in older persons without FTLD. We confirmed the association after controlling for potential confounding due to other age-related pathologies, including AD, Lewy bodies, infarcts, and hippocampal sclerosis. Next, we explored potential functional pathways that may link the TMEM106B risk variants to downstream TDP-43 pathology through DNA methylation, miRNA, or mRNA expression.
METHODS
Participants.
Participants are from 2 community-based clinical pathologic cohort studies of aging, the Religious Orders Study (ROS) and the Rush Memory and Aging Project (MAP).18,19 Participants enroll without known dementia. At the time of these analyses, 1,363 ROS and MAP participants had died and 1,179 had undergone brain autopsies; the autopsy rate exceeds 85%. Of 1,179 autopsied participants, complete TDP-43 pathology had been collected in 620 (ROS = 294; MAP = 326). Notably, there was no statistical difference in age or common neuropathologic burdens between the participants with and without TDP-43 data (table e-1 on the Neurology® Web site at Neurology.org). Of 620, 13 (2%) had FTLD or a related pathology (including 7 with FTLD, 1 with ALS, 2 with progressive supranuclear palsy [PSP], and 3 corticobasal degeneration [CBD]), and were excluded from the analyses. Primary analyses were performed on 544 of 607 participants who also had genotype data. The average age at death was 89.0 years with SD of 6.3 years and range 71.7 to 108.3 years; 365 (67.1%) were female; and average education was 16.1 years (SD 3.6, range 5–28).
Standard protocol approvals, registrations, and patient consent.
Studies were approved by the Institutional Review Board of Rush University Medical Center. Each participant signed informed consent for annual clinical evaluations and organ donation at the time of death.
Uniform neuropathologic evaluation.
Uniform neuropathologic evaluations were conducted by examiners blinded to age and all clinical data. Brains were removed following a standard protocol, weighed, and cut coronally into 1-centimeter slabs. Presence of chronic macroscopic infarcts was assessed using slabs or pictures from both hemispheres, and verified after dissection and histologic review.20 Blocks of predetermined brain regions were dissected from one hemisphere. Six-micrometer sections were stained for hematoxylin & eosin to assess presence of chronic microinfarcts21 and hippocampal sclerosis and α-synuclein immunostain to assess presence of neocortical Lewy bodies.22 Neuropathologic AD diagnosis was based on Braak staging for neurofibrillary tangles and the Consortium to Establish a Registry for Alzheimer's Disease (CERAD) estimate of neuritic plaque density, as recommended by the National Institute on Aging Reagan criteria. AD diagnosis by Reagan criteria required either an intermediate likelihood of AD (i.e., at least Braak stage 3 or 4 and CERAD moderate plaques) or a high likelihood (i.e., at least Braak stage 5 or 6 and CERAD frequent plaques). Neuropathologic diagnoses of FTLD, CBD, and PSP followed standard criteria.23
TDP-43 pathology was assessed in 6 predetermined brain regions of interest: amygdala, hippocampus CA1, dentate gyrus, entorhinal cortex, midtemporal cortex, and midfrontal cortex. Six-micrometer sections were stained for monoclonal antibodies to phosphorylated TDP-43 (pS409/410; 1:100) to assess the counts of TDP-43 cytoplasmic inclusions (both neuronal and glial) in each region. A semiquantitative measure capturing the staging of TDP-43 pathology was used across all the analyses. This measure was rated on 4 levels, including no inclusions; inclusions in amygdala only; inclusions in amygdala as well as entorhinal cortex or hippocampus CA1; and inclusions in amygdala, entorhinal cortex, or hippocampus CA1, and neocortex.
Genetic, epigenetic, and mRNA measures.
DNA was extracted from blood, lymphocytes, or frozen brain tissue. Genome-wide genotyping, quality control pipeline, as well as imputation procedures have been described previously.24 The imputed dosage values of single-nucleotide polymorphisms (SNPs) were used for the primary analyses, and were coded additively in terms of the reference allele. The dosage value captures the level of uncertainty of imputed genotypes at the interrogated sites. We first targeted rs1990622A (allele frequency = 0.58), the index SNP tagging TMEM106B risk variants based on the recent GWAS. Subsequent fine mapping of the locus interrogated a total of 260 additional SNPs that covered both genic as well as 10 kB flanking area of the TMEM106B gene.
Brain DNA methylation, miRNA, and RNA expression data were generated using frozen postmortem dorsolateral prefrontal cortex brain tissue, as previously described.25 Details are in e-Methods.
Statistical analyses.
Individual testing hypotheses (figure 1) were based on the findings in the FTLD literature. We first fit an ordinal logistic regression model with 4-level TDP-43 stage measure as the categorical outcome and rs1990622A the predictor, adjusted for age at death and sex. Here rs1990622A defines the dosage with respect to the SNP's A allele, and was used to estimate the odds ratio (OR) for more advanced TDP-43 pathology with every additional copy of the A allele. Next, we expanded the model by including pathologic indices of AD, chronic macro and micro infarcts, neocortical Lewy bodies, as well as hippocampal sclerosis. The score tests assessed proportional odds assumption for the ordinal logistic regression. To confirm the signal, we interrogated additional SNPs in the TMEM106B locus. Specifically, for each of the 260 SNPs queried, we tested individual SNP association with TDP-43 pathology using an ordinal logistic regression.
Figure 1. Relationship and potential pathways linking TMEM106B variant and transactive response DNA binding protein 43 pathology.

Each line in the figure represents a hypothesis tested in this study. The primary hypothesis is that transmembrane protein 106B (TMEM106B) variants are related to transactive response DNA binding protein 43 (TDP-43) pathology in aging after controlling for other common age-related pathologies. A (1 and 2) indicates the TMEM106B expression pathway; B (1 and 2) indicates granulin expression pathway; and C and D indicate potential epigenetic pathways. Solid lines indicate those relationships that were found to be significant. Dashed lines show relationships that did not reach significance (note miR132 pathway was not significant after controlling for other neurodegenerative pathologies).
Using multiple linear regression models, we examined association of rs1990622 with brain RNA expression of TMEM106B, and separately GRN. In these models, RNA expression level was the outcome and rs1990622 variant the predictor, adjusted for age at death, sex, postmortem intervals, and RNA degradation measure. We also examined association of RNA expression with TDP-43 pathology using similar ordinal logistic regressions mentioned above. Spearman correlation examined the correlation between the TMEM106B and GRN expression.
Finally, we explored potential epigenetic mechanisms that link the TMEM106B risk variant to the downstream TDP-43 pathology. First, we scanned DNA methylation of CpG sites in the GRN locus, and we regressed DNA methylation level of individual CpG sites on rs1990622A dosage, adjusted for age at death, sex, batch, and bisulfite conversion efficiency. Next, we examined cis methylation association with RNA expression by fitting linear regression models with GRN RNA expression as the outcome and methylation of individual CpG sites the predictor. We also tested the hypothesis for association of miR132 with TDP-43 pathology.
RESULTS
TDP-43 pathology in older persons without FTLD.
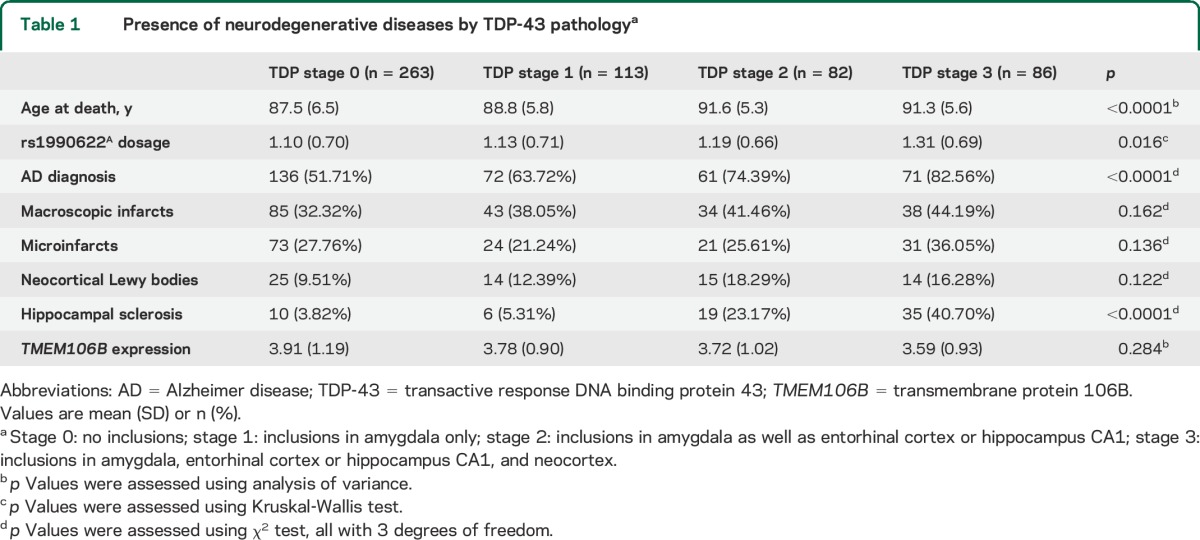
In 544 older persons without FTLD, 263 (48.3%) had no TDP-43 cytoplasmic inclusions; 113 (20.8%) had inclusions in amygdala only; 82 (15.1%) had inclusions in both amygdala and hippocampus CA1 or entorhinal cortex; and in the remaining 86 participants (15.8%), TDP-43 inclusions extended from the mesial temporal lobe to the neocortex. The distribution of TDP-43 pathology was not different between male and female participants. Older age at death was associated with more advanced TDP-43 pathology (p < 0.0001). Chronic infarcts, macroscopic or microscopic, were not associated with TDP-43 pathology, nor were neocortical Lewy bodies. In contrast, AD and hippocampal sclerosis were associated with TDP-43 pathology (table 1).
Table 1.
Presence of neurodegenerative diseases by TDP-43 pathologya
TMEM106B variants and TDP-43 pathology.
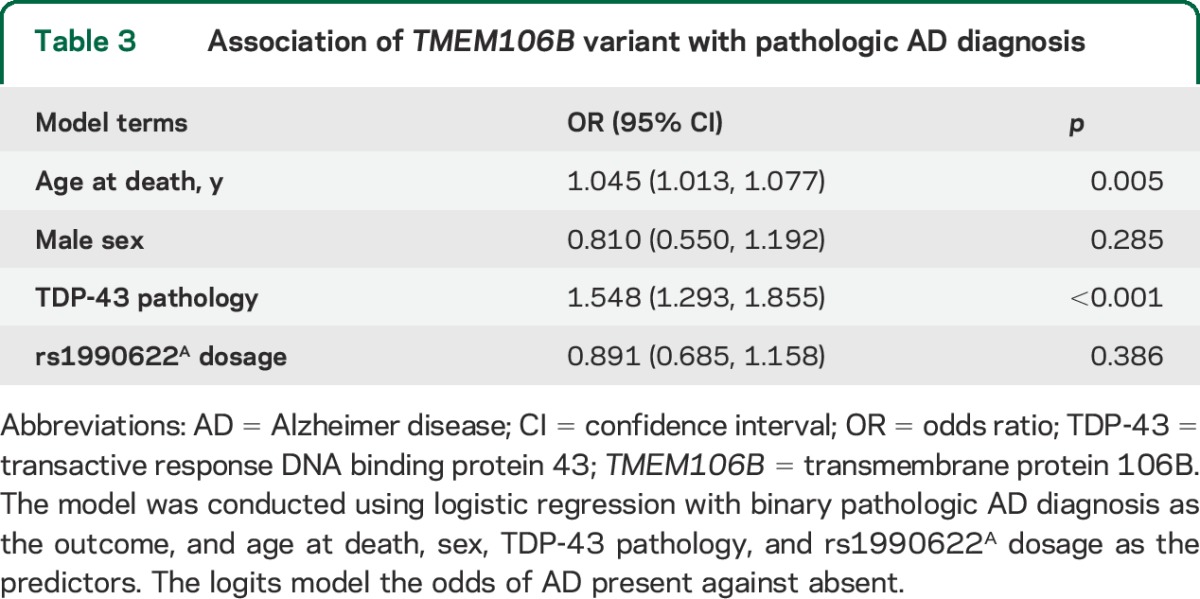
The rs1990622 dosage, coded for the risk allele (A allele), differed by stages of TDP-43 pathology in older persons without FTLD (χ2 = 10.36, df = 3, p = 0.016). In a logistic regression model adjusted for age at death and sex, higher dosage of rs1990622A was associated with more advanced TDP-43 pathology (p = 0.019). Next, we expanded the model by adjusting for common pathologic indices, and the association of rs1990622A with TDP-43 pathology was robustly retained (table 2). Specifically, each additional A allele increased the odds of having more advanced TDP-43 stages by approximately one-third (OR 1.351, 95% confidence interval 1.068–1.709, p = 0.012). Further adjustment for APOE genotype in the model did not change the result. In addition, we did not find evidence of a separate SNP association with pathologic AD diagnosis (table 3).
Table 2.
Association of TMEM106B variant with TDP-43 pathology
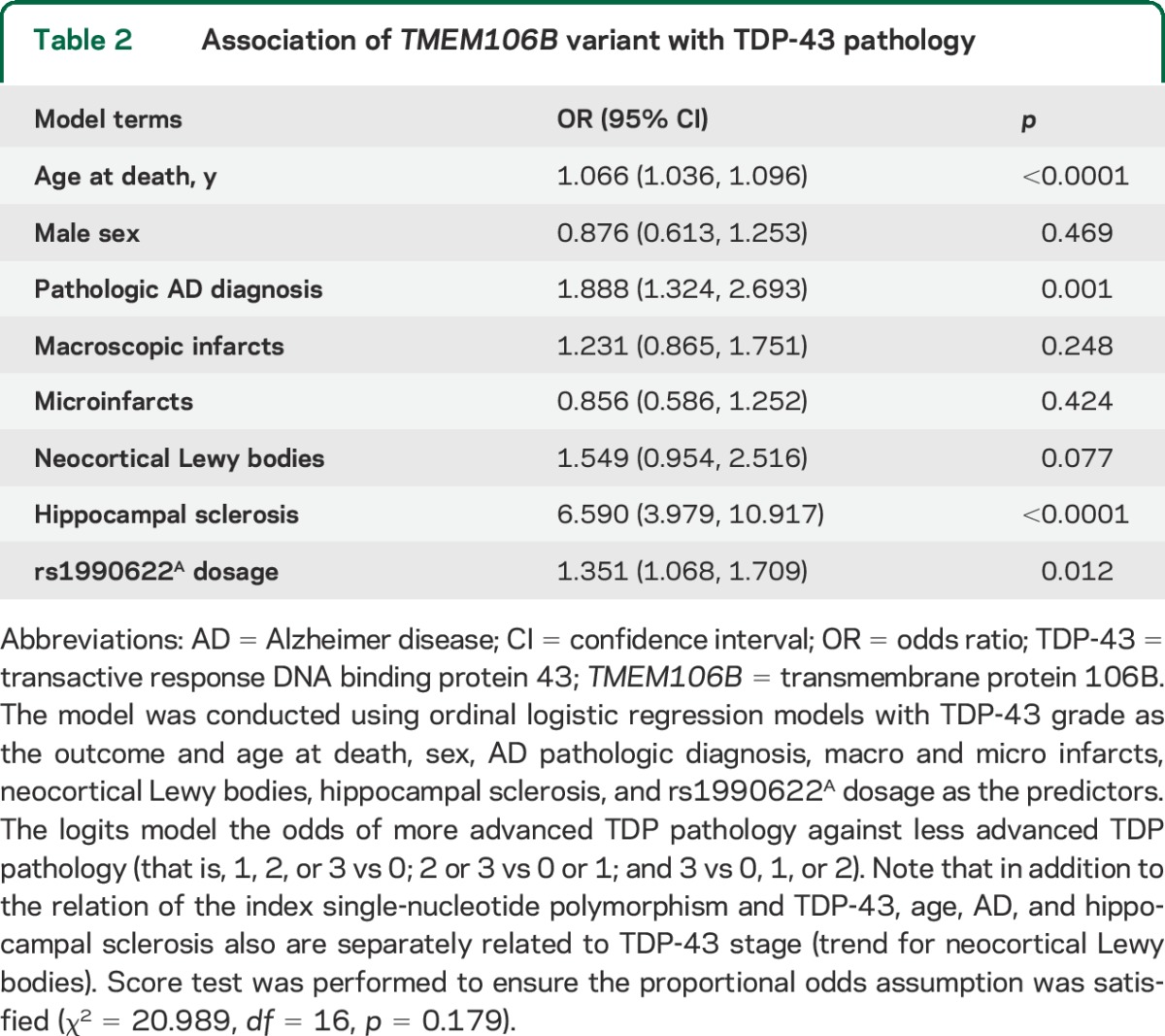
Table 3.
Association of TMEM106B variant with pathologic AD diagnosis
We performed fine mapping by interrogating 260 additional SNPs in TMEM106B (table e-2). The top hit rs6460895C was associated with advanced TDP-43 stages (p = 0.0105), consistent with the result for rs1990622A. The SNP is an intronic variant in linkage disequilibrium of rs1990622, and the Spearman correlation of the 2 SNP dosages was 0.89. Further, a total of 59 SNPs had smaller p values than rs1990622, all of which were highly correlated with rs1990622 such that the pairwise correlations range from 0.86 to 0.99.
TMEM106B variant, RNA expression, and TDP-43 pathology.
Since prior literature suggests that TMEM106B variant influences FTLD susceptibility by regulating GRN level,11 we examined whether rs1990622A was associated with brain RNA expression of TMEM106B and GRN in dorsolateral prefrontal cortex (DLPFC) and subsequently whether expression of these genes was associated with TDP-43 pathology. RNA-Seq data were currently processed in a subset of the sample used in the main analysis (n = 345). As shown in figure 2A, both TMEM106B and GRN were expressed in postmortem brain and were negatively correlated (Spearman correlation = −0.270, p < 0.0001).
Figure 2. Single-nucleotide polymorphism rs1990622, TMEM106B, and GRN expression.

(A) Scatterplot of GRN and transmembrane protein 106B (TMEM106B) expression level. (B) Box plot of TMEM106B expression level by rs1990622AA/AG vs rs1990622GG. (C) Box plot of GRN expression level by rs1990622AA/AG vs rs1990622GG. (D) Bar chart of TMEM106B expression level by transactive response DNA binding protein 43 (TDP-43) stages. (E) Bar chart of GRN expression level by TDP-43 stages.
Higher dosage of rs1990622A was associated with higher level of TMEM106B RNA expression (β = 0.173, p = 0.0235), and the association was independent of other neuropathologies. However, association of TMEM106B expression with odds of TDP-43 pathology did not reach statistical significance. Separately, we did not find direct association of rs1990622A with GRN expression. We explored the genotypic association of the SNP by collapsing rs1990622A dosage into a binary variable for rs1990622GG homozygosity (table e-3). Interestingly, the GRN RNA expression level was lower in rs1990622GG homozygous participants (figure 2C). In a linear regression model adjusted for age at death, sex, postmortem intervals, RNA degradation, and other common neuropathologies, the GRN expression was about 1.5 units higher in participants with rs1990622AA/AG than rs1990622GG (β = 1.536, p = 0.0392). This result was marginally significant considering that the association of rs1990622 with the GRN expression was tested at both allelic and genotypic levels (α = 0.05/2 = 0.025). Interestingly, higher level of GRN expression was associated with higher odds for more advanced TDP-43 stages (OR 1.059, p = 0.0024), and the association was unchanged after adjustment for other neuropathologies. In this reduced sample, the association of rs1990622A with TDP-43 pathology (OR 1.230) was comparable to the result from the larger sample, but the p value was only suggestive (p = 0.1679).
TMEM106B risk variant and GRN DNA methylation.
To investigate whether the association of the TMEM106B risk variant with GRN expression may work through an alteration of methylation levels, we interrogated the 132 CpG sites found in the GRN locus that were sampled by our BeadChip. We first performed multiple linear regressions of DNA methylation on rs1990622A. After correction for multiple testing (α = 0.05/132 = 0.0004), we found 5 CpG sites in the GRN locus where the rs1990622A dosage was associated with a small but significant decrease in methylation (table e-4). Four of these sites were located in the gene body, 3 of which were in an active enhancer (cg20727623, cg00628375, and cg16861818) based on our chromatin map of DLPFC generated from 2 older cognitively nonimpaired subjects with minimal neuropathologies.26 The fourth one (cg13120756) was located next to the GRN transcription start site that is an active conformation in this map.
Next, we examined the cis association of CpGs in the GRN locus with the gene expression (table e-5). The methylation at the 5 CpGs influenced by rs1990622 was not found to be associated with GRN expression, so we could not formally evaluate the possibility that the effect of rs1990622A on GRN expression is mediated through methylation, probably because of our modest sample size. Looking at association of other CpGs with GRN RNA expression, the top site was cg24457026 (p = 0.0003), where methylation was associated with lower level of GRN expression. This CpG is located in the 5′UTR of the GRN gene that is in a strongly transcribed conformation in our map. This result suggests the existence of multiple distinct genomic features that likely influence the level of GRN expression. Notably, the association of TMEM106B variant with GRN RNA expression was slightly attenuated after additional adjustment for methylation at cg24457026 (β = 1.420, p = 0.0549).
miR132 expression and TDP-43 pathology.
TMEM106B is a primary target of miR132, so we examined the association of miR132 expression with TDP-43 pathology. In a logistic regression model adjusted for age at death and sex, higher expression of miRNA was associated with less burden of TDP-43 pathology (OR 0.702, p = 0.0173). Since the miRNA has been reported to be downregulated in AD and FTLD,27,28 we refit the model by further adjusting for other neuropathologies. Indeed, the association of miR132 with TDP-43 pathology was no longer significant after additional neuropathologic indices were added (p = 0.7940).
DISCUSSION
It has been increasingly recognized that pathologic TDP-43 is a common pathology in aging, and contributes to late-life cognitive decline29 and dementia.30 In this study, more than half of our sample showed varying stages of accumulation of TDP-43 cytoplasmic inclusions. The major allele of the TMEM106B index SNP, rs1990622A, which is associated with susceptibility to FTLD-TDP, also increases the risk for accumulation of TDP-43 pathology in older persons without FTLD-TDP. Fine mapping of the locus suggests that top variants, including rs1990622, are mapped onto a single haplotype without a clear candidate causal variant.
TDP-43 pathology is comorbid with other neuropathologies. It has been reported that the TMEM106B variant is associated with hippocampal sclerosis in people with pathologic diagnosed AD, and a separate polymorphism in the ABCC9 gene is also shown to be associated with hippocampal sclerosis.31 In our sample, over 85% of the cases with hippocampal sclerosis have TDP-43 inclusions. Interestingly, we find little evidence for a direct association of rs1990622A with hippocampal sclerosis after adjusting for TDP-43 pathology. As contrast, we show that the association of the TMEM106B risk variant with TDP-43 is independent of AD and hippocampal sclerosis. There are several factors that might contribute to this finding. First, it is unknown whether these 2 pathologies share the same underlying neurobiology, and our results suggest that the TMEM106B variants target specifically at TDP-43, independent of hippocampal sclerosis. Second, hippocampal sclerosis is much less common than TDP-43 pathology, and the sample size in this study might not be sufficient to detect an association with hippocampal sclerosis. Further studies are warranted to disentangle the relationship of TMEM10B variant with TDP-43 and hippocampal sclerosis.
The biological mechanisms underlying the variant association in FTLD remain an active area of investigation. Prior studies on FTLD have proposed a potential pathway such that TMEM106B variants upregulate the TMEM106B expression,6 which subsequently downregulates the GRN expression,11 and GRN protein in general is neuronal protective.32 In our brain sample, we did not find association of TMEM106B expression with TDP-43 pathology; on the other hand, our result shows upregulation of GRN expression in participants who are heterozygous or homozygous for rs1990622A. Subsequently, higher level of GRN expression is related to more advanced TDP-43 pathology. Increased expression of GRN has been reported in patients with ALS.33 A possible explanation is that GRN is overexpressed in response to the neuronal inflammation or degeneration. Such overexpression is hypothesized to be the result of microglial activation.34 Microglia data collection is in progress (n = 122), and these preliminary data suggest a trend for a positive association of activated microglia and GRN expression (data not shown).
Epigenetic factors are also implicated in the pathway from the TMEM106B variants to FTLD-TDP, which include promoter DNA methylation in GRN and miR132/212 cluster. Data from this study suggest that TMEM106B risk variant downregulates DNA methylation in an active enhance region of the GRN locus, and methylation at a strong transcription region in 5′UTR of the GRN gene is associated with a lower level of GRN expression. In addition, our result on miR132 suggests that its association with TDP-43 pathology might be confounded by the role of miR132 in other neuropathologies, potentially AD. Clarification of directionality and elucidating biological mechanisms, including RNA expression and brain epigenetic changes, that potentially link TMEM106B to TDP-43 pathology will require larger numbers and further study.
The study has strengths as well as limitations. Although we excluded persons with a neuropathologic diagnosis of FTLD, the nosology of FTLD continues to evolve and the relationship between TDP-43 of aging and FTLD-TDP may not always be clear. Moreover, early disease and mixed pathologies may obscure an FTLD diagnosis. Regardless of nosology, the current data show that TDP-43, a common and deleterious pathology in aging, is independently related to TMEM106B variants, expanding the significance of this variant and related biological pathways. To our knowledge, this is the first study that investigates and confirms an independent association of TMEM106B variants with TDP-43 pathology in older persons without FTLD. Multilevel genetic, brain epigenetic, and RNA expression data are analyzed in combination with neuropathologic measures collected from hundreds of autopsied participants. These data allow testing the primary hypothesis on genetic variant association with TDP-43, and also provide an opportunity to explore multiple pathways between TMEM106B and TDP-43 pathology in aging. Additional studies are needed to replicate these findings and further elucidate the genetic basis of TDP-43 pathology in aging.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the participants of the Religious Orders Study and the Rush Memory and Aging Project who donated their data and biospecimens.
GLOSSARY
- AD
Alzheimer disease
- ALS
amyotrophic lateral sclerosis
- CBD
corticobasal degeneration
- CERAD
Consortium to Establish a Registry for Alzheimer's Disease
- DLPFC
dorsolateral prefrontal cortex
- FTLD
frontotemporal lobar degeneration
- GWAS
genome-wide association studies
- MAP
Memory and Aging Project
- miRNA
micro RNA
- OR
odds ratio
- PSP
progressive supranuclear palsy
- ROS
Religious Orders Study
- SNP
single-nucleotide polymorphism
- TDP-43
transactive response DNA binding protein 43
- TMEM106B
transmembrane protein 106B
Footnotes
Editorial, page 870
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Drafting/revising the manuscript for content: Drs. Yu, De Jager, Yang, Trojanowski, Bennett, and Schneider. Study concept or design: Drs. Yu, Bennett, and Schneider. Analysis or interpretation of the data: Drs. Yu, De Jager, Yang, Trojanowski, Bennett, and Schneider. Acquisition of data: Drs. De Jager, Bennett, and Schneider. Statistical analysis: Dr. Yu. Study supervision or coordination: Dr. Schneider. Obtaining funding: Drs. Bennett and Schneider.
STUDY FUNDING
Supported by NIH grants P30AG10161, R01AG15819, R01AG17917, R01AG36042, R01AG36836, U01AG46152, and R01AG042210.
DISCLOSURE
L. Yu receives research funding for NIH grants R01AG015819, R01AG017917, R01AG036042, R01AG033678, R01AG038651, U01AG046152, U01AG032984, and U18NS082140. P. De Jager receives research funding for NIH grants R01AG042210, R01AG036836, and R01AG036042. J. Yang receives research funding for NIH grants R01AG036042, R01AG015819, and U01AG046152, and research support from Zinfandel. J. Trojanowski receives research support for NIH grant R01AG10124. D. Bennett serves on the editorial board of Neurology®; has received honoraria for non-industry-sponsored lectures; has served as a consultant to Danone, Inc., Wilmar Schwabe GmbH & Co., Eli Lilly, Inc., Schlesinger Associates, and Geson Lehrman Group; and receives research support for NIH grants P30AG010161, R01AG015819, R01AG017917, R01AG036042, U01AG046152, R01AG039478, R01AG040039, R01NS084965, R01AG022018, P20MD006886, R01AG043617, R01NS078009, R01AG036836, R01NS082416, R01AG038651, R01NS086736, R01AG041797, P01AG014449, U18NS082140, U01AG032984, R01AG042210, R01AG043975, and R01AG034119, and research support from Zinfandel. J. Schneider has received consulting fees or sat on paid advisory boards for AVID radiopharmaceuticals, Eli Lilly Inc., and GE Healthcare; is a monitoring editor of the Journal of Histochemistry and Cytochemistry and on the editorial board of International Journal of Clinical and Experimental Pathology; and receives research funding for NIH grants R01AG042210, R01AG039478, R01AG040039, R01NS084965, R01AG022018, P30AG010161, R01AG015819, R01AG017917, R01AG036042, U01AG046152, R01AG043617, R01AG043379, R01NS078009, R01AG036836, R21ES021290, P01AG014449, and R01AG043975. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006;314:130–133. [DOI] [PubMed] [Google Scholar]
- 2.Amador-Ortiz C, Lin WL, Ahmed Z, et al. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer's disease. Ann Neurol 2007;61:435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nakashima-Yasuda H, Uryu K, Robinson J, et al. Co-morbidity of TDP-43 proteinopathy in Lewy body related diseases. Acta Neuropathol 2007;114:221–229. [DOI] [PubMed] [Google Scholar]
- 4.Nelson PT, Smith CD, Abner EL, et al. Hippocampal sclerosis of aging, a prevalent and high-morbidity brain disease. Acta Neuropathol 2013;126:161–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zarow C, Weiner MW, Ellis WG, Chui HC. Prevalence, laterality, and comorbidity of hippocampal sclerosis in an autopsy sample. Brain Behav 2012;2:435–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van Deerlin VM, Sleiman PM, Martinez-Lage M, et al. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet 2010;42:234–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van der Zee J, Van Langenhove T, Kleinberger G, et al. TMEM106B is associated with frontotemporal lobar degeneration in a clinically diagnosed patient cohort. Brain 2011;134:808–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kovacs GG, Alafuzoff I, Al-Sarraj S, et al. Mixed brain pathologies in dementia: the BrainNet Europe consortium experience. Dement Geriatr Cogn Disord 2008;26:343–350. [DOI] [PubMed] [Google Scholar]
- 9.Fotuhi M, Hachinski V, Whitehouse PJ. Changing perspectives regarding late-life dementia. Nat Rev Neurol 2009;5:649–658. [DOI] [PubMed] [Google Scholar]
- 10.Murray ME, Cannon A, Graff-Radford NR, et al. Differential clinicopathologic and genetic features of late-onset amnestic dementias. Acta Neuropathol 2014;128:411–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rutherford NJ, Carrasquillo MM, Li M, et al. TMEM106B risk variant is implicated in the pathologic presentation of Alzheimer disease. Neurology 2012;79:717–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nicholson AM, Finch NA, Wojtas A, et al. TMEM106B p.T185S regulates TMEM106B protein levels: implications for frontotemporal dementia. J Neurochem 2013;126:781–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Finch N, Carrasquillo MM, Baker M, et al. TMEM106B regulates progranulin levels and the penetrance of FTLD in GRN mutation carriers. Neurology 2011;76:467–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cruchaga C, Graff C, Chiang HH, et al. Association of TMEM106B gene polymorphism with age at onset in granulin mutation carriers and plasma granulin protein levels. Arch Neurol 2011;68:581–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lang CM, Fellerer K, Schwenk BM, et al. Membrane orientation and subcellular localization of transmembrane protein 106B (TMEM106B), a major risk factor for frontotemporal lobar degeneration. J Biol Chem 2012;287:19355–19365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen-Plotkin AS, Unger TL, Gallagher MD, et al. TMEM106B, the risk gene for frontotemporal dementia, is regulated by the microRNA-132/212 cluster and affects progranulin pathways. J Neurosci 2012;32:11213–11227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Banzhaf-Strathmann J, Claus R, Mücke O, et al. Promoter DNA methylation regulates progranulin expression and is altered in FTLD. Acta Neuropathol Commun 2013;1:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bennett DA, Schneider JA, Arvanitakis Z, Wilson RS. Overview and findings from the Religious Orders Study. Curr Alzheimer Res 2012;9:630–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bennett DA, Schneider JA, Buchman AS, Barnes LL, Boyle PA, Wilson RS. Overview and findings from the Rush Memory and Aging Project. Curr Alzheimer Res 2012;9:648–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schneider JA, Bienias JL, Wilson RS, Berry-Kravis E, Evans DA, Bennett DA. The apolipoprotein E epsilon4 allele increases the odds of chronic cerebral infarction [corrected] detected at autopsy in older persons. Stroke 2005;36:954–959. [DOI] [PubMed] [Google Scholar]
- 21.Schneider JA, Wilson RS, Bienias JL, et al. Cerebral infarctions and the likelihood of dementia from Alzheimer's disease pathology. Neurology 2004;62:1148–1156. [DOI] [PubMed] [Google Scholar]
- 22.Schneider JA, Arvanitakis Z, Yu L, Boyle PA, Leurgans SE, Bennett DA. Cognitive impairment, decline and fluctuations in older community-dwelling subjects with Lewy bodies. Brain 2012;135:3005–3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cairns NJ, Bigio EH, Mackenzie IR, et al. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol 2007;114:5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shulman JM, Yu L, Buchman AS, et al. Association of Parkinson disease risk loci with mild parkinsonian signs in older persons. JAMA Neurol 2014;71:429–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bennett DA, Yu L, De Jager PL. Building a pipeline to discover and validate novel therapeutic targets and lead compounds for Alzheimer's disease. Biochem Pharmacol 2014;88:617–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De Jager PL, Srivastava G, Lunnon K, et al. Alzheimer's disease pathology is associated with early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat NeuroSci 2014;17:1156–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wong HK, Veremeyko T, Patel N, et al. De-repression of FOXO3a death axis by microRNA-132 and -212 causes neuronal apoptosis in Alzheimer's disease. Hum Mol Genet 2013;22:3077–3092. [DOI] [PubMed] [Google Scholar]
- 28.Hébert SS, Wang WX, Zhu Q, Nelson PT. A study of small RNAs from cerebral neocortex of pathology-verified Alzheimer's disease, dementia with Lewy bodies, hippocampal sclerosis, frontotemporal lobar dementia, and non-demented human controls. J Alzheimers Dis 2013;35:335–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wilson RS, Yu L, Trojanowski JQ, et al. TDP-43 pathology, cognitive decline, and dementia in Old age. JAMA Neurol 2013;70:1418–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Keage HA, Hunter S, Matthews FE, et al. TDP-43 in the population: prevalence and associations with dementia and age. J Alzheimers Dis Epub 2014 Jun 13. [DOI] [PubMed]
- 31.Nelson PT, Estus S, Abner EL, et al. ABCC9 gene polymorphism is associated with hippocampal sclerosis of aging pathology. Acta Neuropathol 2014;127:825–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Van Damme P, Van Hoecke A, Lambrechts D, et al. Progranulin functions as a neurotrophic factor to regulate neurite outgrowth and enhance neuronal survival. J Cell Biol 2008;181:37–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Malaspina A, Kaushik N, de Belleroche J. Differential expression 14 genes amyotrophic lateral sclerosis spinal cord detected using gridded CDNA arrays. J Neurochem 2001;77:132–145. [DOI] [PubMed] [Google Scholar]
- 34.Ahmed Z, Mackenzie IR, Hutton ML, Dickson DW. Progranulin in frontotemporal lobar degeneration and neuroinflammation. J Neuroinflammation 2007;4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.