

Abstract
Inflammation is associated with several diseases of the prostate including benign enlargement and cancer, but a causal relationship has not been established. Our objective was to characterize the prostate inflammatory microenvironment after infection with a human prostate-derived bacterial strain and to determine the effect of inflammation on prostate cancer progression. To this end, we mimicked typical human prostate infection with retrograde urethral instillation of CP1, a human prostatic isolate of Escherichia coli. CP1 bacteria were tropic for the accessory sex glands and induced acute inflammation in the prostate and seminal vesicles, with chronic inflammation lasting at least 1 year. Compared to controls, infection induced both acute and chronic inflammation with epithelial hyperplasia, stromal hyperplasia, and inflammatory cell infiltrates. In areas of inflammation, epithelial proliferation and hyperplasia often persist, despite decreased expression of androgen receptor (AR). Inflammatory cells in the prostates of CP1-infected mice were characterized at 8 weeks post-infection by flow cytometry, which showed an increase in macrophages and lymphocytes, particularly Th17 cells. Inflammation was additionally assessed in the context of carcinogenesis. Multiplex cytokine profiles of inflamed prostates showed that distinct inflammatory cytokines were expressed during prostate inflammation and cancer, with a subset of cytokines synergistically increased during concurrent inflammation and cancer. Furthermore, CP1 infection in the Hi-Myc mouse model of prostate cancer accelerated the development of invasive prostate adenocarcinoma, with 70% more mice developing cancer by 4.5 months of age. This study provides direct evidence that prostate inflammation accelerates prostate cancer progression and gives insight into the microenvironment changes induced by inflammation that may accelerate tumour initiation or progression.
Keywords: prostate, prostatitis, inflammation, mouse, Escherichia coli
Introduction
Chronic inflammation is associated with increased cancer risk in a number of organs, including the stomach, pancreas, colon, and lung [1]. A variety of epidemiological and histopathological evidence suggests that prostate cancer risk also correlates with inflammation, but a causal relationship has been difficult to establish in human studies [2–6]. One factor contributing to this difficulty is the high prevalence of clinically silent prostatitis. More than 77% of men have histological evidence of prostate inflammation, but less than 10% of these men report symptoms of prostatitis [7]. A second confounding issue is detection bias. Men with symptomatic prostatitis are more likely to be diagnosed with prostate cancer because they are more likely to be treated by a urologist and subsequently screened for cancer [6]. These difficulties have driven an increased focus on animal models of prostate inflammation and prostate cancer [2].
Despite the high prevalence of prostatitis, the cause of this disease remains uncertain in most cases. Many factors have been proposed as the initiating cause of prostate inflammation, including viruses, bacteria, hormones, urinary reflux, and diet [2]. Bacteria can be isolated from only a minority of cases of prostatitis, but when a bacterial species can be cultured from prostatic secretions, the majority of these are Escherichia coli [8]. Numerous studies using non-culture-based techniques have confirmed the presence of E. coli in additional cases of prostatitis by detecting bacterial DNA in inflamed prostates and in corpora amylacea [9–11]. Together, these data indicate that prostate infection by E. coli is an important, and potentially underreported, cause of chronic prostatitis.
Inflammation alters the prostatic microenvironment in multiple ways that may facilitate cancer initiation or progression [2]. Infiltrating leukocytes secrete a variety of cytokines that promote prostate epithelial proliferation [12]. Release of reactive oxygen and nitrogen species can directly damage DNA [13]. Other inflammatory cells, especially macrophages, migrate through the stroma and can secrete proteolytic enzymes that degrade the extracellular matrix and may facilitate invasion or metastasis [14]. A variety of inflammatory cell types have been identified in human proliferative inflammatory atrophy (PIA) and prostate cancer, and have been proposed to mediate many of these changes in the microenvironment. Among these are macrophages and T cells, particularly IL-17-secreting Th17 cells [15–17]. When tested in animal models of prostate and colon cancer, these cell types were found to promote carcinogenesis or tumour progression via STAT3 activation [15,18]. Thus, multiple mechanisms have been postulated to promote cancer initiation or progression due to chronic prostatitis, but the relative contribution of each has not been established.
Animal models have been used to study prostatitis with a variety of methods to induce inflammation, including bacteria, hormone treatment, and immunization [19–22]. Although previous reports describe reactive inflammatory changes and preinvasive mouse prostatic intraepithelial neoplasia (mPIN) in mice with chronic prostatitis, the effect of prostatic inflammation on prostate cancer progression is unknown [19,21,23–25]. We chose to use a recently developed model of bacterial prostatitis using the E. coli isolate CP1. This strain of bacteria differs from other reported bacterial models in that it was isolated from the prostate of a human, and has been shown to induce chronic prostatitis in several mouse strains [26]. Previous analysis of prostatitis induced by CP1 demonstrated tropism for the prostate and induction of persistent inflammation in C57BL/6 J mice, despite the absence of detectable bacteria by culture after 28 days [26]. Because inflammation has been associated with multiple human prostatic diseases, we first characterized the long-term effects of inflammation from a human bacterial isolate on the prostatic epithelium and stroma. Additionally, as chronic inflammation has been linked to multiple cancers, including prostate cancer, we explored the influence of infection-associated inflammation on cancer progression in the Hi-Myc model of prostate cancer [27]. Here we show that CP1 induces chronic inflammation characterized by an influx of macrophages and Th17 lymphocytes, and accelerates cancer progression in Hi-Myc mice. Additionally, we demonstrate distinct cytokine profiles induced by inflammation and cancer.
Materials and methods
Mice
All experimental procedures were approved by the Johns Hopkins Institutional Animal Care and Use Committee (IACUC). Wild-type C57BL/6J and FVB/NJ mice were obtained from Jackson Laboratories (Bar Harbor, ME, USA; Stocks 664 and 1800). FVB-Tg(ARR2/Pbsn-MYC)7Key (Hi-Myc, Strain 01XF5) mice were obtained from NCI Mouse Repository (Frederick, MD, USA). Genotyping was performed using primer sets and protocols recommended by the vendor. Genomic DNA for PCR was isolated from tails.
Bacterial strain and intraurethral inoculation
CP1 is an E. coli strain of the B1 clonal group isolated from the expressed prostatic secretion (EPS) of a patient with chronic prostatitis [26]. Bacterial culture and transurethral inoculation were performed as previously described [26,28]. To infect mice, 10 μl of phosphate-buffered saline containing 1 × 108 cfu CP1 bacteria was introduced into the urethra of anaesthetized mice by catheterization. Sterile saline was introduced in control animals in an identical fashion. All mice were inoculated with a single dose of CP1 at 8 weeks of age. Heat-killed bacteria were heated at 70 °C for 30min. Culture supernatant was prepared by centrifugation followed by 0.2 μm filtration. Lack of viable cells was confirmed for heat-killed bacteria and supernatant by zero colony growth on agar plates.
Histology and immunohistochemistry
At indicated times, prostates were harvested and dissected to separate lobes, fixed in formalin, processed, embedded, sectioned, and stained with haematoxylin and eosin (H&E). Scores were determined by a primary pathologist with post-examination masking. An independent observer repeated the scoring with comprehensive masking. The kappa coefficient for inter-observer agreement was 0.609, with 76% of observations in complete agreement. The weighted kappa coefficient was 0.844, indicating very good agreement. Inflammation and cancer were scored according to established guidelines in a nominal, binary fashion (presence or absence), with prostate lobes scored separately [29,30]. Atrophy and basal/luminal cell hyperplasia were scored in an ordinal fashion, assigning a score of 0–4 according to the percentage of affected tissue in inflamed areas (0 = 0%; 1 = 1–25%; 2 = 26–50%; 3 = 51–75%; 4 = 76–100%). AR and Nkx3.1 loss were scored similarly, with a score assigned by percentage of epithelium with loss of expression in inflamed areas. Ki67, P-STAT3, and Myc were quantified by counting the percentage of IHC-positive nuclei in inflamed areas (300 nuclei per slide, five slides per group). Stromal hyperplasia was quantified by measuring the thickness of stroma (ten measurements per slide, five slides per group).
For immunohistochemistry, slides were deparaffinized and rehydrated before steaming in Target Retrieval Solution (Dako, Carpinteria, CA, USA) or EDTA pH8 (Invitrogen, Carlsbad, CA, USA) for 40min. Endogenous peroxidases were quenched with BLOXALL (Vector Labs, Burlingame, CA, USA), and the slides were blocked for 1 h with Serum-Free Protein Block (Dako). Slides were incubated with antibodies directed against CK8 (Covance, Princeton, NJ, USA; 1E8, 1/2000), CK14 (Covance; PRB-155P, 1/5000), p63 (Sigma, St Louis, MO, USA; 4A4, 1/100), F4/80 (AbD Serotec, Raleigh, NC, USA; CI:A3-1, 1/5000), CD3 (Abcam, Cambridge, MA, USA; ab5690, 1/2000), SMA (Dako; 1A4, 1/1000), Ki67 (Abcam; ab15580, 1/5000), phosphorylated STAT3 (Cell Signaling, Danvers, MA, USA; D3A7, 1/400), Myc (Abcam; Y69, 1/1000), laminin (Sigma; L9393, 1/100), AR (Santa Cruz Biotechnology, Santa Cruz, CA, USA; N-20, 1/500), vimentin (Cell Signaling; D21H3, 1/200), and Nkx3.1 (1/1000) [31]. Staining was visualized with an ImmPRESS Polymer detection kit and ImmPACT DAB (Vector Labs). Internal tissue controls, especially normal prostate, seminal vesicle, and bladder, were used as positive or negative controls as appropriate for the epithelial and mesenchymal markers CK8, CK14, p63, SMA, laminin, AR, vimentin, and Nkx3.1. Lymphoid tissue was used as internal tissue positive controls for F4/80, CD3, Ki67, and P-STAT3. Prostate tissue from Hi-Myc mice was used as a positive control for Myc staining.
Flow cytometry
To obtain prostate-infiltrating lymphocytes, prostate tissue was mechanically disrupted and incubated for 1 h at 37 °C in RPMI media with Liberase DL (Roche Applied Science, Indianapolis, IN, USA) following the manufacturer’s instructions. Lymphocytes were isolated using Ficoll-Paque Premium gradient centrifugation (GE Healthcare, Pittsburgh, PA, USA). For intracellular cytokines, lymphocytes were stimulated for 6 h with PMA (100ng/ml), ionomycin (500ng/ml), and Golgi-stop (BD Biosciences, San José, CA, USA) at 37 °C. Intracellular staining was performed with the Ebioscience permeabilization buffer. The cells were analysed by fluorescence-activated cell sorting (FACS) analysis using a FACSCalibur flow cytometer (BD Biosciences). All antibodies were purchased from BD Biosciences except anti-FoxP3 (eBioscience, San Diego, CA, USA). Staining was performed in seven panels consisting of (1) Ly6G/C (RB6-8C5), B220 (RA3-62B), CD11c (HL3), and CD49b (DX5); (2) Ly6G/C, B220, CD11c, and CD11b (M1/70); (3) γδ TCR (GL3), CD8a (53–6.7), IL-17a (TC11-18H10), and IFNγ (XMG1.2); (4) panel 3 with stimulation; (5) γδ TCR, CD4 (RM4-5), IL17a, and IFNγ; (6) panel 5 with stimulation; and (7) CD4 and FoxP3 (FJK-16s). Gating controls for each panel were prepared with Fluorescence Minus One (FMO) stains of splenocytes. Data were analysed with FlowJo software (Treestar Inc, Ashland, OR, USA).
Cytokine assay
Tissues from saline-treated C57BL/6J, saline-treated FVBN/J, CP1-treated C57BL/6J, saline-treated Hi-Myc, and CP1-treated Hi-Myc mice were harvested at 6 months of age (4 months post-inoculation). The prostate was frozen and stored at −80 °C until the assay. Frozen tissue samples were homogenized in homogenization buffer (50 mM Tris-HCl, pH7.2) containing Na3VO4 and a protease-inhibitor cocktail (Sigma-Aldrich, St Louis, MO, USA) using an OmniTH homogenizer (Omni International, Marietta, GA, USA). Following sonication, the homogenate was centrifuged at 2000 g for 5min. The resulting supernatants were collected as total prostate proteins and protein concentrations were measured using the Bio-Rad Protein Assay (Bio-Rad, Hercules, CA, USA). To quantify cytokine levels, a multiplex mouse 20-plex cytokine immunoassay (Life Technologies, Carlsbad, CA, USA) was used. Analytes measured included GM-CSF, IFNγ, IL-1α, IL-1β, IL-2, IL-4, IL-5, IL-6, IL-10, IL-12, IL-13, IL-17, TNFα, CCL2, CCL3, CXCL1, CXCL9, and CXCL10. Prostate protein samples were run in duplicate as previously described [32]. Analyte concentrations were quantified by fitting using a standard curve and normalized by total protein concentration.
Statistical analysis
All experiments were performed using five or more mice in independent experiments. For comparisons between two groups, Student’s t-test was used. For multiple comparisons, one-way ANOVA with Tukey’s multiple comparisons test was used. Statistical tests were two-sided and p values less than 0.05 were considered statistically significant. Analyses were performed using Prism GraphPad Software. Box and whisker plots indicate minimum and maximum values. Scatter dot plots include mean and error bars indicating one standard deviation.
Results
CP1 induces chronic prostatitis with stromal and epithelial hyperplasia
We first sought to determine the duration and distribution of prostatitis that could be initiated by a single intraurethral exposure to CP1 bacteria. At 8 weeks of age, wild-type C57BL/6J mice were inoculated with CP1 or sterile saline. Bacteria were delivered via a catheter into the proximal urethra (Supplementary Figure 1). Prostate tissue was harvested 8 weeks, 6 months, and 1 year after inoculation and processed for histology. Slides were examined for the presence of inflammation, defined as inflammatory cells within the epithelium or gland lumen, and atrophy, defined as shrunken or dilated glands lined by attenuated epithelial cells with minimal secretory cytoplasm [30]. No in flammation above the expected background was noted in saline-treated control mice at any time point (Figures 1A and 1E). CP1 consistently produced chronic prostatitis, with inflammation present in 89% (34/38) of mice at 8 weeks, 85% (17/20) of mice at 6 months and 40% (4/10) of mice after 1 year. After 8 weeks, mixed inflammatory cells multifocally infiltrated prostate stroma and glands, with reactive epithelial hyperplasia in inflamed glands (Figures 1B, 1F, and 1K) characterized by multiple layers of epithelial cells, tufting, and cribriform changes. At later time points, inflammatory cells were still present, but glands were often lined by attenuated, atrophic epithelium with more prominent stromal hyperplasia (Figures 1C–1D, 1G–1H, and 1K–1L). Inflammation was most common in the anterior prostate, but was common in all lobes (Figure 1I). The majority of mice (71%, 27/38) had inflammation present in two or more lobes after 8 weeks (Figure 1J); however, inflammation was not uniformly distributed, with inflamed and normal glands present in all mice. Viable intact bacteria are required during the initial inoculation to produce this sustained inflammatory response, as neither heat-killed bacteria nor filtered culture supernatant produced any inflammation (Supplementary Figure 2).
Figure 1.
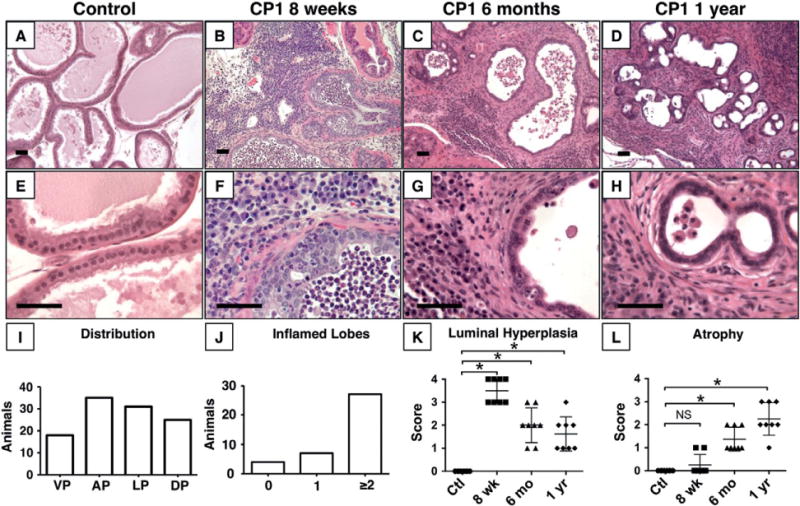
CP1 induces chronic prostatitis. Representative images of prostate glands after control saline treatment (A, E) or CP1-induced inflammation at 8 weeks (B, F), 6 months (C, G), and 1 year (D, H) after infection show sustained inflammation. (I) Distribution of inflammation by individual lobe (N =38). DL = dorsal; AP = anterior; LP = lateral; VP = ventral. (J) Total number of inflamed lobes per mouse (N =38). (K) Luminal hyperplasia score is significantly increased at 8 weeks, 6 months, and 1 year after inoculation compared with saline-treated controls (p < 0.001, n =8 per group). (L) Atrophy score is significantly increased at 6 months and 1 year after inoculation compared with saline-treated controls (p < 0.001, n =8 per group). Scale bar =50 μm.
Consistent with previous reports of other models of chronic prostatitis, CP1 inflammation induces reactive hyperplasia in both basal and luminal compartments [21,23]. Basal cells, marked by immunohistochemical staining for cytokeratin 14 (CK14) or p63, increased from a single, incomplete layer in control mice to a layer often two to three cells thick in areas of inflammation at early time points (Figures 2A–2D). Similarly, the luminal cell layer, marked by CK8, was thickened in inflamed areas (Figures 2E and 2F). At later time points, epithelial hyperplasia was less common, with inflamed glands typically lined by a single layer of attenuated, atrophic epithelial cells with minimal cytoplasm and an incomplete basal cell layer (Figures 1K and 1L and Supplementary Figure 3). In addition to epithelial changes, marked stromal thickening was noted in inflamed glands, especially at later time points (Figures 1C and 1D). Prostate glands in saline-treated controls were surrounded by a thin layer of smooth muscle (Figure 2K), with minimal collagen and few vimentin-positive stromal cells (Supplementary Figure 4). Inflamed glands showed a disruption of the smooth muscle actin (SMA)-positive stromal layer (Figure 2L). The inflamed stroma was significantly thickened by bands of collagen, indicated by blue staining with Masson’s trichrome histochemical stain, and vimentin-positive reactive stromal cells (Supplementary Figure 4).
Figure 2.
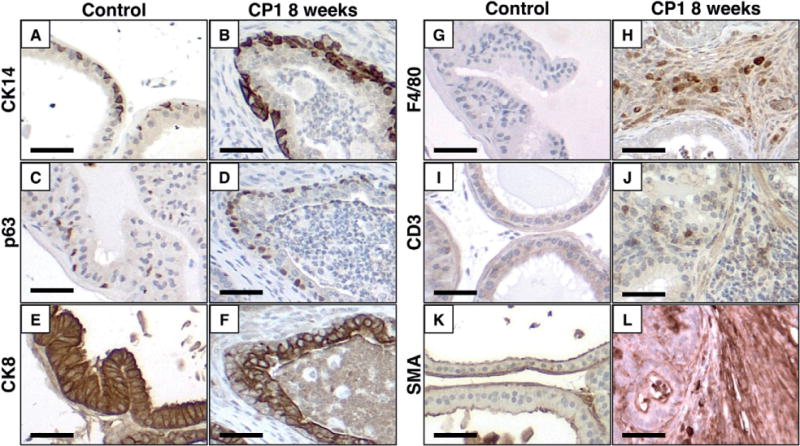
CP1-infected prostates are hyperplastic and infiltrated by immune cells. Representative images of immunohistochemistry comparing saline-treated controls and CP1-infected prostates 8 weeks after infection demonstrate hyperplasia of basal cells (CK14, A, B; p63 C, D), luminal cells (CK8, E, F), and thickened stroma with disruption of the smooth muscle actin-positive layers (SMA, K, L). Inflamed prostates show increased infiltration of macrophages (F4/80, G, H) and T cells (CD3, I, J). Scale bar =50 μm.
In addition to hyperplastic changes, inoculation of CP1 induced infiltration of numerous immune cells. Immunohistochemical analysis of inflamed prostates for the macrophage marker F4/80 showed marked infiltration of the stroma and glands 8 weeks post-infection (Figures 2G and 2H). Similarly, CD3+ T cells were present throughout inflamed areas of the prostates, while rare in controls (Figures 2I and 2J). To further characterize the inflammatory infiltrate, dissociated prostates from wild-type mice (n =5 per group) were analysed by flow cytometry 8 weeks post-infection to quantify inflammatory cells. The absolute number of lymphocytes and macrophages was significantly increased at this time point (Figures 3A–3D). The relative populations of T cells (including CD4+, CD8+, γδ+) and their respective subtypes (IFNγ + or IL-17+), CD4+ FoxP3+ cells (Treg), and CD49b + cells (including NK cells) were quantified. Significant increases in the relative proportion of γδ T cells and CD4+ IL-17+ (Th17) cells were noted in inflamed prostates compared with saline-treated controls (Figure 3).
Figure 3.
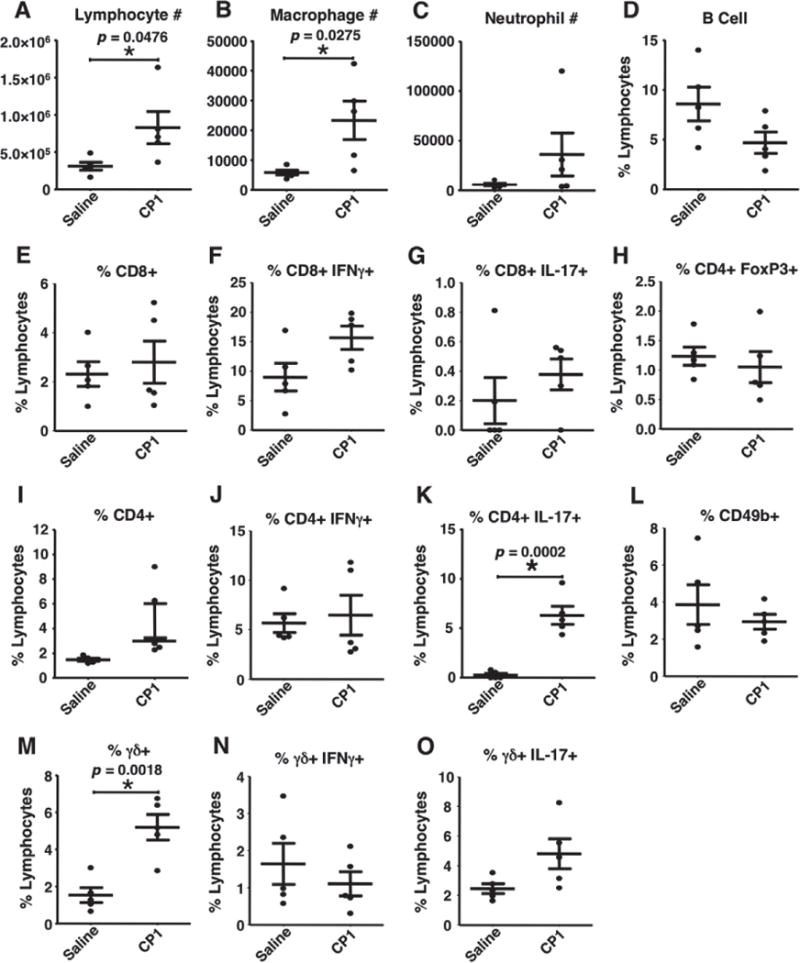
Prostate inflammatory phenotype. Analysis of prostate-infiltrating immune cells by flow cytometry comparing saline-treated controls and CP1-infected wild-type prostates 8 weeks after infection (n =5 per group). Significant differences (p <0.05) were noted in total lymphocyte number, macrophage number, total CD4+ and γδ T cells, and CD4+ IL-17+ T cells.
Chronic prostatitis promotes prostate cancer progression
CP1 inoculation produces marked epithelial and stromal hyperplasia with moderate atypia and occasional cribriform morphology that resembles mPIN, but no definitive foci of invasive cancer were identified at any time point. This indicates that CP1 infection alone is insufficient to initiate invasive cancer formation in wild-type C57BL/6J mice. However, upregulation of multiple oncogenes and pro-oncogenic pathways suggested that CP1-induced inflammation could affect prostate cancer progression. Epithelial proliferation, determined by immunohistochemical staining for Ki67, was dramatically increased at 8 weeks post-infection and remained elevated 1 year post-infection (Figures 4A, 4E, 4I, and 4M). Quantification of the percentage of Ki67-positive epithelial cells showed a significant increase at 8 weeks (9.3 ± 1.8%, p <0.001) and 1 year (10.9 ± 3.2%, p <0.001) compared with controls (0.5 ± 0.3%). In addition to increased proliferation, inflamed prostates showed significantly increased expression of MYC and activation of STAT3 (Figure 4), both oncogenes shown to promote prostate cancer initiation or progression in mice and humans [27,33–35]. Finally, inflammation induced fragmentation of the normally continuous prostate basement membrane, which could facilitate cancer invasion (Figures 4D, 4H, and 4L).
Figure 4.
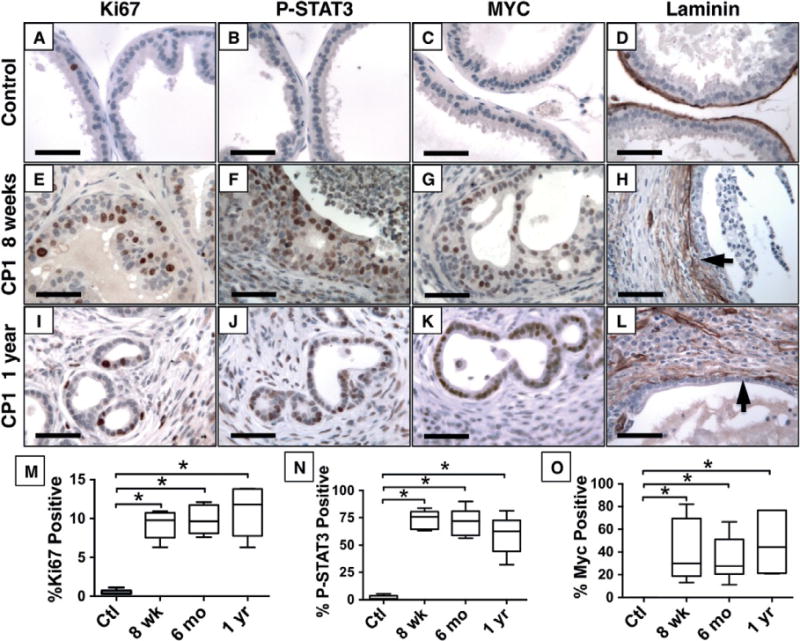
CP1-inflamed epithelium adopts a proliferative and pro-oncogenic phenotype. Immunohistochemistry demonstrates increased proliferation (Ki67, A, E, I), phosphorylated STAT3 (B, F, J) and c-MYC (C, G, K) expression, and disrupted laminin (D, H, L) by CP1-infected mice after 8 weeks (E–H) and 1 year (I–L) compared with saline-treated control mice (A–D). Quantification of the percentage of positive nuclei shows significant increases in Ki67 (M), P-STAT3 (N), and Myc (O) expression (p <0.001, n =8 per group). Scale bar =50 μm.
Similar to previous reports, AR expression and the AR target NKX3.1 are diminished in areas of inflammation (Figure 5) [21]. Luminal cells typically express high levels of nuclear AR and NKX3.1 (Figures 5A and 5E). In castrated mice, AR levels decrease and nuclear localization is lost, with a subsequent decrease in androgen-dependent genes, such as Nkx3.1 (Figures 5B and 5F). In inflamed prostate glands, AR and Nkx3.1 expression are diminished (Figures 5C, 5G, 5I and 5J), but the epithelial cells remain highly proliferative (Figures 5D and 5H).
Figure 5.
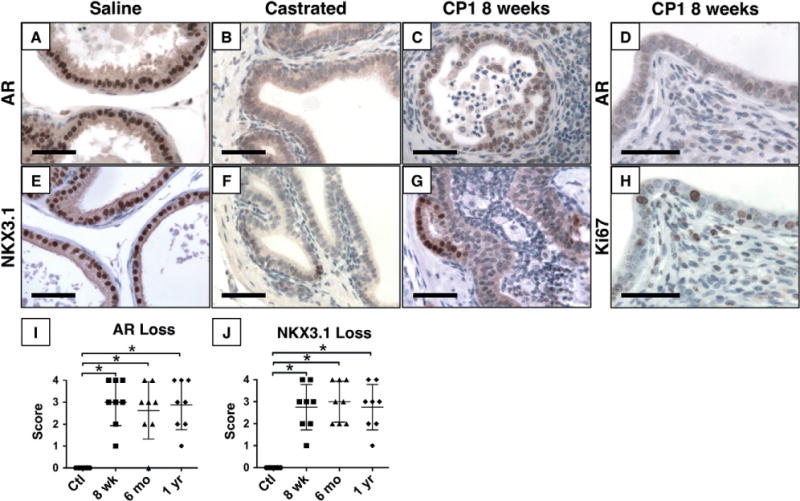
Diminished AR expression and signalling in inflamed epithelium. Immunohistochemistry for AR (A–D) and NKX3.1 (E–G), an androgen regulated protein, shows strong nuclear expression in normal glands (A, E), but significantly decreased expression of both proteins 2 weeks after castration (B, F). Expression of both proteins is decreased to castrate levels in inflamed epithelium 8 weeks post-inoculation (C, G). Despite decreased AR expression, inflamed epithelium remains highly proliferative with numerous Ki67-positive nuclei (D, H). Quantification of the percentage of positive nuclei shows significant loss of AR (I) and NKX3.1 (J) expression (p <0.001, n =8 per group). Scale bar =50 μm.
Because CP1-induced inflammation induces pro-oncogenic epithelial changes, we investigated its role in prostate cancer progression. We inoculated Hi-Myc mice with CP1 or sterile saline at 8 weeks of age. In this model of prostate cancer, mPIN develops at an early age and progresses to invasive cancer by 6 months of age in 100% of animals [36]. At 4.5 months of age (137 ± 2 days), a minority of saline-treated mice had foci of invasive cancer (43%, N =26/60). However, significantly more CP1-treated mice had invasive cancer (73%, N =22/30, p =0.008). Similar to wild-type mice, Hi-Myc mice showed little expression of phosphorylated STAT3 throughout tumourigenesis. In contrast, phosphorylated STAT3 expression was widespread in inflamed areas of CP1-treated Hi-Myc mice (Figure 6). Reactive hyperplasia was present in inflamed areas of Hi-Myc mice, similar to B6 mice at 8 weeks post-inoculation.
Figure 6.
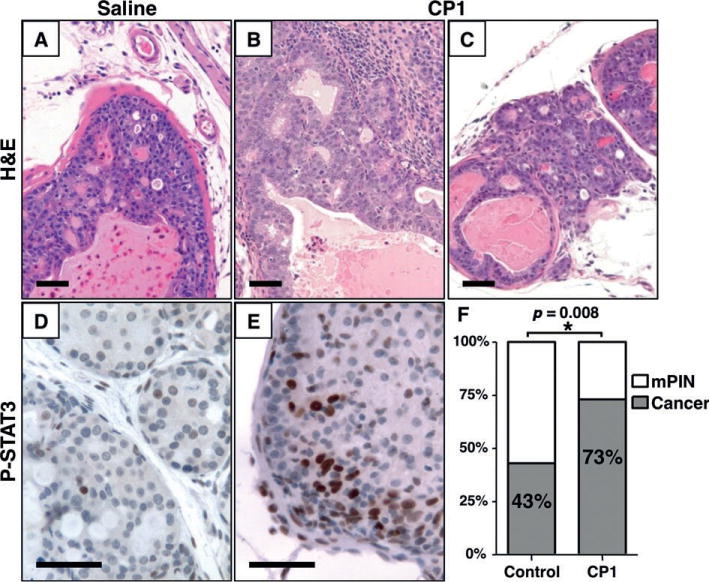
CP1 infection accelerates cancer progression. Representative images of prostates from 4.5-month-old Hi-Myc mice treated with saline (A, D) or CP1 (B, C, E). Immunohistochemistry for phosphorylated STAT3 (D, E) shows activation in CP1-treated, but not saline-treated controls. At 4.5 months of age (F), significantly more mice have invasive carcinoma in the CP1-treated group compared with controls (22/30 versus 26/60, p =0.008). Scale bar =50 μm.
Inflammation and cancer have distinct cytokine profiles
The high incidence of both prostate inflammation and prostate cancer in humans makes it difficult to determine the individual effect of each on cytokine expression and immune cell infiltrates [16]. However, these processes can be separated in mouse models. We employed multiplex assays to compare cytokine levels in saline-treated wild-type mice, CP1-inoculated wild-type mice, saline-treated Hi-Myc mice, and CP1-inoculated Hi-Myc mice, all at 6 months of age (4 months post-infection, Figure 7). Of the 20 cytokines and chemokines included in the multiplex ELISA, 17 had significantly different expression levels among the treatment groups. These cytokines could be separated into distinct profiles of those associated specifically with inflammation, with cancer, or only when inflammation and cancer were present concurrently (Table 1). Cytokines were considered to be associated with inflammation when expression levels were significantly increased in CP1-treated animals compared with wild-type and cancer groups. Cytokines were considered to be associated with cancer when expression levels were significantly increased in Hi-Myc mice compared with wild-type and CP1-treated wild-type mice.
Figure 7.
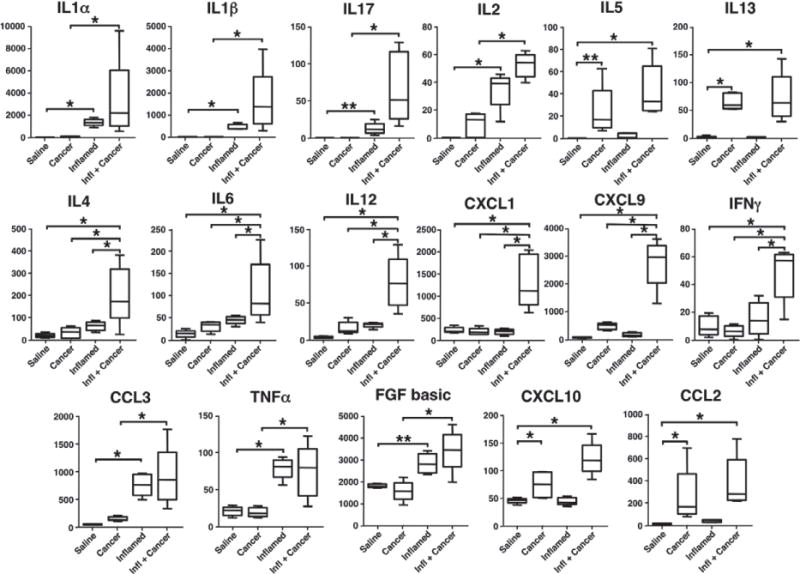
Cytokine expression during inflammation and cancer. Multiplex cytokine profiles of whole prostate lysates from 6-month-old (4 months post-inoculation) saline-treateld wild-type mice (Saline), saline-treated Hi-Myc mice (Cancer), CP1-treated wild-type mice (Inflamed), and CP1-treated Hi-Myc mice (Infl + Cancer). Box plots show median and min/max values of ten mice for saline-treated controls and five mice in other groups. *p <0.01; ** p <0.05.
Table 1.
Inflammation and cancer have distinct cytokine profiles. Cytokines can be grouped according to expression profile. Cytokines associated with inflammation are significantly increased in inflamed prostates, regardless of cancer status. Cytokines associated with cancer are significantly increased in cancer, regardless of inflammation status. Concurrent inflammation and cancer cytokines are significantly increased only when inflammation and cancer are simultaneous
| Cytokines associated with inflammation
|
Cytokines associated with cancer
|
|||
|---|---|---|---|---|
| Inflammation only | Augmented by cancer | Cancer only | Augmented by inflammation | Concurrent inflammation and cancer only |
| CCL3 | IL-1a IL-2 | IL-5 | CXCL10 | IL-4 CXCL1 |
| FGF | IL-1b IL-17 | IL-13 | IL-6 CXCL9 | |
| TNFα | CCL2 | IL-12 IFNγ | ||
Discussion
Inflammation contributes to cancer initiation and progression in a variety of organs, and has been shown to act directly by inducing genetic changes and indirectly by altering the microenvironment through immune cell infiltrate and cytokine expression. Here, we show the first evidence that prostate inflammation accelerates prostate cancer progression and examine the complex inflammatory infiltrate and cytokine milieu associated with inflammation in the prostate glandular microenvironment.
CP1 model of prostatitis
In this report, we utilize the CP1 model of prostatitis, which has proven useful for investigations into mechanisms of prostatitis, chronic pelvic pain, and now prostate cancer. We feel that this model has several distinct advantages. CP1 was isolated from prostatic secretions and thus may more closely model typical human infectious prostatitis. Additionally, some infectious models of prostatitis require direct injection into the prostate or project bacteria into prostate ducts by larger-volume urethral inoculation. CP1 colonizes the mouse prostate after infusion of inoculate of a very small volume into the urethra, thus eliminating the confounding inflammation related to urine reflux or distension of prostate ducts. We show that CP1 is capable of inducing inflammation that persists at least 1 year after a single infusion of bacteria and clearance of the bacteria within 28 days of inoculation [26]. It is not clear if the ability to promote cancer progression is a unique feature of CP1 or if this is a feature of inflammation in general. This will be possible by comparing CP1-induced acceleration of prostate cancer with inflammation induced by other bacteria or other means of inducing prostatitis.
Inflammatory microenvironment
Inflammation induced by CP1 recruits a variety of inflammatory cells, especially macrophages and lymphocytes, to the prostate. One component is a strong Th17 response, indicated both by an increase in CD4+ IL-17+ cells and by an increase in IL-17 production during CP1-induced inflammation. This finding is consistent with previous reports of a strong Th17 response to bacteria [18,37]. However, not all inflammatory models of prostatitis evoke a Th17 response, and the intensity of the Th17 response has been shown to be dependent on the mouse strain and can vary based on environmental factors [38,39]. We hypothesize that accelerated cancer progression in CP1-infected mice is, at least in part, due to activation of the IL-17/IL-6/STAT3 pathway, as this pathway is critical for cancer progression in other organs, and IL-17 has been shown to promote prostate cancer progression in mice [18,40,41]. Although our investigation focused on immune cell subtypes and cytokine expression during inflammation and cancer, one histological hallmark of cancer is invasion through the basement membrane. Stromal remodelling and disruption of the basement membrane, which occurs during inflammation, may provide a purely mechanical advantage for pre-invasive lesions to progress towards malignancy.
Cytokine profiles of inflammation and cancer
Although we anticipated different cytokine expression profiles for infection and cancer, the distinct profile of coincident inflammation and cancer was surprising. This profile may be due to an increase in the intensity of inflammation from the dual stimuli of bacterial infection and cancer or from synergistic interactions of anti-bacterial and anti-tumour inflammation. Cytokine levels during simultaneous inflammation and cancer were higher than either state alone for 11/20 cytokines tested. This increase in inflammatory intensity may activate cytokine expression not present in either state alone. For example, IL-17 is significantly increased during inflammation, but concurrent inflammation and cancer caused an eight-fold increase in mean IL-17 expression compared with inflammation alone. IL-6 expression can be induced by IL-17 activity, and the dramatic increase in IL-17 during simultaneous inflammation and cancer may explain the up-regulation of IL-6 only in this environment [40]. An alternative explanation for the distinct profiles is that the immune responses to infection and cancer initiation are different, and the combination of the two invokes a third distinct immune microenvironment. Inflammation in response to bacterial exposure produces significant tissue damage from reactive oxygen species produced by macrophages and neutrophils. When coupled with the immune response to tumour formation, this may result in a different immune phenotype than either process alone.
If confirmed in human studies, these distinct cytokine profiles could provide a useful diagnostic tool to segregate inflammation and cancer. Although it is more difficult to separate the influence of inflammation from cancer on cytokine expression in humans, we see some correlation between our data and published cytokine data in human studies. For example, TNFα and IL-1α were previously found to be up-regulated in men with chronic prostatitis or chronic pelvic pain syndrome [42]. We have identified a cytokine profile specific for murine prostate cancer (IL-5, IL-13, CCL2). This profile is traditionally associated more with allergic disease than cancer, but IL-13 and CCL2 have been shown to promote prostate cancer cell proliferation and migration [43–45]. Interestingly, the cytokine profiles of CP1-induced inflammation and cancer were mutually exclusive, ie no cytokine was elevated compared with controls in both settings. Although significant immune cell infiltrate is present in Hi-Myc mice with cancer and in mice treated with CP1, this confirms that the microenvironments of the two processes are quite distinct.
Increased proliferation despite decreased androgen receptor expression
Several groups have reported a decrease in AR expression during inflammation [20,21,46]. Here, we show that inflammation induces increased proliferation in luminal epithelial cells despite diminished AR expression. AR has been proposed to function as a growth suppressor in mature luminal cells, but the mechanism driving this programme is unclear from this study [47].
We have demonstrated that chronic prostatitis promotes prostate cancer progression, but this study may be limited by the use of a single bacterial strain and a single model of prostate cancer. Although CP1 was isolated form a human patient, due to the inherent anatomical and physiological differences between mice and humans, we cannot predict that the inflammatory response in the two species will be identical. Furthermore, because we exclusively tested our hypothesis in the Hi-Myc model of prostate cancer, we cannot predict if these findings will apply to other models, such as TRAMP or PTEN deletion. Our study focuses on chronic prostatitis without antibiotic treatment. We have not tested the possibility that antibiotic treatment early in the course of prostatitis is sufficient to prevent acceleration of prostate cancer, but this remains a focus for future studies and may be relevant for translational applications.
Supplementary Material
Figure S1. Catheter placement.
Figure S2. Live bacteria are required to induce inflammation.
Figure S3. Luminal and basal cell hyperplasia partially resolve at later time points.
Figure S4. Inflammation induces reactive stromal hyperplasia.
Acknowledgments
We thank Ms Rebecca Miller and Ms Javaneh Jabbari for excellent technical support, and Dr Xuhang Li and Mr Douglas Adler at the Hopkins Conte Digestive Diseases Research Core for assistance with multiplex cytokine assays. This work was supported by the Prostate Cancer Foundation Norway/Hagen Initiative (EMS), Patrick C Walsh Prostate Cancer Foundation (PJH, EMS), AUA Rising Star (EMS), and HHMI Early Careers Physician Scientist (EMS).
Abbreviations
- AR
androgen receptor
- PIA
proliferative inflammatory atrophy
- PIN
prostatic intraepithelial neoplasia
- PMA
phorbol myristate acetate
- PSA
prostate-specific antigen
Footnotes
No conflicts of interest were declared.
Author contribution statement
BWS, PT, AJS, AR, PJH, CGD, DMB, and EMS designed experiments. BWS, ND, TB, and JG carried out experiments and analysed data. PT, AJS, and CGD provided reagents or materials. All authors were involved in writing the paper and had final approval of the submitted and published versions.
SUPPORTING INFORMATION ON THE INTERNET
The following supporting information may be found in the online version of this article:
References
- 1.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Marzo AM, Platz EA, Sutcliffe S, et al. Inflammation in prostate carcinogenesis. Nature Rev Cancer. 2007;7:256–269. doi: 10.1038/nrc2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Palapattu GS, Sutcliffe S, Bastian PJ, et al. Prostate carcinogenesis and inflammation: emerging insights. Carcinogenesis. 2005;26:1170–1181. doi: 10.1093/carcin/bgh317. [DOI] [PubMed] [Google Scholar]
- 4.Platz EA, De Marzo AM. Epidemiology of inflammation and prostate cancer. J Urol. 2004;171:S36–S40. doi: 10.1097/01.ju.0000108131.43160.77. [DOI] [PubMed] [Google Scholar]
- 5.Cheng I, Witte JS, Jacobsen SJ, et al. Prostatitis, sexually transmitted diseases, and prostate cancer: the California Men’s Health Study. PLoS One. 2010;5:e8736. doi: 10.1371/journal.pone.0008736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nelson WG, De Marzo AM, DeWeese TL, et al. The role of inflammation in the pathogenesis of prostate cancer. J Urol. 2004;172:S6–S11. doi: 10.1097/01.ju.0000142058.99614.ff. discussion S11–S12. [DOI] [PubMed] [Google Scholar]
- 7.Nickel JC, Roehrborn CG, O’Leary MP, et al. The relationship between prostate inflammation and lower urinary tract symptoms: examination of baseline data from the REDUCE trial. Eur Urol. 2008;54:1379–1384. doi: 10.1016/j.eururo.2007.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoon BI, Kim S, Han DS, et al. Acute bacterial prostatitis: how to prevent and manage chronic infection? J Infect Chemother. 2012;18:444–450. doi: 10.1007/s10156-011-0350-y. [DOI] [PubMed] [Google Scholar]
- 9.Yanamandra K, Alexeyev O, Zamotin V, et al. Amyloid formation by the pro-inflammatory S100A8/A9 proteins in the ageing prostate. PLoS One. 2009;4:e5562. doi: 10.1371/journal.pone.0005562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hochreiter WW, Duncan JL, Schaeffer AJ. Evaluation of the bacterial flora of the prostate using a 16S rRNA gene based polymerase chain reaction. J Urol. 2000;163:127–130. [PubMed] [Google Scholar]
- 11.Krieger JN, Riley DE, Vesella RL, et al. Bacterial DNA sequences in prostate tissue from patients with prostate cancer and chronic prostatitis. J Urol. 2000;164:1221–1228. [PubMed] [Google Scholar]
- 12.McDowell KL, Begley LA, Mor-Vaknin N, et al. Leukocytic promotion of prostate cellular proliferation. Prostate. 2010;70:377–389. doi: 10.1002/pros.21071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wiseman H, Halliwell B. Damage to DNA by reactive oxygen and nitrogen species: role in inflammatory disease and progression to cancer. Biochem J. 1996;313:17–29. doi: 10.1042/bj3130017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Comito G, Giannoni E, Segura CP, et al. Cancer-associated fibroblasts and M2-polarized macrophages synergize during prostate carcinoma progression. Oncogene. 2014;33:2423–2431. doi: 10.1038/onc.2013.191. [DOI] [PubMed] [Google Scholar]
- 15.Fang LY, Izumi K, Lai KP, et al. Infiltrating macrophages promote prostate tumorigenesis via modulating androgen receptor-mediated CCL4–STAT3 signaling. Cancer Res. 2013;73:5633–5646. doi: 10.1158/0008-5472.CAN-12-3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sfanos KS, Bruno TC, Maris CH, et al. Phenotypic analysis of prostate-infiltrating lymphocytes reveals TH17 and Treg skewing. Clin Cancer Res. 2008;14:3254–3261. doi: 10.1158/1078-0432.CCR-07-5164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vykhovanets EV, Maclennan GT, Vykhovanets OV, et al. IL-17 expression by macrophages is associated with proliferative inflammatory atrophy lesions in prostate cancer patients. Int J Clin Exp Pathol. 2011;4:552–565. [PMC free article] [PubMed] [Google Scholar]
- 18.Wu S, Rhee KJ, Albesiano E, et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nature Med. 2009;15:1016–1022. doi: 10.1038/nm.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haverkamp JM, Charbonneau B, Crist SA, et al. An inducible model of abacterial prostatitis induces antigen specific inflammatory and proliferative changes in the murine prostate. Prostate. 2011;71:1139–1150. doi: 10.1002/pros.21327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shinohara DB, Vaghasia AM, Yu SH, et al. A mouse model of chronic prostatic inflammation using a human prostate cancer-derived isolate of Propionibacterium acnes. Prostate. 2013;73:1007–1015. doi: 10.1002/pros.22648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khalili M, Mutton LN, Gurel B, et al. Loss of Nkx3.1 expression in bacterial prostatitis: a potential link between inflammation and neoplasia. Am J Pathol. 2010;176:2259–2268. doi: 10.2353/ajpath.2010.080747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Naslund MJ, Strandberg JD, Coffey DS. The role of androgens and estrogens in the pathogenesis of experimental nonbacterial prostatitis. J Urol. 1988;140:1049–1053. doi: 10.1016/s0022-5347(17)41924-0. [DOI] [PubMed] [Google Scholar]
- 23.Kwon OJ, Zhang L, Ittmann MM, et al. Prostatic inflammation enhances basal-to-luminal differentiation and accelerates initiation of prostate cancer with a basal cell origin. Proc Natl Acad Sci U S A. 2014;111:E592–E600. doi: 10.1073/pnas.1318157111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boehm BJ, Colopy SA, Jerde TJ, et al. Acute bacterial inflammation of the mouse prostate. Prostate. 2012;72:307–317. doi: 10.1002/pros.21433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elkahwaji JE, Zhong W, Hopkins WJ, et al. Chronic bacterial infection and inflammation incite reactive hyperplasia in a mouse model of chronic prostatitis. Prostate. 2007;67:14–21. doi: 10.1002/pros.20445. [DOI] [PubMed] [Google Scholar]
- 26.Rudick CN, Berry RE, Johnson JR, et al. Uropathogenic Escherichia coli induces chronic pelvic pain. Infect Immun. 2011;79:628–635. doi: 10.1128/IAI.00910-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ellwood-Yen K, Graeber TG, Wongvipat J, et al. Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell. 2003;4:223–238. doi: 10.1016/s1535-6108(03)00197-1. [DOI] [PubMed] [Google Scholar]
- 28.Thumbikat P, Berry RE, Zhou G, et al. Bacteria-induced uroplakin signaling mediates bladder response to infection. PLoS Pathogens. 2009;5:e1000415. doi: 10.1371/journal.ppat.1000415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ittmann M, Huang J, Radaelli E, et al. Animal models of human prostate cancer: the consensus report of the New York meeting of the Mouse Models of Human Cancers Consortium Prostate Pathology Committee. Cancer Res. 2013;73:2718–2736. doi: 10.1158/0008-5472.CAN-12-4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shappell SB, Thomas GV, Roberts RL, et al. Prostate pathology of genetically engineered mice: definitions and classification. The consensus report from the Bar Harbor meeting of the Mouse Models of Human Cancer Consortium Prostate Pathology Committee. Cancer Res. 2004;64:2270–2305. doi: 10.1158/0008-5472.can-03-0946. [DOI] [PubMed] [Google Scholar]
- 31.Chen H, Bieberich CJ. Structural and functional analysis of domains mediating interaction between NKX-3.1 and PDEF. J Cell Biochem. 2005;94:168–177. doi: 10.1002/jcb.20297. [DOI] [PubMed] [Google Scholar]
- 32.Alex P, Zachos NC, Nguyen T, et al. Distinct cytokine patterns identified from multiplex profiles of murine DSS and TNBS-induced colitis. Inflamm Bowel Dis. 2009;15:341–352. doi: 10.1002/ibd.20753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blando JM, Carbajal S, Abel E, et al. Cooperation between Stat3 and Akt signaling leads to prostate tumor development in transgenic mice. Neoplasia. 2011;13:254–265. doi: 10.1593/neo.101388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gurel B, Iwata T, Koh CM, et al. Nuclear MYC protein overexpression is an early alteration in human prostate carcinogenesis. Mod Pathol. 2008;21:1156–1167. doi: 10.1038/modpathol.2008.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Abdulghani J, Gu L, Dagvadorj A, et al. Stat3 promotes metastatic progression of prostate cancer. Am J Pathol. 2008;172:1717–1728. doi: 10.2353/ajpath.2008.071054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Iwata T, Schultz D, Hicks J, et al. MYC overexpression induces prostatic intraepithelial neoplasia and loss of Nkx3.1 in mouse luminal epithelial cells. PLoS One. 2010;5:e9427. doi: 10.1371/journal.pone.0009427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Osborne LC, Joyce KL, Alenghat T, et al. Resistin-like molecule alpha promotes pathogenic Th17 cell responses and bacterial-induced intestinal inflammation. J Immunol. 2013;190:2292–2300. doi: 10.4049/jimmunol.1200706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ivanov II, Atarashi K, Manel N, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139:485–498. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Quick ML, Wong L, Mukherjee S, et al. Th1–Th17 cells contribute to the development of uropathogenic Escherichia coli-induced chronic pelvic pain. PLoS One. 2013;8:e60987. doi: 10.1371/journal.pone.0060987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang L, Yi T, Kortylewski M, et al. IL-17 can promote tumor growth through an IL-6–Stat3 signaling pathway. J Exp Med. 2009;206:1457–1464. doi: 10.1084/jem.20090207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Q, Liu S, Ge D, et al. Interleukin-17 promotes formation and growth of prostate adenocarcinoma in mouse models. Cancer Res. 2012;72:2589–2599. doi: 10.1158/0008-5472.CAN-11-3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nadler RB, Koch AE, Calhoun EA, et al. IL-1beta and TNF-alpha in prostatic secretions are indicators in the evaluation of men with chronic prostatitis. J Urol. 2000;164:214–218. [PubMed] [Google Scholar]
- 43.Loberg RD, Day LL, Harwood J, et al. CCL2 is a potent regulator of prostate cancer cell migration and proliferation. Neoplasia. 2006;8:578–586. doi: 10.1593/neo.06280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Loberg RD, Ying C, Craig M, et al. CCL2 as an important mediator of prostate cancer growth in vivo through the regulation of macrophage infiltration. Neoplasia. 2007;9:556–562. doi: 10.1593/neo.07307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maini A, Hillman G, Haas GP, et al. Interleukin-13 receptors on human prostate carcinoma cell lines represent a novel target for a chimeric protein composed of IL-13 and a mutated form of Pseudomonas exotoxin. J Urol. 1997;158:948–953. doi: 10.1097/00005392-199709000-00077. [DOI] [PubMed] [Google Scholar]
- 46.Debelec-Butuner B, Alapinar C, Varisli L, et al. Inflammation-mediated abrogation of androgen signaling: an in vitro model of prostate cell inflammation. Mol Carcinog. 2014;53:85–97. doi: 10.1002/mc.21948. [DOI] [PubMed] [Google Scholar]
- 47.Litvinov IV, De Marzo AM, Isaacs JT. Is the Achilles’ heel for prostate cancer therapy a gain of function in androgen receptor signaling? J Clin Endocrinol Metab. 2003;88:2972–2982. doi: 10.1210/jc.2002-022038. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Catheter placement.
Figure S2. Live bacteria are required to induce inflammation.
Figure S3. Luminal and basal cell hyperplasia partially resolve at later time points.
Figure S4. Inflammation induces reactive stromal hyperplasia.