

Abstract
Protein–protein interactions encompass large surface areas, but often a handful of key residues dominate the binding energy landscape. Rationally designed small molecule scaffolds that reproduce the relative positioning and disposition of important binding residues, termed “hotspot residues”, have been shown to successfully inhibit specific protein complexes. Although this strategy has led to development of novel synthetic inhibitors of protein complexes, often direct mimicry of natural amino acid residues does not lead to potent inhibitors. Experimental screening of focused compound libraries is used to further optimize inhibitors but the number of possible designs that can be efficiently synthesized and experimentally tested in academic settings is limited. We have applied the principles of computational protein design to optimization of nonpeptidic helix mimics as ligands for protein complexes. We describe the development of computational tools to design helix mimetics from canonical and noncanonical residue libraries and their application to two therapeutically important protein–protein interactions: p53-MDM2 and p300-HIF1α. The overall study provides a streamlined approach for discovering potent peptidomimetic inhibitors of protein–protein interactions.
Introduction
Protein–protein interactions are often mediated by amino acid residues organized on secondary structures.1 Designed oligomeric ligands that can mimic the array of protein-like functionality at interfaces offer an attractive approach to target therapeutically important interactions.2 Efforts to mimic interfacial α-helices have resulted in three overarching synthetic strategies: helix stabilization, helical foldamers, and helical surface mimetics.3,4 Helix stabilization employs side chain cross-links5,6 or hydrogen-bond surrogates7 to preorganize amino acid residues and initiate helix formation. Helical foldamers are nonnatural oligomers that adopt defined helical conformations;8,9 prominent examples include β-peptide10−12 and peptoid helices.13 Helical surface mimetics utilize conformationally restricted scaffolds with attached functional groups that mimic the topography of α-helical side chains. With the exception of some elegant examples,14−18 surface mimetics typically impart functionality from one face of the helix, while stabilized peptide helices and foldamers are able to reproduce functionality present on multiple faces of the target helix.19 A key advantage of helix surface mimicry is that it affords low molecular weight compounds as modulators of protein interactions.20−25
A recent survey of protein–protein complexes in the Protein Data Bank (PDB) suggests that a significant portion of interface helices use one face to target the binding partner.19,26 This analysis points to the meaningful role that topographical helix mimics can play in affording small molecule inhibitors of protein–protein interactions for which no inhibitors are currently known. The classical examples of helix surface mimics were described by Hamilton et al.27−29 and contained aromatic scaffolds displaying protein-like functionality.3 Inspired by this work, we proposed oligooxopiperazines as a new class of helix mimetics (Figure 1).23 The advantage of oxopiperazine-based scaffolds is that they offer chiral backbones and can be easily assembled from α-amino acids allowing rapid diversification of the scaffold. We were also attracted to the piperazine motif because 2-oxopiperazines and diketopiperazines have a rich history in medicinal chemistry.30−35
Figure 1.
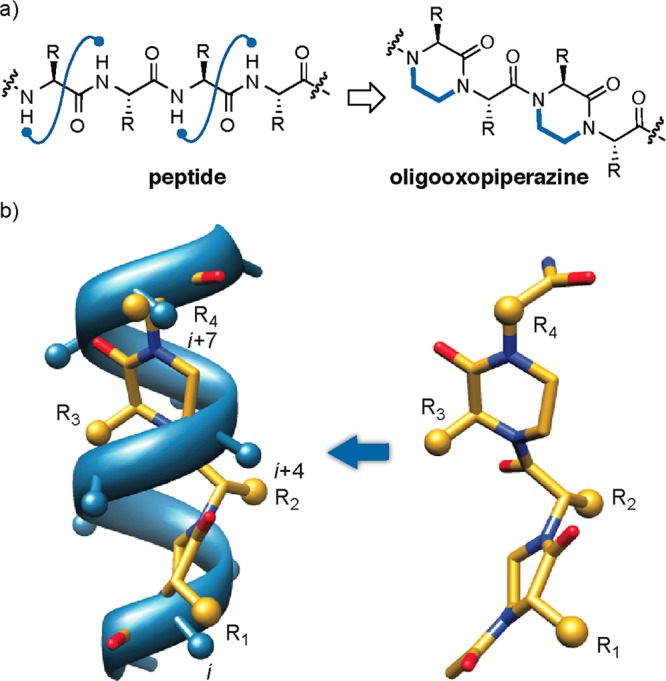
(a) Design of oxopiperazine helix mimetics. (b) Overlay of an 8-mer canonical α-helix and an oxopiperazine dimer (left). Predicted low-energy structure of an oxopiperazine dimer (right). Side chain groups are depicted as spheres.
The potential of oxopiperazine helix mimetics (OHMs) to target protein–protein interactions was recently established in biochemical, cell culture, and in vivo assays.36 We showed that OHMs that mimic a key α-helix from hypoxia inducible factor 1α (HIF1α) can inhibit the interactions of this transcription factor with coactivator p300/CBP. Significantly, the designed compounds downregulate the expression of a specific set of genes and reduce tumor burden in mouse xenograft models. Encouraged by this success, we sought to develop a computational approach to design and optimize oxopiperazine analogues with natural and nonnatural amino acid residues.
The objective of computational molecular design is to reduce the total number of possible designs to a manageable number that can be efficiently synthesized and experimentally tested. For example an oxopiperazine dimer has four variable positions, and assuming a standard library of 17 amino acids (20 canonical amino acids without Cys, Gly and Pro), the total number of possible designs would be >83 500. This calculation does not account for noncanonical amino acids, whose inclusion significantly raises the number of potential designs. Experimentally synthesizing and testing this many designs would be difficult for typical academic laboratories. Computational design offers a means of reducing the number of total designs one must synthesize to obtain potent ligands and streamlines the process of finding a high-affinity binder. Contemporary computational methods for design of PPI inhibitors often emphasize fragment-based screening.37,38 As a complementary approach, peptidomimetic design seeks to graft appropriate side chains on stable synthetic backbones, i.e., helical or β-sheet scaffolds.
We utilized a new computational protocol to develop nanomolar ligands for two different protein–protein interactions. We designed oxopiperazine dimers that mimic the p53 activation domain and HIF1α to develop ligands for Mdm2 and p300/CBP, respectively. The p53–Mdm2 interaction is an attractive target for cancer therapeutics39,40 as well as a model system for evaluating rational design strategies for inhibitor discovery. The activation domain of p53 adopts an α-helical conformation when bound to Mdm2,41 and several classes of stabilized helices and helix mimetics have been shown to target this interaction.20,21,25,42−47 In addition, several potent small molecule inhibitors of this interaction are known and are being evaluated for their in vivo efficacy in advanced preclinical models.48−51 Lastly, a wealth of structural data on the p53–Mdm2 interaction makes it well suited for development of computational strategies52 for ligand optimization.41,53−55
Our second model system for computational design validation is the interaction between HIF1α and coactivators p300/CBP, which mediate transactivation of hypoxia-inducible genes.56,57 The HIF signaling pathway is intimately linked to angiogenesis and metastasis in cancer.58,59 The design of oxopiperazine analogues described previously36 was based on mimicry of natural residues and resulted in submicromolar inhibitors. Here we show that application of the new Rosetta-based peptidomimetic design strategy with noncanonical residues affords compounds that are greater than an order of magnitude more potent than the scaffolds that displayed wild-type residues.
Overall, the study offers a generalized approach for discovering topographical helix mimetics consisting of wild-type and noncanonical residues that can target helical protein–protein interactions with high affinities.
Results and Discussion
Peptidomimetic Design with Rosetta
We investigated a computational approach that combines success in computational protein design60−62 with peptidomimetic scaffolds to develop OHMs as PPI inhibitors. Protein design is the process of predicting an amino acid sequence that will fold into a desired structure or carry out a desired function.60 Computational protein design techniques have made significant strides in recent years.63 A short list of successful applications includes an experimentally validated protein fold not seen in nature,61 redesign of protein–protein and protein–DNA interfaces,64 hyper stabilization of proteins,65 and design of enzymatic and ligand binding activities.62,66−72 We sought to use protein design principles to optimize the affinity of oxopiperazine mimetics using Rosetta (https://www.rosettacommons.org/).73
The basic design protocol in Rosetta uses a fixed backbone target and flexible ligand, with the goal of identifying the set of residues and side chain conformations with the lowest energy (Figure 2). To reduce the computational complexity required to model side chain degrees of freedom, the side chains are represented as “rotamers”, discrete side chain conformations located at the centroids of chi angle clusters, as determined by analyzing experimental protein structures. Recent extensions of the Rosetta framework enable modeling and design of noncanonical amino acids74 on nonnatural scaffolds such as peptoids.75,76 Implementation of oxopiperazine design in Rosetta has been recently described, and the protocols are available on the web (http://rosie.rosettacommons.org).77 Here we expand on our previous Web server implementation by allowing larger rigid-body sampling and designs, which include noncanonical amino acids.
Figure 2.

Key steps in the inhibitor design protocol. The protocol is initiated with identification of hotspot residues at the native interface by computational alanine scans. Positions on the scaffold are identified to mimic hotspot residues, and the scaffold featuring the hotspot mimics is experimentally validated. Computational steps including optimization of the ligand–protein complex conformation and design of hotspot analogues are performed using Rosetta. Top designs are inspected for proper binding of the target interface, and proper designs are experimentally validated.
There were several significant challenges involved in modifying Rosetta to enable modeling and design of oxopiperazine scaffolds. Specifically, we modified Rosetta’s protein centric score function to account for the OHM backbone, we employed recent methods to incorporate noncanonical amino acids in designs, we built core descriptions of oxopiperazine molecules in Rosetta’s internal molecular representation, and last we built methods for conformational sampling that efficiently sample oxopiperazine conformations. We were aided in this endeavor by two key recent developments in the broader Rosetta developers community. A new molecular mechanics-based score function was recently added to Rosetta that does not rely on the protein centric knowledge-based score terms.74 Additionally, a redevelopment of the Rosetta software suite73 has provided key flexibility in the data structures to enable modeling diverse sets of molecules other than proteins and nucleic acids. Finally, we have added new functionality into Rosetta that efficiently samples various oxopiperazine conformations, including a puckering of the oxopiperazine ring.76 This work was supported by quantum mechanical exploration of the backbone conformations to validate backbone energy terms.76
Design of p53/Mdm2 Inhibitors
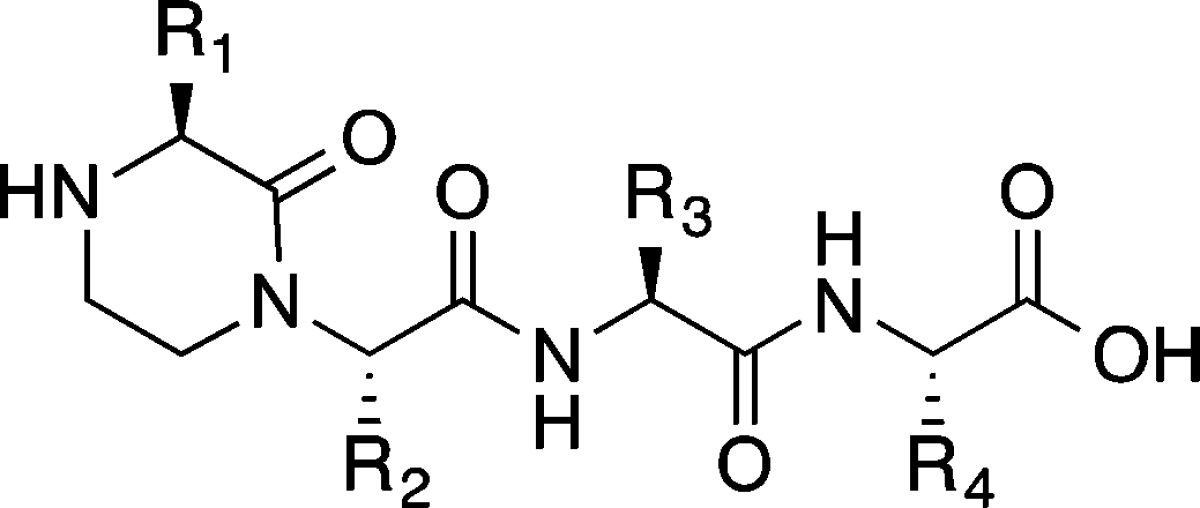
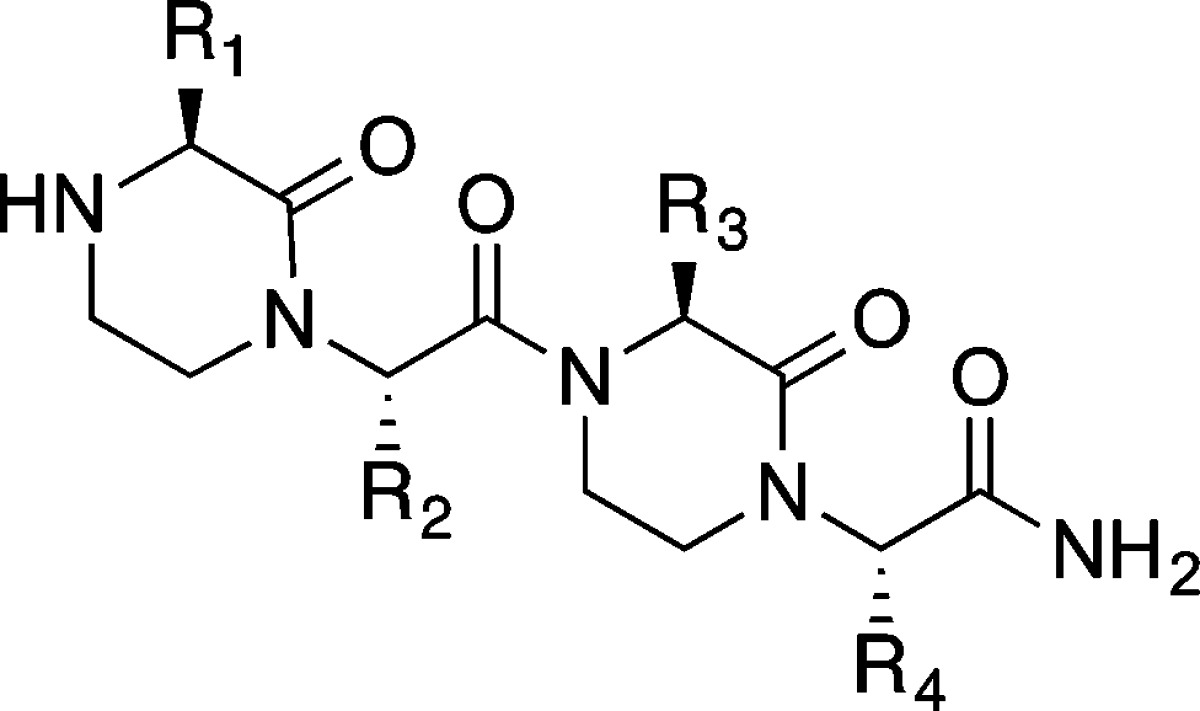
The p53 activation domain targets Mdm2 with three hydrophobic residues Phe19, Trp23, and Leu26 forming key contacts; we began by modeling these residues onto the oxopiperazine scaffold (Figure 1). An oxopiperazine dimer displays four amino acid side chains. Our modeling studies suggest that the first, second, and the fourth side chains, labeled as R1, R2, and R4, respectively, in Figure 1, overlay well on the i, i + 4 and i + 7 side chains of the α-helix. This leaves R3 potentially available for placement of solubilizing groups or small noninteracting side chains, as our preliminary analysis predicted that this residue does not directly contact the receptor. Accordingly, we designed and synthesized mimetic 1 (with the sequence FWAL) and 2 (FWKL), which featured the wild-type residues at the equivalent positions on the nonpeptidic scaffold but alanine or lysine residues at the R3 position (Table 1). OHMs were prepared using a solid phase synthesis methodology as described (Scheme S1).36
Table 1. Design and Mdm2 Binding Properties of Preliminary Oxopiperazine-Derived Helix Mimetics.

| mimetic | R1 | R2 | R3 | R4 | X | Kd (μM)a |
|---|---|---|---|---|---|---|
| 1 | Phe | Trp | Ala | Leu | OH | 65 ± 8 |
| 2 | Phe | Trp | Lys | Leu | OH | ≥200 |
| 3 | Phe | Trp | Leu | Leu | OH | 7.9 ± 0.5 |
| 4 | Phe | Trp | Phe | Leu | OH | 6.9 ± 1.3 |
| 5 | Phe | Trp | Phe | Leu | NH2 | 2.9 ± 0.1 |
| 6 | Phe | Trp | Phe | Lys | NH2 | ≥200 |
| 7 | Lys | Trp | Phe | Leu | NH2 | ≥200 |
| 8 | Phe | Ala | Phe | Leu | NH2 | 64 ± 7 |
Binding affinity for Mdm2 as determined by a competitive fluorescence polarization assay.
We utilized a previously described fluorescence polarization competition assay with a fluorescein-labeled p53 peptide to probe the binding affinity of the mimetics.78,79 Competitive displacement of the p53 peptides provides a strong indication that the designed nonpeptidic ligands are occupying the p53 binding pocket on Mdm2. In this assay, mimetic 1 bound Mdm2 with a dissociation constant, Kd, of 65 μM, while 2 displayed an appreciably lower affinity (Table 1 and Figure 3). To examine the affect of the R3 position on the binding properties, we designed a series of compounds where this position was changed to hydrophobic, anionic, cationic residues. These studies were performed in the context of the dimers (1–4) as well as oxopiperazine monomers linked to uncyclized dipeptides (Table 2). Together these preliminary studies showed that a hydrophobic group such as Leu or Phe at position R3 is preferred. Importantly, comparisons of the dimers 3 (FWLL) and 4 (FWFL) with the monomer-dipeptide sequences 11 and 12 support our hypothesis that cyclization of dipeptides in oxopiperazine rings provides a significant boost to the ability of these helix mimetics to target protein pockets. Ramachandran plots80 derived from quantum mechanical calculations further illustrate the flexibility of the uncyclized derivative as compared to the cyclic dimer (Figure S1).
Figure 3.
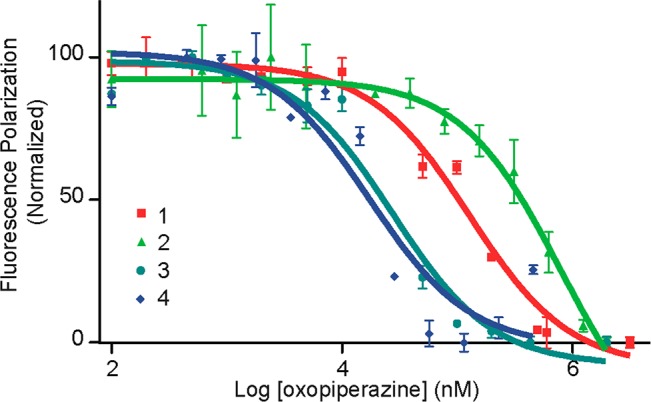
Determination of oxopiperazine analogue binding to His6-tagged Mdm2 by a fluorescence polarization assay. Binding curves for selected compounds in Table 1 are shown.
Table 2. Mdm2 Binding Properties of Oxopiperazine-Dipeptide analogues.
| mimetic | R1 | R2 | R3 | R4 | Kd (μM)a |
|---|---|---|---|---|---|
| 9 | Phe | Trp | Ser | Leu | >400 |
| 10 | Phe | Trp | Asp | Leu | 125 ± 73 |
| 11 | Phe | Trp | Leu | Leu | 94 ± 16 |
| 12 | Phe | Trp | Phe | Leu | 49 ± 10 |
Binding affinity for Mdm2 as determined by a competitive fluorescence polarization assay.
In these preliminary investigations, we also studied the effect of modulating the C-terminal functional group from a carboxylic acid to a carboxamide. Comparison of 4 and 5 illustrates that C-terminal functionalities do not significantly alter the binding profile of the molecules. Mimetic 5 binds Mdm2 with a dissociation constant of roughly 3 μM. Importantly, substitution of the Trp, Phe, and Leu residues at positions R1, R2, and R4, respectively, with alanine or lysine leads to substantial decrease in the binding affinities (5 versus 6–8); these results suggest that the residues in these positions on the dimer are making substantial contacts with the target interface, as expected, from mimickry of p53 Phe19, Trp23, and Leu26 residues within the Mdm2 pocket (Figure 4). We expected that the low micromolar dissociation constants obtained for this new class of helix mimetic scaffold can be further optimized, in keeping with previous studies with p53 mimics which showed that minor changes to contact residues can provide a significant improvement in binding.81 However, we were concerned that cis–trans amide bond isomerization may be contributing to lower affinity. The amide bond linking the R2 residue to the R3 oxopiperazine ring may adopt a trans or a cis conformation. Computational studies suggest that the trans conformation is preferred over the cis conformation by roughly 1.0 kcal/mol or more depending on the identity of the R2 and R3 residues;23 similar to the energy difference observed with proline. The fact that a hydrophobic group is favored over charged residues at the R3 position suggests that this residue may be occupying the Leu26 binding site in Mdm2 as opposed to the R4 residue. This alternative-binding mode would be possible if the cis-amide conformation was accessed in the complex. Mimetic 6 (Table 1) explicitly tests this possibility. If the R4 group is solvent accessible and R3 binds in the Mdm2 hydrophobic pocket, 6 would be expected to bind Mdm2 with a similar affinity as 5, instead of being a rather poor binder as observed. However, we cannot rule out the possibility that both cis and trans conformations contribute to the overall binding affinity. To fully dissect the contribution of the cis and trans conformations, amide bond isosteres where each of the conformations can be controlled will need to be examined in future studies.
Figure 4.
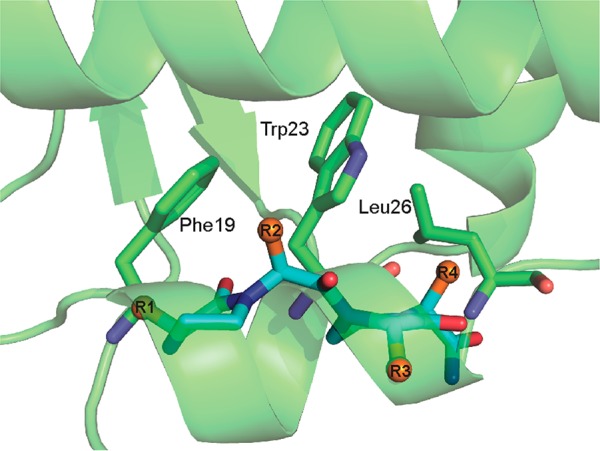
Docking of the oxopiperazine scaffold (cyan) with sidechains shown in orange in p53 binding pocket of Mdm2. Figure shows the relative positioning of the oxopiperazine dimer side chains R1–R4 and p53 hotspot residues Phe19, Trp23, and Leu26 within the protein pocket.
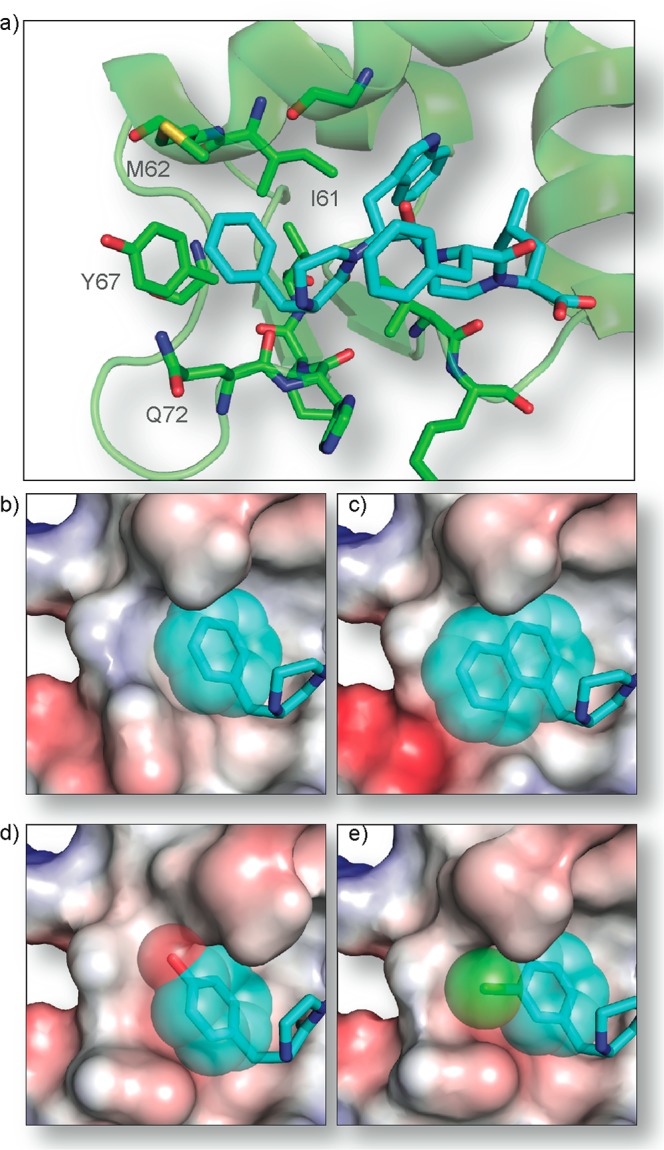
We began our computational design protocol in Rosetta by building a model of 4 and by analyzing the experimental structure activity relationships shown in Table 1. Compound 4 was docked to align with the p53 hotspot residues, and our oxopiperazine docking protocol was used to optimize the rigid-body conformations of the ligand and the protein based on Rosetta’s molecular mechanics energy function (see methods for details). Figures 5a and 8a show that 4 makes several energetically favorable contacts with the Mdm2 interface, suggesting proper mimicry of the p53 hotspot residues. The R1 residue, Phe, of 4 (Figure 5b) is involved in good packing interactions with the Mdm2 interface (residues Ile61, Met62, and Tyr67), including a potential stacking interaction with Tyr67. The R2 residue, Trp (Figure 5c), is well packed in the same pocket as the p53 hotspot Trp23 contacting Mdm2 residues Leu54, Leu57, Gly58, Ile61, Phe86, Phe91, Val93, Ile99, and Ile103. Lastly, the R4 residue, Leu (Figure 5d), also properly mimics the p53 hotspot residue (Leu26), packing well into a pocket formed by several Mdm2 hydrophobic interface residues including Leu54, Val93, His96, Ile99, and Ile103.
Figure 5.
(a) Predicted conformation of Mimetic 4 in Mdm2 pocket. Binding modes of (b) Phe, (c) Trp, and (d) Leu residues (R1, R2, and R4 positions) of 4 are shown.
Figure 8.
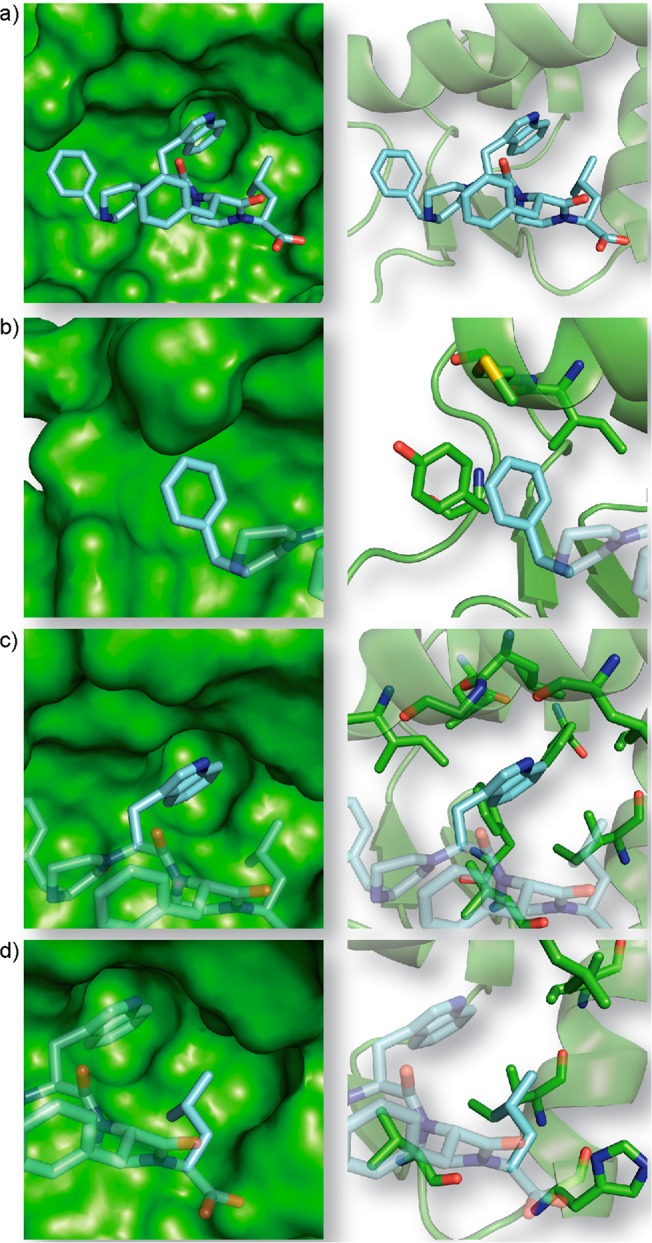
Examination of the N-terminal residue-binding pocket in Mdm2. (a) The phenylalanine residue at the R1 position of 4 (cyan) resides in a flexible pocket consisting of Ile-61, Met62, Tyr67, and Gln72 of Mdm2 (green). (b) Predicted orientations of phenylalanine and analogues (c) naphthylalanine of mimic 15, (d) tyrosine of mimic 16, and (e) 3-chlorophenylalanine of mimic 18. Electrostatic surface of Mdm2 is modeled by Pymol.
Next, we developed an algorithm to predict high-affinity oxopiperazine dimers for Mdm2 using Rosetta and a library of noncanonical amino acids (Table S1). We used the starting conformation of the ligand–Mdm2 complex (developed as in our modeling of compound 5, Figure 5a) as input for Rosetta calculations and designed a two-step iterative protocol consisting of conformation and sequence optimization steps.
The conformation optimization step attempts to find a low-energy conformation between the scaffold and the target protein. During this step, the protocol performs a Monte Carlo search of conformational space making random changes to the rigid-body orientation, oxopiperazine backbone (including ring puckering), and side chain repacking to both the scaffold and target interface. In the sequence optimization step, we make side chain substitutions from a library of both natural and noncanonical amino acids to find the lowest-energy oxopiperazine sequence.
This two-step protocol is repeated for a large number of substitutions and low-energy oxopiperazine sequences (designs), and their 3D models are saved. Low-energy designs were sorted based on calculated binding energy (Figure 6 and Table S2), and the top designs were selected for manual inspection. Manual inspection included verifying that (1) the oxopiperazine scaffold occupied the same pockets as the p53 helix hotspots to ensure inhibition, (2) the conformation entailed good packing among side chains from both sides of interface, and (3) the design was synthetically tractable and likely soluble, etc. (It should be noted that oxopiperazine dimers described here are generally soluble at millimolar concentrations in aqueous solutions.)
Figure 6.
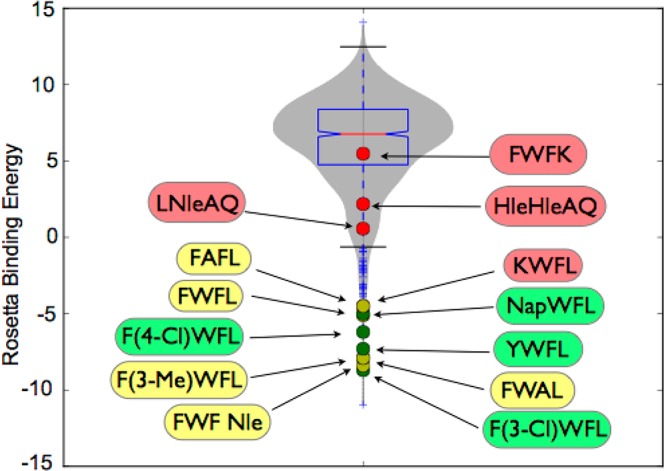
A violin plot showing distribution of the predicted oxopiperazine analogues for their potential to target Mdm2. The binding affinity is expressed as Rosetta binding energy units (REUs). The plot shows the energy scores for the top scoring 1000 designs selected from 30 000 random Rosetta designs (gray violin) as well as experimentally tested designs (dots). The Rosetta score discriminates between good binders (green and yellow label) and weak binders (red label).
The goal of computational peptidomimetic design is to produce a handful of top designs that can be experimentally evaluated. Rosetta predicts a large number of binders for Mdm2 and provides a filtered and ranked list of predicted high-affinity binders composed of natural and noncanonical residues. Figure 6 shows a violin plot in gray indicating the distribution of predicted oxopiperazine ligands spanning the Rosetta binding energy score spectrum. The gray area represents the top 1000 scores from Rosetta’s evaluation of 30 000 designs. This spectrum provides a background on which to compare possible high-affinity Rosetta designs. Figure S2 correlates experimental binding affinity for Mdm2 with Rosetta binding energy score for the subset of sequences we tested using our competition binding assay. Although our goal here is to use Rosetta to suggest designs with high likelihood of success, these data illustrate that Rosetta predictions correlate well with experimental dissociation constants for the oxopiperazines and are competitive with current computational methods.82−85
To show that our Rosetta binding energy protocol enriches for high-affinity binders, selected designs were synthesized and evaluated using the fluorescence polarization competition assay described above. The Rosetta results suggest that the tryptophan residue at position R2 is optimized for that position so we began by synthesizing the variants at each of the other three positions (Table 3 and Figure 7). Mimetic 13 contains a norleucine residue at position R4 in place of the leucine in 5, while compound 14 features a tyrosine group at R3 in place of phenylalanine. Two derivatives, 15 and 16, containing naphthylalanine and tyrosine residues, respectively, at position R1 were synthesized. Binding studies indicate that substitutions at the R3 and R4 positions of dimers do not lead to higher affinity compounds. In contrast, substitutions at the R1 position provided improvements predicted by Rosetta. The naphthyl analogue, 15, binds Mdm2 with a 3-fold higher affinity than 5, while substitution with tyrosine to obtain 16 provides a 400 nM ligand for Mdm2. Based on these results, we prepared and tested two more derivatives of phenylalanine at the R1 position. Mimetic 17 contains a methylated tyrosine group, while 18 features a 3-chloro-phenylalanine residue. Both of these analogues proved to be better than 16. Overall, our designs involving changes at the R1 position yielded a roughly 10-fold improvement over the unoptimized derivative 5.
Table 3. Computationally Predicted Oxopiperazine p53 Mimics and Their Potential to Target Mdm2.

| mimetic | R1 | R2 | R3 | R4 | Kd (μM)a |
|---|---|---|---|---|---|
| 13 | Phe | Trp | Phe | Nle | 2.5 ± 0.5 |
| 14 | Phe | Trp | Tyr | Leu | 3.1 ± 0.2 |
| 15 | Nap | Trp | Phe | Leu | 0.9 ± 0.1 |
| 16 | Tyr | Trp | Phe | Leu | 0.4 ± 0.05 |
| 17 | Tyr(O-Me) | Trp | Phe | Leu | 0.3 ± 0.01 |
| 18 | Phe(3-Cl) | Trp | Phe | Leu | 0.3 ± 0.04 |
| 19 | Phe(3-Me) | Trp | Phe | Leu | 2.6 ± 0.04 |
| 20 | Phe(4-Cl) | Trp | Phe | Leu | 1.3 ± 0.1 |
Binding affinity for Mdm2 as determined by a competitive fluorescence polarization assay.
Figure 7.
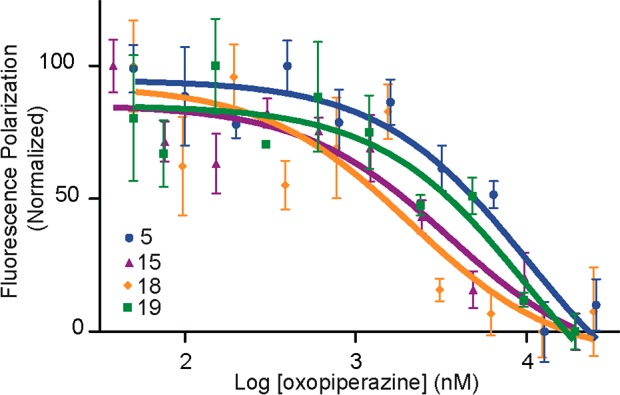
Determination of binding affinity for computationally optimized oxopiperazine analogue binding to His6-tagged Mdm2 by a fluorescence-polarization assay. Binding curves for selected compounds in Table 3 are shown.
Analyses of the minimized complexes show that the Phe residue at the R1 position of 5 is wedged in a pocket formed by Ile61, Met62, Tyr67, and Gln72 of Mdm2. Tyr67 and Gln72 reside on a flexible loop allowing different-sized analogues of Phe to be accommodated in the pocket (Figure 8a). The predicted orientation of the R1 residue for compounds 5, 15, 16, and 18 is shown in Figure 8 and illustrates the plasticity of the pocket. We designed two control compounds, 19 and 20, to investigate the specificity of the pocket for 3-chloro-phenylalanine group. Mimetic 19 contains a bulkier methyl group in place of the chlorine atom, while 20 features the chlorine atom at the 4-position. We find that replacement of the chlorine atom with the methyl group causes an 8-fold decrease in binding affinity and moving it to the para-position on the phenyl ring leads to a 4-fold reduction. These results suggest that the 3-chlorophenyl group makes specific steric and electronic contacts within the pocket, consistent with our computational model of this interaction.
The full list of Rosetta scores and experimental binding affinities for the p53 mimics is included in Table S2. The Rosetta algorithm produced a pool dramatically enriched for high-affinity binders. The lack of a perfect correlation between experimental and computational results within a narrow window of affinities is not surprising in these preliminary studies that represent the first test of Rosetta on a novel backbone that includes noncanonical amino acids. (We find correlation equal to 0.64 and 0.79 for the two interfaces described here.) It is interesting to note that the poor binder, KWFL (7), scored better than expected by Rosetta. Examination of the Mdm2 bound structure of KWFL reveals that the lysine residue does not occupy the p53 Phe19 hotspot pocket, violating our first rule of manual inspection described above (Figure S3). It is not surprising that this compound leads to poor inhibition since the Phe19 pocket offers an important contact for p53. The result underscores the importance of targeting the interaction interface when developing an inhibitor. The algorithm correctly predicts that mimetic 6, in which a lysine group resides in place of leucine, will be a poor binder. We envisage a better correlation in future studies when a larger set of experimental data is available as a training set.82
To confirm that 18 binds to Mdm2 in the p53 binding pocket, we performed 1H–15N HSQC NMR titration experiments with 18 and uniformly 15N-labeled Mdm2. Addition of 18 to 50 μM Mdm2 in Mdm2:18 ratios of 1:0.2 and 1:0.5 provided a concentration-dependent shift in resonances of several Mdm2 residues (Figures 9 and S4). Specifically, addition of 18 leads to shifts in resonances of residues corresponding to the hydrophobic cleft into which the native p53 helix binds. Overall, the NMR results support the Rosetta derived model of the complex.
Figure 9.
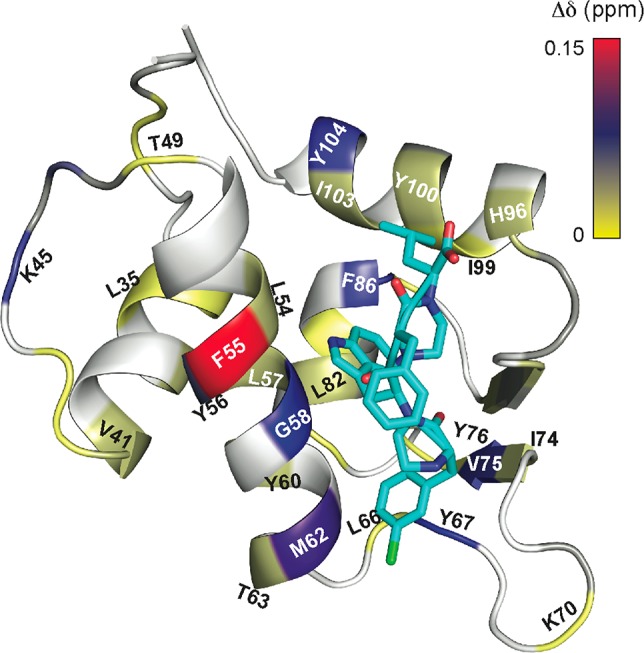
Model depicts the results of a 1H–15N HSQC NMR titration experiment. Mdm2 residues undergoing chemical shift perturbations upon addition of 18 are shown in colors that match the magnitude of the chemical shift change in the scale. Compound 18 is represented in blue. The computationally predicted model of the complex is shown.
Design of HIF1α/p300(CBP) Inhibitors
To further validate the potential of Rosetta oxopiperazine design protocol, we chose to develop inhibitors of a different transcriptional complex. We have recently shown that stabilized peptide helices86 and small molecule oxopiperazine analogues36 that mimic a key helical domain of HIF1α can inhibit hypoxia inducible signaling in cell culture and animal models. The C-terminal domain of HIF1α utilizes two short α-helices to bind to the CH1 domain of p300/CBP (Figure 10). Computational alanine scanning64 studies on the complex reveal that four helical residues from the HIF1α helix816–824 (Leu818, Leu822, Asp823, and Gln824) make close contacts with the CH1 domain of p300/CBP. Three of these residues, Leu818, Leu822, and Gln824, can be mimicked by oxopiperazine dimers consisting of the appropriate building blocks (Figure 10). Based on this analysis, we designed and synthesized analogues of HIF1α to inhibit its binding with p300/CBP. OHM 21 contains projections representing all three wild-type residues from HIF1α: R1 as Leu818, R2 as Leu822, and R4 as Gln824 (Table 4). The R3 position of the oxopiperazine scaffold was not predicted to make contacts with the target protein; an alanine residue was inserted at this position. OHM 22 was designed as a single mutant of 21 with the R2 position substituted with an alanine residue.
Figure 10.

Design of HIF1α mimetics as ligands for p300-CH1. (a) Overlay of HIF1α helix776–826 (in magenta) and OHM 21 (cyan) in complex with CH1 domain of p300/CBP (PDB code 1L8C). The R1, R2 and R4 positions of 21 access the same p300 molecular pockets as Leu818, Leu822, and Gln824 of the HIF1α C-terminal activation domain.
Table 4. Oxopiperazine HIF Mimics Targeting the CH1 Domain of p300/CBP.
| mimetic | R1 | R2 | R3 | R4 | Kd (μM)a |
|---|---|---|---|---|---|
| 21 | Leu | Leu | Ala | Gln | 0.53 ± 0.14 |
| 22 | Leu | Ala | Ala | Gln | >10 |
| 23 | Leu | Nle | Ala | Gln | 0.03 ± 0.01 |
| 24 | Met | Met | Ala | Gln | 0.24 ± 0.04 |
| 25 | Hle | Hle | Ala | Gln | 0.16 ± 0.06 |
Binding affinity for p300-CH1 was determined using an intrinsic tryptophan fluorescence assay.
Results with 21 and 22 have been previously described.36 We found that oxopiperazine 21, consisting of the wild-type residues, bound the CH1 domain of p300 with an affinity of 533 ± 24 nM; whereas, the negative control 22 displayed a very weak affinity for p300-CH1, with Kd value of >10 μM (Table 4 and Figure 11). The binding affinities of OHMs for p300-CH1 domain were evaluated using intrinsic tryptophan fluorescence spectroscopy, as described previously.86,87 Because Trp403 lies in the binding cleft of p300/CBP where a native HIF1α816–824 helix binds, it offers a probe for investigating mimetics of this helix. As part of this earlier study, we also characterized the interaction of OHM 21 with p300-CH1 domain using 1H–15N HSQC NMR titration experiments with the uniformly 15N-labeled CH1. Addition of OHM 21 led to consistent shifts in resonances of residues corresponding to the HIF1α816–824 binding pocket. OHM 21 efficiently downregulated HIF signaling in cell culture at micromolar levels and reduced tumor levels in triple-negative breast cancer cell line MDA-MB-231 mouse xenograft models. Importantly, microarray gene expression profiling data showed that the designed oxopiperazine helix mimetic despite its low molecular weight and a limited number of contacts with the intended target protein shows high specificity on a genome-wide scale.
Figure 11.

Determination of binding affinity for p300-CH1 by a tryptophan fluorescence spectroscopy. Binding curves for compounds 21–25 in Table 4 are shown.
The encouraging results with OHM 21 provide a platform to test the potential of our Rosetta peptidomimetic design strategy to further optimize design of HIF mimetics. Specifically, we wanted to determine if the computational approach could rapidly suggest noncanonical residues that may boost the binding affinity for p300-CH1. We analyzed the p300/OHM 21 binding using our established protocol, with a library of noncanonical amino acids (Table S3). The computational predictions suggested inclusion of longer aliphatic side chains in place of the isobutyl group of leucine would lead to better contacts with the hydrophobic pocket. Specifically, substitution with noncanonical side chains at the R2 position of OHM 21 was predicted to lead to an optimized binder (Figure 12).
Figure 12.

Analysis of the R2 position of HIF OHM mimics in p300-CH1 binding pocket. Space-filling model reveals longer hydrophobic side chains form better packing in the p300-CH1 pocket, natively inhabited by the Leu822 of HIF1α: (a) leucine of 21, (b) alanine of 22, (c) homoleucine of 25, and (d) norleucine of 23.
Figure 13 shows a violin plot for OHMs targeting the CH1 domain. The gray area represents the top 1000 scores from Rosetta’s evaluation of 30 000 designs. The predicted high-affinity designs feature norleucine (Nle) and homoleucine (Hle) residues in place of the wild-type leucine analogues and are substantially lower in energy than the rest of the sequences tested by Rosetta. Substitution of the two leucine residues with methionines is predicted to be less effective than with noncanonical residues, suggesting that space-filling and polarity of side chain groups are necessary for optimal results. Other combinations of homoleucine, norleucine, and leucine residues were also examined (Table S4). To experimentally evaluate the predictions, we prepared three analogues representing top designs in which both leucine groups of 21 were substituted with methionine, norleucine, or homoleucine to obtain OHMs 23–25 (Table 4). Each of theses compounds bound p300 with higher affinity than the parent OHM 21, with OHM 23 providing a 13-fold enhancement in binding affinity (Kd = 30.2 ± 1.87 nM).
Figure 13.

A violin plot showing distribution of the predicted oxopiperazine analogues for their potential to target the CH1 domain of p300/CBP. The binding affinity is expressed as Rosetta binding energy unit (REU). The plot shows the energy scores for the top scoring 1000 designs selected from 30 000 random Rosetta designs (gray violin) as well as experimentally tested designs (dots). The Rosetta score discriminates between good binders (green and yellow label) and weak binders (red label).
We find a strong correlation between the experimental results for p300/CBP and Rosetta predictions (Figure S5 and Table S4) further highlighting the success of the computational design protocol. Since the fluorescence-binding assay uses a native tryptophan residue in the target molecular pocket, it provides a stringent test for the binding site specificity. Characterization of the interaction of OHM 21 with p300-CH1 domain using 1H–15N HSQC NMR titration experiments further confirms the target pocket for the designed analogues.
Cross-Specificities of the Designed Compounds for Mdm2 and p300
Analyses of protein–protein interaction networks suggest that the human interactome consists of hundreds of thousands of different PPIs.88,89 Our own analysis of the high-resolution protein complexes available in the Protein Data Bank reveals that up to 60% of such protein complexes contain an interfacial α-helix.19,26,90 Thus, a central question in the design of helix mimetics as PPI inhibitors pertains to their specificity on the genome-wide scale. We recently probed the specificity of OHM 21, designed to be a transcriptional inhibitor for off-target regulation, using the Affymetrix Human Gene ST 1.0 arrays containing oligonucleotide sequences representing over 28 000 transcripts.36 This compound was found to be remarkably specific given the limited number of contacts it offers.
In the present study, we have computationally designed potent small molecule helix mimetics that feature noncanonical side chains as potential inhibitors of protein–protein interactions. As a preliminary analysis of Rosetta’s ability to predict the specificity of OHMs against unintended targets, we determined the binding affinity of the p53 OHM mimic 18 against p300-CH1 and that of HIF OHM mimics 23 and 25 for Mdm2 (Table 5 and Figure S6). These analogues were chosen because they represent the highest-affinity ligands obtained for their respective targets and contain noncanonical residues. Calculations with the modified version of Rosetta, described above, predict that the p53 mimetic 18 is a poor ligand for p300-CH1 and that HIF mimetics 23 and 25 are not optimal designs for Mdm2. Specifically, the calculated Rosetta binding energy (REU) for OHM 18/p300-CH1 binding is the same as that calculated for 22, a negative control designed for the HIF/p300 interaction (Figure 13). Likewise, Rosetta predicts compounds 23 and 25 to have a high-energy interaction with Mdm2; >6 REU’s when compared to 18 the high-affinity Mdm2 ligand (Figure 6). We confirmed these predictions in experimental binding assays.
Table 5. Cross-Specificities of Oxopiperazine p53 and HIF Mimics Against p300-CH1 and Mdm2.
The binding affinities for p300-CH1 were determined using an intrinsic tryptophan fluorescence assay.
Affinities for Mdm2 were measured using a competitive fluorescence polarization assay with Flu-p53 as a probe.
We tested the binding of oxopiperazine derivatives using the assays described above. As expected the compounds are specific for their cognate receptors (Table 5), with each showing more than 100-fold specificity for the desired protein surface. These results provide support for our hypotheses that the computational strategy developed herein can be used to ultimately predict specificity of the designed peptidomimetics on the genome-wide scale.
Conclusions
Protein–protein interactions are attractive targets for drug design because of their fundamental role in human biology and disease progression. These large interfaces are often dismissed as “undruggable”; however, the past decade has seen emerging methods to inhibit these complexes. Here we describe the potential of small molecule helix mimetics derived from the oxopiperazine scaffold to target protein complexes where one face of the interfacial helix contributes significantly to binding. We find that the affinity of the designed ligands can be enhanced significantly using a combination of computational design and experimental structure–activity relationship data. Central to the present efforts was a novel combination of rational design (i.e., hotspot mimicry) and a new set of Rosetta functionalities for computational design with noncanonical side chains and backbones. We expect that the tools and algorithms developed here will be applicable for targeting PPIs that remain intractable for synthetic inhibition. All computational tools described in this work are freely available to academic researchers via the RosettaCommons (rosettacommons.org). Our efforts show that the principles of computational protein design can be transferred to nonnatural scaffolds featuring noncanonical amino acid residues.
Experimental Section
Docking and Design Protocol in Rosetta
The oxopiperazine dimer scaffold was initially docked by aligning Cβ atoms on the scaffold positions corresponding to hotspot residues on P53 (R1, Phe19, R2, Trp23, R3, Leu26) and HIF1α (R1, Leu818, R2, Leu822, R4, Gln824) using the PDB structure: 1YCR and 1L8C, respectively. The Rosetta relax w/constraints application was run on this initial structure to relieve any clashes that may hinder score analysis. The relaxed complex was then modeled and designed using a protocol developed specifically for oxopiperazine inhibitors. The protocol iterates between a perturbation phase (conformational optimization), attempting to find the lowest-energy conformation of bound ligand and target protein given the current residue identities, and a design phase, which attempts to find residue substitutions including noncanonical analogues that lower the energy given the current conformation. The perturbation phase consists of (a) rigid-body rotation and translation moves, (b) small angle moves of phi and psi, and (c) pucker moves of the oxopiperazine rings. Perturbations were only allowed to the scaffold leaving the target’s backbone fixed. All residues at the interface on both target and ligand were allowed to sample side-chain rotamer space. The design phase consists of residue identity substitutions at positions along the scaffold and rotamer repacking. Substitutions were defined in the Rosetta resfile. Finally, minimization of all degrees of freedom in the complex was performed.
For modeling analysis, we used the same design protocol except residues were fixed to the identities of interest in the Rosetta residue input file (i.e., resfile). Fixing residue identities only allows side chain optimization during the “design” phase. 5000 independent runs (i.e., decoys) were computed for each sequence. For design runs of p53 mimic, substitutions that were allowed included noncanonical amino acids that were derivative of the original hotspot residue (e.g., R1 phenylalanine was designed with 3-methyl-phenylalanine, etc.) See Table S1 for the NCAA_library list. For each position on the scaffold, >10 000 runs were carried out allowing the single position to vary leaving the other positions fixed. This was repeated for each position on the oxopiperazine scaffold. The SVN Revision: 52345 version of Rosetta used was for these studies. For design runs of HIF1α mimic, R1 and R2 were substituted with all hydrophobic noncanonical amino acids in Table S3 except for proline analogues. Detailed protocols including command lines have been previously described76 and can be found in the Supporting Information.
Top designs were selected based on filtering the lowest 5% of total energy decoys and sorting by Rosetta binding energy score. The Rosetta binding energy score was calculated by the equation:
The unbound score was calculated by separating the scaffold from the target receptor, then repacking the side chains and finally calculating the total Rosetta energy of the unbound complex.
Rosetta Binding Discrimination Analysis
A random set of designs against target proteins Mdm2 and p300 were generated from a set of 30 000 Rosetta design runs where all four positions of an oxopiperazine dimer were allowed to vary to any canonical amino acid excluding Cys, Gly, and Pro. The top 1000 of models by total Rosetta score made up the total random set. This random set is shown as a gray histogram (violin plot) in Figures 6 and 13.
The top binding energy score for designs with experimental binding affinities were determined from a set of 5000 decoy structures. As described above, the top 1000 of decoys by total score was then sorted by Rosetta binding energy score, and the lowest Rosetta binding energy score was used.
Quantum Mechanics Calculations
Quantum mechanics calculations were done using the Gaussian 09 (EM64L-G09RevC.01, version date: 2011-09-23) software package.91 An initial optimization using “HF 6-31G(d) Opt SCRF=PCM SCF=Tight” parameters was done for each model structure. The resulting optimized structure was then used for further energy calculations with parameters “B3LYP 6-31G(d) Geom=Check SCRF=PCM SCF=Tight” and “MP2(full) 6-31G(d) Geom=Check SCRF=PCM SCF=Tight”.
Description of Mdm2 Binding Studies
The relative affinities of OHMs for N-terminal His6-tagged Mdm2 (25–117) were determined using fluorescence polarization-based competitive binding assay with fluorescein labeled p53 peptide, Flu-p53. The polarization experiments were performed with a DTX 880 Multimode Detector (Beckman) at 25 °C, with excitation and emission wavelengths at 485 and 535 nm, respectively. All samples were prepared in 96 well plates in 0.1% pluronic F-68 (Sigma). Prior to the competition experiments, the affinity of the Flu-p53 for Mdm2 was determined by monitoring polarization of the fluorescent probe upon binding Mdm2 (Figure S7). For competition binding experiments, appropriate concentrations of the peptides (1 nM–100 μM) were added to the Mdm2-Flu-p53 mixture, and the resulting solution was incubated at 25 °C for 1 h before measuring the degree of dissociation of Flu-p53 by polarization. The binding affinity (KD) values reported for each peptide are the averages of 3–5 individual experiments and were determined by fitting the experimental data to a sigmoidal dose–response nonlinear regression model on GraphPad Prism 5.0.92
Description of p300-CH1 Binding Studies
Relative affinities for of OHMs were determined using a tryptophan fluorescence assay. Spectra were recorded on a QuantaMaster 40 spectrofluorometer (Photon Technology International) in a 10 mm quartz fluorometer cell at 25 °C with 4 nm excitation and 4 nm emission slit widths from 200 to 400 nm at intervals of 1 nm/s. Samples were excited at 295 nm, and fluorescence emission was measured from 200 to 400 nm and recorded at 335 nm. OHM stock solutions were prepared in DMSO. Aliquots containing 1 μL DMSO stocks were added to 400 μL of 1 μM p300-CH1 in 50 mM Tris and 100 mM NaCl (pH 8.0). After each addition, the sample was allowed to equilibrate for 5 min before UV analysis. Background absorbance and sample dilution effects were corrected by titrating DMSO into p300-CH1 in an analogous manner. Final fluorescence is reported as the absolute value of [(F1 – F0)/F1]*100, where F1 is the final fluorescence upon titration, and F0 is the fluorescence of the blank DMSO titration. EC50 values for each peptide were determined by fitting the experimental data to a sigmoidal dose–response nonlinear regression model on GraphPad Prism 5.0, and the dissociation constants, KD, were obtained from equation.
Acknowledgments
This work was financially supported by the National Science Foundation (CHE-1151554 to P.S.A.) and the National Institute for Health (RC4-AI092765, PN2-EY016586, IU54CA143907-01, EY016586-06 to R.B.). B.N.B. thanks the New York University for a Dean’s Dissertation Fellowship. Support from the National Science Foundation (CHE-0958457) in the form of an instrumentation grant is gratefully acknowledged.
Supporting Information Available
Scheme S1, Figures S1–S5, and Tables S1–S4. Compound characterization including NMR spectra and analytical HPLC traces. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
§ These authors contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Jones S.; Thornton J. M. Prog. Biophys. Mol. Biol. 1995, 63, 31. [DOI] [PubMed] [Google Scholar]
- Ko E.; Liu J.; Burgess K. Chem. Soc. Rev. 2011, 40, 4411. [DOI] [PubMed] [Google Scholar]
- Azzarito V.; Long K.; Murphy N. S.; Wilson A. J. Nat. Chem. 2013, 5, 161. [DOI] [PubMed] [Google Scholar]
- Henchey L. K.; Jochim A. L.; Arora P. S. Curr. Opin. Chem. Biol. 2008, 12, 692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafmeister C. E.; Po J.; Verdine G. L. J. Am. Chem. Soc. 2000, 122, 5891. [Google Scholar]
- Harrison R. S.; Shepherd N. E.; Hoang H. N.; Ruiz-Gomez G.; Hill T. A.; Driver R. W.; Desai V. S.; Young P. R.; Abbenante G.; Fairlie D. P. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 11686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patgiri A.; Jochim A. L.; Arora P. S. Acc. Chem. Res. 2008, 41, 1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gellman S. H. Acc. Chem. Res. 1998, 31, 173. [Google Scholar]
- Goodman C. M.; Choi S.; Shandler S.; DeGrado W. F. Nat. Chem. Biol. 2007, 3, 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng R. P.; Gellman S. H.; DeGrado W. F. Chem. Rev. 2001, 101, 3219. [DOI] [PubMed] [Google Scholar]
- Horne W. S.; Gellman S. H. Acc. Chem. Res. 2008, 41, 1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seebach D.; Gardiner J. Acc. Chem. Res. 2008, 41, 1366. [DOI] [PubMed] [Google Scholar]
- Yoo B.; Kirshenbaum K. Curr. Opin. Chem. Biol. 2008, 12, 714. [DOI] [PubMed] [Google Scholar]
- Marimganti S.; Cheemala M. N.; Ahn J. M. Org. Lett. 2009, 11, 4418. [DOI] [PubMed] [Google Scholar]
- Jayatunga M. K. P.; Thompson S.; Hamilton A. D. Bioorg. Med. Chem. Lett. 2014, 24, 717. [DOI] [PubMed] [Google Scholar]
- Thompson S.; Hamilton A. D. Org. Biomol. Chem. 2012, 10, 5780. [DOI] [PubMed] [Google Scholar]
- Thompson S.; Vallinayagam R.; Adler M. J.; Scott R. T. W.; Hamilton A. D. Tetrahedron 2012, 68, 4501. [Google Scholar]
- Jung K.-Y.; Vanommeslaeghe K.; Lanning M. E.; Yap J. L.; Gordon C.; Wilder P. T.; MacKerell A. D.; Fletcher S. Org. Lett. 2013, 15, 3234. [DOI] [PubMed] [Google Scholar]
- Bullock B. N.; Jochim A. L.; Arora P. S. J. Am. Chem. Soc. 2011, 133, 14220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plante J. P.; Burnley T.; Malkova B.; Webb M. E.; Warriner S. L.; Edwards T. A.; Wilson A. J. Chem. Commun. 2009, 5091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaginian A.; Whitby L. R.; Hong S.; Hwang I.; Farooqi B.; Searcey M.; Chen J.; Vogt P. K.; Boger D. L. J. Am. Chem. Soc. 2009, 131, 5564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Restorp P.; Rebek J. Jr. Bioorg. Med. Chem. Lett. 2008, 18, 5909. [DOI] [PubMed] [Google Scholar]
- Tosovska P.; Arora P. S. Org. Lett. 2010, 12, 1588. [DOI] [PubMed] [Google Scholar]
- Buhrlage S. J.; Bates C. A.; Rowe S. P.; Minter A. R.; Brennan B. B.; Majmudar C. Y.; Wemmer D. E.; Al-Hashimi H.; Mapp A. K. ACS Chem. Biol. 2009, 4, 335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. H.; Zhang Q.; Jo S.; Chai S. C.; Oh M.; Im W.; Lu H.; Lim H. S. J. Am. Chem. Soc. 2011, 133, 676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jochim A. L.; Arora P. S. ACS Chem. Biol. 2010, 5, 919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings C. G.; Hamilton A. D. Curr. Opin. Chem. Biol. 2010, 14, 341. [DOI] [PubMed] [Google Scholar]
- Yin H.; Hamilton A. D. Angew. Chem., Int. Ed. 2005, 44, 4130. [DOI] [PubMed] [Google Scholar]
- Orner B. P.; Ernst J. T.; Hamilton A. D. J. Am. Chem. Soc. 2001, 123, 5382. [DOI] [PubMed] [Google Scholar]
- Gante J. Angew. Chem., Int. Ed. Engl. 1994, 33, 1699. [Google Scholar]
- Giannis A.; Kolter T. Angew. Chem., Int. Ed. 1993, 32, 1244. [Google Scholar]
- Herrero S.; Garcia-Lopez M. T.; Latorre M.; Cenarruzabeitia E.; Del Rio J.; Herranz R. J. Org. Chem. 2002, 67, 3866. [DOI] [PubMed] [Google Scholar]
- Kitamura S.; Fukushi H.; Miyawaki T.; Kawamura M.; Konishi N.; Terashita Z.; Naka T. J. Med. Chem. 2001, 44, 2438. [DOI] [PubMed] [Google Scholar]
- Sugihara H.; Fukushi H.; Miyawaki T.; Imai Y.; Terashita Z.; Kawamura M.; Fujisawa Y.; Kita S. J. Med. Chem. 1998, 41, 489. [DOI] [PubMed] [Google Scholar]
- Borthwick A. D.; Davies D. E.; Exall A. M.; Hatley R. J. D.; Hughes J. A.; Irving W. R.; Livermore D. G.; Sollis S. L.; Nerozzi F.; Valko K. L.; Allen M. J.; Perren M.; Shabbir S. S.; Woollard P. M.; Price M. A. J. Med. Chem. 2006, 49, 4159. [DOI] [PubMed] [Google Scholar]
- Lao B. B.; Grishagin I.; Mesallati H.; Brewer T. F.; Olenyuk B. Z.; Arora P. S. Proc. Natl. Acad. Sci. USA 2014, 10.1073/pnas.1402393111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider G.; Fechner U. Nat. Rev. Drug Discovery 2005, 4, 649. [DOI] [PubMed] [Google Scholar]
- Winter A.; Higueruelo A. P.; Marsh M.; Sigurdardottir A.; Pitt W. R.; Blundell T. L. Q. Rev. Biophys. 2012, 45, 383. [DOI] [PubMed] [Google Scholar]
- Vazquez A.; Bond E. E.; Levine A. J.; Bond G. L. Nat. Rev. Drug Discovery 2008, 7, 979. [DOI] [PubMed] [Google Scholar]
- Shangary S.; Wang S. Clin. Cancer Res. 2008, 14, 5318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kussie P. H.; Gorina S.; Marechal V.; Elenbaas B.; Moreau J.; Levine A. J.; Pavletich N. P. Science 1996, 274, 948. [DOI] [PubMed] [Google Scholar]
- Yin H.; Lee G. I.; Park H. S.; Payne G. A.; Rodriguez J. M.; Sebti S. M.; Hamilton A. D. Angew. Chem., Int. Ed. 2005, 44, 2704. [DOI] [PubMed] [Google Scholar]
- Bernal F.; Tyler A. F.; Korsmeyer S. J.; Walensky L. D.; Verdine G. L. J. Am. Chem. Soc. 2007, 129, 2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasan R.; Dias R. L.; Moehle K.; Zerbe O.; Vrijbloed J. W.; Obrecht D.; Robinson J. A. Angew. Chem., Int. Ed. Engl. 2004, 43, 2109. [DOI] [PubMed] [Google Scholar]
- Kritzer J. A.; Lear J. D.; Hodsdon M. E.; Schepartz A. J. Am. Chem. Soc. 2004, 126, 9468. [DOI] [PubMed] [Google Scholar]
- Murray J. K.; Gellman S. H. Biopolymers 2007, 88, 657. [DOI] [PubMed] [Google Scholar]
- Sakurai K.; Schubert C.; Kahne D. J. Am. Chem. Soc. 2006, 128, 11000. [DOI] [PubMed] [Google Scholar]
- Yu S.; Qin D.; Shangary S.; Chen J.; Wang G.; Ding K.; McEachern D.; Qiu S.; Nikolovska-Coleska Z.; Miller R.; Kang S.; Yang D.; Wang S. J. Med. Chem. 2009, 52, 7970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev L. T.; Vu B. T.; Graves B.; Carvajal D.; Podlaski F.; Filipovic Z.; Kong N.; Kammlott U.; Lukacs C.; Klein C.; Fotouhi N.; Liu E. A. Science 2004, 303, 844. [DOI] [PubMed] [Google Scholar]
- Reed D.; Shen Y.; Shelat A. A.; Arnold L. A.; Ferreira A. M.; Zhu F.; Mills N.; Smithson D. C.; Regni C. A.; Bashford D.; Cicero S. A.; Schulman B. A.; Jochemsen A. G.; Guy R. K.; Dyer M. A. J. Biol. Chem. 2010, 285, 10786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y. S.; Graves B.; Guerlavais V.; Tovar C.; Packman K.; To K.-H.; Olson K. A.; Kesavan K.; Gangurde P.; Mukherjee A.; Baker T.; Darlak K.; Elkin C.; Filipovic Z.; Qureshi F. Z.; Cai H.; Berry P.; Feyfant E.; Shi X. E.; Horstick J.; Annis D. A.; Manning A. M.; Fotouhi N.; Nash H.; Vassilev L. T.; Sawyer T. K. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, E3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynes C.; Host H.; Camproux A. C.; Laconde G.; Leroux F.; Mazars A.; Deprez B.; Fahraeus R.; Villoutreix B. O.; Sperandio O. PLoS Comput. Biol. 2010, 6, e1000695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joerger A. C.; Fersht A. R. Annu. Rev. Biochem. 2008, 77, 557. [DOI] [PubMed] [Google Scholar]
- Popowicz G. M.; Dömling A.; Holak T. A. Angew. Chem., Int. Ed. 2011, 50, 2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelsen K.; Jordan J. B.; Lewis J.; Long A. M.; Yang E.; Rew Y.; Zhou J.; Yakowec P.; Schnier P. D.; Huang X.; Poppe L. J. Am. Chem. Soc. 2012, 134, 17059. [DOI] [PubMed] [Google Scholar]
- Hirota K.; Semenza G. L. Crit. Rev. Oncol. Hemat. 2006, 59, 15. [DOI] [PubMed] [Google Scholar]
- Semenza G. L. Nat. Rev. Cancer 2003, 3, 721. [DOI] [PubMed] [Google Scholar]
- Orourke J. F.; Pugh C. W.; Bartlett S. M.; Ratcliffe P. J. Eur. J. Biochem. 1996, 241, 403. [DOI] [PubMed] [Google Scholar]
- Ivan M.; Kondo K.; Yang H. F.; Kim W.; Valiando J.; Ohh M.; Salic A.; Asara J. M.; Lane W. S.; Kaelin W. G. Science 2001, 292, 464. [DOI] [PubMed] [Google Scholar]
- Butterfoss G. L.; Kuhlman B. Annu. Rev. Biophys. Biomol. Struct. 2006, 35, 49. [DOI] [PubMed] [Google Scholar]
- Kuhlman B.; Dantas G.; Ireton G. C.; Varani G.; Stoddard B. L.; Baker D. Science 2003, 302, 1364. [DOI] [PubMed] [Google Scholar]
- Jiang L.; Althoff E. A.; Clemente F. R.; Doyle L.; Rothlisberger D.; Zanghellini A.; Gallaher J. L.; Betker J. L.; Tanaka F.; Barbas C. F.; Hilvert D.; Houk K. N.; Stoddard B. L.; Baker D. Science 2008, 319, 1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripka A. S.; Satyshur K. A.; Bohacek R. S.; Rich D. H. Org. Lett. 2001, 3, 2309. [DOI] [PubMed] [Google Scholar]
- Kortemme T.; Joachimiak L. A.; Bullock A. N.; Schuler A. D.; Stoddard B. L.; Baker D. Nat. Struct. Mol. Biol. 2004, 11, 371. [DOI] [PubMed] [Google Scholar]
- Korkegian A.; Black M. E.; Baker D.; Stoddard B. L. Science 2005, 308, 857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashworth J.; Havranek J. J.; Duarte C. M.; Sussman D.; Monnat R. J.; Stoddard B. L.; Baker D. Nature 2006, 441, 656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahiyat B. I.; Mayo S. L. Science 1997, 278, 82. [DOI] [PubMed] [Google Scholar]
- Harbury P. B.; Plecs J. J.; Tidor B.; Alber T.; Kim P. S. Science 1998, 282, 1462. [DOI] [PubMed] [Google Scholar]
- Joachimiak L. A.; Kortemme T.; Stoddard B. L.; Baker D. J. Mol. Biol. 2006, 361, 195. [DOI] [PubMed] [Google Scholar]
- Rothlisberger D.; Khersonsky O.; Wollacott A. M.; Jiang L.; DeChancie J.; Betker J.; Gallaher J. L.; Althoff E. A.; Zanghellini A.; Dym O.; Albeck S.; Houk K. N.; Tawfik D. S.; Baker D. Nature 2008, 453, 190. [DOI] [PubMed] [Google Scholar]
- Shifman J. M.; Mayo S. L. Proc. Natl. Acad. Sci. U S A 2003, 100, 13274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleishman S. J.; Whitehead T. A.; Ekiert D. C.; Dreyfus C.; Corn J. E.; Strauch E.-M.; Wilson I. A.; Baker D. Science 2011, 332, 816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaver-Fay A.; Tyka M.; Lewis S. M.; Lange O. F.; Thompson J.; Jacak R.; Kaufman K. W.; Renfrew P. D.; Smith C. A.; Sheffler W.; Davis I. W.; Cooper S.; Treuille A.; Mandell D. J.; Richter F.; Ban Y.-E. A.; Fleishman S. J.; Corn J. E.; Kim D. E.; Lyskov S.; Berrondo M.; Mentzer S.; Popovic Z.; Havranek J. J.; Karanicolas J.; Das R.; Meiler J.; Kortemme T.; Gray J. J.; Kuhlman B.; Baker D.; Bradley P. In Methods in Enzymology; Michael L. J., Ludwig B., Eds.; Academic Press: Waltham, MA, 2011; Vol. 487, p 545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renfrew P. D.; Choi E. J.; Bonneau R.; Kuhlman B. PLoS One 2012, 7, e32637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfoss G. L.; Renfrew P. D.; Kuhlman B.; Kirshenbaum K.; Bonneau R. J. Am. Chem. Soc. 2009, 131, 16798. [DOI] [PubMed] [Google Scholar]
- Drew K.; Renfrew P. D.; Craven T.; Butterfoss G. L.; Chou F.-C.; Lyskov S.; Bullock B. N.; Watkins A.; Labonte J. W.; Pacella M.; Kilambi K. P.; Leaver-Fay A.; Kuhlman B.; Gray J. J.; Bradley P.; Kirshenbaum K.; Arora P. S.; Das R.; Bonneau R. PLoS One 2013, 8, e67051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyskov S.; Chou F.-C.; Conchúir S. Ó.; Der B. S.; Drew K.; Kuroda D.; Xu J.; Weitzner B. D.; Renfrew P. D.; Sripakdeevong P.; Borgo B.; Havranek J. J.; Kuhlman B.; Kortemme T.; Bonneau R.; Gray J. J.; Das R. PLoS One 2013, 8, e63906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henchey L. K.; Porter J. R.; Ghosh I.; Arora P. S. ChemBiolChem 2010, 11, 2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight S. M.; Umezawa N.; Lee H. S.; Gellman S. H.; Kay B. K. Anal. Biochem. 2002, 300, 230. [DOI] [PubMed] [Google Scholar]
- Ramachandran G. N.; Sasisekharan V. Adv. Protein Chem. 1968, 23, 283. [DOI] [PubMed] [Google Scholar]
- Garcia-Echeverria C.; Chene P.; Blommers M. J. J.; Furet P. J. Med. Chem. 2000, 43, 3205. [DOI] [PubMed] [Google Scholar]
- Kellogg E. H.; Leaver-Fay A.; Baker D. Proteins 2011, 79, 830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiler J.; Baker D. Proteins 2006, 65, 538. [DOI] [PubMed] [Google Scholar]
- Potapov V.; Cohen M.; Schreiber G. Prot. Eng. Des. Sel 2009, 22, 553. [DOI] [PubMed] [Google Scholar]
- Cheng T.; Li X.; Li Y.; Liu Z.; Wang R. J. Chem. Inf. Model. 2009, 49, 1079. [DOI] [PubMed] [Google Scholar]
- Kushal S.; Lao B. B.; Henchey L. K.; Dubey R.; Mesallati H.; Traaseth N. J.; Olenyuk B. Z.; Arora P. S. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 15602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dial R.; Sun Z. Y.; Freedman S. J. Biochemistry 2003, 42, 9937. [DOI] [PubMed] [Google Scholar]
- Bonetta L. Nature 2010, 468, 851. [DOI] [PubMed] [Google Scholar]
- Venkatesan K.; Rual J.-F.; Vazquez A.; Stelzl U.; Lemmens I.; Hirozane-Kishikawa T.; Hao T.; Zenkner M.; Xin X.; Goh K.-I.; Yildirim M. A.; Simonis N.; Heinzmann K.; Gebreab F.; Sahalie J. M.; Cevik S.; Simon C.; de Smet A.-S.; Dann E.; Smolyar A.; Vinayagam A.; Yu H.; Szeto D.; Borick H.; Dricot A.; Klitgord N.; Murray R. R.; Lin C.; Lalowski M.; Timm J.; Rau K.; Boone C.; Braun P.; Cusick M. E.; Roth F. P.; Hill D. E.; Tavernier J.; Wanker E. E.; Barabasi A.-L.; Vidal M. Nat. Meth. 2009, 6, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jochim A. L.; Arora P. S. Mol. BioSyst. 2009, 5, 924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H. P.; Izmaylov, A. F.; Bloino, J.; Zheng, G.; Sonnenberg, J. L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, J. A., Jr.; Peralta, J. E.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin, K. N.; Staroverov, V. N.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, N. J.; Klene, M.; Knox, J. E.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Zakrzewski, V. G.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Dapprich, S.; Daniels, A. D.; Farkas, Ö.; Foresman, J. B.; Ortiz, J. V.; Cioslowski, J.; Fox, D. J.. Gaussian 09, Revision C.01 ed.; Gaussian, Inc.: Wallingford, CT, 2009.
- Roehrl M. H.; Wang J. Y.; Wagner G. Biochemistry 2004, 43, 16056. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.