

Abstract
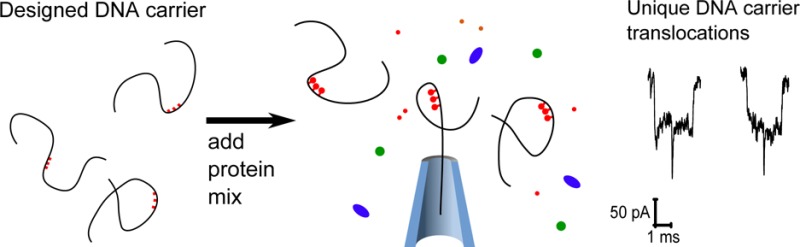
Nanopores are a versatile technique for the detection and characterization of single molecules in solution. An ongoing challenge in the field is to find methods to selectively detect specific biomolecules. In this work we describe a new technique for sensing specific proteins using unmodified solid-state nanopores. We engineered a double strand of DNA by hybridizing nearly two hundred oligonucleotides to a linearized version of the m13mp18 virus genome. This engineered double strand, which we call a DNA carrier, allows positioning of protein binding sites at nanometer accurate intervals along its contour via DNA conjugation chemistry. We measure the ionic current signal of translocating DNA carriers as a function of the number of binding sites and show detection down to the single protein level. Furthermore, we use DNA carriers to develop an assay for identifying a single protein species within a protein mixture.
Introduction
Nanopores have emerged to become an important tool in biophysics and single molecule sensing. The simple detection principle of nanopore sensing is that an analyte modulates the ionic current as it binds or translocates an isolated nanopore. In this paper we focus on the ability of nanopores to sense and analyze protein molecules. Biological pores used for nanopore sensing have typical diameters on the order 1–2 nm, which limits the range of analytes that will freely translocate. For instance, α-hemolysin has a narrowest constriction of 1.4 nm,1 which allows the translocation of single-stranded DNA,2 but globular proteins cannot translocate without unfolding from their native state. Stochastic binding of a protein to a ligand attached to the entrance of a biological nanopore has therefore been used extensively as a method for detection of folded proteins.3−6 Alternatively several strategies based on stochastic blocking of α-hemolysin by single stranded DNA–protein complexes have been reported7 and some proteins can also be detected by their effect on current–voltage curves due to binding to biological pores.8 Denaturation by chemical9 or thermal10 means has been used to unfold globular proteins and permit studies of their translocations through α-hemolysin. Recently techniques of unfolding using high mechanical force with oligonucleotide tethers11,12 or unfoldase13,14 enzymes have been developed which provide new avenues for biological nanopore based protein detection.
Solid-state nanopores can be made with arbitrary dimensions and therefore permit the analysis of proteins that translocate the pore in their native state.15−19 It is also possible to use solid-state nanopores to detect patches of DNA binding proteins randomly attached along a DNA double strand.20−22 A central finding for translocations of single, unbound proteins has been that, at the typical experiment bandwidths used, most proteins pass through too quickly to be measured and only events in the tail of the distribution are detected.23 Recent advances based on thin membranes and high bandwidth amplifiers have shown improvement in resolution.24 However, even with sufficient bandwidth and signal to measure all proteins, it remains a challenge to differentiate single, similarly sized proteins translocating a solid-state nanopore since simple Coulter counter-like measurements without binding do not yield any chemical information.
The addition of a binding motif on a solid-state nanopore to impart selectivity to single molecule protein measurements has been used in several examples. For instance, self-assembly of monolayers can immobilize a single nitriltriacetic acid receptor for stochastic sensing of His-tagged proteins.25 It is also possible to create mobile lipid bilayers on the nanopore and its support surface.26 Protein binding sites are then introduced into the lipids enabling detection at low sample concentrations. However, both these methods require significant engineering of the nanopore and its surface. Also it remains unclear how well they would work for targeting a single protein in an analyte mixture due to the difficulty in separating out translocations due to nontarget molecules.27
In this paper we introduce a versatile approach for specific protein measurement with unmodified solid-state nanopores. We designed carrier DNA molecules 7.2 kbp in length with chemical motifs at tailored positions for binding of one or a few protein molecules of interest. The presence or absence of specific proteins in solution is indicated by studying the characteristic ionic current signatures of these DNA carriers. Using streptavidin as an example protein, we show the measurement of single protein molecules on a specifically designed DNA carrier and develop an assay for detecting streptavidin out of a mixture of four proteins. Finally we show the generic applicability of this system by designing multiple binding locations on the DNA carrier and adapting the binding site chemistry for detection of single antidigoxigenin antibodies.
Materials and Methods
DNA Carrier Synthesis
M13mp18 ssDNA genome was purchased from New England Biolabs. A 39 base nucleotide was hybridized to allow cutting at the EcoRI and BamHI restriction sites. The DNA carrier was then formed by mixing the cut m13mp18 DNA with a 10x molar excess of each binding oligonucleotide and annealing in a buffer containing 10 mM Tris (pH = 8), 1 mM EDTA and 10 mM MgCl2. The excess oligonucleotides were subsequently removed by centrifugation using Amicon Ultra centrifugal filters (see Supporting Information for further details). For atomic force microscopy, DNA samples were adsorbed onto mica using a magnesium containing buffer and tapping mode was used.
Nanopore Fabrication
Glass nanopores were fabricated as previously described.28 Briefly, glass capillaries with inner diameter 0.2 mm and outer diameter 0.5 mm were cleaned in acetone before drying under a stream of nitrogen. Nanopores were made by pulling these capillaries with a laser based puller (Sutter P2000) to form a tip with inner diameter of 15 ± 3 nm estimated by scanning electron microscopy (see Supporting Information). Glass nanopores were integrated into multichannel devices which facilitated experimental throughput.28
Nanopore Measurements
All proteins were purchased in powder form, diluted in 10 mM Tris (pH = 8), 1 mM EDTA before being aliqouted and stored at −20 °C until use. Streptavidin, lysozyme, β-lactoglobulin, β-galactosidase and bovine serum albumin were all purchased from Sigma-Aldrich. Antidigoxigenin antibody (sheep polyclonal) was purchased from Roche Life Sciences. Before each nanopore experiment, the DNA carrier was first incubated with its protein or protein mixture for 30 min before adding to the nanopore reservoir. All nanopore translocation measurements were carried out in a buffer containing either 10 mM Tris (pH = 8), 1 mM EDTA, 4 M LiCl or 10 mM Tris (pH = 8), 1 mM EDTA, 4 M LiCl, 5 mM MgCl2 (we noticed no systematic difference between these two solutions). The applied voltage for all experiments was 600 mV. Ionic currents were recorded and analyzed with custom written Labview programs. All ionic current measurement was performed using an Axopatch 200B (Molecular Devices) with the current signal filtered at 49.9 kHz with an external 8 pole Bessel filter (Frequency Devices) followed by digitization at a sampling frequency of 250 kHz with a 16 bit data acquisition card (National Instruments).
Results
DNA Carrier Design
Our basic design principle was to make a long double strand of DNA which allowed for simple functionalization at selected positions along its contour. To achieve this we followed the strategy used in DNA origami29 where a long, single stranded DNA genome is formed into a desired shape by hundreds of synthesized oligonucleotides. We used the commercially available 7249 base single stranded circular genome of the m13mp18 virus. This circular single strand was cut at the EcoRI and BamHI restriction sites yielding a linear single strand 7228 bases in length. 190 oligonucleotides were designed to hybridize along the length of this 7228 base strand. Each oligonucleotide was 38 bases in length except for the two oligonucleotides at the ends which were each 46 bases long. These two oligonucleotides, at either end, include a four thymine overhang to prevent dimerization. The structure formed is therefore essentially a 7228 bp double strand with nicks on one strand occurring every 38 bases. The sequences of all the oligonucleotides used are given in the Supporting Information. We refer to the double stranded DNA formed in this way as a “DNA carrier” since we use it to selectively drive protein molecules through a solid-state nanopore.
Figure 1a–c shows a schematic and atomic force microscope images of the DNA carrier synthesis. The single stranded m13mp18 genome forms a compact globular shape. After cutting and hybridization of the complementary oligonucleotides, an extended coil-like structure is formed as expected for double stranded DNA. We extensively analyzed the yield of DNA carriers and found that approximately 20% are shorter than expected due to the presence of ∼10% linear m13mp18 DNA before the restriction digestion (Figures S1–S3).
Figure 1.
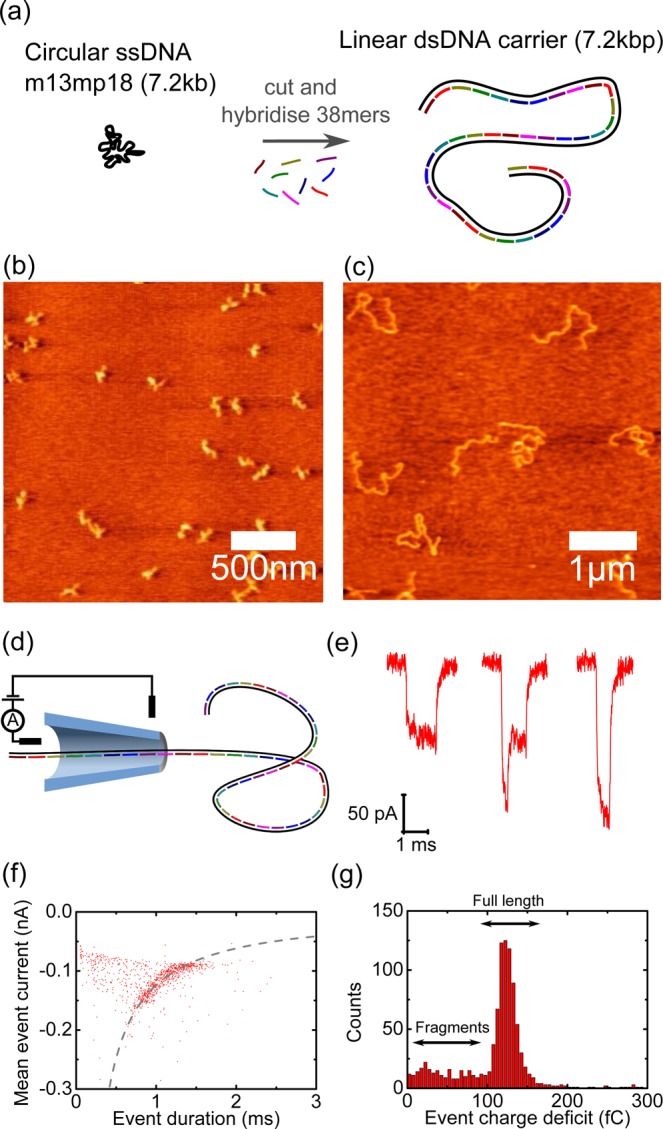
Design, synthesis and nanopore characterization of DNA carriers. (a) Schematic overview–the circular single stranded m13mp18 is cut by restriction enzymes and subsequently mixed with 190 complementary oligonucleotides which hybridize to form a 7.2 kbp DNA double strand which we term a “DNA carrier”. (b) Atomic force microscope image of m13mp18 genomes showing their compact globular shape. (c) Atomic force microscope of synthesized DNA carriers showing the extended coil-like structure. (d) Schematic of DNA carriers translocation through a conical glass nanopore. (e) Typical ionic current blockades showing quantized states due to hairpin configurations. (f) Scatter plot of first 1000 translocations recorded for a glass nanopore. The main band of translocations follows the relationship that the mean event current is inversely proportional to the event duration, which is typical for a single DNA length.30 (g) Histogram of event charge deficit for the 1000 events shown in (f). The main peak at approximately 124 fC is due to the full length 7.2 kbp DNA carriers. The tail of events at smaller values is attributed to fragments.31
Initially, we investigated the ionic current signals due to translocations of the double stranded DNA carrier through solid-state nanopores. All nanopore experiments were performed using conical shaped glass nanopores with final opening diameters of 15 ± 3 nm (mean ± s.d.) (Figures S4–S6 and Table S1). We use glass nanopores due to their ease of manufacture and low levels of high frequency noise.28,32 A salt concentration of 4 M LiCl was used to reduce the DNA translocation velocity.33 Figure 1e shows typical ionic current translocations when 1 nM of DNA carrier is added to the sample reservoir and a potential of +600 mV is applied. We measure multilevel ionic current blockades, which are in line with previous observations for double stranded DNA translocations.30,34,35 These events are caused by the hairpin conformations a DNA molecule can take as it passes through the nanopore. We compared the DNA carrier translocations to a 7 kbp DNA plasmid fragment and both gave similar statistics for the percentage of folded translocations (Figures S7 and S8). Therefore, the 38 base interval nicks on one strand of the DNA carrier do not create a significant difference compared to DNA with a phosphate backbone without regular breaks.
Analysis of Proteins Bound to DNA Carrier
Our DNA carrier design allows for easy and highly controlled positioning of functional motifs along the DNA carrier contour at 38 bp (13 nm) spacings. These motifs can be attached at the 5′ or 3′ end of any of the 38mer oligonucleotides which form one strand of the double helix. As an initial proof of concept, we studied the binding of streptavidin at designed positions along the DNA carrier. Streptavidin is a tetrameric protein which binds strongly to biotin36 with a dissociation constant KD on the order 10–14 M. We created designs where 5, 3, and 1 of the oligonucleotides at the center of the DNA carrier were functionalized with biotin (Figure 2a–c). The biotin group was attached to three thymine nucleotides so that it slightly protruded from the DNA carrier double helix. In the 5B and 3B designs, each biotin plus three thymine motif was separated by 38 bp (∼13 nm) along the DNA carrier to avoid one streptavidin linking to two biotins (streptavidin having a diameter of ∼6 nm16). Streptavidin was added at a significant excess of five times compared to the number of biotin modified oligonucleotides to avoid multimerization of DNA carriers.
Figure 2.

Tailoring the number of binding sites on DNA carriers. (a), (b) and (c) show schematics of DNA carrier designs with 5, 3, and 1 biotin groups after incubation with streptavidin. For each design three typical translocation events are shown in (d), (e) and (f). Only events beginning and ending with one DNA double strand were selected. (g) The size and duration of the current spike in the center was measured relative to the double stranded DNA level. (h) Normalized histograms of the current change ΔI for the three designs. Each graph shows three lines which are independent nanopores. The minimum (threshold) ΔI was set to 40 pA. The total number of detected protein current spikes are 407 (5B), 288 (3B) and 166 (1B).
The three designs called 5B, 3B and 1B (containing 5, 3, and 1 biotin sites respectively) were incubated with the five times excess of streptavidin for 30 min before adding the samples for analysis by electrophoretic translocations through a glass nanopore. The final DNA carrier concentration in the nanopore reservoir was 1–4 nM meaning that nearly all binding sites will be occupied for KD ∼ 10–14 M. Crucially, DNA carrier events could be easily distinguished from translocations of unbound streptavidin since the event duration is significantly larger giving a much larger event charge deficit (Figures S10–S12). An event sorting algorithm was used to select translocations of only the full length DNA carrier (based on ECD as in Figure 1g) and also where the DNA carrier passed without a hairpin at the beginning or end of the event (Figures S13–S15). This excludes nearly all translocations with DNA folds, as quantified later in Figure 3, a small background remains after this algorithm and gives ∼5% false positives. For these selected translocations which are primarily unfolded DNA carriers, we expect a signal in the center of the translocation due to the centrally located streptavidin binding site. Indeed for all three designs we observe a characteristic current spike close to the center of the translocation event corresponding to the passage of the protein (typical events shown in Figure 2d–f).
Figure 3.
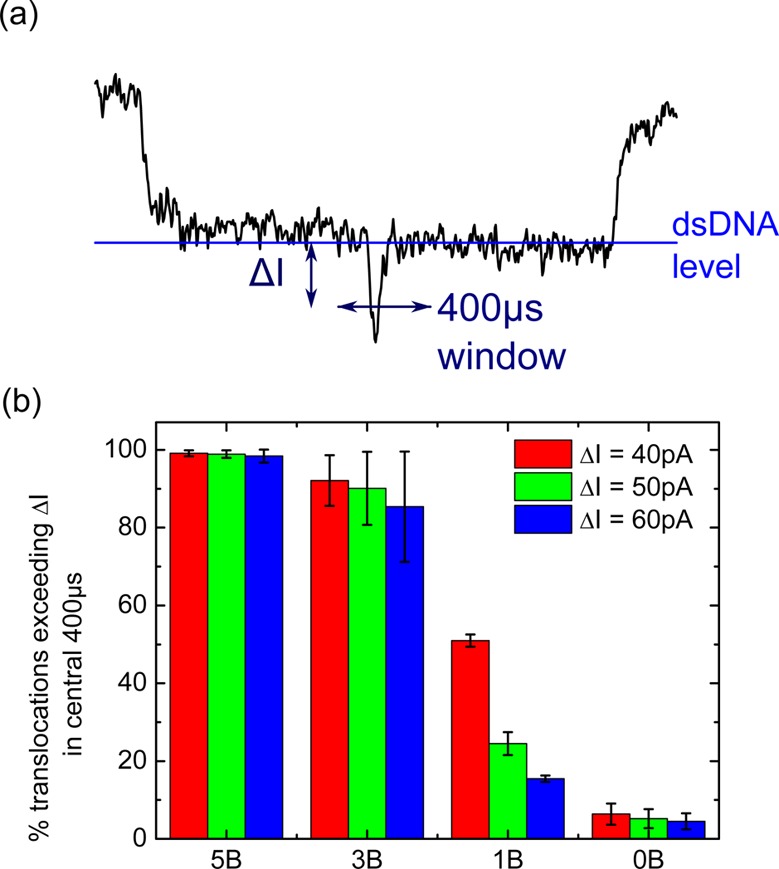
Analysis of detection efficiency for different numbers of bound proteins. (a) A 400 μs window is created at the center of the translocation. The threshold ΔI (from the one double strand DNA level) is varied, and in (b) the percentage of translocations exceeding ΔI in the 400 μs window is calculated. The percentage is shown for designs 5B, 3B, 1B and 0B with 5, 3, 1, and 0 streptavidin attached, respectively. Error bars are the standard deviation from N independent nanopores with N = 3 (for 5B and 1B), N = 5 (for 3B) and N = 6 (for 0B) (raw values given in Tables S2–S5).
The amplitude of the protein signal increases as we increase the number of protein binding sites (Figure 2h). Glass nanopores have a conical shape and behave similarly to a cylindrical nanopore with length of a few hundred nanometers. The increase in current change with increasing number of streptavidin binding sites can therefore be explained as a direct consequence of the higher exclusion volume for ions. Our results show we do not resolve independent spikes from each individual protein when the spacing is only 38 bp (13 nm). However, we will show later that binding sites separated by ∼600 nm yield easily resolvable independent current spikes (see Figure 5a).
Figure 5.
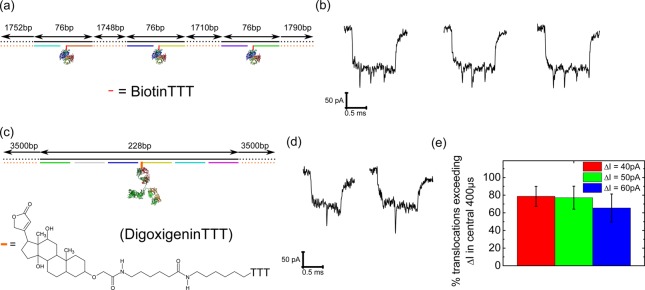
Adaptable binding site positions and chemistry. (a) DNA carrier design with three biotins separated at approximately one-quarter intervals along the DNA carrier. (b) Translocations after incubation of design (a) with streptavidin showing three spikes at approximately one-quarter, two quarters and three-quarters of the total translocation time. (c) Design of a DNA carrier with a digoxigenin tag at the central position and bound to an antidigoxigenin antibody. (d) Typical translocations after incubation with antidigoxigenin showing the presence of a current spike in the center. (e) Percentage of translocation exceeding ΔI from one DNA strand level (as in Figure 3). Error bars show the standard deviation from three independent nanopores with raw values given in Table S10.
The percentage of DNA carrier translocations showing a central current spike can vary depending on how many streptavidin binding sites are designed. We therefore developed a method for quantifying the detection efficiency for the 5B, 3B and 1B designs. For each DNA carrier translocation, beginning and ending with a current level indicating a single DNA double strand, we created a 400 μs window which was ±200 μs either side of the center of the event (Figure 3a). We then determined whether the current exceeded a threshold ΔI in this time window with ΔI measured relative to the dsDNA level. The minimum threshold was set to 40 pA to give a sufficient signal-to-noise ratio for our automated detection routine. In Figure 3b we evaluate the percentage of DNA carrier translocations which show a current spike in the central 400 μs with magnitude greater than ΔI.
When there are no biotin groups on the DNA carrier design, we measure a background of ∼5% translocations showing a false positive protein spike. These can be attributed to translocations where the DNA has two folds in the center or more complicated folding events. It is important to note that these constitute only a small proportion of the total events in agreement with previous analysis for translocations of double strand DNA through similar sized nanopores.35 The designs with five streptavidin and three streptavidin have means of 99 and 92%, respectively, for the number of detectable current events at 40 pA threshold. Furthermore, the percentage of detectable current spikes does not decrease significantly as the threshold is increased. This strongly suggests that we nearly always detect a protein signal when we have 5 or 3 streptavidin attached. For the 1B design where only a single streptavidin is attached we measure a protein signal in 51% of translocations at 40 pA threshold. This percentage decreases strongly as we increase the threshold to 50 and 60 pA.
The low detection efficiency for a single bound streptavidin on the DNA carrier could be due to the presence of DNA carriers without streptavidin attached or a current signal that is too small to pass the threshold. The number of unbound biotin sites should be very low due to the high biotin–streptavidin affinity. Therefore, we believe that the low percentage of detected current spikes for the one streptavidin design is due to the low signal-to-noise ratio for a single streptavidin protein. This detection efficiency could possibly be improved by using ultrathin membranes which give higher current signals compared to glass nanopores.37
Detection of Streptavidin from Protein Mixtures
So far our results show the ability to design and measure protein attachment on the DNA carrier. As described above, the ionic current signatures due to translocation of the DNA carrier are significantly different to streptavidin that we can use the event charge deficit to easily distinguish the two. Previous analysis19 shows that a range of other globular proteins have similarly low levels of event charge deficit compared to streptavidin. Because of the possibility to impart binding specificity on the DNA carriers, we reasoned that it should be possible to accurately detect and thus identify a bound analyte against a background of different translocating proteins.
In order to test this hypothesis we made two protein mixtures using proteins which have been characterized previously with nanopores.19 Mixture 1 (mix1) contained streptavidin, β-lactoglobulin, β-galactosidase and lysozyme. Mixture 2 (mix2) contained bovine serum albumin (BSA), β-lactoglobulin, β-galactosidase and lysozyme. Mix2 therefore acts as a control with streptavidin replaced by BSA. We tested incubations of the 3B DNA carrier (featuring three biotin sites at the center) with either mix1 or mix2 (Figure 4b). In all mixture experiments the final concentration in the nanopore reservoir was 15 nM of each protein and 1 nM of DNA carrier. The 3B design was chosen as a compromise between a design that needed relatively few binding sites and one that had a good detection efficiency (as shown in Figure 3). We also performed separate controls where a DNA carrier with no biotin modifications (0B) was incubated under the same conditions (with mix1 or mix2) before being translocated through a nanopore.
Figure 4.

An assay for selective protein detection on a DNA carrier. (a) Events that begin and end with the one DNA double strand level are selected. If the current exceeds 50 pA from the baseline one DNA strand level within a central 400 μs window, the event is labeled positive for protein detection. Left shows an example of a positive translocation, right shows an example of a negative translocation. (b) Two DNA carrier designs used for experiment and controls. Three biotin (3B) design is as in Figure 3 with 3 biotin tags at the center. No modifications (0B) design has no biotin groups. Each design was incubated with one of two mixtures: mix1 contained the target streptavidin protein and mix2 contained BSA as a substitute control. A high percentage of threshold crossing events is only observed for the correct combination of binding site (biotin) and target protein (streptavidin). Error bars are the standard deviation from four independent nanopores (raw data is given in Tables S6–S9).
Translocations were recorded in these four cases (3B+mix1, 3B+mix2, 0B+mix1, 0B+mix2) and each case was repeated four times with separate nanopores to determine experimental consistency. As before, DNA carrier events were selected based on event charge deficit and those translocations beginning and ending with the one DNA level. On the basis of our detection efficiency analysis in Figure 3, we set a threshold of 50 pA in a 400 μs central window for positive protein detection.
We observe a significantly higher percentage of threshold crossing events only when biotin functionalized DNA carrier and streptavidin are present in the mixture (Figure 4b). The mean number of positive detections for the correct combination of biotin functionalized DNA carrier and streptavidin is 87%. In the control samples we consistently measure a mean of approximately 10% false positives. We anticipate that further optimization of the detection algorithm could help to decrease the number of false negatives and false positives. Nevertheless, these results demonstrate that the unique ionic current signature of the DNA carrier can be used for selectively identifying the presence of a target protein in a mixture.
Adaptability of Binding Site Position and Chemistry
As a simple demonstration of the possibilities of the DNA carrier approach we present two further examples of DNA carrier designs. First we designed a DNA carrier with three oligonucleotides functionalized with biotin groups at approximately one-quarter (∼600 nm) intervals along the DNA carrier. After incubation with five times excess of streptavidin, the structure was translocated through a nanopore. Figure 5b shows three translocations where we clearly resolve three separated spikes during the translocation. The spikes occur at approximately equal time points as expected from the design. Multiple protein markers on the same DNA molecule provide reference points which could help for probing DNA velocity during an individual translocation rather than single protrusions of DNA which was recently demonstrated.38
As a second demonstration of the adaptability of DNA carriers we chose a different chemical tag for the selective sensing of antibodies. A DNA carrier was modified with a digoxigenin and three thymine motif covalently attached to the 38mer oligonucleotide at the central position (Figure 5c). This DNA carrier was incubated with antidigoxigenin antibodies which are known to form a high affinity interaction with digoxigenin39 with KD ∼ 1 nM.40 The final concentration in the nanopore reservoir was 3 nM of DNA carriers and 8 nM of antidigoxigenin so that a high proportion (∼85%) of DNA carriers should have an antibody bound assuming equilibrium and KD = 1 nM. We again observe current spikes at the center of the translocation due to the protein (Figure 5d). The current spike is typically larger than that for a single streptavidin which is consistent with the higher molecular weight of the antidigoxigenin (∼150 kDa compared to ∼60 kDa for streptavidin) and results in a higher detection efficiency compared to designs binding a single streptavidin (Figure 5e).
Discussion
In this paper we have shown a method for fabricating DNA strands designed for the selective detection of proteins with solid-state nanopores. The ability to rationally create DNA strands in this way has great potential for simple solid-state nanopore sensing of specific proteins. In particular we have shown how information can be encoded in the DNA structure: in this paper we incorporated protein binding sites at specific locations along the length of the DNA. We have then used the ability of solid-state nanopores to scan for individual molecules attached on a long double strand of DNA which has previously been shown for detecting fluorophores41 and PNA.42 Single stranded DNA–protein complexes have also been used for protein detection methods with α-hemolysin based on monitoring stochastic blocking events.7 Our method is distinguished from this since we use translocation measurements through large solid-state nanopores that allow the proteins to pass in their native state. Therefore, our technique could enable larger binding sites like aptamers,43−45 small peptides or antibody fragments46−48 to be readily used. There are also simple routes open to multiplex measurements for example by attaching different protein binding sites at different positions along the DNA strand.
The protein measurement system described here has several potential advantages over existing solid-state nanopore based methods for selective protein detection. First, it does not rely on surface modification of a nanopore but rather we do our engineering in the design of the DNA carrier in solution. Second, in our method the affinity between the binding site on the DNA carrier and its target protein can be high. In contrast, protein sensing by stochastic binding at the mouth of a nanopore requires that the binding strength must be adequately tuned to give characteristic on–off times on the order of milliseconds to seconds.
Another potential advantage is that the transport of the protein attached to the DNA carrier becomes dominated by the electrokinetics of the DNA. This means that the detection does not need to be optimized for different proteins (dependent on eg charge, diffusion coefficient) but rather will be dependent on the DNA transport which has been extensively characterized in previous literature. For instance one improvement that could be made, based on known DNA transport, is to apply a salt gradient across the nanopore which can significantly enhance the DNA capture rate and enable high throughput at pM concentration.49 Finally we have also demonstrated the potential to measure a specific protein from mixtures, which represents an important step toward the goal of specific solid-state nanopore detection in complex samples for diagnostics. The compatibility of this system with more complex samples will require addressing potential background due to DNA binding proteins and DNA nucleases.
Acknowledgments
We thank Vivek Thacker and Nadanai Laohakunakorn for critical reading of this manuscript. N.A.W.B. was supported by an EPSRC Doctoral Prize Award. U.F.K. acknowledges support by an ERC starting grant, PassMembrane 261101.
Supporting Information Available
Further materials and methods details on DNA carrier synthesis, nanopore fabrication and characterization, automated ionic current analysis programs, protein only translocations and data set statistics. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare the following competing financial interest(s): The results in this publication are contributing to a patent application.
Supplementary Material
References
- Song L.; Hobaugh M. R.; Shustak C.; Cheley S.; Bayley H.; Gouaux J. E. Science 1996, 274, 1859. [DOI] [PubMed] [Google Scholar]
- Kasianowicz J. J.; Brandin E.; Branton D.; Deamer D. W. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 13770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Movileanu L.; Howorka S.; Braha O.; Bayley H. Nat. Biotechnol. 2000, 18, 1091. [DOI] [PubMed] [Google Scholar]
- Xie H.; Braha O.; Gu L.-Q.; Cheley S.; Bayley H. Chem. Biol. 2005, 12, 109. [DOI] [PubMed] [Google Scholar]
- Rotem D.; Jayasinghe L.; Salichou M.; Bayley H. J. Am. Chem. Soc. 2012, 134, 2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheley S.; Xie H.; Bayley H. Chembiochem. 2006, 7, 1923. [DOI] [PubMed] [Google Scholar]
- Kasianowicz J. J.; Henrickson S. E.; Weetall H. H.; Robertson B. Anal. Chem. 2001, 73, 2268. [DOI] [PubMed] [Google Scholar]
- Halverson K. M.; Panchal R. G.; Nguyen T. L.; Gussio R.; Little S. F.; Misakian M.; Bavari S.; Kasianowicz J. J. J. Biol. Chem. 2005, 280, 34056. [DOI] [PubMed] [Google Scholar]
- Oukhaled G.; Mathé J.; Biance A.-L.; Bacri L.; Betton J.-M.; Lairez D.; Pelta J.; Auvray L. Phys. Rev. Lett. 2007, 98, 158101. [DOI] [PubMed] [Google Scholar]
- Payet L.; Martinho M.; Pastoriza-Gallego M.; Betton J.-M.; Auvray L.; Pelta J.; Mathé J. Anal. Chem. 2012, 84, 4071. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Larrea D.; Bayley H. Nat. Nanotechnol. 2013, 8, 288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen C. B.; Rodriguez-Larrea D.; Bayley H. Nat. Biotechnol. 2014, 32, 179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nivala J.; Marks D. B.; Akeson M. Nat. Biotechnol. 2013, 31, 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nivala J.; Mulroney L.; Li G.; Schreiber J.; Akeson M. ACS Nano 2014, 8, 12365. [DOI] [PubMed] [Google Scholar]
- Pedone D.; Firnkes M.; Rant U. Anal. Chem. 2009, 81, 9689. [DOI] [PubMed] [Google Scholar]
- Firnkes M.; Pedone D.; Knezevic J.; Döblinger M.; Rant U. Nano Lett. 2010, 10, 2162. [DOI] [PubMed] [Google Scholar]
- Han A.; Schürmann G.; Mondin G.; Bitterli R. A.; Hegelbach N. G.; de Rooij N. F.; Staufer U. Appl. Phys. Lett. 2006, 88, 093901. [Google Scholar]
- Fologea D.; Ledden B.; McNabb D. S.; Li J. Appl. Phys. Lett. 2007, 91, 539011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W.; Bell N. A. W.; Hernández-Ainsa S.; Thacker V. V.; Thackray A. M.; Bujdoso R.; Keyser U. F. ACS Nano 2013, 7, 4129. [DOI] [PubMed] [Google Scholar]
- Kowalczyk S. W.; Hall A. R.; Dekker C. Nano Lett. 2010, 10, 324. [DOI] [PubMed] [Google Scholar]
- Hall A. R.; van Dorp S.; Lemay S. G.; Dekker C. Nano Lett. 2009, 9, 4441. [DOI] [PubMed] [Google Scholar]
- Smeets R. M. M.; Kowalczyk S. W.; Hall A. R.; Dekker N. H.; Dekker C. Nano Lett. 2009, 9, 3089. [DOI] [PubMed] [Google Scholar]
- Plesa C.; Kowalczyk S. W.; Zinsmeester R.; Grosberg A. Y.; Rabin Y.; Dekker C. Nano Lett. 2013, 13, 658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin J.; Henley R. Y.; Muthukumar M.; Rosenstein J. K.; Wanunu M. Biophys. J. 2014, 106, 696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei R.; Gatterdam V.; Wieneke R.; Tampé R.; Rant U. Nat. Nanotechnol. 2012, 7, 257. [DOI] [PubMed] [Google Scholar]
- Yusko E. C.; Johnson J. M.; Majd S.; Prangkio P.; Rollings R. C.; Li J.; Yang J.; Mayer M. Nat. Nanotechnol. 2011, 6, 253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howorka S.; Siwy Z. Chem. Soc. Rev. 2009, 38, 2360. [DOI] [PubMed] [Google Scholar]
- Bell N. A. W.; Thacker V. V.; Hernández-Ainsa S.; Fuentes-Perez M. E.; Moreno-Herrero F.; Liedl T.; Keyser U. F. Lab Chip 2013, 13, 1859. [DOI] [PubMed] [Google Scholar]
- Rothemund P. W. K. Nature 2006, 440, 297. [DOI] [PubMed] [Google Scholar]
- Li J.; Gershow M.; Stein D.; Brandin E.; Golovchenko J. A. Nat. Mater. 2003, 2, 611. [DOI] [PubMed] [Google Scholar]
- Mihovilovic M.; Hagerty N.; Stein D. Phys. Rev. Lett. 2013, 110, 028102. [DOI] [PubMed] [Google Scholar]
- Steinbock L. J.; Bulushev R. D.; Krishnan S.; Raillon C.; Radenovic A. ACS Nano 2013, 7, 11255. [DOI] [PubMed] [Google Scholar]
- Kowalczyk S. W.; Wells D. B.; Aksimentiev A.; Dekker C. Nano Lett. 2012, 12, 1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbock L. J.; Otto O.; Chimerel C.; Gornall J.; Keyser U. F. Nano Lett. 2010, 10, 2493. [DOI] [PubMed] [Google Scholar]
- Storm A.; Chen J.; Zandbergen H.; Dekker C. Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys. 2005, 71, 051903. [DOI] [PubMed] [Google Scholar]
- Holmberg A.; Blomstergren A.; Nord O.; Lukacs M.; Lundeberg J.; Uhlén M. Electrophoresis 2005, 26, 501. [DOI] [PubMed] [Google Scholar]
- Wanunu M.; Dadosh T.; Ray V.; Jin J.; McReynolds L.; Drndić M. Nat. Nanotechnol. 2010, 5, 807. [DOI] [PubMed] [Google Scholar]
- Plesa C.; Loo N.; Van Ketterer P.; Dietz H.; Dekker C. Nano Lett. 2015, 15, 732. [DOI] [PubMed] [Google Scholar]
- Kessler C. Mol. Cell. Probes 1991, 5, 161. [DOI] [PubMed] [Google Scholar]
- Janissen R.; Berghuis B. A.; Dulin D.; Wink M.; van Laar T.; Dekker N. H. Nucleic Acids Res. 2014, 42, e137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally B.; Wanunu M.; Meller A. Nano Lett. 2008, 8, 3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer A.; Wanunu M.; Morrison W. Nano Lett. 2010, 10, 738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song S.; Wang L.; Li J.; Fan C.; Zhao J. Trends Anal. Chem. 2008, 27, 108. [Google Scholar]
- Cho E. J.; Lee J.-W.; Ellington A. D. Annu. Rev. Anal. Chem. 2009, 2, 241. [DOI] [PubMed] [Google Scholar]
- Lu Y.; Liu J. Curr. Opin. Biotechnol. 2006, 17, 580. [DOI] [PubMed] [Google Scholar]
- Niemeyer C. M. Angew. Chem., Int. Ed. Engl. 2010, 49, 1200. [DOI] [PubMed] [Google Scholar]
- Saccà B.; Niemeyer C. M. Chem. Soc. Rev. 2011, 40, 5910. [DOI] [PubMed] [Google Scholar]
- Douglas S. M.; Bachelet I.; Church G. M. Science 2012, 335, 831. [DOI] [PubMed] [Google Scholar]
- Wanunu M.; Morrison W.; Rabin Y.; Grosberg A. Y.; Meller A. Nat. Nanotechnol. 2010, 5, 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.