

Supplemental Digital Content is available in the text.
Key Words: scalable clinical-grade vector manufacture, γ-retroviral vector, fixed-bed bioreactor, high vector titers, high vector yields
Abstract
The successful genetic engineering of patient T cells with γ-retroviral vectors expressing chimeric antigen receptors or T-cell receptors for phase II clinical trials and beyond requires the large-scale manufacture of high-titer vector stocks. The production of retroviral vectors from stable packaging cell lines using roller bottles or 10- to 40-layer cell factories is limited by a narrow harvest window, labor intensity, open-system operations, and the requirement for significant incubator space. To circumvent these shortcomings, we optimized the production of vector stocks in a disposable fixed-bed bioreactor using good manufacturing practice–grade packaging cell lines. High-titer vector stocks were harvested over 10 days, representing a much broader harvest window than the 3-day harvest afforded by cell factories. For PG13 and 293Vec packaging cells, the average vector titer and the vector stocks’ yield in the bioreactor were higher by 3.2- to 7.3-fold, and 5.6- to 13.1-fold, respectively, than those obtained in cell factories. The vector production was 10.4 and 18.6 times more efficient than in cell factories for PG13 and 293Vec cells, respectively. Furthermore, the vectors produced from the fixed-bed bioreactors passed the release test assays for clinical applications. Therefore, a single vector lot derived from 293Vec is suitable to transduce up to 500 patients cell doses in the context of large clinical trials using chimeric antigen receptors or T-cell receptors. These findings demonstrate for the first time that a robust fixed-bed bioreactor process can be used to produce γ-retroviral vector stocks scalable up to the commercialization phase.
Generation of large-scale, high-titer, clinical-grade retroviral viral vector stocks under current good manufacturing practice is a prerequisite for the implementation of phase I/II clinical trials using cell engineering approaches. Previous studies from our laboratory established a large-scale clinical-grade retroviral vector production platform using 10-layer cell factories,1 which currently supports multiple phase I clinical trials.2–4 Nonetheless, limitations in incubator space and the number of 10-layer cell factories that operators can handle per production run renders further scaling up difficult. In addition, the optimal harvest window for vector stocks in 10 tray-cell factories is confined to 3 days due to the rapid decline of vector titer in static culture. To overcome those limitations and to meet the increasing demand for clinical-grade vector stocks, it is imperative to establish new vector production platforms that are robust, scalable, and practical to handle.
The Pall iCELLis nano system is a scalable, disposable bioreactor that combines the advantages of single-use technologies with those of a fixed-bed. Its compact design not only eliminates the need for microcarriers, but also the requirement for a large footprint. Moreover, it allows the initiation of a perfusion mode whenever needed. The fixed-bed is packed with custom microfiber carriers which allows the biomass immobilized on the carrier to grow to a very high cell density. A built-in magnetic drive impeller facilitates the circulation of culture medium. Culture media passes through the bedding in the upward direction and falls as a thin-film down the outer wall of the fixed-bed where it takes up oxygen that is fed into the bioreactor. The levels of CO2, oxygen, and pH, as well as agitation speed and gas flow are continuously measured and recorded, and can be regulated through its multichannel controller. This fixed-bed bioreactor was originally developed to produce human and veterinary viral vaccines from MDBK and Vero cells as well as monoclonal antibodies (Pall, personal written communications). We therefore investigated this system for large-scale clinical-grade vector production using the 293Vec and PG13 packaging cell lines that we currently use for the production of clinical grade vector stocks in our phase I clinical trials. The growth of the 293Vec and PG13 vector producers and the characteristics of the viral vector stocks generated from 293Vec and PG13 producers were tested, in the 0.53 m2 (40 mL C1 compaction), the 1.07 m2 (40 mL C2 compaction), the 2.67 m2 (200 mL C1 compaction), and the 5.33 m2 (200 mL C2 compaction) bioreactors. We found that the 200 mL C1 bioreactor platform was 10 to 20 times more efficient than the 10-layer cell factories in the production of clinical-grade vectors. Moreover, the vector stocks generated from the fixed-bed bioreactors passed a range of release tests, allowing the qualification of these vector stocks for phase I/II clinical trials. The improved production efficiency and the safety profiles of the vector stocks produced in the fixed-bed bioreactor make this bioreactor a unique system for scalable clinical-grade vector production up to 30 L per run.
MATERIALS AND METHODS
Cells Lines and Culture Conditions
The PG13 packaging line was derived from a genetically engineered PG13 cell clone expressing an anti-CD19 chimeric antigen receptor (CAR).5–7 293Vec-GP packaging cell lines were derived from a genetically engineered 293Vec cell clone expressing anti-PSMA CAR.8,9 Both cell lines were maintained in Dulbecco’s modified Eagle’s medium (Life Technologies), containing 10% heat-inactivated fetal bovine serum (Gemini) and 2 mM of glutamine (Life Technologies).
iCELLis Nano Fixed-Bed Bioreactor Culture
The Pall Life Sciences iCELLis nano bioreactors, 40 mL C1, 40 mL C2, 200 mL C1, and 200 mL C2, were used in the configuration as proposed by the manufacturer. For the 40 mL C1 and 40 mL C2 bioreactors, a working volume of 800 mL in the bioreactor housing was used during testing. For the 200 mL C1 and the 200 mL C2 bioreactors, a working volume of 800 mL for the bioreactor housing was used for the first 2 to 3 days. An additional 2000 mL perfusion loop was connected to the system for a total of 2800 mL cultivation volume. All the bioreactors with microfiber fixed-bed were autoclaved before cell inoculation. The parameters for all our test runs were pH of 7.4, O2 at 50% air saturation, temperature of 37°C, an inoculation agitation of 1000 rpm for 5 hours, followed by a culture agitation of 800 to 850 rpm. These parameters were recorded and analyzed using the JUMO PCA and PCC program. The cell numbers in the fixed-bed bioreactors were determined daily by nuclei enumeration. Lactate levels were measured daily using Lactate Pro test strips and test meter (ARKRAY Inc., Kyoto, Japan). Glucose levels were determined using an Accu-Chek blood glucose testing system (Roche Diagnostics, Indianapolis, IN).
Cell Number Determination
The number of cells attached to the microfibers was determined daily by counting cell nuclei using crystal violet staining after cell lysis. Briefly, 2 microfiber carrier strips from the top of the fixed-bed were taken out and lysed with 0.5 mL lysis buffer A for 1 minute. The reaction was stopped by adding 0.5 mL of stabilizing buffer B (Chemometec). Nuclei from the lysed cells were counted using a hemocytometer.
Staining of the Cells Attached to the Microfibers
Microfiber strips taken from the top of the fixed-bed were incubated with 0.4% crystal violet in 20% ethanol for 2 minutes and then rinsed in deionized water once. Images of the cells on the microfibers were taken using Nikon Microscope and camera.
Endotoxin Detection
Endotoxin levels were determined in the last harvest of the viral vector stocks derived from the 293Vec and the PG13 packaging cells in the runs performed with the iCELLis Nano 200 mL C1 bioreactors. The FDA-approved Endosafe Portable Test System (Charles River Laboratories) was used according to the manufacturer’s recommendations.
Rapid Mycoplasma Testing
Mycoplasma detection was performed on the last harvest of viral vector stocks derived from the 293Vec and the PG13 packaging cell lines in the test runs performed with the iCELLis Nano 200 mL C1 bioreactors using the MycoAlert Mycoplasma Detection Kit, following manufacturer’s recommendations (Cambrex).
T-cell Transduction
T cells selected and activated with CD3/CD28 Dynabeads (Invitrogen) from healthy donors were transduced with vector stocks harvested from iCELLis Nano bioreactors or cell factories at various dilutions using T cells at 1×106 per mL. Transduction efficiency was determined by FACS analysis using FITC conjugated anti-CD3 (invitrogen), biotin-conjugated goat anti-mouse antibody followed by PE-labeled streptavidin (Becton Dickinson) for CAR.
Determination of Vector Titer by Real-Time Polymerase Chain Reaction Analysis
Genomic DNA was extracted from transduced T cells. Quantification of the SFG-1928z vector copy number (from PG13 packaging cells) or SFG-TP28z vector copy number (from 293Vec packaging cells) was determined by real-time polymerase chain reaction as previously described.6 Titers of the vector stocks were calculated using the following formulation. Vector titer=Vector copy number×Number of cells at time of transduction×Dilution factor of the vector/volume of culture at time of transduction.
Detection of Gibbon Ape Leukemia Virus Envelope by PCR for Testing of RCR
The presence of DNA sequences encoding the gibbon ape leukemia virus (GaLV) envelope was tested in the genomic DNA extracted from T lymphocytes transduced with the final harvests of the 200 mL C1 bioreactors for both 293Vec and PG13 packaging cell lines. The primer sequences specific for the GaLV envelope, and the positive control for this method was the same as previously described.6
Southern Blot Analysis
10 μg of Genomic DNA extracted from untransduced T lymphocytes or T lymphocytes transduced with vector stocks harvested from the 200 mL C1 bioreactors runs were digested with Nhe I endonuclease. A 32P-labeled probe specific for the vector sequence was prepared using a Random primer labeling system (GE), probes were subsequently purified using a Microspin G-50 column (GE). Southern blot analysis was performed using PerfectHyb hybridation buffer (Sigma) following manufacturer’s recommendations.
RESULTS
Comparison of the Fixed-Bed Bioreactor to Roller Bottles and 10-Layer Cell Factories
A range of surface areas, including 0.53 m2 (40 mL Nano C1 compaction), 1.07 m2 (40 mL C2 compaction), 2.67 m2 (200 mL C1), and 5.33 m2 (200 mL C2) depending on the fixed-bed volume and the custom carrier compaction level are available. The footprint of the fixed-bed bioreactors is the same regardless of the various fixed-bed volumes and compaction levels with a docking footprint of 200×120×200 mm (D×H×W). The 200 mL C2 bioreactor has a surface area equivalent to 64 roller bottles (850 mL) and 8.5 ten-layer cell factories (CF-10) (Table S1, Supplemental Digital Content 2, http://links.lww.com/JIT/A368).
Growth of 293Vec and PG13 Packaging Cells in Fixed-Bed Bioreactors
To investigate the usage of the fixed-bed bioreactors for retroviral vector production, we first tested the growth of 293Vec packaging cells in the 40 mL C1 bioreactor using 800 mL of working volume without a perfusion loop. 293Vec cells were inoculated at 50,000 cells/cm2 with an agitation rate of 1000 rpm during the inoculation phase, followed by an agitation rate of 800 rpm during the cultivation phase. Cells were batch-fed daily with 800 mL of fresh medium. Samples were taken from the bioreactor to determine glucose and lactate levels on a daily basis. The number of cells attached to the microfibers was determined by nuclei enumeration as described in the Materials and methods section. The number of cells that did not attach to the microfibers was determined in the cultivation media by trypan blue exclusion. The 293Vec packaging cells showed continuous growth during the 10-day culture period. More than 99% of the cells were attached to the microfibers, whereas <1% of the cells were detected in suspension. 293Vec packaging cells reached a cell density of 560,000 cells/cm2 in the 40 mL C1 bioreactor by day 10 (Fig. 1A). We subsequently tested the same 293Vec cell line in the 200 mL C1 bioreactor under the same conditions, except for the addition of a 2000 mL perfusion loop starting on day 3. We found that the growth of 293Vec packaging cells in the 200 mL C1 bioreactor was even more robust. Cell density reached 975,000/cm2 on day 10 and 1,180,000 cells/cm2 by the end of the testing on day 11 (Fig. 1A).
FIGURE 1.
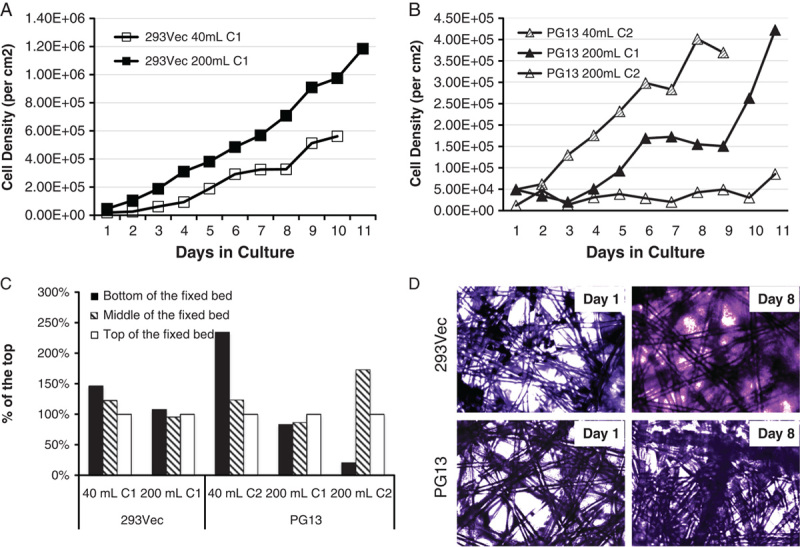
Growth and distribution of 293Vec and PG13 packaging cells in the iCELLis fixed-bed bioreactors. A, Growth of 293Vec packaging cells in 40 mL C1 and 200 mL C1 bioreactors. B, Growth of PG13 packaging cells in 200 mL C1, 40 ml C2, and 200 mL C2 bioreactors. C, Distribution of 293Vec packaging cells in the 40 mL C1 and 200 mL C1 bioreactor fixed-bed, and distribution of PG13 packaging cells in the 200 mL C1, 40 ml C2, and 200 mL C2 bioreactor fixed-bed. D, Attachment and growth of 293Vec and PG13 packaging cells on microfiber in bioreactor 1 day and 8 days postinoculation.
Encouraged by the results with the 293Vec cell line, we tested the PG13 packaging cell in the 40 mL C2, 200 mL C1, and 200 mL C2 bioreactors. A perfusion loop was added on day 2 for the 200 mL C1 and 200 mL C2 bioreactors, but not for the 40 mL C2 bioreactor. The 40 mL C2 bioreactor has twice the microfiber content as the 40 mL C1 bioreactor, and thus provides a surface area of 1.07 m2, with the same 2 cm fixed-bed height. In the 40 mL C2 bioreactor, the PG13 packaging cell line showed continuous growth during the 9-day culture period and reached 400,000 cells/cm2 at day 8 (Fig. 1B). We next tested the PG13 packaging cell line in 200 mL C1 bioreactor. Because of a lack in oxygen supply during the 200 mL C1 run on day 2 and day 3 of the culture (Fig. S1B, Supplemental Digital Content 1, http://links.lww.com/JIT/A367), the cell growth of the PG13 cells was delayed from day 5 to day 8. However, the cell growth subsequently recovered, and cell density reached 421,000 cells/cm2 by day 10 (Fig. 1B). We further tested the 200 mL C2 bioreactor using PG13 packing cells line. The 200 mL C2 bioreactor has twice the microfiber content compared with the 200 mL C1 bioreactor with the same 10 cm fixed-bed height, which gives a total surface area of 5.33 m2 (Table S1, Supplemental Digital Content 2, http://links.lww.com/JIT/A368). We hypothesized that the larger surface area of 200 mL C2 bioreactor would allow further expansion of the PG13 packaging cells, and the production of higher titer vector stocks. However, in the 200 mL C2 bioreactor, the PG13 packaging cells failed to grow to high cell density. By the end of the test run on day 10, the cell density was approximately 85,000 cells/cm2 (Fig. 1B), which was much lower than the cell density in the 200 mL C1 bioreactor. These observations did not support a direct scaling up scheme from the 40 mL C2 bioreactor to the 200 mL C2 bioreactor for the PG13 packaging cell line. Nonetheless, the 200 mL C1 bioreactor supported a robust growth pattern for both 293Vec and PG13 packaging cells.
In addition to the cell density, we evaluated the cell distribution in the fixed-bed bioreactors by sampling microfibers at the top, in the middle and at the bottom of the bed after disassembling the bioreactor at the end of each test run. With the same inoculation and cultivation agitation, both 293Vec and PG13 packaging cells were distributed evenly in both the 40 mL and the 200 mL C1 bioreactors. However, in the 200 mL C2 bioreactors, the distribution of the PG13 cells was uneven throughout the fixed-bed (Fig. 1C). Similar uneven distribution of cells was observed in the 40 mL C2 bioreactor (Fig. 1C). These findings suggest that the compaction factor of 2 was too high and prevented the distribution of the cells in the bioreactor with the inoculation agitation speed of 1000 rpm. As higher agitation speed would result in high shear stress on cells, which was not recommended by the manufacturer, we therefore focused on the 200 mL C1 bioreactors.
In line with rapid increase of cell density for both 293Vec and PG13 cell lines in the 200 mL C1 bioreactors, crystal violet staining of the microfibers sampled from the bioreactor showed expansion of the cells in the junctions between fibers (Fig. 1D). The lactate level increased rapidly from 2 to 19 mM in the 293Vec 200 mL C1 run, and from 2 to 23 mM in the PG13 200 mL C1 run, whereas glucose in the medium was rapidly consumed (data not shown). These observations demonstrated that both PG13 and 293Vec packaging cells tolerated high lactate concentration and that the 200 mL C1 bioreactor could support optimal cell growth.
Comparison of Large-scale Clinical-grade Vector Production in 200 mL C1 Bioreactor and in Six 10-Layer Cell Factories
To further evaluate the feasibility of large-scale clinical-grade vector production using the 200 mL C1 bioreactor, we harvested the viral vector stocks from the 200 mL C1 PG13 and the 293Vec packaging runs described previously on a daily basis for titration and release testing purposes. The experimental parameter settings, including time, pH, CO2 and O2 level, agitation speeds, and gas flow were all digitally recorded during the entire runs with the multichannel process controller. The run for the 293Vec packaging cell ran smoothly as indicated in Figure S1A (Supplemental Digital Content 1, http://links.lww.com/JIT/A367). However, we encountered a problem with the oxygen supply with the PG13 packaging cell run on day 2 and day 3 as recorded in real-time in Figure S1B (Supplemental Digital Content 1, http://links.lww.com/JIT/A367). The real-time monitoring feature of fixed-bed bioreactor allows for the identification of potential problems during the prolonged culture period.
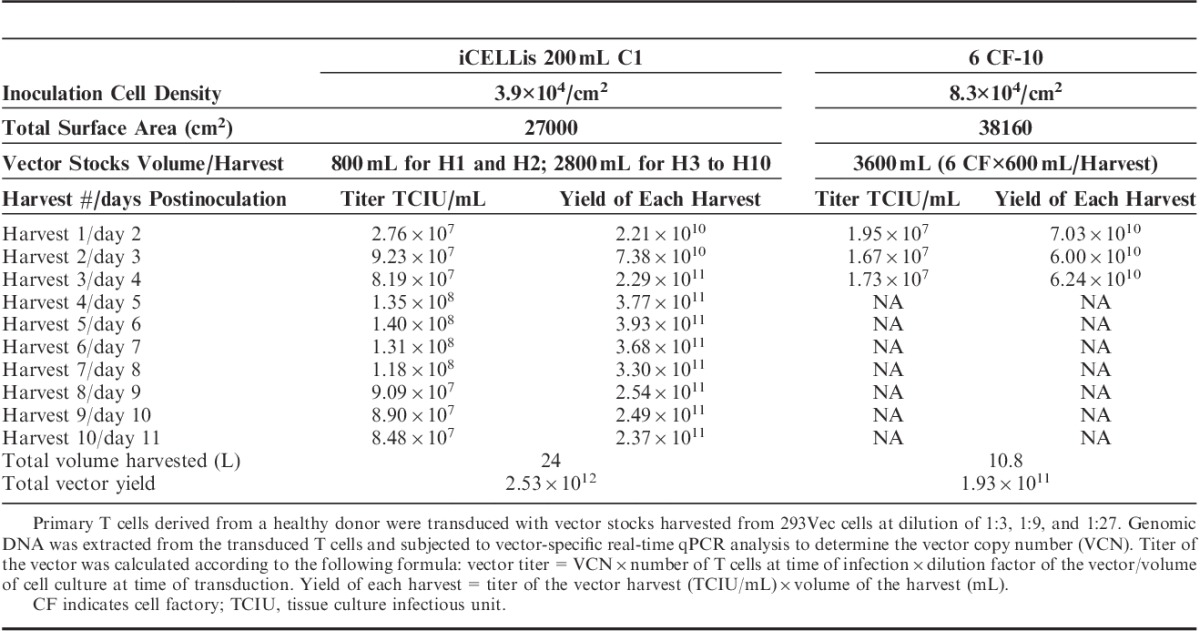
The titers of the viral vector stocks produced in the fixed-bed bioreactors were determined by transduction of primary T lymphocytes and compared with the titers of the vector stocks generated using the 10-layer cell factories. With a dilution factor of 27 (vol/vol) corresponding to a multiplicity of infection (MOI) of 3.0 to 5.2, all vector stocks harvested from day 2 to 11 during the 293Vec 200 mL C1 bioreactor run effectively transduced primary T cells in the range of 74% to 90%, which was similar or higher than the transduction efficiency of 71% obtained with the vector harvested from the 10-layer cell factories (Figs. 2A, B). A linear correlation between the MOI and the transduction efficiency was observed for all vector harvests derived from both 293Vec and PG13 cells (Fig. 2B). The titer and the yield obtained with the fixed-bed 200 mL C1 bioreactors and with the 10-layer cell factories are summarized in Table 1. Total tissue culture infectious unit (TCIU) from the 10 harvests of the 293Vec vector stocks reached 2.5×1012, whereas the total TCIU from the 3 harvests of the 10-layer cell factories reached 1.9×1011. Considering the surface area of the six 10-layer cell factories is 1.4-fold that of the surface area of the 200 mL C1 bioreactor (Table 1), the production efficiency in the 200 mL C1 bioreactor is 18.6 times that of the 10-layer cell factories for the 293Vec packaging cells.
FIGURE 2.
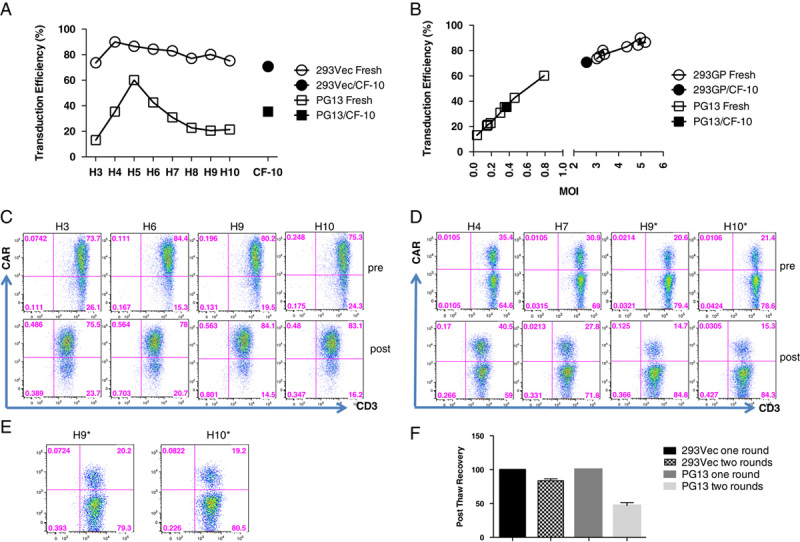
Transduction efficiency in primary T cells and vector stocks stability. A, Primary T cells were transduced with vector stocks derived from 293Vec cells (harvest 3 to 10 at 1:27 dilution), and with vector stocks derived from PG13 cells (harvest 3 to 10 at 1:4 dilution) both produced in the 200 mL C1 bioreactor, as described in materials and methods section. Transduction efficiency was determined by FACS analysis 72 hours posttransduction. Control vector stocks derived from a 10-layer cell factory (CF-10) production were included for both 293Vec and PG13 cells. B, Linear correlation between the transduction efficiency and the multiplicity of infection for vector stocks derived from 293Vec and PG13 packaging cells using the 200 mL C1 bioreactor. C, Stability of the vector stocks derived from 293Vec cells upon long-term storage at −80°C. Short-term-storage (pre) and 1-year poststorage (post) vector stocks were used to transduce primary T cells (harvests 3, 6, 9, and 10 at 1:27 dilution). Transduction efficiency was determined by FACS analysis 72 hours posttransduction. D, Stability of the vector stocks derived from PG13 packaging cells upon long-term storage at −80°C. Short-term-storage (pre) and 10 months poststorage (post) vector stocks were used to transduce primary T cells (harvests 4, 7, 9*, and 10* at 1:4 dilution). Transduction efficiency was determined by FACS analysis 72 hours posttransduction. *Harvest 9 and 10 are vector stocks harvested in serum-free media. E, T-cell transduction efficiency using serum-free harvest 9 and 10 vector stocks from PG13 packing cells at 1:2 dilution upon 10-month storage at −80°C. F, Vector stability upon multiple rounds of freeze-thaw cycles. Vector stocks derived from either 293Vec cells (harvests 5 and 6) produced in 200 mL C1 bioreactor and PG13 cells (harvest 2, pooled harvests 3 and 4, pooled harvests 5 and 6, and pooled harvests 7 and 8) produced in the 40 mL C2 bioreactor were frozen at −80°C and then thawed. The titers of the vector stocks that underwent 2 rounds of freeze-thaw cycles were compared with that of the same vectors that underwent 1 round of freeze-thaw cycle.
TABLE 1.
Comparison of Vector Production Yields Derived From 293Vec Cells Using Either 200 mL C1 Bioreactor or Six 10-Layer Cell Factories
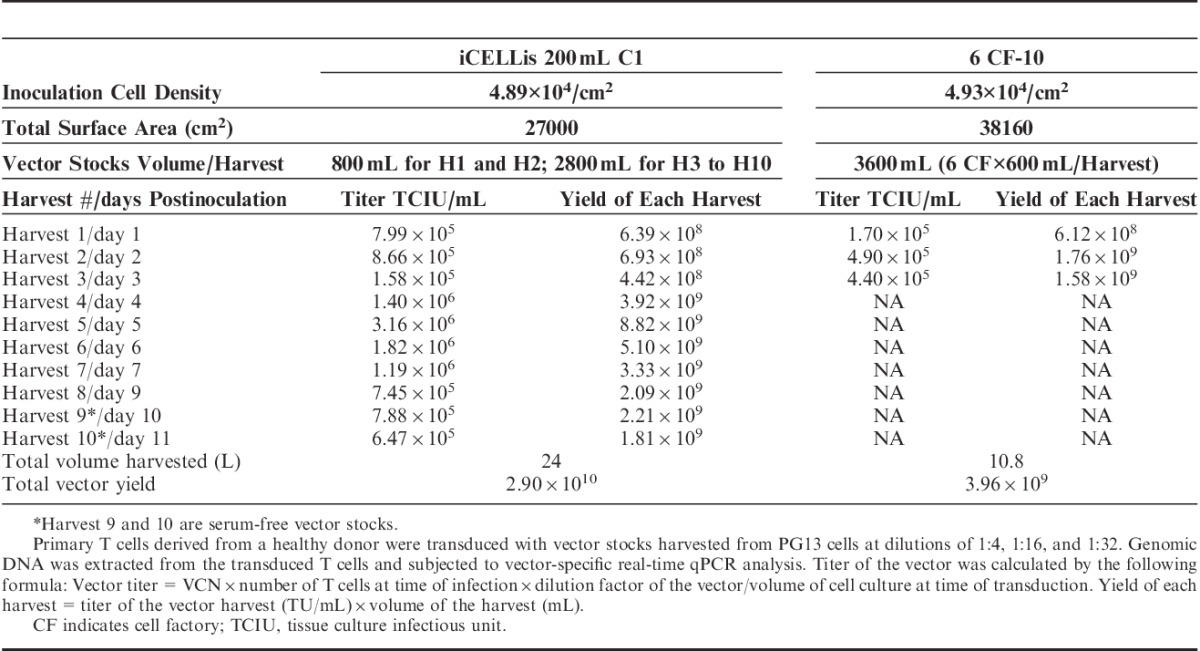
Consistently, we observed that the PG13 packaging line also continuously generated high-titer vector stocks throughout the testing period from harvest 1 to harvest 10 in the 200 mL C1 bioreactor. For the last 2 harvests (harvest 9 and harvest 10), serum-free medium was used to test the feasibility of generating serum-free vector stocks. The titer of the vector peaked at 3.2×106 TCIU/mL in the 200 mL C1 bioreactor on day 5, whereas the maximum titer of 4.9×105 TCIU/mL in the 10-layer cell factories on day 2 is approximately 6-fold lower. The titer of the serum-free vector stocks from the last 2 serum-free harvests also demonstrated high titers of 7.88×105 and 6.47×105 TCIU/mL, respectively (Table 2). Titer of the harvest on day 3 was the lowest, coinciding with the oxygen supply error, which suggests that optimized oxygen concentration is critical for the generation of high-titer vector stocks. Altogether, for the PG13 cells, the production efficiency in the 200 mL C1 bioreactor system was 10.4 times that of the 10-layer cell factories, and the average titer of the vector stock increased up to 3.2-fold.
TABLE 2.
Comparison of Vector Production Yields Derived From PG13 Cells Using Either 200 mL C1 Bioreactor or Six 10-Layer Cell Factories
Stability of the Vector Stocks Upon Long-term Storage
Our laboratory and others previously demonstrated that vector stocks generated in the 10-layer cell factories are stable up to 3 to 9 years at −80°C.1,10 We tested the stability of the vector stocks derived from PG13 cells after 1 year of storage and after 10 months of storage for the 293Vec packaging cells. Harvests 3, 6, 9, and 10 from 293Vec cells and harvests 4, 7, 9, and 10 from PG13 cells were titrated on primary T lymphocytes. All the vector stock harvests containing 10% of serum maintained the same titer (Figs. 2C, D). However, the titer of the vector stocks harvested in serum-free medium decreased. At the dilution factor of 4 (vol/vol) corresponding to a MOI in the range of 0.04 to 0.79, the transduction efficiency of serum-free harvest 9 (H9*) and harvest 10 (H10*) from PG13 cells was much lower than that of the same vector stocks before storage at −80°C (Fig. 2D). Only at a lower dilution factor of 2, the transduction efficiency of the serum-free vector stocks was comparable to that of the same vector before storage (Fig. 2E). Therefore, the titer of the serum-free vector dropped approximately 50% after 1-year storage, suggesting than the serum-free vector stocks are less stable than the serum-containing vector stocks.
Stability of the Vector Stocks Upon Multiple Freeze and Thaw Cycles
Previous studies showed that the half-life of the GaLV-enveloped vector was approximately 7.5 hours in DMEM with 10% serum at 37°C and about 54 hours at 4°C.11 An alternative approach to 4°C storage of the viral stocks was therefore needed during the 10-day harvest of the vector stocks from the 200 mL C1 bioreactor. To this end, we tested the stability of the vector stocks harvested with serum-containing medium upon multiple freeze and thaw cycles. After an additional freeze and thaw cycle, we observed a modest 16% decrease of the titer of the vector stocks derived from the 293Vec packaging cell line, whereas a more dramatic decrease of 53% of the titer of vector stocks derived from the PG13 packaging cell line was found (Fig. 2F). The titer of the vector stocks derived from the 293Vec cells is much higher than that of the PG13 cells (Tables 1 and 2), suggesting that vector stocks with higher initial titers are more resilient to repeated freeze and thaw cycles. Alternatively, vectors stocks produced in the human 293Vec packaging cells may be more stable during cryopreservation than stocks derived from the murine PG13 packaging cells.
Vector Stocks Derived From Fixed Bioreactors are Qualitatively Equivalent to Vector Stocks Derived From Cell Factories
To demonstrate the suitability of the fixed-bed bioreactor for the production of clinical-grade vector stocks, we performed an array of release tests on the final harvests of each of the 200 mL C1 bioreactor runs for the 293Vec cells and the PG13 cells (Table 3). Both vector stocks were sterile, with either undetectable or low levels of endoxin observed, and were devoid of mycoplasma and replication-competent retrovirus as demonstrated by both GaLV RCR PCR testing or Marker-Rescue cell culture assay (Fig. 3A, Table 3). Vector integrity was also tested using Southern blotting analysis. As expected, a single vector-specific band of 3.6 Kb was detected in T lymphocytes transduced with the vector stocks from 293Vec packaging cells, and a single vector-specific band of 3.5 kb was detected in T lymphocytes transduced with the vector stocks from the PG13 packaging cells (Fig. 3B). The observed safety profiles of the vector stocks generated in the fixed-bed bioreactor as well as the improved production efficiency suggested this bioreactor system may be ideal for the production of large-scale clinical-grade retroviral vectors.
TABLE 3.
Release Tests Performed on the Last Harvest of the Vector Stocks Derived From 293Vec and PG13 Cells Produced in the 200 mL C1 Bioreactor
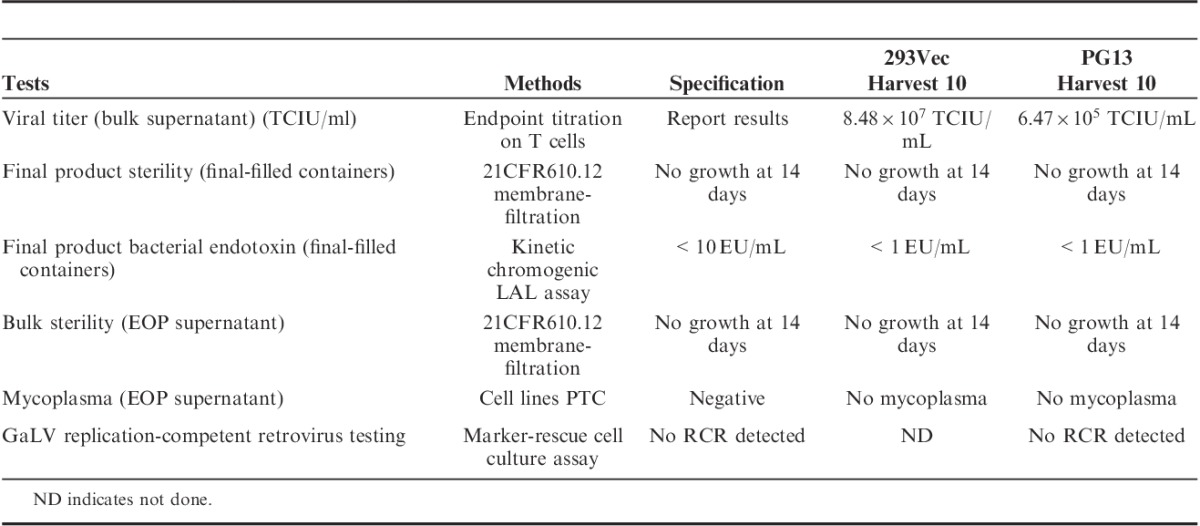
FIGURE 3.
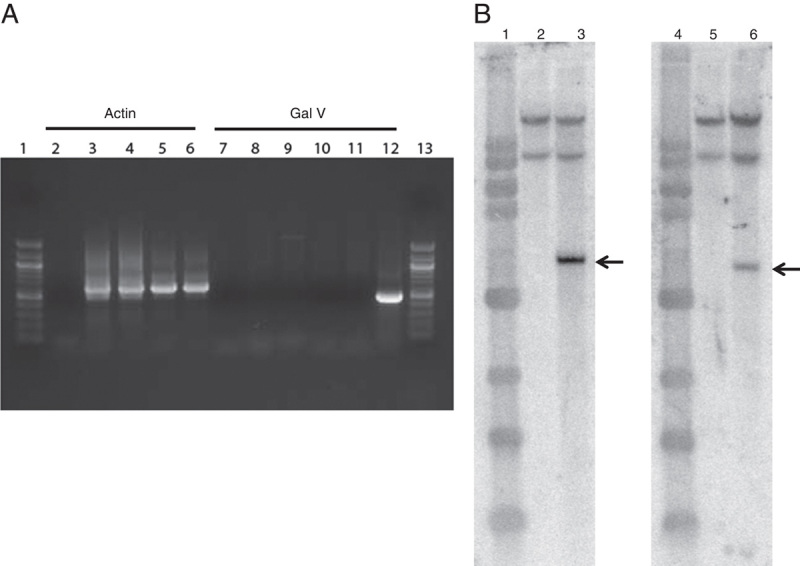
GaLV RCR assay using PCR and vector integrity assay by Southern blot analysis. A, Genomic DNA extracted from untransduced or transduced T lymphocytes were examined for the GaLV envelope using specific primers for PCR as previously described.6 Lanes 1 and 13, 100 bp DNA ladder; lanes 2 and 7, no template control; lanes 3 and 8, 5 and 10, untransduced T cells (UT); lanes 4 and 9, transduced with harvest 10 derived from 293Vec cells (SFG-TP28z vector); lanes 6 and 11, transduced with harvest 10 derived from PG13 cells (SFG-1928z vector); lane 12: positive control (6 copies GaLV env plasmid). B, Genomic DNA extracted from untransduced or transduced T lymphocytes were digested and hybridized with 32P-labeled specific probes as described in material and methods. Lanes 1 and 4, 1 kb DNA ladder; lanes 2 and 5, untransduced T cells; lane 3, transduced T cells with harvest 10 derived from 293Vec cells (SFG-TP28z vector); lane 6, transduced T cells with harvest 10 derived from PG13 cells (SFG-1928z vector). Arrows indicate the expected integrated vector size.
DISCUSSION
Retroviral vectors are used in a large number of clinical trials for gene transfer-based adoptive cell therapies.12,13 It is therefore desirable to develop a robust platform for the production of vector stocks to support the increasing demand of clinical-grade vectors. In this study, we investigated the usage of a fixed-bed bioreactor for the generation of scalable clinical-grade vector stocks. The production efficiencies for both the 293Vec and the PG13 packaging cell lines are significantly increased in the 200 mL C1 bioreactor when compared with the six 10-layer cell factories. The 200 mL C1 bioreactor not only supported the production of higher titer vector stocks, but also extended the time window for vector harvesting (Tables 1 and 2). We observed a linear scalability when comparing the 40 mL C1 bioreactor to the 200 mL C1 bioreactor (Fig. 1A and data not shown). However, there was no linear scalability observed between the 200 mL C1 bioreactor and the 200 mL C2 bioreactor (Fig. 1B). The larger surface area gained by higher fiber density in the 200 mL C2 bioreactor was offset by the uneven distribution of cells (Fig. 1C), slower cell expansion (Fig. 1B), as well as a reduced vector harvest window, which resulted in decreased production efficiency (data not shown). More recently, bioreactors with compaction factor of 2 were discontinued and replaced with a bioreactor with compaction factor of 1.5. It will be of interest to test whether the C1.5 bioreactors outperform the C1 bioreactors in the context of our experimental settings. Nonetheless, we have demonstrated in this study that the 200 mL C1 bioreactor is a robust and economical vector production platform when compared with the 10-layer cell factories.
During our production runs, we observed a direct correlation between the oxygen level, the cell growth rate, and the vector titers. Disruption of the oxygen supply on day 2 during the PG13 production run in the 200 mL C1 bioreactor led to a transient 5-fold decrease in vector titer on day 3 (Table 2) concomitant with a delay in cell expansion (Fig. 1B). It is interesting to note that, the cell growth and the vector titer recovered shortly after restoration of the oxygen supply. These observations emphasize the importance of maintaining optimal oxygen level in the generation of high-titer vector stocks. The real-time monitoring of the critical parameters by the bioreactor controller made it possible to identify and resolve potential problems in a timely fashion.
To address the intrinsic short half-life of the GaLV-envelope11,14 and the broad harvest window of 10 days, we tested the effect of 2 rounds of freeze-thaw cycles on vector titer. A modest drop of 16% in vector titer was observed for high-titer vector stocks derived from the 293Vec packaging cell line, whereas a more profound 53% drop in vector titer was observed for the relatively lower titer vector stocks generated from the PG13 packaging cell line (Fig. 2F, Tables 1 and 2). As the vectors produced from the 293Vec and the PG13 cells are both pseudotyped with the GaLV envelope,9,15 and express vectors with similar design and size,7,8 our results strongly suggest that higher titer vector stocks are more stable upon freeze-thaw. It is also possible that the human 293 membrane components of the virions may be sturdier than their counterparts derived from the murine PG13 cells. Nonetheless, given the fact that vector stocks from both 293Vec and PG13 cells passed the required release tests, the 293Vec cells appear to be the packaging cell line of choice based upon its higher vector titer (Table 3, Fig. 2).
Merten and colleagues have compared different bioreactor systems, such as the CellCube system, the Fibra-Cel fixed-bed basket reactor, and the Cytodex beads stir-tank Cellift reactor. This group also reported that the Fibra-Cel fixed-bed basket reactor system demonstrates the highest productivity.14 It will be interesting to compare the Fibra-Cel fixed-bed system and the Pall Life Sciences iCELLis system in our experimental setting. The iCELLis bioreactor presents the advantage of scalability. Commercial scale bioreactors with surface areas of 66 to 500 m2 are available in a small footprint with real-time process control.
On the basis of our results, we are designing a production and harvest scheme for the manufacturing of large-scale clinical-grade retroviral vectors using the 200 mL C1 bioreactor (Fig. S2, Supplemental Digital Content 3, http://links.lww.com/JIT/A369). To initiate vector manufacturing, a vial of cells is thawed and passaged in T flasks until sufficient number of cells are accumulated to inoculate the 200 mL bioreactor (1 to 2 passages). Cells in the bioreactor are batch-fed with 800 mL of medium for 2 days with perfusion starting from day 2 with an additional 2000 mL of medium. The cells are subsequently batch-fed with a total of 2800 mL from day 3 to day 11. Viral stocks are collected on a daily basis and stored at −80°C. After determining the vector titer, high-titer vector harvests are pooled and aliquoted in suitable-sized cryocontainers. On the basis of our current CAR T-cell manufacturing process,2,6,16 a single vector lot derived from either PG13 or 293Vec manufactured in the 200 mL C1 bioreactor is suitable to transduce at least 70 to 500 patient T-cell doses, respectively. This scale is amply sufficient for most phase I and II clinical trials. Our results further suggest that the industrial scale of these bioreactors with surface areas of 66 to 500 m2 could accommodate vector production for thousands of patients as required for the commercialization phase of genetically modified T cells expressing either CARs or T-cell receptors.
Many important parameters are involved in vector production, such as the choice of packaging cell line,17,18 vector design,19,20 type of bioreactor,14 medium additives,21 serum concentration,22,23 temperature,15,24 perfusion rate during the cultivation,14 and timing for vector harvest.15 Our data demonstrate for the first time that the Pall Life Sciences iCELLis fixed-bed bioreactor is a robust platform for scalable clinical-grade retroviral vector production. Ongoing studies will build upon the iCELLis bioreactor experience to identify other important parameters for optimizing large-scale clinical-grade retroviral vector production, such as vector production in serum-free media or with serum replacement supplement, as well as the addition of stabilizing agents that would increase the stability of vector stocks harvested in serum-free media. The implementation of and further improvements to this platform will ease the process and reduce the cost of manufacturing genetically modified cells for adoptive cell therapy applications.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Matt Kremer at Pall Life Sciences for providing MSKCC the opportunity to test the iCELLis Nano bioreactors in the context of this application. They also thank Manuel Caruso (Université Laval, QC) for providing the 293Vec parental cell line.
The authors also thank Matt Kremer and Mark Bonyhadi for critically reviewing the manuscript.
CONFLICT Of INTERESTS/FINANCIAL DISCLOSURES
Supported by the NCI P30 CA08748, P50 CA086438 and Starr Foundation Tri-Institutional Stem Cell Initiative.
I.R. is a scientific cofounder of Juno Therapeutics and a consultant for Juno Therapeutics. All the remaining authors have declared that there are no financial conflicts of interest with regard to this work.
Footnotes
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Website, www.immunotherapy-journal.com.
X.W. and I.R.: designed the research plan; X.W., M.O., J.Q., T.W, G.H.: performed research; S.B. contributed to quality control testing; X.W., I.R., and M.S.: wrote the manuscript.
REFERENCES
- 1.Przybylowski M, Hakakha A, Stefanski J, et al. Production scale-up and validation of packaging cell clearance of clinical-grade retroviral vector stocks produced in cell factories. Gene Therapy. 2006;13:95–100. [DOI] [PubMed] [Google Scholar]
- 2.Brentjens RJ, Riviere I, Park JH, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118:4817–4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brentjens RJ, Davila ML, Riviere I, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5:177ra138:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davila ML, Bouhassira DC, Park JH, et al. Chimeric antigen receptors for the adoptive T cell therapy of hematologic malignancies. Int J Hematol. 2014;99:361–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miller AD, Garcia JV, von Suher N, et al. Construction and properties of retrovirus packaging cells based on gibbon ape leukemia virus. J Virol. 1991;65:2220–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hollyman D, Stefanski J, Przybylowski M, et al. Manufacturing validation of biologically functional T cells targeted to CD19 antigen for autologous adoptive cell therapy. J Immunother. 2009;32:169–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brentjens RJ, Santos E, Nikhamin Y, et al. Genetically targeted T cells eradicate systemic acute lymphoblastic leukemia xenografts. Clin Cancer Res. 2007;13:5426–5435. [DOI] [PubMed] [Google Scholar]
- 8.Maher J, Brentjens RJ, Gunset G, et al. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta/CD28 receptor. Nat Biotechnol. 2002;20:70–75. [DOI] [PubMed] [Google Scholar]
- 9.Ghani K, Wang X, de Campos-Lima PO, et al. Efficient human hematopoietic cell transduction using RD114- and GALV-pseudotyped retroviral vectors produced in suspension and serum-free media. Hum Gene Ther. 2009;20:966–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lamers CH, van Elzakker P, Luider BA, et al. Retroviral vectors for clinical immunogene therapy are stable for up to 9 years. Cancer Gene Ther. 2008;15:268–274. [DOI] [PubMed] [Google Scholar]
- 11.Schilz AJ, Kuhlcke K, Fauser AA, et al. Optimization of retroviral vector generation for clinical application. J Gene Med. 2001;3:427–436. [DOI] [PubMed] [Google Scholar]
- 12.Suerth JD, Schambach A, Baum C. Genetic modification of lymphocytes by retrovirus-based vectors. Curr Opin Immunol. 2012;24:598–608. [DOI] [PubMed] [Google Scholar]
- 13.Rivat C, Santilli G, Gaspar HB, et al. Gene therapy for primary immunodeficiencies. Hum Gene Ther. 2012;23:668–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Merten OW, Cruz PE, Rochette C, et al. Comparison of different bioreactor systems for the production of high titer retroviral vectors. Biotechnol Prog. 2001;17:326–335. [DOI] [PubMed] [Google Scholar]
- 15.Reeves L, Smucker P, Cornetta K. Packaging cell line characteristics and optimizing retroviral vector titer: the National Gene Vector Laboratory experience. Hum Gene Ther. 2000;11:2093–2103. [DOI] [PubMed] [Google Scholar]
- 16.Davila ML, Riviere I, Wang X, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6:224ra225:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rigg RJ, Chen J, Dando JS, et al. A novel human amphotropic packaging cell line: high titer, complement resistance, and improved safety. Virology. 1996;218:290–295. [DOI] [PubMed] [Google Scholar]
- 18.Forestell SP, Dando JS, Chen J, et al. Novel retroviral packaging cell lines: complementary tropisms and improved vector production for efficient gene transfer. Gene Ther. 1997;4:600–610. [DOI] [PubMed] [Google Scholar]
- 19.Palu G, Parolin C, Takeuchi Y, et al. Progress with retroviral gene vectors. Rev Med Virol. 2000;10:185–202. [DOI] [PubMed] [Google Scholar]
- 20.Mangeot PE, Negre D, Dubois B, et al. Development of minimal lentivirus vectors derived from simian immunodeficiency virus (SIVmac251) and their use for gene transfer into human dendritic cells. J Virol. 2000;74:8307–8315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Olsen JC, Sechelski J. Use of sodium butyrate to enhance production of retroviral vectors expressing CFTR cDNA. Hum Gene Ther. 1995;6:1195–1202. [DOI] [PubMed] [Google Scholar]
- 22.Shen BQ, Clarke MF, Palsson BO. Kinetics of retroviral production from the amphotropic PsiCRIP murine producer cell line. Cytotechnology. 1996;22:185–195. [DOI] [PubMed] [Google Scholar]
- 23.Gerin PA, Gilligan MG, Searle PF, et al. Improved titers of retroviral vectors from the human FLYRD18 packaging cell line in serum- and protein-free medium. Hum Gene Ther. 1999;10:1965–1974. [DOI] [PubMed] [Google Scholar]
- 24.McTaggart S, Al-Rubeai M. Effects of culture parameters on the production of retroviral vectors by a human packaging cell line. Biotechnol Prog. 2000;16:859–865. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.