

Abstract
Cannabinoid receptors are a family of G-protein coupled receptors that are involved in a wide variety of physiological processes and diseases. One of the key regulators that are unique to cannabinoid receptors is the cannabinoid receptor interacting proteins (CRIPs). Among them CRIP1a was found to decrease the constitutive activity of the cannabinoid type-1 receptor (CB1R). The aim of this study is to gain an understanding of the interaction between CRIP1a and CB1R through using different computational techniques. The generated model demonstrated several key putative interactions between CRIP1a and CB1R, including those involving Lys130 of CRIP1a.
Ever since their discovery in the mid-1980s, cannabinoid receptors have been receiving increasing attention as their roles in an expanding array of vital human physiological processes are elucidated. For example, roles in regulation of motivation, motor function, memory, appetite and energy homeostasis, pain perception, immune function, gastrointestinal and cardiovascular function, and bone mass maintenance have all been attributed to cannabinoid receptors. These receptors represent an important class of the G protein-coupled receptor (GPCR) superfamily.1 Currently, this class is comprised of two subtypes, the cannabinoid 1 receptor (CB1R) and cannabinoid 2 receptor (CB2R), although other targets of some cannabinoic ligands have been described.2 Of the two subtypes, CB1R is the major subtype expressed in neuronal cells, while it is also co-expressed to a lesser extent with CB2R in immune cells and other peripheral tissues.3 Cannabinoid receptors are endogenously activated by the lipid-derived endocannabinoid ligands, anandamide and 2-arachidonoyl glycerol (2-AG), among others. CB1R signaling and regulation have biomedical relevance because CB1Rs are involved in a wide range of diseases, including substance abuse disorders (they are a major target of Δ9-tetrahydrocannabinol, the main psychoactive constituent in marijuana) and neurodegenerative diseases such as Parkinson's, Alzheimer's, Huntington's diseases, cancer, obesity, inflammatory bowel disease, and neuropathic and inflammatory pain.4-7
The CB1R signals mainly through the activation of G proteins of the Gi/o family, which inhibit adenylyl cyclases and regulate ion channels, including calcium and potassium channels.8 Evidence also exists that in certain cell types CB1Rs can stimulate adenylyl cyclase via Gs, which can induce receptor-mediated Ca2+ fluxes and stimulate phospholipases.3 Moreover, stimulation of CB1Rs results in the phosphorylation and activation of mitogen-activated protein kinases (MAPKs) that regulate nuclear transcription factors.9 In recent years, it has become evident that CB1Rs also interact with various non-G-protein GPCR-interacting proteins that can modulate CB1R function.10 For example, CB1Rs are regulated through mechanisms similar to those of other GPCRs, such as GPCR kinases and β-arrestins. In addition, CB1Rs have the ability to form homo- and hetero-dimers/oligomers, resulting in altered pharmacological properties, which might contribute to the diverse pharmacological effects of cannabinoids observed in various tissues.3 However, one mechanism that appears to be unique to CB1Rs is related to their binding to CRIP1a and CRIP1b, the cannabinoid receptor interacting proteins.11
CRIP1a/b are globular proteins that were first discovered by the Lewis group when they observed that the deletion of the CB1R C-terminus resulted in delaying the time required to peak Ca2+ current inhibition, augmented the tonic CB1R-mediated inhibition of Ca2+ currents, and promoted the ability of CB1R to sequester G-proteins.12,13 These findings suggested that the C-terminal tail could be serving as an auto-inhibitor. Searching for additional proteins that might be involved in regulating CB1R's activity, they used the CB1R distal C-terminus as bait in a yeast two-hybrid screen, and identified two proteins: CRIP1a and CRIP1b. Later, CRIP1a was shown to bind to a GST-labeled CB1R-C-terminal tail fusion protein and also to co-immunoprecipitate with CB1R, although no interaction of CRIP1a with the CB2R has been observed.11
CRIP1a and CRIP1b are generated by alternative splicing of the Cnrip gene, which is located on chromosome 2 in humans.11 CRIP1a is most highly expressed in the brain, and its homologs are found throughout the vertebrates. Interestingly, CRIP1a was shown to selectively reverse basal, but not CB1R agonist-induced, inhibition of voltage-gated Ca2+ channels when co-transfected with CB1R in superior cervical ganglion neurons, which suggests that CRIP1a inhibits constitutive CB1R activity.11 Supporting this interpretation, the ability of the CB1R inverse agonist rimonabant (SR141716A) to stimulate basal Ca2+ channel activity in CB1R-transfected neurons was eliminated by co-expression of CRIP1a.11
Interestingly, CRIP1a possesses a palmitoylation site and a C-terminus PDZ class I ligand. The palmitoylation site may play a role in localizing CRIP1a to the plasma membrane.11 The PDZ ligand domain may play several roles: 1) allowing CRIP1a to interact with other proteins, act as a scaffolding site, and/or enabling the formation of heterodimers between CB1Rs and other receptors; and 2) potentially modulating the localization, desensitization, or internalization of CB1Rs.10,14
CRIP1a may also be involved in the balance between neuroprotection and degeneration. Katona's group employed quantitative PCR to compare the levels of CB1R and CRIP1a mRNA in epileptic and healthy postmortem human hippocampal tissue. Reduced levels of both CRIP1a and CB1R mRNA were found in sclerotic hippocampi.15 Alternatively, CRIP1a mRNA was found to be elevated following kainic acid-induced seizures in rats.16 Both reports suggest that CRIP1a plays a role in modulating CB1R function in the pathogenesis or neuroadaptive response to epilepsy. Moreover, in a model of glutamate excitoxicity in cultured cortical neurons, virally-mediated expression of CRIP1a inhibited the protective effects of a cannabinoid agonist while conferring a protective effect to an antagonist.17 In addition to its putative roles in the brain, CRIP1a is presynaptically expressed along with CB1Rs in the retina, and the Cnrip1 gene exhibits hypermethylation in a subset of colorectal carcinomas and adenomas.18-20
While none are definitive, the findings above suggest potentially important functions of CRIP1a in multiple physiological systems, yet very little is known about the exact mechanisms by which CRIP1a binds to the CB1R, which is crucial to understanding the regulation of CB1R signaling. In this report, we apply a palette of complementary computational techniques, including homology modeling, Ab initio and protein threading, to generate all atom molecular models for CRIP1a. Then, using protein-protein docking methods, the resulting CRIP1a model is docked to the C-terminus of the CB1R to generate a model for the CRIP1a-CB1R interaction.
A general workflow for building the CRIP1a-CB1R molecular model is shown in Scheme 1. First, the secondary structure pattern of CRIP1a was predicted. Then, three different methods were used to build 3D models for CRIP1a (homology modeling, Ab initio and protein threading). Hydrogen atoms were added to the CRIP1a models followed by energy minimization. Next, all models were evaluated with multiple scoring paradigms in order to choose the best model to carry forward into succeeding stages. The best such model for CRIP1a was then docked to the C-terminus of the CB1R, after which the resulting models for the CRIP1a-CB1R complex were energy minimized, clustered and finally evaluated to determine the most reliable protein-protein interaction model.
Scheme 1.

General scheme of the CRIP1a-CB1R interaction model construction.
CRIP1a secondary structure pattern was predicted using the amino acid primary sequence by several algorithms, including: HMMSTR, SSPRO 4, CDM, JNET, SABLE, PORTER, NetSurfP, SPINE X and PSIPRED.21-29 A consensus secondary structure was generated using the GeneSilico Metaserver.30 For the prediction of CRIP1a structural class and fold type, the 1D protein structure prediction software from Kurgan's lab, which predicts structural class and fold type information from the primary sequence of a protein, was used.31,32 Predictions by the majority of algorithms suggest that CRIP1a is composed almost exclusively of ß-sheets and loops, which make CRIP1a a member of all ß proteins class (Figure 1). This was confirmed by the structural class prediction algorithm 1D, which also predicted CRIP1a is of the Concanavalin A-like lectins/glucanases all ß strands sandwich fold.31,32
Figure 1.

Secondary Structure prediction of CRIP1a by several algorithms, consensus is shown in red.
Several algorithms were used for generating sequence alignments: BLAST and the GeneSilico Metaserver, which encompasses a combination of different homology and protein threading methods for template searches and sequence alignment, e.g., COMA, HHBLITS, Profile Comparer, FFAS, HHSEARCH, pGenTHREADER, Phyre, Pcons5, consens3d, jmbrank and sp3.30,33-45 For prediction of distant homologues, protein threading algorithms were used, which are pDomTHREADER, I-TASSER, RaptorX, LOMETS (LOcal MEta-Threading-Server); and MUSTER (MUlti-Sources ThreadER).41,46-54 The BLAST search identified the amino acid sequences of proteins that are closely related to CRIP1a.33 However, all such identified proteins were of low homology relative to CRIP1a (data not shown). Additional algorithms with higher sensitivity were used to detect distant homologs to CRIP1a, which yielded template crystal structures from 111 different alignments of 74 different protein chains. Each algorithm has its own scoring method, so it is difficult to directly compare their results. However, it was clear that most of the generated alignments were in the lowest range of their respective algorithm's scoring scales. Only pDomTHREADER, an algorithm generally regarded as precise and sensitive in discriminating superfamilies, yielded two alignments in the high range. pDomTHREADER combines information from both sequence and structure to produce domain alignments. The highest scoring alignment thus produced was for PDB ID 1DS6 Chain B, (human Rho-specific guanine nucleotide dissociation inhibitor 2, RhoGDI 2), which covers amino acids 1-152 of CRIP1a's 164 amino acids, i.e., all but the last of CRIP1a's ß-sheet (see Figure 2).
Figure 2.

Alignment of CRIP1a with RhoGDI 2 (PDB ID 1DS6 Chain B), the best scoring template. The RhoGDI 2 secondary structure and the predicted (consensus) secondary structure for CRIP1a are shown in red above and below the sequence alignment, respectively S: β-sheet and H: helix.
RhoGDI, an all ß protein of sandwich fold type, plays an important role in G-protein coupled receptor (GPCR) signaling, including that of CB1R.55 The pDomTHREADER analysis also suggested CRIP1a to be somewhat homologous to RhoGDI 1, implying the possibility of CRIP1a sharing a similar function with the RhoGDIs. RhoGDI decreases the activity of Rho by preventing guanine nucleotide exchange and membrane association. RhoGDI may also act as a positive regulator for Rho activities by providing spatial restriction, guidance and availability signals to effectors, functions that are essential for the correct targeting and regulation of these effectors.55 RhoGDI 2 has an identity of 15.9% to the CRIP1a sequence, which is quite low; however, pDomTHREADER uses the primary sequence of a protein to infer distant relationships to other protein families that are not detectable by simple percentage identity. These relationships often suggest common function and can often provide templates for the construction of high quality 3D structural models.41
While other alignments with higher percentage identity were found, they had low scores and did not cover as large a fraction of the CRIP1a sequence. For example, 1AOZ (ascorbate oxidase) chain A and 1AYO (alpha 2-macroglobulin) chain A both have 20% identity with CRIP1a. Their alignment scores, however, were lower than that of RhoGDI 2 and did not provide templates for several of CRIP1a's ß-sheets. A complete list of the algorithms used and their resulting suggested templates can be found in Supporting Information (Table S1).
Based on the produced alignments, we used MODELLER to generate CRIP1a models and ranked them according to the DOPE score.56 The highest scoring model was that based on the RhoGDI 2 template (Figure 3a). The constructed model for CRIP1a follows the predicted secondary structure; however residues 153-162 were removed since they exceeded the extents of the template. Thus, to produce a complete model, residues 153-162 were re-modeled using the next best scoring template – fibrinogen-binding protein SdrG (PDB ID 1R17) chain B. After energy minimizing and scoring this model, it had the highest score of all generated models (Figure 3b). To ensure no major modeling defects, a Ramachandron plot was generated by MOLPROBITY for the model and was found to be within the acceptable limits (Figure 4).57
Figure 3.

Best ranking CRIP1a model using RhoGDI 2 as a template (PDB ID 1DS6 chain B). (a) 3D structure constructed for CRIP1a β -sheets are in yellow and loops are shown in green. (b) Superimposition of backbone atoms of CRIP1a model (red) and the RhoGDI 2 crystal structure (blue).
Figure 4.

Ramachandron plots for the best full model of CRIP1a generated using RhoGDI 2 as a template (generated by MOLPROBITY).57
The QUARK algorithm for Ab initio protein folding and structure prediction was used to generate models using replica-exchange Monte Carlo simulation guided by an atomic-level knowledge-based force field.58 Furthermore, hybrid methods combining Ab initio with other techniques were used: Bhageerath (incorporating bioinformatics tools) and Robetta's algorithm (incorporating comparative models of protein domains).59-63
The various models generated by Ab initio methods, all of which were all ß proteins, were evaluated using the DOPE method. The DOPE score of a protein can be viewed as a conformational energy that measures the relative stability of a conformation with respect to other conformations of the same protein. It can be used to choose the best model out of a set of predicted model structures for a particular protein sequence. Because the DOPE energy is not normalized based on the size of the protein, the absolute score for a protein is not meaningful, but the relative energies of different conformations are useful in model evaluation.56 According to DOPE scoring, models built using Ab initio methods were found to have high scores (Supporting Information Table S2); however, none of them had a score better than the model generated using RhoGDI 2 as a template. It is interesting to note that the best scoring model of this set was generated by the hybrid bioinformatics/Ab initio Bhageerath algorithm.
According to Niehaus et al., the last 9 amino acids on the C-terminus of CB1R are the minimum residues required for CRIP1a binding.11 PEP-FOLD, which is a de novo approach for prediction of peptide structures from amino acid sequences, was used to build the 3D structure of this 9 amino acid peptide (Figure 5).64-71
Figure 5.

The last 9 amino acids of the CB1R C-terminus with sequence STDTSAEAL.
Protein-protein docking was performed using the HADDOCK (High Ambiguity Driven DOCKing) algorithm. 72 The site on CRIP1a where the CB1R C-terminus binds is unknown, so this 9 amino acid peptide was docked to all possible sites encompassing amino acids 34-110 of CRIP1a. This range was chosen because these amino acids are common between CRIP1a and CRIP1b and thus most likely critical for CB1R binding.11 Docking results were individually inspected, after which high scoring models were passed into the refinement step.
All docked poses were refined with FireDock73,74 (Fast Interaction Refinement in a molecular Docking) algorithm and then rescored using the HINT75-81 forcefield that describes and quantifies all interactions in the biological environment by exploiting the interaction information implicit in LogPo/w (the partition coefficient for 1-octanol/water solute transfer). We call HINT a “natural” force field because it is based on empirical energetic terms that are defined by real experiments, and thus encodes interaction types including Coulombic, hydrogen bond and hydrophobic interactions expected to be found between molecules in the biological environment. It is a free energy force field that includes solvation/desolvation and entropy in addition to the other enthalpic terms.75-81 The HINT score (HTOTAL) is a double sum over all atom-atom pairs of the product (bij) of the hydrophobic atom constants (ai, partial log Poctanol/water) and atomic solvent accessible surface areas (Si) for the interacting atoms, mediated by a function of the distance between the atoms:
| (1) |
where Rij is a simple exponential function, e−r, rij is an adaptation of the Lennard-Jones function, and Tij is a logic function assuming +1 or −1 values, depending on the polar (Lewis acid or base) nature of interacting atoms.80-83
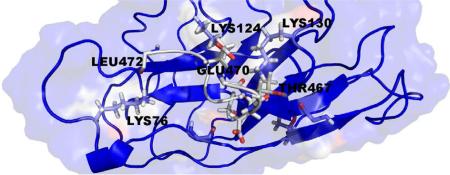
The best scoring CRIP1a-CB1R model according to HINT is shown in Figure 6. The 9 amino acids of the CB1R C-terminus (Figure 5) are mostly polar; therefore, polar interactions are the dominant type of interaction between CRIP1a and CB1R. Most notably, the model suggests that hydrogen bonds are formed between the CB1R backbone carbonyl oxygens of Ser464 and Thr465 and Aap466 with the terminal amine in Lys130 from CRIP1a, between one of the CB1R carboxyl oxygens of Asp466 and the phenolic oxygen of Tyr85 from CRIP1a, and between the Thr467 hydroxyl group from CB1R and the backbone carbonyl oxygen of Asn61 from CRIP1a. The model suggests that Lys130 from CRIP1a has a quite significant role in the interaction between CRIP1a and CB1R (Figure 7). Also, there are some hydrophobic interactions, for example, between Val67 from CRIP1a and Ala471 from CB1R. The complete HINT score interaction analysis is reported in Supporting Information (Table S3). This CRIP1a-CB1R interaction model supports the assertion that CRIP1a might be responsible for blocking the coupling of CB1R to specific Gi/o proteins that are responsible for the tonic inhibition of Ca2+ channels, but not to other Gi/o proteins that could inhibit Ca2+ channels in response to agonist activation.11 The concept of CB1R differential G-protein coupling and the subsequent selective signal transduction mechanisms was previously discussed by Anavi-Goffer et al., where they found through Gi/o protein reconstitution experiments that the combination of CB1R and Gαi3(C351G) significantly enhanced the tonic inhibition of Ca2+ channels, while CB1R and GαoA(C351G) abolished the tonic inhibition of Ca2+ channels.84 An earlier study of Nie et al. also reinforces the differential coupling concept: they showed that D164N point mutation of CB1R blocked tonic inhibition of Ca2+ channels, whereas agonist-dependent Ca2+ channel inhibition was not affected.13 The possibility that CRIP1a blocks the coupling of CB1R to specific Gi/o proteins without affecting the binding of other Gi/o proteins may provide an explanation for the finding that CRIP1a selectively blocks basal CB1R modulation of Ca2+ channel activity, but not CB1R agonist-induced modulation of this activity.11
Figure 6.

Best Docked model for CRIP1a-CB1R complex. CRIP1a is shown in blue and the 9 C-terminal amino acids of CB1R are shown as a space filling model.
Figure 7.

Key interactions between the CB1R and CRIP1a as predicted by the model. (a) Model showing CRIP1a (grey) bound to CB1R (blue). (b) 2D representation of the key interactions between CRIP1a (black) and CB1R (blue).
The electrostatic properties of the CRIP1a-CB1R complex interface was evaluated using Adaptive Poisson-Boltzmann Solver (APBS) tools plug-in for PyMOL.85-87 This tool uses APBS to solve the Poisson-Boltzmann equation (PBE) to assess electrostatic properties.86 All images were created using PyMOL.87 As can be seen in figure 8, the electrostatic complementarity analysis reveals the high complementarity in both CRIP1a and CB1R interfaces.
Figure 8.

Complementarity of electrostatic potentials at the interface of the predicted CRIP1a-CB1R complex. In the middle, CRIP1a and CB1R C-terminus are aligned in a pre-complex position to better show the spatial complementarity of electrostatic potentials of the molecules. Positive potential is shown as blue and negative potential is shown as red. To the left, CRIP1a is shown; the CB1R binding site is enclosed in a back dashed rectangle. To the right, the binding interface of the C-terminus of CB1R is shown. It can be clearly seen the complementarity of electrostatic potentials at the interface of the complex, as the area of the positive potential (blue) on CRIP1a faces the negative potential (red) on CB1R C-terminus and vice versa.
In conclusion, previous work has explored the effects of binding CRIP1a to CB1R; however, this interaction has never been examined at the atomic level, as there are no available X-ray crystal structures for the complex or either of the interacting proteins. Here, we used three different types of computational techniques, comparative, Ab initio and protein threading, to build 3D models of the CRIP1a protein. The best scoring model was obtained through protein threading using RhoGDI 2 as a template. RhoGDI 2, another effector with a significant role in G-protein coupled receptor signaling (including CB1R), is an all ß protein of the sandwich fold type, which is the same fold predicted for CRIP1a. Also modeled was the 9 amino acid C-terminal end peptide of CB1R. The peptide was docked as a ligand to the best scored model of CRIP1a, resulting in mostly favorable polar interactions. The energetics and binding mode analyses suggest that Lys130 of CRIP1a may play a significant role in this interaction. Our model may be used to guide the design of future site-directed mutagenesis experiments. Understanding the structures of these proteins and, particularly, their interactions will form the foundation for understanding mechanisms of CB1R regulation in the CNS, and may also lead to advances in drug development for the treatment of disorders involving modulation of CB1R activity.
Supplementary Material
Acknowledgements
This work was supported by R21-DA025321 from the National Institute on Drug Abuse and a Mutli-School Award from the A.D. Williams Fund of VCU with support from UT1-TR000058 from NIH's National Center for Advancing Translational Research.
Abbreviations
- CB1R
cannabinoid 1 receptor
- CB2R
cannabinoid 2 receptor
- Cnrip1
cannabinoid receptor interacting protein gene
- CRIP1a
cannabinoid receptor interacting protein 1a
- CRIP1b
cannabinoid receptor interacting protein 1b
Footnotes
Supplementary Material
Table S1: Algorithms used for detection of templates for CRIP1a with their results. Table S2: Generated models arranged according to their calculated DOPE scores from lowest score to highest score (lower scores indicate better models). Table S3: Full HINT table for the highest score CRIP1a-CB1R complex model. Supplementary data associated with this article can be found, in the online version.
The authors declare no competing financial interest.
References
- 1.Mackie K. J. Neuroendocrinol. 2008;20(Suppl. 1):10. doi: 10.1111/j.1365-2826.2008.01671.x. [DOI] [PubMed] [Google Scholar]
- 2.Pertwee RG, Howlett AC, Abood ME, Alexander SP, Di Marzo V, Elphick MR, Greasley PJ, Hansen HS, Kunos G, Mackie K, Mechoulam R, Rossm RA. Pharmacol. Rev. 2010;62:588. doi: 10.1124/pr.110.003004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Turu G, Hunyady L. J. Mol. Endocrinol. 2010;44:75. doi: 10.1677/JME-08-0190. [DOI] [PubMed] [Google Scholar]
- 4.Thakur GA, Tichkule R, Bajaj S, Makriyannis A. Expert Opin. Ther. Pat. 2009;19:1647. doi: 10.1517/13543770903436505. [DOI] [PubMed] [Google Scholar]
- 5.Oesch S, Gertsch J. J. Pharm. Pharmacol. 2009;61:839. doi: 10.1211/jpp/61.07.0002. [DOI] [PubMed] [Google Scholar]
- 6.Lee HK, Choi EB, Pak CS. Curr. Top. Med. Chem. 2009;9:482. doi: 10.2174/156802609788897844. [DOI] [PubMed] [Google Scholar]
- 7.Miller LK, Devi LA. Pharmacol. Rev. 2011;63:461. doi: 10.1124/pr.110.003491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hudson BD, Hébert TE, Kelly ME. Mol. Pharmacol. 2010;77:1. doi: 10.1124/mol.109.060251. [DOI] [PubMed] [Google Scholar]
- 9.Howlett AC. In: Cannabinoids. Handbook of Experimental Pharmacology. Pertwee RG, editor. Springer; New York City: 2005. pp. 53–80. [DOI] [PubMed] [Google Scholar]
- 10.Smith TH, Sim-Selley LJ, Selley DE. Br. J. Pharmacol. 2010;160:454. doi: 10.1111/j.1476-5381.2010.00777.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Niehaus JL, Liu Y, Wallis KT, Egertová M, Bhartur SG, Mukhopadhyay S, Shi S, He H, Selley DE, Howlett AC, Elphick MR, Lewis DL. Mol. Pharmacol. 2007;72:1557. doi: 10.1124/mol.107.039263. [DOI] [PubMed] [Google Scholar]
- 12.Nie J, Lewis DL. Neuroscience. 2001;107:161. doi: 10.1016/s0306-4522(01)00335-9. [DOI] [PubMed] [Google Scholar]
- 13.Nie J, Lewis DL. J. Neurosci. 2001;21:8758. doi: 10.1523/JNEUROSCI.21-22-08758.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Howlett AC, Blume LC, Dalton GD. Curr. Med. Chem. 2010;17:1382. doi: 10.2174/092986710790980023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ludányi A, Eross L, Czirják S, Vajda J, Halász P, Watanabe M, Palkovits M, Maglóczky Z, Freund TF, Katona I. J. Neurosci. 2008;28:2976. doi: 10.1523/JNEUROSCI.4465-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bojnik E, Turunçm E, Armağan G, Kanıt L, Benyhe S, Yalçın A, Borsodi A. Epilepsy Res. 2012;99:64. doi: 10.1016/j.eplepsyres.2011.10.020. [DOI] [PubMed] [Google Scholar]
- 17.Stauffer B, Wallis KT, Wilson SP, Egertová M, Elphick MR, Lewis DL, Hardy LR. Neurosci. Lett. 2011;503:224. doi: 10.1016/j.neulet.2011.08.041. [DOI] [PubMed] [Google Scholar]
- 18.Hu SS, Arnold A, Hutchens JM, Radicke J, Cravatt BF, Wager-Miller J, Mackie K, Straiker A. J. Comp. Neurol. 2010;518:3848. doi: 10.1002/cne.22429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oster B, Thorsen K, Lamy P, Wojdacz TK, Hansen LL, Birkenkamp-Demtröder K, Sørensen KD, Laurberg S, Orntoft TF, Andersen CL. Int. J. Cancer. 2011;129:2855. doi: 10.1002/ijc.25951. [DOI] [PubMed] [Google Scholar]
- 20.Lind GE, Danielsen SA, Ahlquist T, Merok MA, Andresen K, Skotheim RI, Hektoen M, Rognum TO, Meling GI, Hoff G, Bretthauer M, Thiis-Evensen E, Nesbakken A, Lothe RA. Mol. Cancer. 2011;10:85. doi: 10.1186/1476-4598-10-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bystroff C, Thorsson V, Baker D. J. Mol. Biol. 2000;301:173. doi: 10.1006/jmbi.2000.3837. [DOI] [PubMed] [Google Scholar]
- 22.Cheng J, Randall AZ, Sweredoski MJ, Baldi P. Nucleic Acids Res. 2005;33:W72. doi: 10.1093/nar/gki396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheng H, Sen TZ, Kloczkowski A, Margaritis D, Jernigan RL. Polymer. 2005;46:4314. doi: 10.1016/j.polymer.2005.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cuff JA, Barton GJ. Proteins. 1999;40:502. doi: 10.1002/1097-0134(20000815)40:3<502::aid-prot170>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 25.Adamczak R, Porollo A, Meller J. Proteins. 2005;59:467. doi: 10.1002/prot.20441. [DOI] [PubMed] [Google Scholar]
- 26.Pollastri G, McLysaght A. Bioinformatics. 2005;21:1719. doi: 10.1093/bioinformatics/bti203. [DOI] [PubMed] [Google Scholar]
- 27.Petersen B, Petersen TN, Andersen P, Nielsen M, Lundegaard C. BMC Struct. Biol. 2009;9:51. doi: 10.1186/1472-6807-9-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Faraggi E, Zhang T, Yang Y, Kurgan L, Zhou Y. J. Comput. Chem. 2012;33:259. doi: 10.1002/jcc.21968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jones DT. J. Mol. Biol. 1999;292:195. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- 30.Kurowski MA, Bujnicki JM. Nucleic Acids Res. 2003;31:3305. doi: 10.1093/nar/gkg557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen K, Kurgan LA. Bioinformatics. 2007;23:2843. doi: 10.1093/bioinformatics/btm475. [DOI] [PubMed] [Google Scholar]
- 32.Chen K, Kurgan LA, Ruan JJ. Comput. Chem. 2008;29:1596. doi: 10.1002/jcc.20918. [DOI] [PubMed] [Google Scholar]
- 33.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Nucleic Acids Res. 1997;25:3389. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rychlewski L, Jaroszewski L, Li W, Godzik A. Protein Sci. 2000;9:232. doi: 10.1110/ps.9.2.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Biegert A, Soding J. Proc. Natl. Acad. Sci. USA. 2009;106:3770. doi: 10.1073/pnas.0810767106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Margelevicius M, Laganeckas M, Venclovas C. Bioinformatics. 2010;26:1905. doi: 10.1093/bioinformatics/btq306. [DOI] [PubMed] [Google Scholar]
- 37.Remmert M, Biegert A, Hauser A, Söding J. Nat. Methods. 2011;9:173. doi: 10.1038/nmeth.1818. [DOI] [PubMed] [Google Scholar]
- 38.Madera M. Bioinformatics. 2008;24:2630. doi: 10.1093/bioinformatics/btn504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jaroszewski L, Li Z, Cai XH, Weber C, Godzik A. Nucleic Acids Res. 2011;39(Web Server issue):W38. doi: 10.1093/nar/gkr441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Söding J. Bioinformatics. 2005;21:951. doi: 10.1093/bioinformatics/bti125. [DOI] [PubMed] [Google Scholar]
- 41.Lobley A, Sadowski MI, Jones DT. Bioinformatics. 2009;25:1761. doi: 10.1093/bioinformatics/btp302. [DOI] [PubMed] [Google Scholar]
- 42.Kelley LA, Sternberg MJ. Nat. Protoc. 2009;4:363. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 43.Lundström J, Rychlewski L, Bujnicki J, Elofsson A. Protein Sci. 2001;10:2354. doi: 10.1110/ps.08501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wallner B, Fang H, Elofsson A. Proteins. 2003;53(Suppl. 6):534. doi: 10.1002/prot.10536. [DOI] [PubMed] [Google Scholar]
- 45.Wallner B, Elofsson A. Bioinformatics. 2005;21:4248. doi: 10.1093/bioinformatics/bti702. [DOI] [PubMed] [Google Scholar]
- 46.Roy A, Kucukural A, Zhang Y. Nat. Protoc. 2010;5:725. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peng J, Xu J. Proteins. 2011;79(Suppl. 10):161. doi: 10.1002/prot.23175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peng J, Xu J. Proteins. 2011;79:1930. doi: 10.1002/prot.23016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Källberg M, Wang H, Wang S, Peng J, Wang Z, Lu H, Xu J. Nat. Protoc. 2012;7:1511. doi: 10.1038/nprot.2012.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Peng J, Xu J. Bioinformatics. 2010;26:i294. doi: 10.1093/bioinformatics/btq192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhao F, Peng J, Xu J. Bioinformatics. 2010;26:i310. doi: 10.1093/bioinformatics/btq193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao F, Peng J, Debartolo J, Freed KF, Sosnick TR, Xu J. J. Comput. Biol. 2010;17:783. doi: 10.1089/cmb.2009.0235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu S, Zhang Y. Nucleic Acids Res. 2007;35:3375. doi: 10.1093/nar/gkm251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu S, Zhang Y. Proteins. 2008;72:547. doi: 10.1002/prot.21945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dovas A, Couchman JR. Biochem. J. 2005;390(Pt 1):1. doi: 10.1042/BJ20050104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shen MY, Sali A. Protein Sci. 2006;15:2507. doi: 10.1110/ps.062416606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. Acta. Crystallogr. D Biol. Crystallogr. 2010;66(Pt 1):12. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xu D, Zhang Y. Protein. 2012;80:1715. doi: 10.1002/prot.24065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shenoy SR, Jayaram B. Curr. Protein Pept. Sci. 2010;11:498. doi: 10.2174/138920310794109094. [DOI] [PubMed] [Google Scholar]
- 60.Jayaram B, Bhushan K, Shenoy SR, Narang P, Bose S, Agrawal P, Sahu D, Pandey V. Nucleic Acids Res. 2006;34:6195. doi: 10.1093/nar/gkl789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Narang P, Bhushan K, Bose S, Jayaram B. J. Biomol. Struct. Dyn. 2006;23:385. doi: 10.1080/07391102.2006.10531234. [DOI] [PubMed] [Google Scholar]
- 62.Narang P, Bhushan K, Bose S, Jayaram B. Phys. Chem. Chem. Phys. 2005;7:2364. doi: 10.1039/b502226f. [DOI] [PubMed] [Google Scholar]
- 63.Chivian D, Kim DE, Malmstrom L, Bradley P, Robertson T, Murphy P, Strauss CEM, Bonneau R, Rohl CA, Baker D. Proteins. 2003;53(Suppl. 6):524. doi: 10.1002/prot.10529. [DOI] [PubMed] [Google Scholar]
- 63a.A. Camproux AC, Tuffery P, Chevrolat JP, Boisvieux JF, Hazout S. Protein Eng. 1999;12:1063. doi: 10.1093/protein/12.12.1063. [DOI] [PubMed] [Google Scholar]
- 64.Camproux AC, Gautier R, Tufféry P. J. Mol. Biol. 2004;339:591. doi: 10.1016/j.jmb.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 65.Tufféry P, Guyon F, Derreumaux P. J. Comput. Chem. 2005;26:506. doi: 10.1002/jcc.20181. [DOI] [PubMed] [Google Scholar]
- 66.Tufféry P, Derreumaux P. Proteins. 2005;61:732. doi: 10.1002/prot.20698. [DOI] [PubMed] [Google Scholar]
- 67.Maupetit J, Derreumaux P, Tufféry P. J. Comput. Chem. 2010;31:726. doi: 10.1002/jcc.21365. [DOI] [PubMed] [Google Scholar]
- 68.Maupetit J, Tufféry P, Derreumaux P. Proteins. 2007;69:394. doi: 10.1002/prot.21505. [DOI] [PubMed] [Google Scholar]
- 69.Kaur H, Garg A, Raghava GP. Protein Pept. Lett. 2007;14:626. doi: 10.2174/092986607781483859. [DOI] [PubMed] [Google Scholar]
- 70.Beaufays J, Lins L, Thomas, Brasseur R. J. Pept. Sci. 2012;18:17. doi: 10.1002/psc.1410. [DOI] [PubMed] [Google Scholar]
- 71.de Vries SJ, van Dijk M, Bonvin AM. Nat. Protoc. 2010;5:883. doi: 10.1038/nprot.2010.32. [DOI] [PubMed] [Google Scholar]
- 72.Andrusier N, Nussinov R, Wolfson HJ. Proteins. 2007;69:139. doi: 10.1002/prot.21495. [DOI] [PubMed] [Google Scholar]
- 73.Mashiach E, Schneidman-Duhovny D, Andrusier N, Nussinov R, Wolfson HJ. Nucleic Acids Res. 2008;36:W229. doi: 10.1093/nar/gkn186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fornabaio M, Spyrakis F, Mozzarelli A, Cozzini P, Abraham DJ, Kellogg GE. J. Med. Chem. 2004;47:4507. doi: 10.1021/jm030596b. [DOI] [PubMed] [Google Scholar]
- 75.Spyrakis F, Cozzini P, Bertoli C, Marabotti A, Kellogg GE, Mozzarelli A. BMC Struct. Biol. 2007;7:4. doi: 10.1186/1472-6807-7-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Marabotti A, Spyrakis F, Facchiano A, Cozzini P, Alberti S, Kellogg GE, Mozzarelli A. J. Comput. Chem. 2008;29:1955. doi: 10.1002/jcc.20954. [DOI] [PubMed] [Google Scholar]
- 77.Burnett JC, Kellogg GE, Abraham DJ. Biochemistry. 2000;39:1622. doi: 10.1021/bi991724u. [DOI] [PubMed] [Google Scholar]
- 78.Amadasi A, Spyrakis F, Cozzini P, Abraham DJ, Kellogg GE, Mozzarelli A. J. Mol. Biol. 2006;358:289. doi: 10.1016/j.jmb.2006.01.053. [DOI] [PubMed] [Google Scholar]
- 79.Kellogg GE, Abraham DJ. Eur. J. Med. Chem. 2000;35:651. doi: 10.1016/s0223-5234(00)00167-7. [DOI] [PubMed] [Google Scholar]
- 80.Sarkar A, Kellogg GE. Curr. Top. Med. Chem. 2010;10:67. doi: 10.2174/156802610790232233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Levitt M. J. Mol. Biol. 1983;168:595. doi: 10.1016/s0022-2836(83)80304-0. [DOI] [PubMed] [Google Scholar]
- 82.Levitt M, Perutz MF. J. Mol. Biol. 1988;201:751. doi: 10.1016/0022-2836(88)90471-8. [DOI] [PubMed] [Google Scholar]
- 83.Anavi-Goffer S, Fleischer D, Hurst DP, Lynch DL, Barnett-Norris J, Shi S, Lewis DL, Mukhopadhyay S, Howlett AC, Reggio PH, Abood ME. J. Biol. Chem. 2007;282:25100. doi: 10.1074/jbc.M703388200. [DOI] [PubMed] [Google Scholar]
- 84.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Proc. Natl. Acad. Sci. USA. 2001;98:10037. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lerner MG, Carlson HA. APBS plugin for PyMOL. University of Michigan; Ann Arbor: 2006. [Google Scholar]
- 86.The PyMOL Molecular Graphics System, Version 1.2r3pre. Schrödinger, LLC; [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.