

Abstract
Hypoxia stimulates pulmonary hypertension (PH) in part by increasing the proliferation of pulmonary vascular wall cells. Recent evidence suggests that signaling events involved in hypoxia-induced cell proliferation include sustained nuclear factor-kappaB (NF-κB) activation, increased NADPH oxidase 4 (Nox4) expression, and downregulation of peroxisome proliferator-activated receptor gamma (PPARγ) levels. To further understand the role of reduced PPARγ levels associated with PH pathobiology, siRNA was employed to reduce PPARγ levels in human pulmonary artery smooth muscle cells (HPASMC) in vitro under normoxic conditions. PPARγ protein levels were reduced to levels comparable to those observed under hypoxic conditions. Depletion of PPARγ for 24 - 72 hours activated mitogen-activated protein kinase, ERK 1/2, and NF-κB. Inhibition of ERK 1/2 prevented NF-κB activation caused by PPARγ depletion indicating that ERK 1/2 lies upstream of NF-κB activation. Depletion of PPARγ for 72 hours increased NF-κB-dependent Nox4 expression and H2O2 production. Inhibition of NF-κB or Nox4 attenuated PPARγ depletion-induced HPASMC proliferation. Degradation of PPARγ depletion-induced H2O2 by PEG-catalase prevented HPASMC proliferation and also ERK 1/2 and NF-κB activation and Nox4 expression indicating that H2O2 participates in feed-forward activation of above signaling events. Contrary to the effects of PPARγ depletion, HPASMC PPARγ overexpression reduced ERK 1/2 and NF-κB activation, Nox4 expression and cell proliferation. Taken together these findings provide novel evidence that PPARγ plays a central role in the regulation of the ERK1/2-NF-κB-Nox4-H2O2 signaling axis in HPASMC. These results indicate that reductions in PPARγ caused by pathophysiological stimuli such as prolonged hypoxia exposure are sufficient to promote the proliferation of pulmonary vascular smooth muscle cells observed in PH pathobiology.
Keywords: PPARγ, NF-κB, ERK 1/2, Nox4, pulmonary hypertension, pulmonary artery smooth muscle cell
Introduction
Peroxisome proliferator-activated receptors (PPARs) are members of the nuclear hormone receptor superfamily of ligand-activated transcription factors that play important roles in cell metabolism, growth, differentiation, and inflammation via regulation of a large number of gene networks [1, 2]. Three PPAR isoforms, α, β, and γ are expressed in tissue-specific patterns. Upon activation by endogenous or exogenous ligands, PPARs form heterodimers with the 9-cis retinoic acid receptor (RXR-α) and bind to peroxisome proliferator response elements (PPRE) in the promoter regions of target genes to stimulate their expression [3, 4]. Activation of PPARγ can also cause transrepression of other pro-inflammatory transcription factors [5]. Because PPARγ is expressed in numerous cells including pulmonary vascular endothelial and smooth muscle cells, the goal of the current study was to further explore the role of PPARγ in pulmonary vascular smooth muscle cell function [6, 7].
Pulmonary hypertension (PH) is characterized by increases in pulmonary artery pressure and pulmonary vascular resistance that cause significant morbidity and mortality [8]. Growing evidence supports the role of PPARγ in pulmonary vascular regulation. PPARγ activation with exogenous synthetic thiazolidinedione ligands attenuates PH and pulmonary vascular remodeling in several experimental models of PH [9-13], whereas loss of PPARγ expression is associated with PH. Expression of PPARγ is reduced in the lungs of rodents with PH caused by chronic hypoxia [11, 13]. Reduced PPARγ expression has also been observed in the vascular lesions of patients with idiopathic pulmonary arterial hypertension, and in a rat model of severe PH caused by treatment with hypoxia and a VEGF receptor antagonist [14]. Levels of PPARγ are attenuated in pulmonary artery endothelial cells isolated from patients with idiopathic pulmonary arterial hypertension [15]. Furthermore, targeted and constitutive genetic ablation of PPARγ from endothelial [16] or vascular smooth muscle cells [17] is associated with the development of spontaneous PH in mice. Taken together, these reports suggest that activation of PPARγ attenuates pulmonary vascular dysfunction and PH whereas reductions in PPARγ contribute to PH pathogenesis.
Hypoxia reduces PPARγ expression and activity via activation of oxidative stress signals [13]. Hypoxia increases Nox4 expression in the pulmonary vasculature [18], and Nox4-derived H2O2 reduces PPARγ expression and activity in PASMC [19], and H2O2 similarly reduces PPARγ in endothelial cells in vitro [20]. Hypoxia activates both mitogen-activated protein kinases that regulate PPARγ transcriptional activity and the pro-inflammatory transcription factor, NF-κB [21, 22]. For example, hypoxia increases Nox4 expression in HPASMC by stimulating NF-κB p65 binding to the Nox4 promoter [23]. Recent findings from our laboratory demonstrate that hypoxia induces ERK-mediated-NF-κB activation, Nox4 expression, H2O2 generation and PPARγ downregulation in HPASMCs and that Nox4-derived H2O2 is in turn required for ERK 1/2 activation suggesting the existence of cyclic signaling cascades underlying chronic hypoxia-induced derangements in pulmonary vascular wall cells [19]. Although these studies clarify mechanisms involved in hypoxia-induced reductions in PPARγ expression, the downstream signaling events attributable to PPARγ downregulation are not well defined. Therefore, the current study explores the ability of reductions in PPARγ to stimulate proliferative signaling mechanisms associated with hypoxia-induced PH pathobiology.
Our findings demonstrate that loss of PPARγ is sufficient to promote HPASMC proliferation through ERK1/2-NF-κB-Nox4 dependent H2O2 generation. Taken together with previous reports, these findings further emphasize the importance of PPARγ in pulmonary vascular cell biology and elucidate mechanistic pathways by which stimuli that reduce PPARγ stimulate derangements in PASMC function. We postulate that sustained activation of these pathways caused by PPARγ downregulation contributes to PH pathobiology. Strategies targeting suppression or reversal of these pathways may preserve PPARγ function in the pulmonary vascular wall and provide a novel therapeutic strategy in PH.
Materials and Methods
Reagents
The ERK 1/2 inhibitor (PD98059) and PEG-catalase were purchased from Calbiochem (La Jolla, CA) and Sigma-Aldrich (St. Louis, MO), respectively. Antibodies against phospho-(Thr202/Tyr204)-ERK 1/2, total ERK 1/2, and phospho-(Ser536)-NF-κB were purchased from Cell Signaling Technology (Beverly, MA). Antibodies against PPARγ, total NF-κB, IκBα, Nox4, and actin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibody against PGC-1α was purchased from Millipore (Billerica, MA). Antibody against GAPDH was purchased from Sigma-Aldrich (St. Louis, MO). All other materials were purchased from VWR Scientific Corp. (Gaithersburg, MD) and Fisher Scientific (Pittsburg, PA). The Nox4 inhibitor, GKT137831 was obtained through a material transfer agreement from GenKyoTex (Geneva, Switzerland).
Cell Culture and siRNA transfections
Human pulmonary artery smooth muscle cells (HPASMC) were purchased from Lonza (Basel, Switzerland). HPASMC monolayers (passages 3-4) were grown at 37°C in a 5% CO2 atmosphere in culture media (SmGM-2, Lonza) containing 2% fetal calf serum, growth factors, and antibiotics as previously reported [19]. Upon reaching 50-60% confluency, the cells were transfected with 50-100 nM non-targeting siRNA (control siRNA) or siRNA targeting human PPARγ using Dharmafect transfection reagent (Dharmacon, Waltham, MA) for 12 hours. Cells were then washed with serum-free media and recovered for 24 - 72 hours in complete growth media under normoxic conditions (21% O2, 5% CO2) at 37°C in a cell culture incubator.
Overexpression of PPARγ in HPASMC
HPASMC monolayers were grown at 37°C in a 5% CO2 atmosphere in culture media (SmGM-2, Lonza) containing 5% fetal calf serum, growth factors, and antibiotics. Human PPARγ in adenovirus (Ad-hPPARγ) or Ad-GFP (Vector Biolabs, Philadelphia, PA) were applied to cells at 3-28 MOI (Ad-GFP was applied at 3 MOI and Ad-PPARγ was applied at 28 MOI) for 4 hours in 2% FBS media. Media were replaced with fresh SmGM-2 media, and HPASMC were cultured for 72 hrs.
Cell proliferation assays
HPASMC proliferation was determined with a quantitative colorimetric assay employing dimethylthiazol (MTT assay; ATCC) as described earlier [19]. Briefly, cells transfected with non-targeting control siRNA (si-Con) or with siRNA against PPARγ (si-PPARγ) were treated with or without PEG-catalase (1000 u/ml) during the last 24 hours of the 72 hour recovery period. The cells were then incubated with the MTT reagent for 4 hours. The mitochondrial reductase present in living cells reduces MTT to purple formazan, which is detected by spectrophotometry. Samples were then analyzed using an ELISA plate reader (λ = 570 nm), and values from treated cells were normalized to values from corresponding control cells. To assess proliferation by cell counting, HPASMCs were counted using a hemocytometer and cell viability was determined by Trypan blue exclusion assay as described previously [24]. We previously reported that MTT assays and cell counting methods produced similar results in hypoxia-exposed pulmonary vascular wall cells [24].
Quantitative real-time PCR (qRT-PCR)
Total RNA was isolated using RNeasy Mini Kit (Qiagen, Valencia, CA), and RNA was quantified by Nanodrop spectrophotometry (Thermo Scientific, Wilmington, DE). cDNA was prepared using the iScript cDNA synthesis kit (Bio-Rad, Hercules, CA). Quantitative PCR was performed to assess the expression level of PPARγ1 RNA using primers based on human PPARγ1 mRNA sequence: F, 5′-gtggc cgcag atttg aaaga ag-3′ and R, 5′-tgtca accat ggtca tttcg-3′. Real-time PCR was performed using iQ SYBR Green Supermix and the iCycler real-time PCR detection system (Bio-Rad). Amplicon expression in each sample was normalized to 9s RNA levels. The relative abundance of target mRNA in each sample was calculated using ΔΔCT methods (Applied Biosystems, Carlsbad, CA).
Western blot analysis
HPASMC protein lysates were resolved by SDS-PAGE and subjected to Western blot analysis for PPARγ, phospho-ERK 1/2, phospho-NF-κB/p65, Nox4 or PGC-1α and normalized to their respective total forms (non-phosphorylated) or β-actin or GAPDH as appropriate. Relative levels of immunoreactive proteins were quantified using the Licor or the ChemiDoc XRS imaging systems and Quantity One software (Bio-Rad Laboratories).
Amplex Red H2O2 assay
H2O2 production was measured with Amplex Red Hydrogen Peroxide/Peroxidase Assay Kit (Invitrogen, Molecular Probes, Eugene, OR). The assay is based on the detection of H2O2 which reacts with 1:1 stoichiometry with Amplex® Red reagent in combination with horseradish peroxidase to produce highly fluorescent resorufin red. H2O2 released from HPASMC was quantified by fluorometric detection on a plate reader (λex = 590 nm and λem = 560 nm), followed by plotting sample values against a standard curve containing known concentrations of H2O2. The concentration of H2O2 was normalized to the total protein concentration of each sample.
Statistical analysis
When comparing more than 2 groups, data were analyzed using analysis of variance (ANOVA). Post hoc analysis using the Student Neuman Keuls test was employed to detect differences between individual groups. In studies comparing only two experimental groups, data were analyzed with Student's t-test to determine significance of treatment effects. The level of statistical significance was taken as p<0.05.
Results
Depletion of PPARγ activates ERK 1/2 in HPASMC
Hypoxia promotes proliferation of HPASMC through reduction in PPARγ levels via upstream activation of ERK 1/2 followed by activation of NF-κB, Nox4 expression and H2O2 generation [19, 23]. In the current study, we further hypothesized that reduced PPARγ levels in HPASMCs promote sustained NF-κB activation and cell proliferation associated with chronic hypoxia. To address this possibility, siRNA was used to deplete PPARγ levels in HPASMCs under normoxic conditions (21% O2, 5% CO2). To mimic alterations in HPASMC caused by exposure to hypoxia for 72 hours, HPASMC were transfected with control or PPARγ siRNA for 24 or 72 hours. As shown in Figure 1A, PPARγ mRNA levels were significantly reduced 72 hours following transfection with PPARγ siRNA. Furthermore, compared to control siRNA, treatment with PPARγ siRNA significantly reduced PPARγ protein levels by approximately 25 to 50% at both 24 and 72 hours transfection duration (Figs. 1B and C). These levels of PPARγ depletion are comparable in magnitude to those observed in hypoxia-exposed HPASMC [19]. The impact of PPARγ protein depletion with siRNA was confirmed by immunoblotting for its target gene, PGC-1α (peroxisome proliferator-activated receptor-gamma coactivator) [Fig 1C].
Figure 1. siRNA-mediated knockdown of PPARγ.
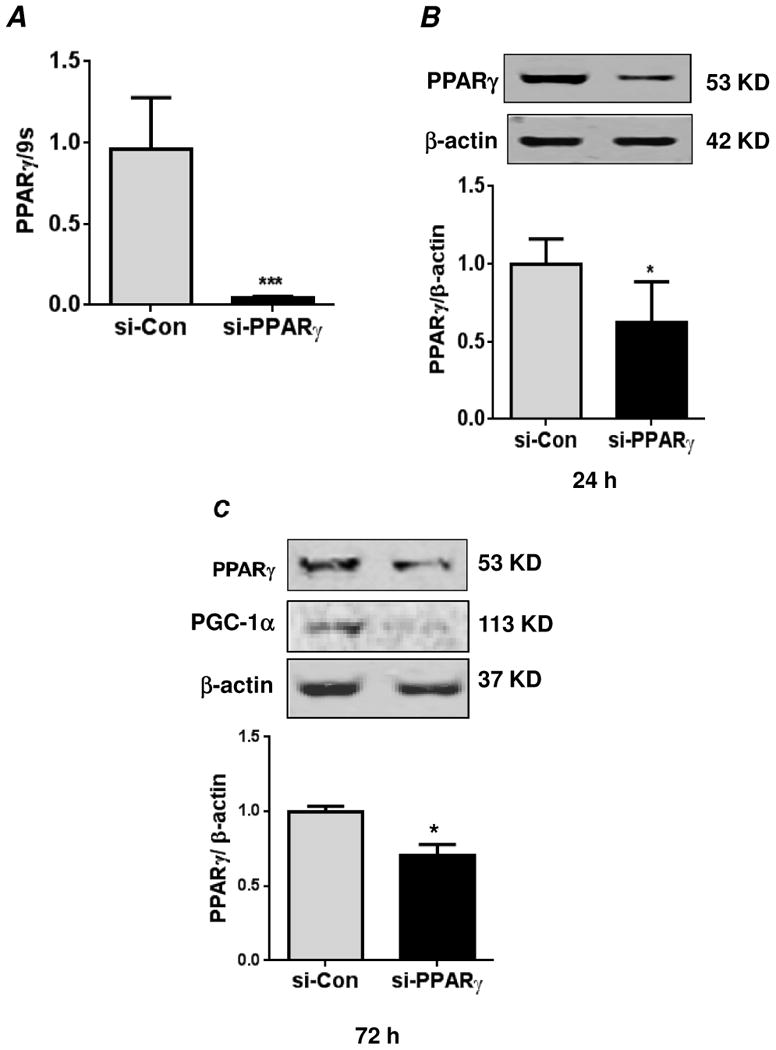
(A) Human pulmonary artery smooth muscle cells (HPASMC) were transfected with control siRNA (si-Con) or PPARγ siRNA (si-PPARγ) under normoxic conditions (21% O2 and 5% CO2 at 37°C) for 72 hours. RNA was isolated, and PPARγ mRNA levels were determined by qRT-PCR. The mRNA levels were normalized to 9S ribosomal RNA. Each bar represents the mean ± SEM of levels of PPARγ mRNA relative to 9S ribosomal RNA in the same sample expressed as fold-change versus control. n=6, ***p<0.001. In related experiments, HPASMC were transfected with control siRNA or PPARγ siRNA under normoxic conditions for 24 hours (B) or 72 hours (C). Cells were lysed and analyzed for PPARγ protein levels or its target gene, PGC-1α by western blot. The PPARγ protein levels were normalized with β-actin levels. Each bar represents the mean ± SEM of levels of PPARγ protein relative to β-actin in the same sample expressed as fold-change versus control. n=3, *p<0.05.
Because hypoxia reduced PPARγ levels and activated HPASMC ERK 1/2 [19], the effect of PPARγ depletion on ERK 1/2 activation was examined. PPARγ depletion enhanced ERK 1/2 activation detected as Thr202/Tyr204 phosphorylation at both 24 and 72 hours of siRNA transfection indicating that PPARγ downregulation promotes early and sustained MAPK activation (Figs. 2A and B). Compared to untransfected cells, neither transfection reagent alone nor si-Con significantly altered phospho-ERK 1/2 levels (data not shown).
Figure 2. PPARγ knockdown promotes HPASMC ERK 1/2 activation.
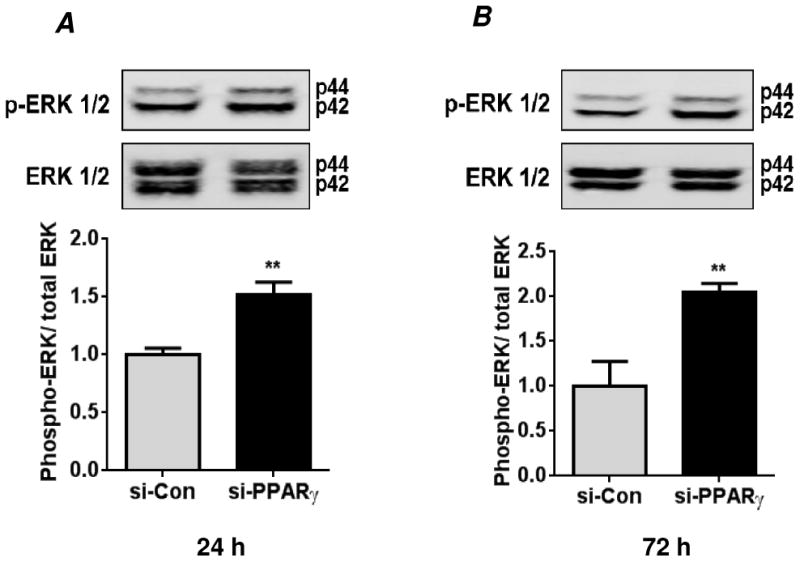
HPASMC were transfected with control siRNA (si-Con) or PPARγ siRNA (si-PPARγ) under normoxic conditions for 24 hours (A) or 72 hours (B). Cells were lysed and analyzed by western blot for activation of extracellular regulated kinase (ERK)-1/2 using an anti-phospho-(Thr202/Tyr204)-ERK1/2 antibody. The phospho-ERK 1/2 levels were normalized with total ERK 1/2 levels. Each bar represents levels of phospho-ERK 1/2 relative to total ERK 1/2 in the same sample expressed as fold-change versus control. n=3, **p<0.01.
PPARγ depletion promotes ERK 1/2-mediated activation of NF-κB in HPASMC
PPARγ depletion for 72 hours stimulated degradation of IκBα, an NF-κB pathway inhibitory protein (Fig. 3A). In addition to IκBα degradation, a critical determinant of NF-κB activation is its phosphorylation at serine residue(s) (Ser536, 276, 529, 311) required for its complete transactivation [25-28]. We previously reported that hypoxia reduces PPARγ and stimulates phosphorylation of NF-κB p65 at Ser536 in HPASMC [19]. Therefore the effect of normoxic PPARγ depletion on the phosphorylation status of NF-κB p65 at Ser536 was examined. Compared to control cells, PPARγ depletion promoted phosphorylation of NF-κB p65 at both 24 and 72 hours of siRNA transfection (Figs. 3B and C). Thus, consistent with ERK 1/2 activation, PPARγ depletion promotes both early and sustained activation of NF-κB. Since hypoxia promotes NF-κB activation in HPASMC through the activation of ERK 1/2 [19], the possibility that ERK 1/2 could also mediate PPARγ depletion-induced NF-κB activation was examined. As observed in Figure 3C, pharmacological inhibition of ERK 1/2 using PD98059 during the last 24 hours of the 72 hours transfection period significantly attenuated phosphorylation of NF-κB p65 in PPARγ depleted cells.
Figure 3. PPARγ knockdown in HPASMC promotes ERK 1/2-mediated activation of NF-κB.
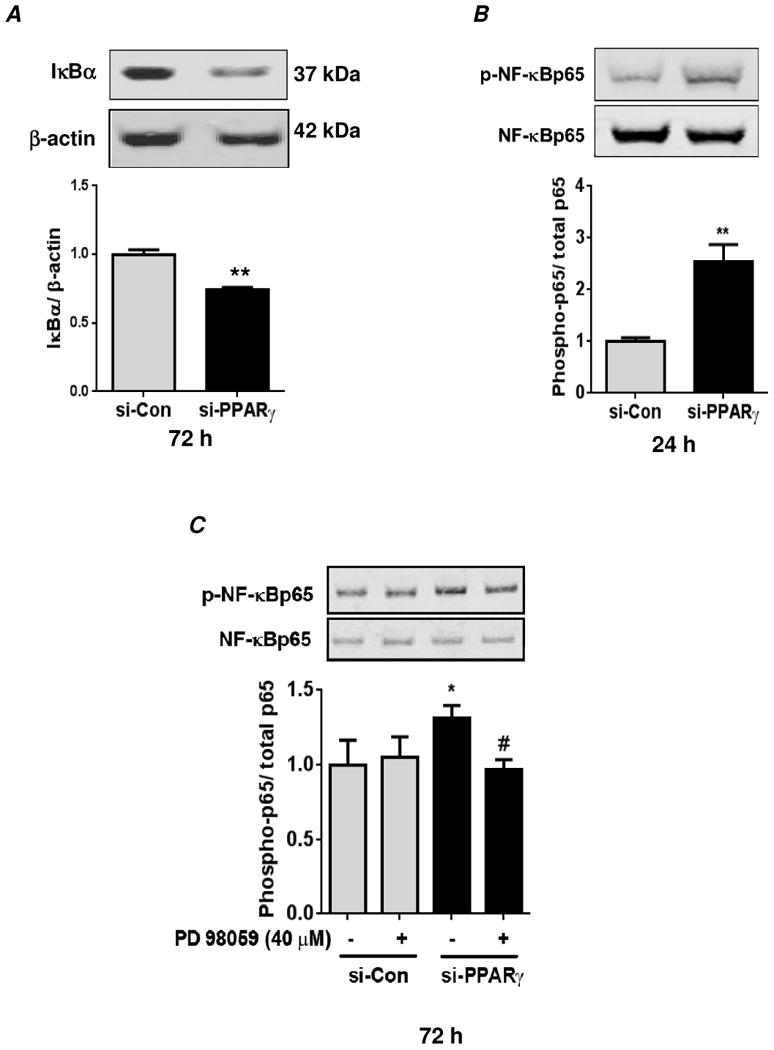
HPASMC were transfected with control siRNA (si-Con) or PPARγ siRNA (si-PPARγ) under normoxic conditions for 72 hours (A) or 24 hours (B). Cells were lysed and analyzed by western blot for IκBα (A) or phospho-p65 using an anti-phospho-(Ser536)-NF-κBp65 antibody (B). In (A), the levels of IκBα were normalized with β-actin. Each bar represents mean ± SEM IKBα levels relative to β-actin in the same sample expressed as fold-change versus control. n=3, **p<0.01. In (B), phospho-NF-κBp65 levels were normalized with total NF-κBp65 levels. Each bar represents mean ± SEM of phospho-NF-κBp65 levels relative to total NF-κBp65 in the same sample expressed as fold-change versus control. n=3-6, **p<0.01. (C) Cells were transfected with siRNA as described above, and treated with the ERK 1/2 inhibitor, PD98059 (40 μM) or vehicle (DMSO) during the final 24 hours of the 72 hour transfection period. Cells were lysed and analyzed for phospho-NF-κBp65 by western blot as described above. Each bar represents mean ± SEM phospho-NF-κBp65 levels relative to total NF-κBp65 in the same sample expressed as fold-change versus control. n=3, *p<0.05 vs si-Con; #p<0.05 vs vehicle treated si-PPARγ.
PPARγ depletion promotes NF-κB-dependent Nox4 expression and Nox4-dependent H2O2 generation in HPASMC
We previously reported that exposure to hypoxia for 72 hours reduced PPARγ expression and stimulated Nox4 expression through an NF-κB- dependent pathway in HPASMC [19, 23]. To further examine the role of PPARγ depletion alone in these signaling events, the current study examined HPASMC after 72 hours of treatment with si-PPARγ. Consistent with NF-κB activation (Figs. 3A-C), a significant increase in Nox4 protein expression was observed in PPARγ depleted cells compared to untreated or si-Con cells (Fig. 4A). Furthermore, pharmacological inhibition of NF-κB using CAPE (caffeic acid phenethyl ester) compound attenuated Nox4 protein expression caused by PPARγ depletion (Fig. 4A) suggesting that NF-κB regulates Nox4 expression in this response. Because Nox4 is a major source of hypoxia-induced H2O2 generation in HPASMC [15, 19, 23], the effect of PPARγ depletion on H2O2 generation was examined. Consistent with increases in Nox4 expression levels, PPARγ depletion for 72 hours caused a significant increase in HPASMC H2O2 generation. Evidence that Nox4 upregulation increased HPASMC H2O2 generation was provided by studies using a novel Nox4 inhibitor, GKT137831. Treatment with GKT137831 significantly attenuated H2O2 generation caused by PPARγ depletion (Fig. 4B).
Figure 4. PPARγ knockdown promotes NF-κB-dependent Nox4 expression and Nox4-derived H2O2 generation in HPASMC.
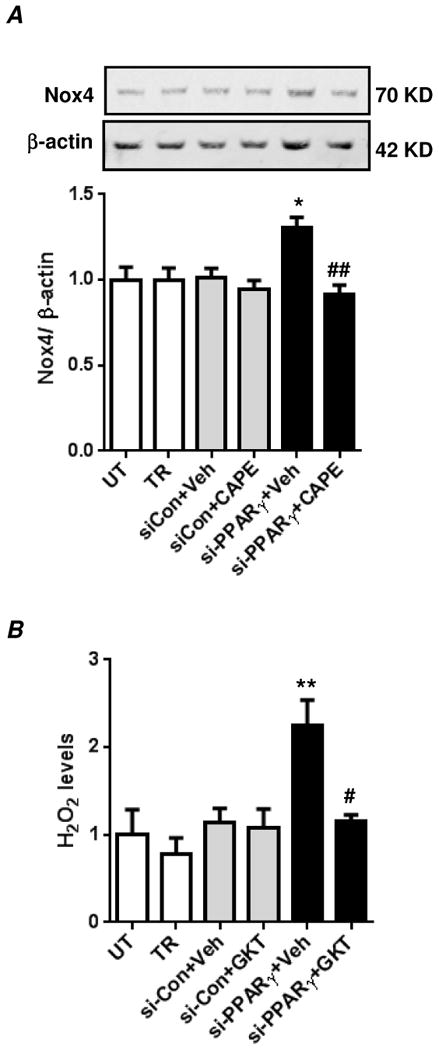
(A) HPASMC were untransfected (UT) or transfected with transfection reagent alone (TR), control siRNA (si-Con) or PPARγ siRNA (si-PPARγ) under normoxic conditions for 72 hours. Cells were treated with CAPE compound (10 μM) or equivalent volume of DMSO (vehicle) during the final 24 hours of the 72 hours transfection period. Cells were lysed and analyzed by western blot for Nox4 protein expression. Nox4 levels were normalized with β-actin levels. Each bar represents mean ± SEM Nox4 relative to β-actin in the same sample expressed as fold-change versus UT. n=6-7, *p<0.05 compared to UT, ##p<0.01 compared to PPARγ siRNA transfected cells that were treated with DMSO. (B) HPASMC were transfected as above under normoxic conditions for 72 hours. Cells were treated with GKT137831 (20 μM) or equivalent volume of DMSO (vehicle) during the final 24 hours of the 72 hour transfection period, and H2O2 concentration was measured with the Amplex Red assay. Each bar represents mean ± SEM H2O2 concentration as fold-change versus UT. n=6-7, **p<0.01 compared to UT; #p<0.05 compared to si-PPARγ+Veh.
PPARγ depletion promotes HPASMC proliferation
Given the requirement for H2O2 in hypoxia-induced proliferation of pulmonary vascular wall cells [19], we next examined whether PPARγ depletion promotes proliferation of HPASMC via an H2O2-dependent mechanism. Determination of HPASMC proliferation by MTT assay revealed that PPARγ depletion increases HPASMC proliferation and that degradation of H2O2 by PEG-catalase treatment during the final 24 hours of the 72 hour transfection period attenuated proliferation of PPARγ-depleted cells (Fig. 5A). Cell proliferation caused by PPARγ depletion was also verified by cell counting which produced results similar to those observed with the MTT assay (Fig. 5B). Consistent with these observations, pharmacological inhibition of NF-κB or Nox4 significantly attenuated HPASMC proliferation caused by PPARγ depletion (Fig. 5C).
Figure 5. PPARγ knockdown promotes HPASMC proliferation via an NF-κB-Nox4-H2O2-dependent mechanism.
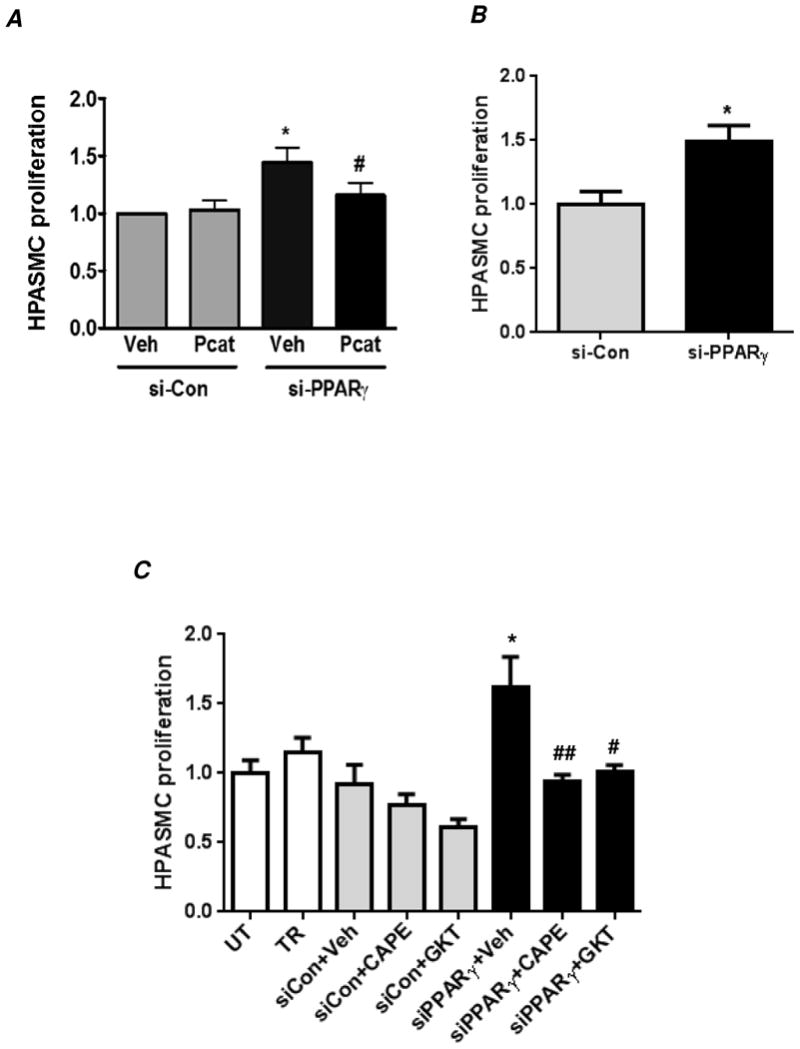
HPASMC were untransfected (UT) or transfected with transfection reagent alone (TR), control siRNA (si-Con) or PPARγ siRNA (si-PPARγ) under normoxic conditions for 72 hours. Selected HPASMC were treated with PEG-catalase (1000 U/ml) (A), CAPE (10 μM) or GKT137831 (20 μM) (C) or with vehicle (Veh, DMSO) during the last 24 hours of the 72 hour transfection period. Cell proliferation was determined by MTT assay. (B) Cell proliferation upon PPARγ depletion was also verified by manual cell counting method. In (A), each bar represents mean ± SEM HPASMC proliferation as fold-change versus control. n=3, *p<0.05 compared to Veh/si-Con; #p<0.05 compared to Veh/si-PPARγ. In (B), each bar represents mean ± SEM HPASMC proliferation (cells/ml) as fold-change versus control. n=3, *p<0.05 compared to si-Con. In (C), each bar represents mean ± SEM HPASMC proliferation as fold-change versus UT. n=5-7, **p<0.01 compared to UT; # ##p<0.05 and 0.01, respectively, compared to si-PPARγ+Veh.
PPARγ depletion-induced H2O2 production mediates feed-forward ERK 1/2 and NF-κB activation and Nox4 expression
To further understand the role of H2O2 generated by PPARγ depletion in HPASMC signaling, HPASMC transfected with si-Con or siPPARγ were treated with PEG-catalase (Pcat) to degrade H2O2 or with DMSO as a control vehicle during the final 24 hours of the 72 hours transfection period. Immunoblots of cell lysates revealed that Pcat attenuated ERK 1/2 and NF-κB activation and Nox4 protein expression caused by PPARγ depletion suggesting that H2O2 mediates sustained activation of these signaling events in HPASMC proliferation (Fig. 6A-C).
Figure 6. Production of H2O2 caused by PPARγ knockdown is required for sustained ERK 1/2 and NF-κB activation as well as Nox4 expression.
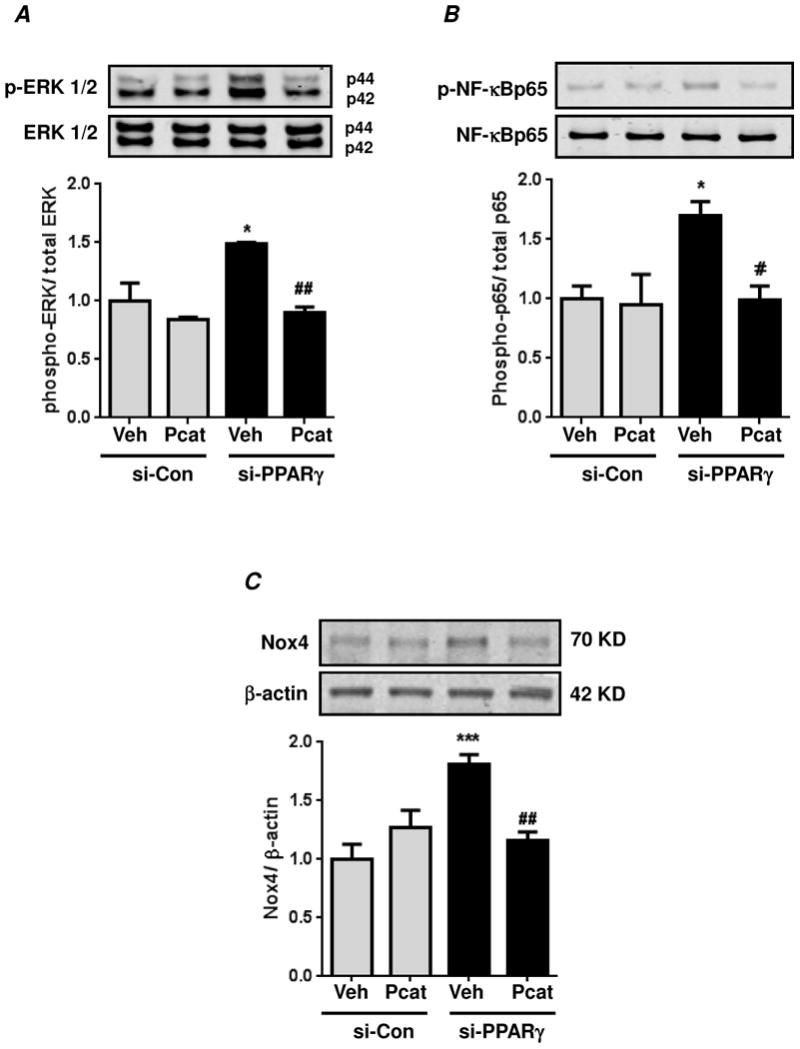
HPASMC were transfected with control siRNA (si-Con) or PPARγ siRNA (si-PPARγ) under normoxic conditions for 72 hours. During final 24 hours of transfection, cells were treated with PEG-catalase (Pcat, 1000 U/ml) or with DMSO as control vehicle (Veh). Cell lysates were immunoblotted for phospho-ERK 1/2 (A) phospho-p65 (B) or Nox4 (C). In (A), each bar represents mean ± SEM phospho-ERK 1/2 relative to total ERK 1/2 in the same sample expressed as fold-change versus control. n=3, *p<0.05 vs si-Con; ##p<0.01 vs vehicle treated si-PPARγ. In (B), each bar represents mean ± SEM phospho-NF-κBp65 relative to total NF-κBp65 in the same sample expressed as fold-change versus control. n=3, *p<0.05 vs si-Con; #p<0.05 vs vehicle treated si-PPARγ. In (C), each bar represents mean ± SEM Nox4 relative to β-actin in the same sample expressed as fold-change versus control. n=6, ***p<0.001 vs si-Con; ##p<0.01 vs vehicle treated si-PPARγ.
Overexpression of PPARγ inhibits ERK 1/2 and p65 activation, Nox4 protein expression and HPASMC proliferation
To provide proof-of-concept for the regulatory role of PPARγ in these signaling pathways, reciprocal experiments were performed defining the impact of PPARγ overexpression on ERK 1/2 and p65 activation and Nox4 protein expression in HPASMC. As expected, adenoviral-mediated PPARγ overexpression in HPASMC reduced basal ERK and p65 activation, Nox4 expression and cell proliferation (by manual cell counting) compared to control cells (Figs. 7A-D). Cell proliferation was also verified by MTT assay which yielded similar results (data not shown).
Figure 7. PPARγ overexpression reduces basal ERK 1/2 and NF-κB activation and Nox4 protein expression and HPASMC proliferation.
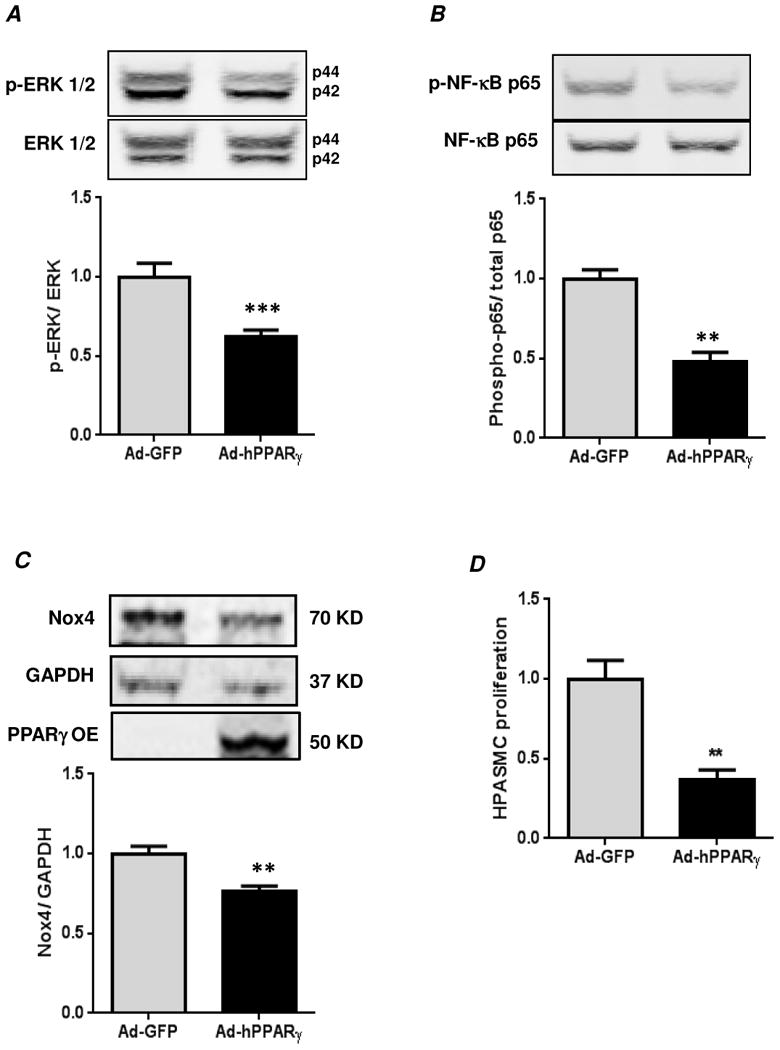
Confluent HPASMC monolayers were transfected with human PPARγ in adenovirus (Ad-hPPARγ) or Ad-GFP (Vector Biolabs, Philadelphia, PA) as described in Materials and Methods. Cells were lysed and immunoblotted for phospho-ERK 1/2 (A), phospho-p65 (B), or Nox4 (C). In (D), HPASMC proliferation was measured by manual cell counting method. In (A), each bar represents mean ± SEM phospho-ERK 1/2 relative to total ERK 1/2 in the same sample expressed as fold-change versus Ad-GFP. n=4, ***p<0.001. In (B), each bar represents mean ± SEM phospho-p65 relative to total p65 in the same sample expressed as fold-change versus Ad-GFP. n=4, **p<0.01. In (C), each bar represents mean ± SEM Nox4 relative to GAPDH in the same sample expressed as fold-change versus Ad-GFP. n=4, **p<0.01. Overexpression of PPARγ (PPARγ-OE) was confirmed by immunoblotting. The image was captured at a lower intensity to highlight the expression of exogenously introduced PPARγ. In (D), each bar represents mean ± SEM HPASMC proliferation (cells/ml) expressed as fold-change versus Ad-GFP. n=4, **p<0.01. Similar results were obtained by MTT assay (data not shown).
Discussion
Excessive proliferation of pulmonary vascular wall cells with increases in pulmonary vascular remodeling and resistance are crucial hallmarks of pulmonary hypertension (PH) pathogenesis. The nuclear hormone receptor, PPARγ plays an important role in normal vascular function [7, 12, 15]. PPARγ levels are reduced in lungs of various rodent models of hypoxia-induced PH and in vascular cells and lesions of patients with idiopathic pulmonary hypertension [11, 13-15]. However, the contribution of reduced PPARγ levels to the pathobiology of PH continues to be defined. We recently demonstrated that hypoxia reduced PPARγ protein and stimulated HPASMC proliferation via sustained activation of ERK 1/2 and NF-κB, which increased Nox4 protein expression and H2O2 generation [19]. To better understand the contributions of altered PPARγ expression to vascular cell signaling and proliferation, the current study employed siRNA to reduce PPARγ in normoxic HPASMC and investigate downstream pathways that cause HPASMC proliferation.
Exposure to hypoxia (1% O2) for 72 hours reduced HPASMC PPARγ protein levels by approximately 50% [19]. To mimic this observation, the current study employed siRNA to induce comparable reductions in PPARγ protein levels under normoxic conditions (Figs. 1B and C). The increase in HPASMC proliferation caused by PPARγ depletion (1.34 fold) was similar to that observed under hypoxic conditions (1.5 fold) [19]. The MAP kinase, ERK1/2, is required for cell proliferation and growth [29], and increased ERK1/2 activity mediates PPARγ phosphorylation and proteosomal degradation leading to decreased PPARγ expression and transcriptional activity [30, 31]. Using HPASMC, we previously demonstrated that hypoxic ERK 1/2 activation was required for downregulation of PPARγ expression and transcriptional activity [19]. Further, neither inhibition of p38 nor JNK altered hypoxia-induced reductions in PPARγ expression and activity [19]. Therefore, in the current study, we focused on ERK 1/2 activation during PPARγ depletion. Our findings demonstrate that 24 or 72 hrs of PPARγ depletion is sufficient to induce ERK 1/2 activation (Fig. 2). Taken together, these findings suggest that potential feed forward cycles of PPARγ depletion and ERK1/2 activation contribute to altered smooth muscle cell phenotypes.
Hypoxia also activated NF-κB p65 which binds to the Nox4 promoter to stimulate H2O2 generation and HPASMC proliferation [19, 23]. In the current study, siRNA-mediated loss of PPARγ at both 24 and 72 hrs caused a significant increase in p65 phosphorylation (Fig. 3), which indicates that PPARγ depletion engages upstream kinases to mediate p65 phosphorylation and NF-κB activation. Based on the observed ERK 1/2 activation (Fig. 2), we addressed the effect of ERK 1/2 inhibition on p65 phosphorylation caused by PPARγ depletion. Indeed pharmacological inhibition of ERK 1/2 with PD98059 significantly attenuated p65 phosphorylation caused by PPARγ depletion (Fig. 3C) suggesting that ERK 1/2 lies upstream of p65 to mediate this effect.
Previous evidence demonstrated that Nox4 constitutes an important downstream target of hypoxic signaling. Hypoxia stimulated ERK 1/2 and NF-κB activation to increase Nox4 expression in HPASMC, and Nox4-derived H2O2 reduced PPARγ expression and promoted HPASMC proliferation [19, 23]. In addition, pharmacological inhibition of Nox4 attenuated experimental PH and reductions in PPARγ in vitro and in vivo [15]. The current study therefore sought to determine if reductions in PPARγ were not only sufficient to activate signaling and transcriptional pathways such as ERK1/2 and NF-κB, respectively but also capable of modulating the expression of Nox4. Our results demonstrate that PPARγ depletion not only stimulated ERK 1/2 and p65 activation but also increased Nox4 protein expression, H2O2 generation, and HPASMC proliferation (Figs. 4 and 5). Our observations with GKT137831 are consistent with our previous findings in which Nox4 inhibition attenuated hypoxia-induced HPASMC proliferation [15]. GKT137831 is a novel Nox1/Nox4 inhibitor that is potent and orally bioavailable [32]. This compound was employed in the current study due to its ability to inhibit Nox4 activity, which is selectively up-regulated in the pulmonary vasculature under hypoxic conditions [18] and associated with hypoxia-induced HPASMC proliferation [23, 33], vascular remodeling, and pulmonary hypertension [13, 18].
Previously we reported that hypoxia-induced H2O2 production promoted cyclic ERK 1/2 activation and Nox4 expression in HPASMC [19]. Similar observations with PPARγ depletion in the current study (Fig. 6A-C) suggest that, under hypoxic conditions, H2O2 production remains sustained not only due to hypoxia-induced signaling events but also likely due to hypoxic-reductions in PPARγ levels. Such a sustained level of H2O2 is crucial for maintaining sustained ERK and NF-κB activation and Nox4 expression thereby contributing to PH pathology associated with chronic hypoxia. Taken together, the downregulation of ERK 1/2, p65 and Nox4 and reductions in cell proliferation caused by overexpression of PPARγ (Fig. 7) provides novel proof-of-concept on the regulatory role of PPARγ and is consistent with previous reports that the PPARγ ligand, rosiglitazone, attenuated hypoxia-induced HPASMC Nox4 expression [23]. The findings in the current study are consistent with the postulate that reductions in SMC PPARγ generate proliferative mediators in the pulmonary vasculature.
Previous work has suggested that targeted smooth muscle cell PPARγ depletion promotes PH in vivo. Mice with constitutive smooth muscle-targeted PPARγ depletion had elevated right ventricular systolic pressure (RVSP), right ventricular hypertrophy (RVH), and muscularization of small pulmonary arteries under normoxic conditions [17]. The current results provide novel insights into mechanisms by which loss of PPARγ function may contribute to PASMC proliferation and PH pathobiology. These results are also consistent with previous reports that endothelial-targeted loss of vascular PPARγ function enhanced aortic NF-κB activity [34]. PPARγ depletion also increased basal and stimulated systemic vascular SMC proliferation [35]. The current findings demonstrate that constitutive PPARγ function limits the activity of pulmonary vascular wall signaling cascades that lead to the upregulation of proliferative pathways including Nox4. The current findings also support the previously reported mutually repressive relationship between PPARγ and NF-κB in vascular wall cells [19, 23]. Hypoxia downregulates PPARγ by post-transcriptional mechanisms via upregulation of miRNA27a [36] and via post-translational mechanisms by activating NF-κB which induces PPARγ suppression [19]. Although hypoxia-induced ERK activation is partly dependent on Nox4-derived H2O2 production [19], the exact signaling mechanisms upstream of ERK 1/2 are not well defined. However, based on evidence that chronic hypoxia increased calcium influx [37-39] and on our unpublished observations that the calcium-dependent kinase, Pyk2 is involved in hypoxic ERK 1/2 activation in HPASMC, we speculate that hypoxic increases in calcium play a proximal role in stimulating these proximal signaling events. As a master regulatory switch in metabolic function, alterations in PPARγ may also contribute to metabolic derangements in pulmonary vascular wall cells to mediate the glycolytic, apoptosis-resistant, hyperproliferative pulmonary vascular cell phenotype associated with the pathobiology of PH. As illustrated in Figure 8, we postulate that derangements in PPARγ play a critical role in pulmonary vascular smooth muscle cell proliferation. Targeted interruption of these feed-forward signaling cascades could provide novel therapeutic strategies to attenuate sustained signaling derangements and alterations in gene expression and redox signaling that promote pulmonary vascular cell phenotypic alterations and PH pathogenesis.
Figure 8. Schematic representation of ERK1/2-p65-Nox4-mediated pathways of cell proliferation in HPASMC caused by PPARγ depletion.
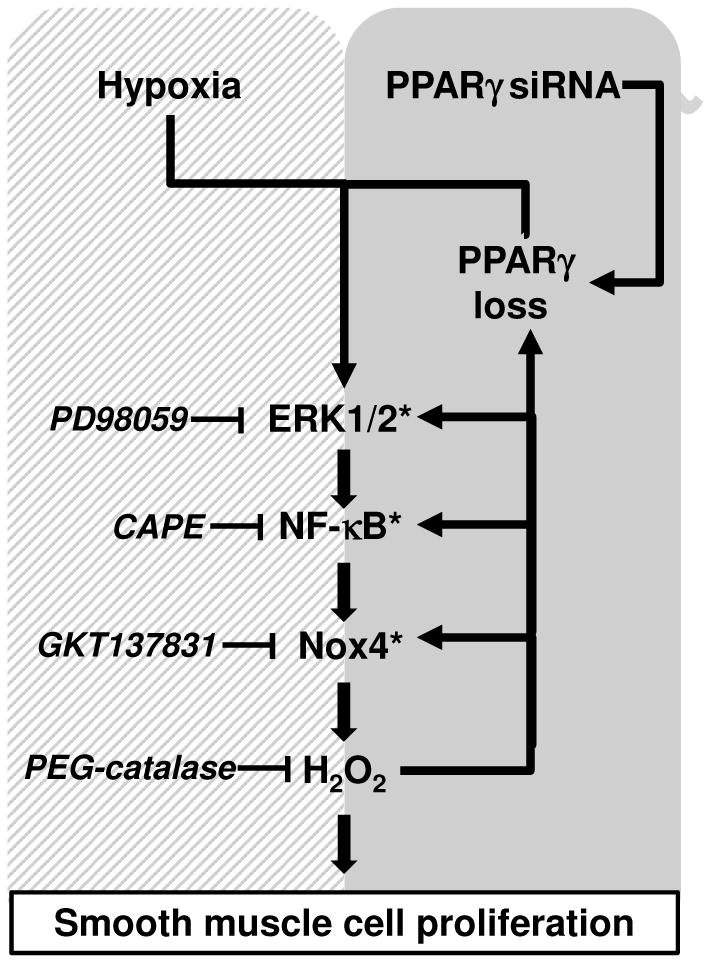
We previously demonstrated that hypoxia promotes HPASMC proliferation and PPARγ downregulation via an ERK1/2-NF-κB p65-Nox4-dependent mechanism [19] (hatched box). The findings in the current study (gray box) demonstrate that loss of PPARγ is sufficient to activate ERK1/2 and increase NF-κB p65 activity, Nox4 expression, H2O2 production, and HPASMC proliferation whereas PPARγ overexpression attenuated ERK1/2, NF-κB, and Nox4 (denoted by asterisks). These findings indicate that PPARγ provides constitutive inhibition of ERK, NF-κB, and Nox4 signaling pathways critical in PASMC proliferation. These findings further emphasize that targeting PPARγ may provide a useful therapeutic strategy to attenuate PASMC proliferation and pulmonary vascular remodeling.
Highlights.
Reduced PPARγ expression is sufficient to activate pathophysiological signaling mechanisms in pulmonary artery smooth muscle cells (PASMC).
PPARγ depletion promotes ERK 1/2-dependent NF-κB activation and NF-κB-dependent Nox4 expression.
PPARγ-depletion stimulates PASMC proliferation by increasing Nox4-derived H2O2.
These observations demonstrate that reductions in PPARγ caused by hypoxia or other pathophysiological stimuli are sufficient to promote alterations in PASMC signaling and proliferation that can contribute to the pathobiology of pulmonary hypertension.
Acknowledgments
This manuscript is based on work that was supported in part by Merit Review funding from the Department of Veterans Affairs Research, Veterans Health Administration, Office of Research and Development (1I01BX001910 to CMH), by NIH grant R01HL102167 (CMH and RLS), and by American Heart Association Scientist Development Grants (KMB and BYK). The contents reported herein do not represent the views of the Department of Veterans Affairs or the United States Government.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the productionprocess errors may be discovered which could affect the content, and all legal disclaimersthat apply to the journal pertain.
References
- 1.Picard F, Auwerx J. PPAR(gamma) and glucose homeostasis. Annu Rev Nutr. 2002;22:167–197. doi: 10.1146/annurev.nutr.22.010402.102808. [DOI] [PubMed] [Google Scholar]
- 2.Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M, Evans RM. PPARγ signaling and metabolism: the good, the bad and the future. Nat Med. 2013;19:557–566. doi: 10.1038/nm.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bardot O, Aldridge TC, Latruffe N, Green S. PPAR-RXR heterodimer activates a peroxisome proliferator response element upstream of the bifunctional enzyme gene. Biochem Biophys Res Commun. 1993;192:37–45. doi: 10.1006/bbrc.1993.1378. [DOI] [PubMed] [Google Scholar]
- 4.Tontonoz P, Hu E, Spiegelman BM. Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell. 1994;79:1147–1156. doi: 10.1016/0092-8674(94)90006-x. [DOI] [PubMed] [Google Scholar]
- 5.Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, Rose DW, Willson TM, Rosenfeld MG, Glass CK. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature. 2005;437:759–763. doi: 10.1038/nature03988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Calnek DS, Mazzella L, Roser S, Roman J, Hart CM. Peroxisome proliferator-activated receptor gamma ligands increase release of nitric oxide in endothelial cells. Arterioscler Thromb Vasc Biol. 2003;23:52–57. doi: 10.1161/01.atv.0000044461.01844.c9. [DOI] [PubMed] [Google Scholar]
- 7.Green DE, Sutliff RL, Hart CM. Is peroxisome proliferator-activated receptor gamma (PPARγ) a therapeutic target for the treatment of pulmonary hypertension? Pulmonary Circulation. 2011;1:33–47. doi: 10.4103/2045-8932.78101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest. 2008;118:2372–2379. doi: 10.1172/JCI33452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crossno JT, Jr, Garat CV, Reusch JE, Morris KG, Dempsey EC, McMurtry IF, Stenmark KR, Klemm DJ. Rosiglitazone attenuates hypoxia-induced pulmonary arterial modeling. Am J Physiol Lung Cell Mol Physiol. 2007;292:L885–897. doi: 10.1152/ajplung.00258.2006. [DOI] [PubMed] [Google Scholar]
- 10.Hansmann G, Wagner RA, Schellong S, Perez VA, Urashima T, Wang L, Sheikh AY, Suen RS, Stewart DJ, Rabinovitch M. Pulmonary arterial hypertension is linked to insulin resistance and reversed by peroxisome proliferator-activated receptor-gamma activation. Circulation. 2007;115:1275–1284. doi: 10.1161/CIRCULATIONAHA.106.663120. [DOI] [PubMed] [Google Scholar]
- 11.Kim EK, Lee JH, Oh YM, Lee YS, Lee SD. Rosiglitazone attenuates hypoxia-induced pulmonary arterial hypertension in rats. Respirology. 2010;15:659–668. doi: 10.1111/j.1440-1843.2010.01756.x. [DOI] [PubMed] [Google Scholar]
- 12.Matsuda Y, Hoshikawa Y, Ameshima S, Suzuki S, Okada Y, Tabata T, Sugawara T, Matsumura Y, Kondo T. Effects of peroxisome proliferator-activated receptor gamma ligands on monocrotaline-induced pulmonary hypertension in rats. Nihon Kokyuki Gakkai Zasshi. 2005;43:283–288. [PubMed] [Google Scholar]
- 13.Nisbet RE, Bland JM, Kleinhenz DJ, Mitchell PO, Walp ER, Sutliff RL, Hart CM. Rosiglitazone attenuates chronic hypoxia-induced pulmonary hypertension in a mouse model. Am J Respir Cell Mol Biol. 2010;42:482–490. doi: 10.1165/rcmb.2008-0132OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ameshima S, Golpon H, Cool CD, Chan D, Vandivier RW, Gardai SJ, Wick M, Nemenoff RA, Geraci MW, Voelkel NF. Peroxisome proliferator-activated receptor gamma (PPARgamma) expression is decreased in pulmonary hypertension and affects endothelial cell growth. Circ Res. 2003;92:1162–1169. doi: 10.1161/01.RES.0000073585.50092.14. [DOI] [PubMed] [Google Scholar]
- 15.Green DE, Murphy TC, Kang BY, Kleinhenz J, Szyndralewiez C, Page P, Sutliff RL, Hart CM. The NOX4 inhibitor, GKT137831, attenuates hypoxia-induced pulmonary vascular cell proliferation. Am J Respir Cell Mol Biol. 2012;47:718–726. doi: 10.1165/rcmb.2011-0418OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guignabert C, Alvira CM, Alastalo TP, Sawada H, Hansmann G, Zhao M, Wang L, El-Bizri N, Rabinovitch M. Tie2-mediated loss of peroxisome proliferator-activated receptor-{gamma} in mice causes PDGF-receptor {beta}-dependent pulmonary arterial muscularization. Am J Physiol Lung Cell Mol Physiol. 2009;297:L1082–L1090. doi: 10.1152/ajplung.00199.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hansmann G, de Jesus Perez VA, Alastalo TP, Alvira CM, Guignabert C, Bekker JM, Schellong S, Urashima T, Wang L, Morrell NW, Rabinovitch M. An antiproliferative BMP-2/PPARgamma/apoE axis in human and murine SMCs and its role in pulmonary hypertension. J Clin Invest. 2008;118:1846–1857. doi: 10.1172/JCI32503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mittal M, Roth M, Konig P, Hofmann S, Dony E, Goyal P, Selbitz AC, Schermuly RT, Ghofrani HA, Kwapiszewska G, Kummer W, Klepetko W, Hoda MA, Fink L, Hanze J, Seeger W, Grimminger F, Schmidt HH, Weissmann N. Hypoxia-dependent regulation of nonphagocytic NADPH oxidase NOX4 in the pulmonary vasculature. Circ Res. 2007;101:258–267. doi: 10.1161/CIRCRESAHA.107.148015. [DOI] [PubMed] [Google Scholar]
- 19.Lu X, Bijli KM, Ramirez A, Murphy TC, Kleinhenz J, Hart CM. Hypoxia downregulates PPARγ via an ERK 1/2-NF-κB-Nox4-dependent mechanism in human pulmonary artery smooth muscle cells. Free Radic Biol Med. 2013;63:151–160. doi: 10.1016/j.freeradbiomed.2013.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blanquicett C, Kang BY, Ritzenthaler JD, Jones DP, Hart CM. Oxidative stress modulates PPAR gamma in vascular endothelial cells. Free Radic Biol Med. 2010;48:1618–1625. doi: 10.1016/j.freeradbiomed.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Du J, Xu R, Hu Z, Tian Y, Zhu Y, Gu L, Zhou L. PI3K and ERK-induced Rac1 activation mediates hypoxia-induced HIF-1α expression in MCF-7 breast cancer cells. PLoS One. 2011;6:e25213. doi: 10.1371/journal.pone.0025213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hung HF, Wang BW, Chang H, Shyu KG. The molecular regulation of resistin expression in cultured vascular smooth muscle cells under hypoxia. J Hypertens. 2008;26:2349–2360. doi: 10.1097/HJH.0b013e328311fa30. [DOI] [PubMed] [Google Scholar]
- 23.Lu X, Murphy TC, Nanes MS, Hart CM. PPARγ regulates hypoxia-induced Nox4 expression in human pulmonary artery smooth muscle cells through NF-κB. Am J Physiol Lung Cell Mol Physiol. 2010;299:L559–566. doi: 10.1152/ajplung.00090.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kang BY, Kleinhenz JM, Murphy TC, Hart CM. The PPARγ ligand rosiglitazone attenuates hypoxia-induced endothelin signaling in vitro and in vivo. Am J Physiol Lung Cell Mol Physiol. 2011;301:L881–L891. doi: 10.1152/ajplung.00195.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghosh S, Hayden MS. New regulators of NF-κB in inflammation. Nat Rev Immunol. 2008;8:837–848. doi: 10.1038/nri2423. [DOI] [PubMed] [Google Scholar]
- 26.Rahman A, Fazal F. Blocking NF-kappaB: An inflammatory issue. Proc Am Thorac Soc. 2011;8:497–503. doi: 10.1513/pats.201101-009MW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bijli KM, Rahman A. NF-κB signaling in endothelium. In: Aird WC, editor. Endothelial biomedicine. New York: Cambridge University Press; 2007. pp. 784–795. [Google Scholar]
- 28.Chen LF, Greene WC. Shaping the nuclear action of NF-κB. Nat Rev Mol Cell Biol. 2004;5:392–401. doi: 10.1038/nrm1368. [DOI] [PubMed] [Google Scholar]
- 29.Berra E, Pages G, Pouyssegur J. MAP kinases and hypoxia in the control of VEGF expression. Cancer Metastasis Rev. 2000;19:139–145. doi: 10.1023/a:1026506011458. [DOI] [PubMed] [Google Scholar]
- 30.Hauser S, Adelmant G, Sarraf P, Wright HM, Mueller E, Spiegelman BM. Degradation of the peroxisome proliferator-activated receptor gamma is linked to ligand-dependent activation. J Biol Chem. 2000;275:18527–18533. doi: 10.1074/jbc.M001297200. [DOI] [PubMed] [Google Scholar]
- 31.Burgermeister E, Chuderland D, Hanoch T, Meyer M, Liscovitch M, Seger R. Interaction with MEK causes nuclear export and downregulation of peroxisome proliferator-activated receptor gamma. Mol Cell Biol. 2007;27:803–817. doi: 10.1128/MCB.00601-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aoyama T, Paik YH, Watanabe S, Laleu B, Gaggini F, Fioraso-Cartier L, Molango S, Heitz F, Merlot C, Szyndralewiez C, Page P, Brenner DA. Nicotinamide adenine dinucleotide phosphate oxidase in experimental liver fibrosis: GKT137831 as a novel potential therapeutic agent. Hepatology. 2012;56:2316–2327. doi: 10.1002/hep.25938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ismail S, Sturrock A, Wu P, Cahill B, Norman K, Huecksteadt T, Sanders K, Kennedy T, Hoidal J. Nox4 mediates hypoxia-induced proliferation of human pulmonary artery smooth muscle cells: the role of autocrine production of transforming growth factor-{beta}1 and insulin-like growth factor binding protein-3. Am J Physiol Lung Cell Mol Physiol. 2009;296:L489–L499. doi: 10.1152/ajplung.90488.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kleinhenz JM, Kleinhenz DJ, You S, Ritzenthaler JD, Hansen JM, Archer DR, Sutliff RL, Hart CM. Disruption of endothelial peroxisome proliferator-activated receptor-gamma reduces vascular nitric oxide production. Am J Physiol Heart Circ Physiol. 2009;297:H1647–1654. doi: 10.1152/ajpheart.00148.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meredith D, Panchatcharam M, Miriyala S, Tsai YS, Morris AJ, Maeda N, Stouffer GA, Smyth SS. Dominant-negative loss of PPARγ function enhances smooth muscle cell proliferation, migration and vascular remodelling. Atherioscler Thromb Vasc Biol. 2009;29:465–471. doi: 10.1161/ATVBAHA.109.184234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kang BY, Park KK, Green DE, Bijli KM, Searles CD, Sutliff RL, Hart CM. Hypoxia mediates mutual repression between microRNA-27a and PPARγ in the pulmonary vasculature. PLoS One. 2013;8:e79503. doi: 10.1371/journal.pone.0079503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin MJ, Leung GP, Zhang WM, Yang XR, Yip KP, Tse CM, Sham JS. Chronic hypoxia-induced upregulation of store-operated and receptor-operated Ca2+ channels in pulmonary arterial smooth muscle cells: a novel mechanism of hypoxic pulmonary hypertension. Circ Res. 2004;95:496–505. doi: 10.1161/01.RES.0000138952.16382.ad. [DOI] [PubMed] [Google Scholar]
- 38.Tang C, To WK, Meng F, Wang Y, Gu Y. A role of receptor-operated Ca2+ entry in human pulmonary artery smooth muscle cells in response to hypoxia. Physiol Res. 2010;59:909–918. doi: 10.33549/physiolres.931875. [DOI] [PubMed] [Google Scholar]
- 39.Hou X, Chen J, Luo Y, Liu F, Xu G, Gao Y. Silencing of STIM1 attenuates hypoxia-induced PASMCs proliferation via inhibition of the SOC/Ca2+/NFAT pathway. Resp Res. 2013;14:2–10. doi: 10.1186/1465-9921-14-2. [DOI] [PMC free article] [PubMed] [Google Scholar]