

Abstract
Pex1 and Pex6 are Type-2 AAA+ ATPases required for the de-novo biogenesis of peroxisomes. Mutations in Pex1 and Pex6 account for the majority of the most severe forms of peroxisome biogenesis disorders in humans. Here we show that the ATP-dependent complex of Pex1 and Pex6 from S. cerevisiae is a heterohexamer with alternating subunits. Within the Pex1/Pex6 complex, only the D2 ATPase ring hydrolyzes ATP, while nucleotide binding in the D1 ring promotes complex assembly. ATP hydrolysis by Pex1 is highly coordinated with that of Pex6. Furthermore, Pex15, the membrane anchor required for Pex1/Pex6 recruitment to peroxisomes inhibits the ATP-hydrolysis activity of Pex1/Pex6.
Keywords: Peroxisome, AAA+ ATPase, Pex1, Pex6, Pex15
Introduction
Peroxisomes are ubiquitous, small organelles that perform a variety of metabolic reactions in eukaryotic cells [1], typically including the β-oxidation of long-chain fatty acids and the breakdown of the metabolic by-product hydrogen peroxide. Unlike most other organelles, peroxisomes are not essential, and if lost, they can be generated de novo within the cell. In humans, defects in peroxisome biogenesis cause a spectrum of disorders (PBDs) including Zellweger Syndrome and Infantile Refsum disease [2]. The outcomes and severity of these disorders reflect where the genetic mutation impinges on peroxisome formation. While many of the genes required for peroxisome biogenesis have been identified and are mostly conserved throughout eukaryotes [3], the mechanisms involved in peroxisome biogenesis remain unclear.
Current models of peroxisome biogenesis posit that peroxisomal membrane proteins transit through the endoplasmic reticulum (ER) before they are packaged into pre-peroxisomal vesicles [4, 5]. These vesicles fuse to form peroxisomes competent for the import of peroxisomal matrix proteins [6], which are synthesized and folded in the cytoplasm [7]. These matrix proteins display one of two peroxisomal targeting signals that are recognized by two corresponding shuttle receptors, Pex5 or the Pex7/Pex18/Pex21 complex [8–14]. The cargo-bound receptors dock on the peroxisomal membrane through interactions with the Pex13/Pex14/Pex17 docking complex [15–17] and import the matrix protein using an unidentified mechanism that results in the partial insertion of the receptor into the peroxisome membrane. The receptors are subsequently ubiquitinated by the E2 ubiquitin conjugating enzymes Pex4 and Ubc4 and the peroxisome specific E3 RING ligases, Pex2/Pex10/Pex12 [18–23]. The AAA+ ATPases Pex1 and Pex6 then extract the ubiquitinated receptors from the peroxisome membrane [24]. Depending on the type of ubiquitination signal, extracted receptors are either recycled for another round of import or degraded by the 26S proteasome [23, 25–27].
Mutations in the AAA+ ATPases Pex1 and Pex6 are the most common cause of the more severe forms of PBDs [28, 29], yet their role in peroxisome biogenesis remains poorly understood. Pex1 and Pex6 are Type-2 AAA+ ATPases, which contain two conserved ATPase domains, termed D1 and D2, preceded by an N-terminal domain (NTD) that interacts with substrates and adaptor proteins. These AAA+ ATPases typically assemble as hexamers, in which the nucleotide binding pockets form at the interfaces between neighboring subunits in the ring. This architecture allows for highly coordinated nucleotide hydrolysis in the hexamer [30–33], which drives conformational changes that mechanically process protein substrates. Different Type-2 AAA+ ATPases, however, utilize different mechanisms to convert the energy of ATP hydrolysis into the mechanical force specific to their function in the cell [34]. For example, NSF dissociates SNARE complexes required for vesicle fusion through large conformational changes in its N-terminal domain [35], while ClpB disaggregates proteins by processive translocation through its central pore [36].
Two distinct roles have been proposed for Pex1 and Pex6 in peroxisome biogenesis based on the known functions of the homologs NSF (Sec18 in yeast) in vesicle fusion and p97 (Cdc48 in yeast) in the extraction of ubiquitinated proteins from the ER membrane prior to degradation by the 26S proteasome. In the first role, Pex1 and Pex6 function in peroxisome membrane fusion during peroxisome assembly, analogous to NSF in the secretory pathway [37]. Pre-peroxisomal vesicle fusion has been observed to be Pex1/Pex6 dependent in vitro and in vivo [6, 38]. In the second role, Pex1 and Pex6 extract the ubiquitinated shuttle receptors from the peroxisome membrane in a mechanism similar to that of p97 in ER- associated degradation (ERAD). Accordingly, purified yeast Pex1 and Pex6 have been observed to extract Pex5 from membrane fractions in an ATP-dependent manner. This process requires Pex15, the membrane anchor for Pex1 and Pex6 [24].
Based on their interaction in cell lysates and when purified as recombinant proteins [39–41], Pex1 and Pex6 appear to form a complex. The formation of this complex is ATP dependent and requires functional nucleotide-binding sites in at least one of the ATPase domains [40–42]. However, the architecture of the complex and the relative contributions of Pex1 and Pex6 to the overall activity remain unknown. Speculative models of the Pex1/Pex6 assembly include a hexamer of alternating subunits, a hexamer composed of a Pex1 trimer and three Pex6 monomers, and a complex formed from two interacting homohexamers of Pex1 and Pex6 [41, 43]. Other examples for heteromeric AAA+ ATPases are the mitochondrial mAAA+ protease, composed of Yta10 and Yta12, and the base subcomplex of the 26S proteasome, containing six distinct ATPases Rpt1-Rpt6. In both cases, mutations in individual subunits have differential effects on ATP-hydrolysis activity and substrate processing [44, 45], indicating distinct roles of subunits within the heterohexamer.
Here we show that Pex1 and Pex6 assemble as a heterohexamer with alternating subunits. Pex1 and Pex6 have distinct N-terminal domains; the N-terminal domain of Pex1 localizes above the ATPase barrel, while the Pex6 N-terminal domain is in an equatorial position. There are no major conformational changes in these N-terminal domains when the subunits of the Pex1/Pex6 complex adopt different nucleotide states. The robust ATPase activity of the heterohexamer derives from a highly coordinated hydrolysis in the D2 domains of Pex1 and Pex6. Pex1/Pex6 binding to the membrane anchor Pex15 inhibits ATP hydrolysis, likely by interfering with the activity of Pex1.
Results
Sequence analysis predicts that Pex1 and Pex6 have the archetypal domain structure of Type-2 AAA+ ATPases, with an N-terminal domain followed by two ATPase domains, termed D1 and D2 (Figure 1A). The typical ATPase binding pocket contains three characteristic motifs: the Walker A and Walker B motifs essential for nucleotide binding and hydrolysis, respectively, and the arginine residue, termed the R-finger, which is located at the subunit interface and essential for ATP hydrolysis in the neighboring subunit [46]. In the case of Pex1 and Pex6, however, the catalytic residues of the Walker A, Walker B, and R-finger motifs are only fully present in the D2 domains (Figure 1A). In the D1 domain, both Pex1 and Pex6 have substitutions in the Walker B motif known to impair nucleotide hydrolysis in other ATPases [46]. The Walker A motif of Pex6’s D1 domain also contains substitutions in the conserved glycines, making it unclear if the Pex6 D1 domain can bind ATP. In contrast, the Walker A of Pex1 D1 is fully archetypal. Since the nucleotide binding pocket is formed between subunits, with one subunit contributing the Walker A and B motif and the other contributing the R-finger, it is critical to assess whether Pex1 and Pex6 form homomeric or heteromeric ATP-binding sites to understand how these endogenous substitutions affect the ATPase activity of the complex.
Figure 1.
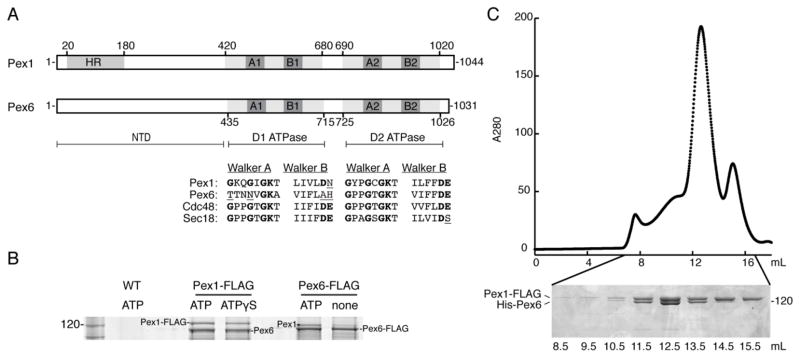
A) Schematic representation of Pex1 and Pex6 from S. cerevisiae. Both are Type-2 AAA+ ATPases with an N-terminal domain (NTD) and two ATPase domains, D1 and D2. HR: N-terminal region homologous to the N-domain of p97 and NSF. A1 and A2: Walker A motif (GxxGxGKT) in the D1 or D2 ATPase domain. B1 and B2: Walker B motif (ϕϕϕϕDE, ϕ: hydrophobic amino acid) in the D1 or D2 ATPase domain. The alignment shows the conservation of the Walker A and Walker B motifs in ScPex1, ScPex6, ScCdc48, and ScSec18. Poorly conserved residues are underlined.
B) Endogenous Pex1 and Pex6 from S. cerevisiae depend on the presence of nucleotide to form a complex. Pex6 co-immunoprecipitated with FLAG-tagged Pex1 in the presence of ATP or ATPγS. Pex1 co-immunoprecipitated with FLAG-tagged Pex6 in the presence of ATP, but this association is diminished when no nucleotide is present. A mock co-immunoprecipitation using the parent wild-type strain with untagged Pex1 and Pex6 served as a control.
C) Recombinant Pex1-FLAG and His-Pex6 purified as a stoichiometric complex from E. coli. Pex1-FLAG and His-Pex6 were co-expressed in BL21* E. coli and subsequently purified by Ni-NTA agarose, anti-FLAG affinity resin, and size exclusion chromatography. The Superose 6 elution profile and SDS-PAGE analysis of the fractions show that the main peak contains both Pex1-FLAG and His-Pex6, and a minor peak represents a smaller homo-oligomer of Pex1-FLAG.
Using an affinity purification of Pex1-FLAG or Pex6-FLAG tagged at their genomic locus in S. cerevisiae, we verified that Pex1 and Pex6 interact in vivo in an ATP-dependent manner (Figure 1B). Both Pex1 and Pex6 co-immunoprecipitated with their FLAG-tagged counterpart when ATP or ATPγS was present. However, in the absence of nucleotide the association of Pex1 with Pex6-FLAG was substantially diminished. The untagged protein was identified by SDS-PAGE based on the size difference between tagged and untagged versions, and its identity confirmed by mass spectrometry (data not shown).
To further interrogate the interaction between Pex1 and Pex6, the recombinant proteins were co-expressed and co-purified from E. coli. A heteromeric complex of Pex1-FLAG and His-Pex6 was tandem-affinity purified using Ni-NTA agarose and α-FLAG affinity resin. Size-exclusion chromatography on the eluate resulted in two peaks: the most abundant and larger species contained both Pex1-FLAG and His-Pex6, while the smaller second peak contained mostly Pex1-FLAG (Figure 1C).
The purified Pex1-FLAG/His-Pex6 complex showed robust ATP hydrolysis with a Km of 0.7 mM ATP and a Vmax of 6700 ATP/hexamer/min (Figure 2A). The smaller size-exclusion peak for Pex1-FLAG, which is consistent with the previously reported homo-oligomer of Pex1 [41, 43], contained less than 10% of the ATPase activity of the Pex1/Pex6 complex. Since the two peaks were incompletely resolved by size-exclusion chromatography, we attribute the activity in the Pex1-FLAG peak to residual contamination by the Pex1/Pex6 complex and conclude that the Pex1 homo-oligomer has minimal ATP-hydrolysis activity.
Figure 2.

A) The recombinant Pex1-FLAG/His-Pex6 complex is an active ATPase with a Km of 0.7 mM ATP and Vmax of 6700 ATP/hexamer/min.
B) The cytosolic domain of Pex15, tPex15, inhibits the ATPase activity of Pex1-FLAG/His-Pex6 with an apparent KD of 185 nM.
The observed ATPase rate for the isolated Pex1/Pex6 complex is considerable, and whether Pex1/Pex6 would sustain such high ATP turnover in vivo, especially in the absence of substrate, is unclear. It is conceivable that interacting proteins modulate this ATPase rate in the cell. To test the potential effect of Pex15, the anchor for Pex1/Pex6 at the peroxisome membrane, we purified its cytoplasmic domain (tPex15) and measured Pex1/Pex6 ATP hydrolysis in the presence of increasing tPex15 concentrations. We found that tPex15 represses the ATPase activity of Pex1/Pex6 by 87%, with an apparent Kd of 185 nM (Figure 2B). Previous studies showed that Pex15 binds the N-terminal domain and D1 domain of Pex6 [40, 47]. In a heterohexamer with multiple Pex6 subunits one would thus expect multiple Pex15 binding sites. However, we observed no indication for cooperative binding (n = 0.9) or binding to several non-equivalent sites. A monomer of Pex15 might therefore inhibit the entire Pex1/Pex6 complex, either by contacting several subunits simultaneously or by binding to a single subunit that affects coordinated ATP hydrolysis in the hexamer.
Having confirmed that the recombinant Pex1/Pex6 complex is an active ATPase that interacts with known in-vivo binding partners, we performed negative-stain electron microscopy to further understand its stoichiometry and architecture. The Pex1/Pex6 particles were identifiable by the two stacked hexameric rings that are a characteristic feature of Type-2 AAA+ ATPases (Figure 3A, Supplementary Figure 1 and 2). Axial views of the heterohexamer show hook-shaped extensions that give the complex a pseudo 3-fold symmetry (Figure 3A, Supplementary Figure 2). The resulting triangular shape is consistent with an alternating heterohexamer of Pex1 and Pex6 with disparate N-terminal domains. To confirm that Pex1 and Pex6 alternate within the heterohexamer, we replaced the N-terminal His tag on Pex6 with the 42-kDa maltose binding protein (MBP). In the resulting class averages, the additional density for the MBP tag was readily apparent at the apices of the Pex1/Pex6 triangular complex (Figure 3B, asterisks). Although the MBP tag was flexible, aligning the Pex1/Pex6 hexamer and averaging the variable density of the MBP tag clearly shows that the MBP tag rotates around an attachment site on the hook-shaped extension (Figure 3B, Supplementary Movie 1). Therefore, alternating subunits of Pex1 and Pex6 constitute the heterohexamer, and the N-terminal domain of Pex6 forms hook-shaped extension protrudes from the ATPase rings.
Figure 3.

A) 2D class averages from negative-stain electron microscopy of recombinant Pex1-FLAG/His-Pex6 in the presence of 3 mM ATP.
B) Three representative class averages of Pex1-FLAG/MBP-Pex6 show extra density for MBP at the apices of the heterohexamer, consistent with an attachment to the extended N-terminal domain. 2D classes were aligned and averaged to show the flexible attachment of the MBP tag to the N-terminal extensions of Pex6 in the heterohexamer.
C) The 3D reconstruction of the Pex1-FLAG/His-Pex6 heterohexamer at 23-Å resolution. D1 and D2 mark the individual ATPase rings. The top view depicts the D1 ring and NTDs (left), and the bottom view shows the D2 ring (right). In the top view, the hash marks delineate the “dimer” interfaces in the “trimer of dimer” arrangement.
D) The density of truncated NSF (EMDB: 2041) constituting only the D1 and D2 domains was docked into the 3D reconstruction of Pex1/Pex6 to emphasize the density of the Pex1/Pex6 N-terminal domains.
E) The Pex1/Pex6 D2 ATPase ring exhibits the canonical architecture of the small α-helical AAA+ subdomain interacting with the large AAA+ subdomain of the counter-clockwise-neighboring subunit. Shown are homology models for the Pex1 and Pex6 D2 domains docked into the density of the Pex1/Pex6 D2 ATPase ring.
A three-dimensional (3D) reconstruction of Pex1-FLAG/His-Pex6 complex was obtained at 23 Å resolution without imposing any symmetry (Figure 3C). In this reconstruction, the D1 and D2 ATPase domains of Pex1 and Pex6 form two stacked rings (Figure 3C, side view) with aligned axial pores (Figure 3C, top and bottom views). The EM density of truncated NSF comprising only the D1 and D2 domains [48] matches the Pex1 and Pex6 D1 and D2 rings in size and circumference (Figure 3D), illustrating that the density above and equatorial to this barrel represents the N-terminal domains. Homology models of Pex1 and Pex6 D2 ATPase domains aligned to the structure of p97 fit well within the D2 ATPase ring, demonstrating the canonical position of the small α-helical AAA+ subdomain interacting with the large AAA+ subdomain of the neighboring subunit (Figure 3E). We also observed connecting density between the D2 large AAA+ subdomain and the D1 ring above, whose individual ATPase domains are more difficult to define due to the extensive interaction with the N-terminal domains.
The N-terminal domains divide the hexamer into a “trimer of dimers” (Figure 3C, hash marks in top view). Within each “dimer”, there are interactions between the N-terminal domains, whereas these domains are separated by a large gap from the neighboring subunits in the ring. Therefore, only the D1 and D2 ATPase domains form the interface between the “dimers”. The most striking feature of the N-terminal domains is the hook-shaped extension that descends from above the D1 ring to make substantial co-planar contacts with the D1 ATPase domain of the same subunit (Figure 3C, side view). The localization of the N-terminal MBP tag on Pex6 in the 2D images illustrates that this extension is most likely the N-terminal domain of Pex6. A distinct globular moiety above the adjacent Pex1 ATPase subunit is most likely part of the Pex1 N-terminal domain. These observations are summarized by a model for the localization of Pex1 and Pex6 within the 3D reconstruction (Figure 4). The 23-Å resolution of the reconstruction does not allow an exact delineation between the globular N-terminal domain of Pex1 and the interacting top portion of the hook-shaped N-terminal domain of Pex6.
Figure 4.
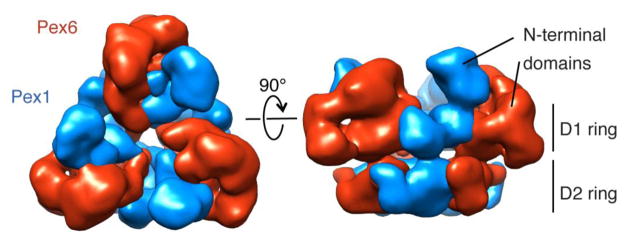
A) A proposed model showing the outlines for Pex1 and Pex6 in the segmented heterohexamer.
Our model suggests that the interface between Pex1 and its clockwise Pex6 neighbor primarily relies on the Pex1 ATP binding pockets, as the respective N-terminal domains are separated by a large gap (Figure 3C, hash marks in top view). Interestingly, Pex1 contains a well-conserved Walker A nucleotide-binding motif not only in D2, but also in D1, allowing the nucleotide-dependent stabilization of the Pex1-Pex6 interface. In contrast, the D1 ATP-binding pocket at the other subunit interface is formed by the degenerated Walker A and Walker B motifs of Pex6’s D1 domain and the poorly conserved R-finger of Pex1’s D1 domain. This interface between Pex6 and its clockwise Pex1 neighbor is buttressed by the contacting N-terminal domains, which may reinforce subunit interactions in the absence of a well-formed D1 nucleotide-binding pocket. In support of this model, we found that a mutation of the conserved Walker-A lysine in the D1 domain of Pex1, but not Pex6, dramatically affected the recovery of fully assembled recombinant hexamer (Supplementary Figure 3). Compared to the ATPase rate of wild-type Pex1/Pex6, this Pex1 D1 Walker A mutant hexamer retained 70% activity. Due to potential defects in assembly and stability of this mutant hexamer, the activity represents a lower bound and could be even closer to the wild-type value. Based on these findings we conclude that the D1 domain of Pex1 is important for heterohexamer assembly, but not for nucleotide hydrolysis.
Previous studies of NSF observed dramatic conformational changes in the position of the N-terminal domain depending on the nucleotide state from an “up” position above the D1 ring to a “down” position co-planar with the D1 ring. The “up” position has also been observed in the presence of substrate and adaptor proteins, leading to the proposal that this nucleotide-dependent conformational change exerts a mechanical force on the substrate [35, 49]. For p97, the N-terminal domain is also highly mobile, changing its position in a nucleotide-dependent manner from an equatorial arrangement alongside the ATPase barrel to above the D1 ring. This latter “up” orientation appears to be stabilized by binding of some of the p97 adaptor proteins [50–53]. To determine whether the Pex1/Pex6 complex undergoes similar conformational changes during ATP hydrolysis, we performed negative-stain electron microscopy in the presence of saturating levels of ATPγS or ADP. There were no major rearrangements of the N-terminal domains between the ATP, ATPγS, and ADP-bound states (Figure 5). However, we do observe several changes between the ADP- and ATPγS-bound structures, including a rotation of the D2 ring relative to the D1 ring (Supplementary Movie 2), rearrangements of the D1 and D2 nucleotide binding sites, (Figure 5), and an increase in density in the central pore just above the D2 ring for the ATPγS-bound complex. The 3D reconstruction of the complex with saturating ATP shows a relative orientation of the D1 and D2 rings that resembles the ATPγS state, but the nucleotide binding pockets are less defined, indicative of structurally varying nucleotide states occurring during hydrolysis. The observed asymmetry in D2 ring suggests the presence of different nucleotide states in the hexamer and that Pex1/Pex6 hydrolyzes ATP by a non-concerted mechanism, similar to related ATPases [30, 31, 54, 55].
Figure 5.

A comparison of the 3D reconstructions for ADP-, ATPγS-, and ATP-bound Pex1/Pex6 heterohexamer at 17, 23, and 23 Å resolution, respectively. A) Side-views of Pex1/Pex6 reveal no large changes in the N-terminal domain conformation. B) Vertical cross-sections at the axial pore show increased density in the D2-ring pore for the ATPγS-bound state. C) Horizontal cross-sections through the D1 ATPase domains. D) Horizontal cross-section through the D2 ATPase domains.
A central question in understanding the detailed mechanisms of heterohexameric AAA+ enzymes is how distinct subunits contribute to motor function and coordinate their activities. To determine how Pex1 and Pex6 ATPase activity is coordinated in the hexamer, we introduced Walker B mutations in the D2 domains of Pex1 and Pex6. A glutamate-to-glutamine mutation in the Walker B motif hinders the coordination of a catalytic water molecule and prevents ATP hydrolysis, but not ATP binding [56]. In the Pex1 and Pex6 D2 Walker B double mutant, we observed no ATP-hydrolysis activity (Figure 6A, Pex1-WB/Pex6-WB). Given the robust ATPase rate we observed for the Pex1 D1 Walker-A mutant and the endogenous deleterious substitutions in the D1 active sites of wild-type Pex1 and Pex6, this result is consistent with the D2 ATPase domains accounting for all of the ATP hydrolysis in Pex1/Pex6.
Figure 6.

A) A comparison of ATPase activities for wild-type Pex1-FLAG/His-Pex6 and its variants with mutations in the D2 domains. The schematic below the graph indicates the D2 mutations made in the context of the heterohexamer, with Pex1 represented in light gray and Pex6 in dark gray. The ATP-binding sites are colored green when wild type and red when mutated. WB: Mutation of Glu to Asn in the Walker B motif, leading to inhibition of ATP hydrolysis. RK: Mutation of Arg to Lys in the R-finger, a residue contributed to the neighboring ATP-binding site, which inhibits ATP hydrolysis when mutated.
B) The cytoplasmic domain of Pex15 inhibits the ATPase activity of the wild-type Pex1/Pex6 complex, but not the Pex1-WB/Pex6 mutant.
C) The cytoplasmic domain of Pex15 co-immunoprecipitates with both wild-type and the D2 Walker B mutant Pex1/Pex6 complex. The immunoprecipitation was performed on purified proteins using the FLAG tag on Pex1.
Interestingly, when only Pex6 D2 contained the Walker B mutation, the complex was also completely ATPase deficient, despite the presence of three wild-type ATPase pockets in Pex1 (Figure 6A, Pex1/Pex6-WB). On the other hand, when Pex1 D2 contained the Walker B mutation and only the Pex6 D2 domains were functional, we observed 20% of Pex1/Pex6 wild-type ATPase activity (Figure 6A, Pex1-WB/Pex6). Thus, Pex1 is completely inhibited when Pex6 is trapped in a permanently ATP-bound state, whereas Pex6 maintains some hydrolysis activity despite an ATP-bound state in Pex1. Consequently, there appears to be a stronger coordination of Pex1 with Pex6, than of Pex6 with Pex1.
The mitochondrial mAAA protease, consisting of alternating Yta10 and Yta12 subunits, exhibits a similar behavior in subunit communication. A Walker B mutation in Yta12 completely abrogates ATPase activity, while the same mutation in Yta10 yields ~30% of wild-type activity. Suppressor screens revealed that the loss of ATPase activity in the Yta12 Walker B mutant could be overcome through a mutation in the Yta10 R-finger [44], suggesting that Yta10 is inhibited through its R-finger by the ATP-bound state of Yta12. To determine whether a similar mechanism is used for communication between Pex1 and Pex6, we mutated the D2 R-finger of Pex1 in addition to the D2 Walker B motif in Pex6 (Figure 6A, Pex1-WB/Pex6-RK), and vice versa. Unlike Yta10/Yta12, the R-finger mutation did not increase the ATPase activity, but rather further abrogated ATP hydrolysis. Therefore, the ATP-hydrolysis activity of Pex1/Pex6 may be coordinated by a different mechanism.
Intriguingly, the ATPase rate of the Pex1-WB/Pex6 mutant is very similar to the rate observed for wild-type Pex1/Pex6 at saturating concentrations of tPex15. To determine whether tPex15 binding to Pex1/Pex6 specifically inhibits ATP hydrolysis in Pex1, we tested for any additional inhibitory effects of tPex15 on the Pex1-WB/Pex6 mutant. Unlike for wild-type Pex1/Pex6, we did not observe any inhibition of Pex1-WB/Pex6 by tPex15 (Figure 6B). Since tPex15 co-immunoprecipitates with both wild-type Pex1/Pex6 and Pex1-WB/Pex6-WB (Figure 6C), it is unlikely that the lack of inhibition is due to a defect in tPex15 binding. Therefore, we hypothesize that tPex15 inhibits the ATPase activity of Pex1/Pex6 by affecting hydrolysis in the Pex1 subunits.
Discussion
Previous studies have conclusively demonstrated that Pex1 and Pex6 form a complex in vivo, and that the formation of this complex is ATP dependent [39–42]. Here, we show that the Pex1/Pex6 complex is a heterohexamer composed of alternating subunits (1-6-1-6-1-6). This architecture dictates that the nucleotide binding sites are themselves heteromeric, forming at the interfaces between Pex1 and Pex6 within the complex. By utilizing mutations in the signature ATPase motifs, we found that all of the ATPase activity in the Pex1/Pex6 heterohexamer derives from the D2 domains, while nucleotide binding in the D1 domain is required for efficient complex assembly.
Interestingly, we found that the ATP-hydrolysis activity of Pex1 is strongly coordinated with that of Pex6; mutations preventing hydrolysis by Pex6 completely inhibit hydrolysis by Pex1. This inhibition of one subunit by a Walker B mutation in an adjacent subunit was previously observed for the mitochondrial mAAA protease Yta10/Yta12. However, unlike the mitochondrial ATPases, the inhibition of Pex1 by Pex6-WB could not be overcome by mutations in the Pex1 R-finger, indicating that the coordination of Pex1 and Pex6 is mediated through a different mechanism than the one utilized within the Yta10/Yta12 complex.
We observed approximately 20% of wild-type activity for the Pex1-WB/Pex6 complex. However, we do not assume that Pex6 contributes only 20% to the total ATPase activity of the wild-type complex. The strong coupling of ATP hydrolysis of Pex1 with that of Pex6 suggests that Pex6 fires prior to Pex1, which would lead to a balanced contribution of subunits to the overall ATPase activity of the complex. The hydrolysis activity of the Pex1-WB/Pex6 mutant may be limited to 20% for two main reasons. One could be the partial inhibition of Pex6 by a hydrolysis-dead Pex1 neighbor, and the other may be related to the total number of ATP-loadable sites in the Pex1/Pex6 hexamer. Several previous studies on related AAA+ enzymes suggest that a closed ring topology allows only four of the six nucleotide-binding sites to be occupied [31, 54, 55]. Indeed, we observed asymmetry in the nucleotide binding sites in saturating conditions of ATP, ATPγS, and ADP. Assuming a similar behavior for Pex1-WB/Pex6, the three hydrolysis-deficient Pex1 D2 sites would preferably be ATP-bound, leaving only a single Pex6 site in the D2 ring available for ATP binding and hydrolysis.
We found that binding of Pex1/Pex6 to the cytoplasmic domain of its membrane anchor Pex15 inhibits ATP hydrolysis by up to 87% at saturating conditions, leading to a residual ATPase activity that is similar to that of Pex1-WB/Pex6. These data suggest that Pex15 inhibits the ATPase activity of Pex1, and not Pex6. Previous studies have shown that Pex15 and its human homolog Pex26 interact directly with the N-terminal domain and D1 domain of Pex6 [40, 47], and only indirectly with Pex1. Pex15 binding could allosterically affect Pex1 while directly binding only to Pex6, but, given the observed proximity of the Pex6 N-terminal domain and the small α-helical AAA+ subdomain of the Pex1 D2 ATPase, it is also possible that Pex15 could simultaneously interact with the Pex6 N-terminal domain and the Pex1 D2 ATPase.
In vivo, Pex1 and Pex6 are distributed between the cytoplasm and the peroxisome membrane, to which they are recruited by Pex15 [47, 57]. Our data indicate that the heterohexamer localized at the peroxisome membrane and bound to Pex15 has a lower ATP-hydrolysis rate due to Pex15-mediated inhibition. We hypothesize that this regulated inhibition of Pex1/Pex6 by Pex15 may allow for substrate engagement at the peroxisomal membrane and subsequent accelerated processing after Pex1/Pex6 dissociates from Pex15 and the membrane.
Surprisingly, we observed no major nucleotide-dependent conformational changes in the N-terminal domains of Pex1/Pex6 akin to those reported for p97 and NSF. Large variations in the position of the N-terminal domains have prompted speculations that p97 and NSF exert mechanical force on externally-bound substrates through hydrolysis-driven conformational changes in those domains, rather than using a mechanism of substrate threading through the central pore. While we observed no changes in the position of the N-terminal domains, we cannot rule out that the engagement of an ubiquitinated Pex5 substrate or the presence of an adaptor protein such as Pex15 or the de-ubiquitinase Ubp15 might stabilize such alternative N-terminal domain conformations in Pex1/Pex6. An alternative model proposes that Type-2 AAA+ ATPases unfold substrates via translocation through the central pore [32, 58–60]. For Pex1/Pex6, the D1 ring is not only ATPase-deficient, but also lacks well-conserved pore loops to engage and translocate a substrate in response to hydrolysis-driven conformational changes. The substrate would therefore have to enter far enough into the central pore of Pex1/Pex6 to contact the pore loops of the D2 ring. In fact, for several other Type-2 AAA+ ATPases that process substrates through the central pore the D2 domain has been found to be more important than D1 in driving translocation [58, 60].
Substrate engagement and processing by Pex1/Pex6 still remain unresolved. The only known substrate of Pex1/Pex6 is the ubiquitinated cargo receptor Pex5, which Pex1/Pex6 extracts from peroxisomal membranes in an ATP-dependent manner. Ubiquitination of Pex5 is required for its ATP-dependent extraction [18] and could possibly mediate the interaction with Pex1/Pex6, either directly or through adaptor proteins. A potential ubiquitin-binding site within the complex is the N-terminal domain of Pex1, which shares structural similarity with the ubiquitin-interacting motif in Ufd1 and the N-domain of p97 [61, 62]. However, besides this structural similarity, there is no direct evidence of an ubiquitin-binding site within Pex1 or Pex6, and indirect binding through an adaptor protein such as Ubp15 remains another possibility.
Pex1 and Pex6, as well as their substrates and interacting proteins, are conserved from yeast to humans. We therefore expect that our results regarding the architecture of the Pex1/Pex6 heterohexamer and the contributions of individual ATPase domains to nucleotide hydrolysis will be directly applicable to the human system. To fully understand the effects of Pex1 and Pex6 mutations and eventually tackle new therapeutic approaches for peroxisome biogenesis disorders, future work will have to address how Pex1/Pex6 engages with adaptor proteins and processes its substrates. In addition, increased understanding of the Pex1 and Pex6 mechanism may help elucidate the common principles of the related AAA+ ATPases NSF and p97, which are essential for cell viability and therefore less tractable for mutational studies in vivo.
Materials and Methods
Genomic tagging of Pex1 and Pex6
Pex1 and Pex6 were tagged with a C-terminal 3xFLAG tag at their genomic locus using a pFA6A-3xFLAG-KANMX6 cassette as a template. The addition of the 3xFLAG tag was confirmed by junction PCR and the up-regulation of the FLAG-tagged protein during growth on oleic acid as the sole carbon source. The genotype of the resulting strains are W303 MATa ura3-1 his3-11 trp1-1 leu2-3 leu2-112 can1-100 PEX1::PEX1-3xFLAG (KanMX) and W303 MATa ura3-1 his3-11 trp1-1 leu2-3 leu2-112 can1-100 PEX6::PEX6-3xFLAG (KanMX). The 3xFLAG tag had no effect on the localization of GFP-PTS1.
FLAG immunoprecipitation
The WT, Pex1-FLAG, and Pex6-FLAG strains were pre-cultured in YPD for 3 days. On the day of the growth, each strain was inoculated into 1 L of YPD at an OD 600 of 0.15 and grown at 30 degrees C to an OD 600 of ~1. The cultures were centrifuged at 5000 × g, and the pellets resuspended in 2 L of YNO (5 g peptone, 3 g yeast extract, 1.4 mL oleic acid, 2 mL Tween-40, 5 g KH2PO4 per liter). After growth overnight at 30 degrees C, the cultures were centrifuged at 5000 × g, washed once with water, and resuspended in 1 mL per gram of wet weight in IP buffer (60 mM HEPES pH 7.6, 50 mM NaCl, 50 mM KCl, 10% glycerol, 10 mM MgCl2, 0.5 mM EDTA) with proteasome inhibitors. The resuspended cultures were frozen and stored at −80 degrees C before lysis by a SPEX 6870 Freezer/Mill at 15 cps.
For the anti-FLAG immunoprecipitation, Triton X-100 and PMSF were added to the thawed samples to a final concentration of 1% v/v TritonX-100 and 1 mM PMSF. The Pex1-FLAG and Pex6-FLAG samples were split, diluted to equal protein concentration, and nucleotide was added to the following concentrations: 5 mM ATP with ATP regeneration buffer (0.05 mg/mL creatine phosphokinase, 5 mg/mL creatine phosphate), or 0.4 mM ATPγS, or no nucleotide. Each sample was incubated with anti-FLAG M2 affinity resin, washed several times with IP buffer with the appropriate nucleotide, and eluted with FLAG peptide. The FLAG-tagged protein and bound proteins were resolved by SDS-PAGE.
Purification of Pex1 and Pex6
Pex1-FLAG and His-Pex6 wildtype and mutant complexes were co-expressed in BL21* E. coli from the pETDuet and pCOLADuet vectors. The expression strain was grown in DYT (16 g tryptone, 10 g yeast extract, 5 g NaCl) and appropriate antibiotics at 30°C and induced at OD600=0.6–0.9 with 0.3 mM IPTG before overnight incubation at 18°C. The E. coli were harvested at 6000×g for 20 minutes at 4 degrees, and the pellet was resuspended in Ni_A buffer (25 mM HEPES pH 7.6, 100 mM NaCl, 100 mM KCl, 10% glycerol, 10 mM MgCl2, 0.5 mM EDTA, 20 mM imidazole) with benzonase, lysozyme (0.2 mg/mL), and protease inhibitors and frozen at −80°C. Cells were lysed by sonication at 90 mAmps with 15-second pulses on, 90 seconds off for a total of 120 seconds on. Cell debris and unlysed cells were pelleted at 30,000×g and the supernatant was transferred to Falcon tubes containing 5 ml of pre-washed Ni-NTA agarose. The cell lysate and agarose were incubated with gentle rocking for 1–2 hours at 4°C before the agarose was batch washed with 2×50 mL washes of Ni_A with 0.5 mM ATP. After the batch washes, the agarose was poured into a gravity flow column and washed until the flowthrough contained no protein, as judged by a Bradford assay. The bound protein was then eluted with Ni_A with 0.5 mM ATP and 500 mM imidazole, and the elution was added to re-suspended and pre-washed anti-FLAG affinity resin (Sigma) for 2 hours of batch binding at 4°C. After two hours, the anti-FLAG affinity resin was poured into a gravity flow column, and washed with 50 mL of Ni_A with 0.5 mM ATP. The bound protein was eluted with Ni_A with 0.5 mM ATP and 0.3 ug/mL FLAG peptide and concentrated on a 100 MWCO spin concentrator before snap-freezing in N2 (l). To separate the Pex1-FLAG/His-Pex6 hexamer from other oligomers, the concentrated FLAG elution was loaded on a Superose6 size exclusion column equilibrated in GF buffer (60 mM HEPES pH 7.6, 50 mM NaCl, 50 mM KCl, 10% glycerol, 10 mM MgCl2, 0.5 mM EDTA) with 0.5 mM ATP and 1 mM DTT. The concentration of protein was determined by a Bradford assay. The amino acid changes in the ATPase motifs of Pex1 and Pex6 are as follows: Pex1-Walker B D2 (E798Q), Pex1-RK D2 (R852K), Pex6-WB D2 (E832Q), Pex6-RK D2 (R889K).
For His-Pex1-FLAG/MBP-Pex6, we followed the same protocol through the elution from the Ni-NTA agarose, at which point the elution was batch bound to amylose resin rather than anti-FLAG affinity resin and eluted with Ni_A with 0.5 mM ATP and 10 mM maltose.
To purify the cytoplasmic domain of Pex15 (aa 1-327), we replaced the transmembrane domain with a FLAG-6xHis tag and, followed the same purification protocol as for Pex1/Pex6, but without ATP in the buffer.
ATPase assays
The ATPase activity of the wildtype and mutant Pex1/Pex6 complexes were monitored using an ATP/NADH coupled enzyme assay. In this assay, the regeneration of hydrolyzed ATP is coupled to the oxidation of NADH, which is measured at 340 nm [63]. 1X reaction mixture consists of (3 U ml−1 pyruvate kinase, 3 U ml−1 lactate dehydrogenase, 1 mM NADH and 7.5 mM phosphoenol pyruvate). The assays were performed in either a 96 well plate using a SpectraMAX 190 plate reader or a cuvette using an Agilent 8453 UV-Vis spectrometer over 900 seconds with 10 second time points. For saturating conditions, we used 5 mM ATP. In all of the ATPase assays, we used 5 nM Pex1/Pex6 to stay in the linear range of the assay for the entire duration of the experiment. Measurements at up to 100 nM Pex1/Pex6 showed no concentration dependence of ATPase activity and thus confirmed the stability of the hexamer at 5 nM. To monitor the effect of tPex15, Pex1/Pex6 and varying concentrations of tPex15 were pre-incubated before the addition of reaction mixture.
Negative Stain Electron Microscopy
Sample preparation
Pex1-FLAG/His-Pex6 samples were diluted to final concentration of 22 nM in GF buffer lacking glycerol, but supplemented with different nucleotides (3 mM ADP, ATP and ATPγS) and 1 mM TCEP for EM studies. The sample was incubated on ice for 5 minutes in the presence of ATP, or 15 minutes in the presence of ADP or ATPγS to allow for nucleotide exchange immediately prior to negative staining. 4 μl of sample was applied to freshly plasma-cleaned 400 mesh Cu-Rh maxtaform grids (Electron Microscopy Sciences) that had been coated with a thin layer of carbon. After incubating for 1 minute, excess protein was wicked off with a filter paper (Whatman No. 1) and the grid was immediately inverted and placed on 50 μl droplet of 2% (w/v) uranyl formate solution. After 30 s, excess stain was wicked off from the grid by touching the edge with filter paper. This staining step was repeated three times for thorough embedding of the sample, and the grids were air dried after the last blotting step. A majority of the particles exhibited a preferential orientation on the carbon support using this staining method, yielding mostly end-on projections. To overcome this issue, we pretreated the plasma-cleaned grids by placing 5 μl of 0.1 % (w/v) poly-L-lysine hydrobromide (Polysciences) onto the carbon surface for 90 s, followed by two washes with 10μl drops of water. After the grids dried, Pex1/6 samples were applied and stained as described above. This treatment provided additional lateral views of the complexes. His-Pex1-FLAG/MBP-Pex6 grids were prepared in similar manner as described above.
Data acquisition
Data for all the samples were acquired on a Tecnai Spirit (FEI) transmission electron microscope, operating at 120 keV, using the Leginon automated data acquisition system [64]. Micrographs were acquired at a nominal magnification of 52,000X on an F416 CMOS 4K×4K camera (TVIPS) with a pixel size of 2.05 Å/pixel at the specimen level using an electron dose of 20 electrons/Å2, with a defocus range from 0.3 μm to 1.5 μm.
Image Processing
694, 825, and 1,340 micrographs were collected for Pex1-FLAG/His-Pex6 in presence of 3mM ADP, ATP, and ATPγS, respectively. The Appion image processing pipeline [65] was used for processing of micrographs and initial 2D analyses. CTFFindv3 [66] was used for determining the contrast transfer function (CTF) of each micrograph, and particles were selected from micrographs using Difference of Gaussians (DoG)-based automated particle picker [67]. Phases for each micrograph were corrected using EMAN [68] and particles were extracted using a 160 × 160 pixel box. Individual particles were normalized by eliminating pixels with values above or below 4.5σ of the mean pixel value using the normalization function in the XMIPP package [69]. For faster computation the particles were binned by a factor of two. Initial stacks of 86,824, 74,533 and 65,904 particles for data collected in presence of 3mM ADP, ATP, and ATPγS, respectively, were obtained.
Each of the extracted particle datasets was subjected to five rounds of iterative multivariate statistical analysis (MSA) [70], and multi-reference alignment (MRA) in Appion to remove any erroneously picked non-particle features and aggregates. This resulted in final stacks containing 57,538, 53,837 and 41,476 particles for 3 mM ADP, ATP, and ATPγS datasets, respectively. Reference-free 2D alignment and classification were done for each dataset using the ISAC [71] program within the EMAN2/SPARX software package [72, 73].
An initial 3D model was generated from the ISAC 2D class averages using the “e2initialmodel.py” function in the EMAN2 software package, with c3 symmetry imposed. This initial model was low-pass filtered to 60Å resolution and used as a starting point for 3D classification of each particle dataset into five classes using Relion [74]. After 25 iterations of 3D classification, 23,989, 11,946, and 10,371 particles (for the ADP, ATP, and ATPγS datasets, respectively) belonging to well-resolved 3D class averages were used for further refinement by projection matching in Relion. The reported resolutions of the final refinements by Gold Standard Fourier Shell Correlation at a cutoff of 0.143 were 17.26, 23.4, and 23.4 Å for the 3 mM ADP, ATP, and ATPγS datasets, respectively (Supplementary Figure 2).
870 micrographs were acquired for His-Pex1-FLAG/MBP-Pex6 sample, from which 67,216 particles were extracted using a box size of 192 pixels. These particles had preferred orientation on the carbon support, even after poly-L-lysine treatment, yielding only end-on projections. This particle stack was further binned down by a factor of two and reference-free 2D classification was performed using ISAC as described earlier.
Homology modeling
Homology models for the D1 and D2 ATPase domains of Saccharomyces cerevisiae Pex1 and Pex6 were obtained using the Phyre2 server [75]. The p97 crystal structure (PDB ID: 3CF1 [76]) was used as a template for model building. It was fitted into individual ATPase densities, and the appropriate homology models were aligned (Figure 3E). All rigid-body fitting of atomic models into EM density, as well as generation of figures and movies depicting 3D reconstructions were performed using UCSF Chimera [77].
The 3D density maps for the ADP, ATP, and ATPγS reconstructions have been deposited at the Electron Microscopy Data Bank (EMDB), with reference codes EMD-6253, EMD-6255, and EMD-6254, respectively.
Supplementary Material
Supplementary Figure 1. Raw micrographs of Pex1/Pex6 in the presence of different nucleotides as labeled. On the left are micrographs showing end-on orientation of particles on carbon surface. On the right are micrographs from poly-L-lysine pre-treated grids, where the particles primarily adsorb to the carbon surface on their sides.
Supplementary Figure 2. A) Forward projections from negative stain reconstructions of Pex1/Pex6 complex in presence of ADP, ATPγS, and ATP in the upper panels, with matching reference-free 2D class averages shown in the bottom panels.
B) Gold Standard FSC plots for the 3D reconstructions of Pex1/Pex6, reporting resolutions of ~17Å for the ADP-bound structure, and ~23Å for ATPγS- and ATP-bound structures, using a 0.143 cutoff.
C) Angular distribution of particles contributing to 3D reconstructions, with the diameter of each sphere related to the number of particles at that Euler angle (larger spheres indicate more particles.
Supplementary Figure 3. A) The D1 Walker A mutants of Pex-FLAG/His-Pex6 were purified by a sequential Ni-NTA affinity purification and α-FLAG affinity resin. The presented images of a Coomassie-stained SDS PAGE shows the Ni-NTA elution, FLAG flowthrough, and FLAG elution for each mutant. The two mutant hexamers were purified side by side, with similar wash and elution volumes.
B) The Pex1-WA1/Pex6 mutant has an assembly defect, resulting in a depleted peak at the expected elution volume for intact Pex1/Pex6 complex (~12.5 mL, compare to Pex1/Pex6-WA1 mutant).
Supplementary Movie 1. Flexible attachment of MBP to the amino terminal end of Pex6.
Supplementary Movie 2. Comparison between ADP-, ATPγS-, and ATP-bound structures of Pex1/Pex6 showing the structural variations between the different nucleotide-bound states.
Highlights.
Peroxisome biogenesis depends on the activity of the Pex1/Pex6 AAA+ ATPase
EM reveals that Pex1/Pex6 is a heterohexamer with alternating subunits
Pex1 and Pex6 are highly coordinated in their ATP-hydrolysis activity
The membrane anchor Pex15 inhibits the Pex1/Pex6 ATPase activity
Insights into the motor frequently mutated in human peroxisome biogenesis disorders
Acknowledgments
We thank the members of the Martin lab for helpful discussion. B.M.G. acknowledges support from the Miller Institute for Basic Research in Science, University of California, Berkeley. A.M. acknowledges support from the Searle Scholars Program, start-up funds from the University of California Berkeley Molecular and Cell Biology Department, the US National Institutes of Health (grant R01-GM094497), the US National Science Foundation CAREER Program (NSF-MCB- 1150288). This work was also supported by the Damon Runyon Cancer Research Foundation (DFS-#07-13), the Pew Scholars program, the Searle Scholars program, and NIH grant DP2 EB020402-01 to G.C.L.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tabak HF, Braakman I, van der Zand A. Peroxisome formation and maintenance are dependent on the endoplasmic reticulum. Annu Rev Biochem. 2013;82:723–44. doi: 10.1146/annurev-biochem-081111-125123. [DOI] [PubMed] [Google Scholar]
- 2.Weller S, Gould SJ, Valle D. Peroxisome biogenesis disorders. Annu Rev Genomics Hum Genet. 2003;4:165–211. doi: 10.1146/annurev.genom.4.070802.110424. [DOI] [PubMed] [Google Scholar]
- 3.Schluter A, Fourcade S, Ripp R, Mandel JL, Poch O, Pujol A. The evolutionary origin of peroxisomes: an ER-peroxisome connection. Mol Biol Evol. 2006;23:838–45. doi: 10.1093/molbev/msj103. [DOI] [PubMed] [Google Scholar]
- 4.Hoepfner D, Schildknegt D, Braakman I, Philippsen P, Tabak HF. Contribution of the endoplasmic reticulum to peroxisome formation. Cell. 2005;122:85–95. doi: 10.1016/j.cell.2005.04.025. [DOI] [PubMed] [Google Scholar]
- 5.Lam SK, Yoda N, Schekman R. A vesicle carrier that mediates peroxisome protein traffic from the endoplasmic reticulum. Proc Natl Acad Sci U S A. 2010;107:21523–8. doi: 10.1073/pnas.1013397107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van der Zand A, Gent J, Braakman I, Tabak HF. Biochemically distinct vesicles from the endoplasmic reticulum fuse to form peroxisomes. Cell. 2012;149:397–409. doi: 10.1016/j.cell.2012.01.054. [DOI] [PubMed] [Google Scholar]
- 7.Lazarow PB, Fujiki Y. Biogenesis of peroxisomes. Annu Rev Cell Biol. 1985;1:489–530. doi: 10.1146/annurev.cb.01.110185.002421. [DOI] [PubMed] [Google Scholar]
- 8.Gould SJ, Keller GA, Hosken N, Wilkinson J, Subramani S. A conserved tripeptide sorts proteins to peroxisomes. J Cell Biol. 1989;108:1657–64. doi: 10.1083/jcb.108.5.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Subramani S. Protein import into peroxisomes and biogenesis of the organelle. Annu Rev Cell Biol. 1993;9:445–78. doi: 10.1146/annurev.cb.09.110193.002305. [DOI] [PubMed] [Google Scholar]
- 10.Brocard C, Kragler F, Simon MM, Schuster T, Hartig A. The tetratricopeptide repeat-domain of the PAS10 protein of Saccharomyces cerevisiae is essential for binding the peroxisomal targeting signal-SKL. Biochem Biophys Res Commun. 1994;204:1016–22. doi: 10.1006/bbrc.1994.2564. [DOI] [PubMed] [Google Scholar]
- 11.Terlecky SR, Nuttley WM, McCollum D, Sock E, Subramani S. The Pichia pastoris peroxisomal protein PAS8p is the receptor for the C-terminal tripeptide peroxisomal targeting signal. EMBO J. 1995;14:3627–34. doi: 10.1002/j.1460-2075.1995.tb00032.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rehling P, Marzioch M, Niesen F, Wittke E, Veenhuis M, Kunau WH. The import receptor for the peroxisomal targeting signal 2 (PTS2) in Saccharomyces cerevisiae is encoded by the PAS7 gene. EMBO J. 1996;15:2901–13. [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang JW, Lazarow PB. Peb1p (Pas7p) is an intraperoxisomal receptor for the NH2-terminal, type 2, peroxisomal targeting sequence of thiolase: Peb1p itself is targeted to peroxisomes by an NH2-terminal peptide. J Cell Biol. 1996;132:325–34. doi: 10.1083/jcb.132.3.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Purdue PE, Yang X, Lazarow PB. Pex18p and Pex21p, a novel pair of related peroxins essential for peroxisomal targeting by the PTS2 pathway. J Cell Biol. 1998;143:1859–69. doi: 10.1083/jcb.143.7.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Albertini M, Rehling P, Erdmann R, Girzalsky W, Kiel JA, Veenhuis M, et al. Pex14p, a peroxisomal membrane protein binding both receptors of the two PTS-dependent import pathways. Cell. 1997;89:83–92. doi: 10.1016/s0092-8674(00)80185-3. [DOI] [PubMed] [Google Scholar]
- 16.Girzalsky W, Rehling P, Stein K, Kipper J, Blank L, Kunau WH, et al. Involvement of Pex13p in Pex14p localization and peroxisomal targeting signal 2-dependent protein import into peroxisomes. J Cell Biol. 1999;144:1151–62. doi: 10.1083/jcb.144.6.1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stein K, Schell-Steven A, Erdmann R, Rottensteiner H. Interactions of Pex7p and Pex18p/Pex21p with the peroxisomal docking machinery: implications for the first steps in PTS2 protein import. Mol Cell Biol. 2002;22:6056–69. doi: 10.1128/MCB.22.17.6056-6069.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Platta HW, El Magraoui F, Schlee D, Grunau S, Girzalsky W, Erdmann R. Ubiquitination of the peroxisomal import receptor Pex5p is required for its recycling. J Cell Biol. 2007;177:197–204. doi: 10.1083/jcb.200611012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Platta HW, El Magraoui F, Baumer BE, Schlee D, Girzalsky W, Erdmann R. Pex2 and pex12 function as protein-ubiquitin ligases in peroxisomal protein import. Mol Cell Biol. 2009;29:5505–16. doi: 10.1128/MCB.00388-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Platta HW, Hagen S, Reidick C, Erdmann R. The peroxisomal receptor dislocation pathway: to the exportomer and beyond. Biochimie. 2014;98:16–28. doi: 10.1016/j.biochi.2013.12.009. [DOI] [PubMed] [Google Scholar]
- 21.van der Klei IJ, Hilbrands RE, Kiel JA, Rasmussen SW, Cregg JM, Veenhuis M. The ubiquitin-conjugating enzyme Pex4p of Hansenula polymorpha is required for efficient functioning of the PTS1 import machinery. EMBO J. 1998;17:3608–18. doi: 10.1093/emboj/17.13.3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Williams C, van den Berg M, Geers E, Distel B. Pex10p functions as an E3 ligase for the Ubc4p-dependent ubiquitination of Pex5p. Biochem Biophys Res Commun. 2008;374:620–4. doi: 10.1016/j.bbrc.2008.07.054. [DOI] [PubMed] [Google Scholar]
- 23.Williams C, van den Berg M, Sprenger RR, Distel B. A conserved cysteine is essential for Pex4p-dependent ubiquitination of the peroxisomal import receptor Pex5p. J Biol Chem. 2007;282:22534–43. doi: 10.1074/jbc.M702038200. [DOI] [PubMed] [Google Scholar]
- 24.Platta HW, Grunau S, Rosenkranz K, Girzalsky W, Erdmann R. Functional role of the AAA peroxins in dislocation of the cycling PTS1 receptor back to the cytosol. Nat Cell Biol. 2005;7:817–22. doi: 10.1038/ncb1281. [DOI] [PubMed] [Google Scholar]
- 25.Kragt A, Voorn-Brouwer T, van den Berg M, Distel B. The Saccharomyces cerevisiae peroxisomal import receptor Pex5p is monoubiquitinated in wild type cells. J Biol Chem. 2005;280:7867–74. doi: 10.1074/jbc.M413553200. [DOI] [PubMed] [Google Scholar]
- 26.Platta HW, Girzalsky W, Erdmann R. Ubiquitination of the peroxisomal import receptor Pex5p. Biochem J. 2004;384:37–45. doi: 10.1042/BJ20040572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kiel JA, Emmrich K, Meyer HE, Kunau WH. Ubiquitination of the peroxisomal targeting signal type 1 receptor, Pex5p, suggests the presence of a quality control mechanism during peroxisomal matrix protein import. J Biol Chem. 2005;280:1921–30. doi: 10.1074/jbc.M403632200. [DOI] [PubMed] [Google Scholar]
- 28.Geisbrecht BV, Collins CS, Reuber BE, Gould SJ. Disruption of a PEX1-PEX6 interaction is the most common cause of the neurologic disorders Zellweger syndrome, neonatal adrenoleukodystrophy, and infantile Refsum disease. Proc Natl Acad Sci U S A. 1998;95:8630–5. doi: 10.1073/pnas.95.15.8630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reuber BE, Germain-Lee E, Collins CS, Morrell JC, Ameritunga R, Moser HW, et al. Mutations in PEX1 are the most common cause of peroxisome biogenesis disorders. Nat Genet. 1997;17:445–8. doi: 10.1038/ng1297-445. [DOI] [PubMed] [Google Scholar]
- 30.Martin A, Baker TA, Sauer RT. Rebuilt AAA + motors reveal operating principles for ATP-fuelled machines. Nature. 2005;437:1115–20. doi: 10.1038/nature04031. [DOI] [PubMed] [Google Scholar]
- 31.Carroni M, Kummer E, Oguchi Y, Wendler P, Clare DK, Sinning I, et al. Head-to-tail interactions of the coiled-coil domains regulate ClpB activity and cooperation with Hsp70 in protein disaggregation. Elife. 2014;3:e02481. doi: 10.7554/eLife.02481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wendler P, Shorter J, Snead D, Plisson C, Clare DK, Lindquist S, et al. Motor mechanism for protein threading through Hsp104. Mol Cell. 2009;34:81–92. doi: 10.1016/j.molcel.2009.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Olivares AO, Nager AR, Iosefson O, Sauer RT, Baker TA. Mechanochemical basis of protein degradation by a double-ring AAA+ machine. Nat Struct Mol Biol. 2014;21:871–5. doi: 10.1038/nsmb.2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brunger AT, DeLaBarre B. NSF and p97/VCP: similar at first, different at last. FEBS Lett. 2003;555:126–33. doi: 10.1016/s0014-5793(03)01107-4. [DOI] [PubMed] [Google Scholar]
- 35.Chang LF, Chen S, Liu CC, Pan X, Jiang J, Bai XC, et al. Structural characterization of full-length NSF and 20S particles. Nat Struct Mol Biol. 2012;19:268–75. doi: 10.1038/nsmb.2237. [DOI] [PubMed] [Google Scholar]
- 36.Weibezahn J, Tessarz P, Schlieker C, Zahn R, Maglica Z, Lee S, et al. Thermotolerance requires refolding of aggregated proteins by substrate translocation through the central pore of ClpB. Cell. 2004;119:653–65. doi: 10.1016/j.cell.2004.11.027. [DOI] [PubMed] [Google Scholar]
- 37.Titorenko VI, Chan H, Rachubinski RA. Fusion of small peroxisomal vesicles in vitro reconstructs an early step in the in vivo multistep peroxisome assembly pathway of Yarrowia lipolytica. J Cell Biol. 2000;148:29–44. doi: 10.1083/jcb.148.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Titorenko VI, Rachubinski RA. Peroxisomal membrane fusion requires two AAA family ATPases, Pex1p and Pex6p. J Cell Biol. 2000;150:881–6. doi: 10.1083/jcb.150.4.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kiel JA, Hilbrands RE, van der Klei IJ, Rasmussen SW, Salomons FA, van der Heide M, et al. Hansenula polymorpha Pex1p and Pex6p are peroxisome-associated AAA proteins that functionally and physically interact. Yeast. 1999;15:1059–78. doi: 10.1002/(SICI)1097-0061(199908)15:11<1059::AID-YEA434>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 40.Tamura S, Yasutake S, Matsumoto N, Fujiki Y. Dynamic and functional assembly of the AAA peroxins, Pex1p and Pex6p, and their membrane receptor Pex26p. J Biol Chem. 2006;281:27693–704. doi: 10.1074/jbc.M605159200. [DOI] [PubMed] [Google Scholar]
- 41.Saffian D, Grimm I, Girzalsky W, Erdmann R. ATP-dependent assembly of the heteromeric Pex1p-Pex6p-complex of the peroxisomal matrix protein import machinery. J Struct Biol. 2012;179:126–32. doi: 10.1016/j.jsb.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 42.Faber KN, Heyman JA, Subramani S. Two AAA family peroxins, PpPex1p and PpPex6p, interact with each other in an ATP-dependent manner and are associated with different subcellular membranous structures distinct from peroxisomes. Mol Cell Biol. 1998;18:936–43. doi: 10.1128/mcb.18.2.936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fujiki Y, Miyata N, Matsumoto N, Tamura S. Dynamic and functional assembly of the AAA peroxins, Pex1p and Pex6p, and their membrane receptor Pex26p involved in shuttling of the PTS1 receptor Pex5p in peroxisome biogenesis. Biochem Soc Trans. 2008;36:109–13. doi: 10.1042/BST0360109. [DOI] [PubMed] [Google Scholar]
- 44.Augustin S, Gerdes F, Lee S, Tsai FT, Langer T, Tatsuta T. An intersubunit signaling network coordinates ATP hydrolysis by m-AAA proteases. Mol Cell. 2009;35:574–85. doi: 10.1016/j.molcel.2009.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beckwith R, Estrin E, Worden EJ, Martin A. Reconstitution of the 26S proteasome reveals functional asymmetries in its AAA+ unfoldase. Nat Struct Mol Biol. 2013;20:1164–72. doi: 10.1038/nsmb.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hanson PI, Whiteheart SW. AAA+ proteins: have engine, will work. Nat Rev Mol Cell Biol. 2005;6:519–29. doi: 10.1038/nrm1684. [DOI] [PubMed] [Google Scholar]
- 47.Birschmann I, Stroobants AK, van den Berg M, Schafer A, Rosenkranz K, Kunau WH, et al. Pex15p of Saccharomyces cerevisiae provides a molecular basis for recruitment of the AAA peroxin Pex6p to peroxisomal membranes. Mol Biol Cell. 2003;14:2226–36. doi: 10.1091/mbc.E02-11-0752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moeller A, Zhao C, Fried MG, Wilson-Kubalek EM, Carragher B, Whiteheart SW. Nucleotide-dependent conformational changes in the N-Ethylmaleimide Sensitive Factor (NSF) and their potential role in SNARE complex disassembly. J Struct Biol. 2012;177:335–43. doi: 10.1016/j.jsb.2011.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhao M, Wu S, Zhou Q, Vivona S, Cipriano DJ, Cheng Y, et al. Mechanistic insights into the recycling machine of the SNARE complex. Nature. 2015 doi: 10.1038/nature14148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang X, Shaw A, Bates PA, Newman RH, Gowen B, Orlova E, et al. Structure of the AAA ATPase p97. Mol Cell. 2000;6:1473–84. doi: 10.1016/s1097-2765(00)00143-x. [DOI] [PubMed] [Google Scholar]
- 51.Bebeacua C, Forster A, McKeown C, Meyer HH, Zhang X, Freemont PS. Distinct conformations of the protein complex p97-Ufd1-Npl4 revealed by electron cryomicroscopy. Proc Natl Acad Sci U S A. 2012;109:1098–103. doi: 10.1073/pnas.1114341109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ewens CA, Panico S, Kloppsteck P, McKeown C, Ebong IO, Robinson C, et al. The p97-FAF1 protein complex reveals a common mode of p97 adaptor binding. J Biol Chem. 2014;289:12077–84. doi: 10.1074/jbc.M114.559591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.DeLaBarre B, Brunger AT. Complete structure of p97/valosin-containing protein reveals communication between nucleotide domains. Nat Struct Biol. 2003;10:856–63. doi: 10.1038/nsb972. [DOI] [PubMed] [Google Scholar]
- 54.Hersch GL, Burton RE, Bolon DN, Baker TA, Sauer RT. Asymmetric interactions of ATP with the AAA+ ClpX6 unfoldase: allosteric control of a protein machine. Cell. 2005;121:1017–27. doi: 10.1016/j.cell.2005.05.024. [DOI] [PubMed] [Google Scholar]
- 55.Yakamavich JA, Baker TA, Sauer RT. Asymmetric nucleotide transactions of the HslUV protease. J Mol Biol. 2008;380:946–57. doi: 10.1016/j.jmb.2008.05.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Erzberger JP, Berger JM. Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu Rev Biophys Biomol Struct. 2006;35:93–114. doi: 10.1146/annurev.biophys.35.040405.101933. [DOI] [PubMed] [Google Scholar]
- 57.Matsumoto N, Tamura S, Fujiki Y. The pathogenic peroxin Pex26p recruits the Pex1p-Pex6p AAA ATPase complexes to peroxisomes. Nat Cell Biol. 2003;5:454–60. doi: 10.1038/ncb982. [DOI] [PubMed] [Google Scholar]
- 58.Hinnerwisch J, Fenton WA, Furtak KJ, Farr GW, Horwich AL. Loops in the central channel of ClpA chaperone mediate protein binding, unfolding, and translocation. Cell. 2005;121:1029–41. doi: 10.1016/j.cell.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 59.Mogk A, Schlieker C, Strub C, Rist W, Weibezahn J, Bukau B. Roles of individual domains and conserved motifs of the AAA+ chaperone ClpB in oligomerization, ATP hydrolysis, and chaperone activity. J Biol Chem. 2003;278:17615–24. doi: 10.1074/jbc.M209686200. [DOI] [PubMed] [Google Scholar]
- 60.Kress W, Mutschler H, Weber-Ban E. Both ATPase domains of ClpA are critical for processing of stable protein structures. J Biol Chem. 2009;284:31441–52. doi: 10.1074/jbc.M109.022319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shiozawa K, Maita N, Tomii K, Seto A, Goda N, Akiyama Y, et al. Structure of the N-terminal domain of PEX1 AAA-ATPase. Characterization of a putative adaptor-binding domain. J Biol Chem. 2004;279:50060–8. doi: 10.1074/jbc.M407837200. [DOI] [PubMed] [Google Scholar]
- 62.Park S, Isaacson R, Kim HT, Silver PA, Wagner G. Ufd1 exhibits the AAA-ATPase fold with two distinct ubiquitin interaction sites. Structure. 2005;13:995–1005. doi: 10.1016/j.str.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 63.Norby JG. Coupled assay of Na+,K+-ATPase activity. Methods Enzymol. 1988;156:116–9. doi: 10.1016/0076-6879(88)56014-7. [DOI] [PubMed] [Google Scholar]
- 64.Suloway C, Pulokas J, Fellmann D, Cheng A, Guerra F, Quispe J, et al. Automated molecular microscopy: the new Leginon system. J Struct Biol. 2005;151:41–60. doi: 10.1016/j.jsb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 65.Lander GC, Stagg SM, Voss NR, Cheng A, Fellmann D, Pulokas J, et al. Appion: an integrated, database-driven pipeline to facilitate EM image processing. J Struct Biol. 2009;166:95–102. doi: 10.1016/j.jsb.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mindell JA, Grigorieff N. Accurate determination of local defocus and specimen tilt in electron microscopy. J Struct Biol. 2003;142:334–47. doi: 10.1016/s1047-8477(03)00069-8. [DOI] [PubMed] [Google Scholar]
- 67.Voss NR, Yoshioka CK, Radermacher M, Potter CS, Carragher B. DoG Picker and TiltPicker: software tools to facilitate particle selection in single particle electron microscopy. J Struct Biol. 2009;166:205–13. doi: 10.1016/j.jsb.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ludtke SJ, Baldwin PR, Chiu W. EMAN: semiautomated software for high-resolution single-particle reconstructions. J Struct Biol. 1999;128:82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- 69.Sorzano CO, Marabini R, Velazquez-Muriel J, Bilbao-Castro JR, Scheres SH, Carazo JM, et al. XMIPP: a new generation of an open-source image processing package for electron microscopy. J Struct Biol. 2004;148:194–204. doi: 10.1016/j.jsb.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 70.Ogura T, Iwasaki K, Sato C. Topology representing network enables highly accurate classification of protein images taken by cryo electron-microscope without masking. J Struct Biol. 2003;143:185–200. doi: 10.1016/j.jsb.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 71.Yang Z, Fang J, Chittuluru J, Asturias FJ, Penczek PA. Iterative stable alignment and clustering of 2D transmission electron microscope images. Structure. 2012;20:237–47. doi: 10.1016/j.str.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hohn M, Tang G, Goodyear G, Baldwin PR, Huang Z, Penczek PA, et al. SPARX, a new environment for Cryo-EM image processing. J Struct Biol. 2007;157:47–55. doi: 10.1016/j.jsb.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 73.Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, et al. EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol. 2007;157:38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 74.Scheres SH. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol. 2012;180:519–30. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc. 2009;4:363–71. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 76.Davies JM, Brunger AT, Weis WI. Improved structures of full-length p97, an AAA ATPase: implications for mechanisms of nucleotide-dependent conformational change. Structure. 2008;16:715–26. doi: 10.1016/j.str.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 77.Goddard TD, Huang CC, Ferrin TE. Visualizing density maps with UCSF Chimera. J Struct Biol. 2007;157:281–7. doi: 10.1016/j.jsb.2006.06.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Raw micrographs of Pex1/Pex6 in the presence of different nucleotides as labeled. On the left are micrographs showing end-on orientation of particles on carbon surface. On the right are micrographs from poly-L-lysine pre-treated grids, where the particles primarily adsorb to the carbon surface on their sides.
Supplementary Figure 2. A) Forward projections from negative stain reconstructions of Pex1/Pex6 complex in presence of ADP, ATPγS, and ATP in the upper panels, with matching reference-free 2D class averages shown in the bottom panels.
B) Gold Standard FSC plots for the 3D reconstructions of Pex1/Pex6, reporting resolutions of ~17Å for the ADP-bound structure, and ~23Å for ATPγS- and ATP-bound structures, using a 0.143 cutoff.
C) Angular distribution of particles contributing to 3D reconstructions, with the diameter of each sphere related to the number of particles at that Euler angle (larger spheres indicate more particles.
Supplementary Figure 3. A) The D1 Walker A mutants of Pex-FLAG/His-Pex6 were purified by a sequential Ni-NTA affinity purification and α-FLAG affinity resin. The presented images of a Coomassie-stained SDS PAGE shows the Ni-NTA elution, FLAG flowthrough, and FLAG elution for each mutant. The two mutant hexamers were purified side by side, with similar wash and elution volumes.
B) The Pex1-WA1/Pex6 mutant has an assembly defect, resulting in a depleted peak at the expected elution volume for intact Pex1/Pex6 complex (~12.5 mL, compare to Pex1/Pex6-WA1 mutant).
Supplementary Movie 1. Flexible attachment of MBP to the amino terminal end of Pex6.
Supplementary Movie 2. Comparison between ADP-, ATPγS-, and ATP-bound structures of Pex1/Pex6 showing the structural variations between the different nucleotide-bound states.