

Abstract
Mass spectrometry (MS) has become a powerful and widely utilized tool in the investigation of protein thiol chemistry, biochemistry, and biology. Very early biochemical studies of metabolic enzymes have brought to light the broad spectrum of reactivity profiles that distinguish cysteine thiols with functions in catalysis and protein stability from other cysteine residues in proteins. The development of MS methods for the analysis of proteins using electrospray ionization (ESI) or matrix-assisted laser desorption/ionization (MALDI) coupled with the emergence of high-resolution mass analyzers have been instrumental in advancing studies of thiol modifications, both in single proteins and within the cellular context. This article reviews MS instrumentation and methods of analysis employed in investigations of thiols and their reactivity toward a range of small biomolecules. A selected number of studies are detailed to highlight the advantages brought about by the MS technologies along with the caveats associated with these analyses.
1. Introduction
Mass spectrometry (MS) is an essential analytical technology with applications to virtually all areas of life sciences and beyond. Though the basic physical and chemical principles of MS have been known since the 19th century [1], this technology has been applied to biological sciences for only the past several decades following the development of devices that enable the transfer of polar molecules like proteins, nucleic acids, and metabolites from liquid and solid states to the gas phase through the process of ionization. This leap in engineering was achieved by John Fenn (USA) and Koichi Tanaka (Japan) who were awarded the Nobel Prize in Chemistry in 2002 for the developments of electrospray ionization (ESI) and soft laser desorption (SLD), respectively. ESI is today ubiquitous in studies of biomolecules, and the development of SLD facilitated the later application of the previously known matrix-assisted laser desorption/ionization (MALDI) to the analysis of proteins, also used widely to this day [2, 3].
Over the following years the efforts of countless researchers have expanded the applications of MS to the sequencing of proteins, identification of metabolites, confirmation of gene sequences, studies of how nucleic acids and proteins are modified following their biosynthesis, the detection of non-covalent protein complexes [4], and even measurement of intact microorganisms such as viruses [5]. The utility of MS in sequencing and identifying proteins was established in 1993 with the first report of peptide mass fingerprinting, and MS-based relative quantification of proteins and peptides was achieved several years later with the use of isotope-coded affinity tags (ICAT) [6, 7]. Beginning with the development of ICAT, which leverages the reactivity of biotin-iodoacetyl reagents with cysteine thiols, isotope labeling has been a hallmark of quantification by MS and underlies the principle of approaches like stable isotope labeling by amino acids in cell culture (SILAC) [8], tandem mass tags (TMT) [9], and isobaric tags for relative and absolute quantification (iTRAQ) [10], among others. Today, the analysis of proteins and peptides by MS provides a wealth of information enabling investigations of protein structure, reactivity, and function. Moreover, the use of MS in the field of proteomics has led to the identification of new proteins, quantification of protein expression and post-translational modifications (PTMs), elucidation of components in complex protein assemblies, and verification of genome sequences (i.e., proteogenomics).
The dynamic post-translational modification (PTM) of proteins is particularly significant in the regulation of biological processes, and has led to the establishment of specialized proteomics fields focused on identifying and quantifying selective PTMs, such as phosphorylation, glycosylation, methylation, and others. Modifications of cysteine thiols are also increasingly recognized as critical regulatory switches controlling protein function. However, methods for investigating Cys thiol PTMs in single recombinant proteins and in the whole cellular context are less robust than for other PTMs and are marked by significant caveats to be considered to ensure accurate results. Investigations of thiol PTMs are challenged by the labile nature of modified cysteines, often requiring derivatization to provide a stable, detectable moiety and to prevent subsequent reactions, including as artifacts during cell lysis and sample processing. Thorough parallel control experiments are required to address these experimental challenges and are noted throughout this review.
The thiol proteome is the target of numerous diverse oxidative modifications derived from the interactions of Cys residues with reactive nitrogen, oxygen, and sulfur species, leading to over 15 modifications (Table 1). Many oxidized Cys products are reversible and/or subsequently react with other cysteine-derived species to yield more thermodynamically stable products. Several modifications are generally considered to be irreversible, however, for some, enzyme-catalyzed repair mechanisms are known [11, 12]. A particular example, described in more detail later in this review, is the reduction of peroxiredoxin (Prx) sulfinic acid to the active thiol form catalyzed by sulfiredoxin (Srx) in the presence of ATP, Mg2+, and the thioredoxin/thioredoxin reductase (Trx/TrxR) system. The lifespans of reversible thiol modifications are controlled by local protein structure and the microenvironment wherein the modified Cys resides. Factors affecting the microenvironment include 1) variations of oxidant and antioxidant levels within the surrounding area and 2) the nearby conformation and intrinsic chemistry (nucleophilicity, acidity, basicity) of adjacent amino acids. Together, these physical and chemical environments influence the chemical stability of the modified Cys, requiring a careful optimization of sample preparation prior to MS analysis, the details of which are discussed in the following sections.
Table 1.
Oxidation states and delta masses for cysteine oxidation products
| Species | Functional Group | Oxidation State | Reversibility | ΔMass (Da, R=H) |
|---|---|---|---|---|
| Thiol |
|
−2 | N/A | N/A |
| S-Palmitate |

|
−2 | + | +224.2140 |
| Thiyl radical |
|
−1 | + | −1.0078 |
| Intramolecular disulfide |

|
−1 | + | −2.0156 |
| Mixed disulfide |
|
−1 | + | +305.0682 (RS= GS) |
| Perthiol |
|
−1 | + | +31.9721 |
| Thiosulfate |

|
−1 | + | +78.9496 |
| Sulfenic acid |
|
0 | + | +15.9949 |
| Sulfenamide |
|
0 | + | +15.0109 |
| Sulfenyl halide |
|
0 | + | +33.9611 (X= Cl) +77.9105 (X= Br) |
| S-Nitrosothiol |
|
0 | + | +28.9902 |
| Thiosulfinate |

|
+1 | + | +321.0631 (RS= GS) |
| Sulfinamide |

|
+2 | − | +31.0058 |
| Sulfinic acid |

|
+2 | + | +31.9899 |
| Thiosulfonate |

|
+3 | + | +337.0580 (RS= GS) |
| Sulfonic acid |

|
+4 | − | +47.9848 |
| Sulfonamide |

|
+4 | − | +47.0008 |
2. Basics of MS for Investigation of Proteins and Peptides
This section provides a brief overview of the principles of MS and basic components of MS instrumentation. Modern MS instrumentation is characterized by parameters like ease of use, versatility, mass accuracy, sensitivity, and ease of automation. The components of mass spectrometers will be discussed briefly in light of the MS-based approaches most commonly used to study thiol modifications in proteins and peptides, which are summarized in Table 1. For additional details, the reader is referred to other articles on this topic ([13] and others).
Sample preparation and separation
The analysis of proteins and peptides often requires clean samples devoid of detergents and salts, which interfere with ionization. In particular, Na+ and K+ salts provide strong ion pairing in positive ion mode and can lead to signal fractionation and ion suppression. For these reasons, samples are often desalted before MS analysis or prepared in volatile buffers like ammonium acetate and ammonium bicarbonate, which are readily lost as ammonia, acetic acid, carbon dioxide, and water during the desolvation process [18]. The analysis of single biomolecules is routinely performed by direct infusion following desalting, but more complex mixtures require one or more dimensions of separation prior to MS analysis. Preparative scale methods for sample fractionation include HPLC and gel electrophoresis, and often require subsequent processing steps to prepare samples for MS. Alternatively, analytical separation is carried out using modules interfaced to MS; HPLC (and related nanoLC instruments) and capillary electrophoresis (CE) are both used in this manner. LC approaches used in proteomics applications include separation on the basis of differential polarity (e.g., reverse-phase liquid chromatography (RPLC) [14] and hydrophilic interaction liquid chromatography (HILIC) [15, 16]) or by charge (ion exchange). Recent LC advances allow for 2-dimensional separations, often ion exchange directly followed by RPLC. Additionally, separation of ions based on their gas phase mobility has emerged as a useful analytical tool [Ion Mobility MS, IMS-MS] (see [17, 18] for recent in-depth reviews). In IMS-MS, ions migrate through a uniform electric field toward a detector, where ions with a larger surface area undergo an increased number of friction-inducing collisions with inert gas relative to smaller ions slowing their motion. Experimental IMS data is compared to known protein architectures, and coupled with MS data, provides valuable structural information [19].
Ionization sources
ESI/NSI VS MALDI
Proteomics applications rely most often on MALDI and ESI ion sources, which provide “softer” means of ionization compared to electron ionization or chemical ionization methods. Using MALDI, ions are introduced to the mass spectrometer by UV laser-induced ionization of a matrix containing the sample, facilitating sample ionization. MALDI sources often generate +1 charged ions (referred to as [M+H]+ or [M+Na]+), which for proteins and peptides typically result in very large m/z ratios. More recently, MALDI has been applied to the spatial analysis of tissue samples in a method named MALDI imaging MS [20] in which tissue slices are treated with MALDI matrix and irradiated in small, specific regions of interest, producing a mass spectrum for each set of x,y-coordinates within a tissue section. As an in-depth discussion of MALDI imaging MS is beyond the scope of this review, the reader is directed to a recent article on this approach [21].
ESI sources provide more direct ionization, where an analyte solution is introduced into the source by a fused silica or stainless steel capillary across which a voltage is applied. A mist of charged droplets is produced from which solvent is evaporated, decreasing droplet size, leading to droplet burst and the emergence of many small gaseous ions [22]. Unlike MALDI, ESI provides ions of higher charge states, resulting in more complex mass spectra but reduced m/z ratios, allowing compatibility with a wider range of MS analyzers. Nanospray ionization (NSI, also abbreviated nanoESI) was later developed on the principle of ESI but uses decreased flow rates (nL/min) and spray voltage, requiring less sample without a loss in sensitivity. By nature of lower analyte velocity and decreased capillary diameter, the droplets produced by NSI are remarkably smaller than those produced by ESI: 1–2 μm in diameter for ESI and < 200 nm in diameter for NSI [18]. NSI requires the use of nanoLC instrumentation that separates the analytes on columns typically made of 50–150 μm ID fused silica, containing 10–250 mm bed length packed with desired media for the separation of targeted analytes and using flow rates below 1 μL/min, typically between 200 and 400 nL/min.
Mass analyzers
Mass analyzers routinely utilized in proteomics applications include quadrupoles, time-of-flight (TOF), ion traps, and the newer orbitrap analyzers. The relative performance of mass analyzers is related to parameters such as scan speed, mass accuracy, resolution, acquisition range, and tandem analysis capabilities. Time of flight (TOF) mass analyzers measure the time required for ions to travel from the source to the detector and offer a high upper limit of detection (~10,000 m/z), making them particularly well-suited for studies of intact proteins, especially with MALDI sources. Acquisition of tandem mass spectra (MS/MS) using the TOF analyzer requires the use of a quadrupole or additional TOF mass filter (Q-TOF, TOF-TOF). In contrast, ion traps use magnetic and electric fields to capture and eject ions by ascending m/z ratio. Compared to triple quadrupoles, ion traps can be less sensitive for quantification, but are smaller, less costly, and also serve as collision cells for tandem MS fragmentation [22]. Additionally, ion traps, when used in tandem MS, are restricted in that their lower limit of detection is one-third of the precursor ion m/z ratio (e.g., isolation and fragmentation of a 600 m/z ion produces many daughter ions; only those > 200 m/z are observed). Developed within the past 15 years, the orbitrap is a pulsed mass analyzer that uses static electric fields to trap and analyze ions, offering increased sensitivity and resolution along with high mass accuracy [23].
Since MALDI, ESI, and NSI methods of ionization are not sufficient to fragment most molecules, the acquisition of m/z ratios for all ions entering the detector provides the MS1 spectrum (of precursor ions), from which high resolution mass, molecular formula, and in some cases, relative quantification, may be obtained. Information regarding peptide sequence, structure elucidation, and site resolution of modifications requires tandem MS analysis in which a precursor ion is isolated and fragmented to produce numerous fragment (or daughter) ions. Isolation of precursor ions is performed by quadrupoles or ion traps, and fragmentation is induced most commonly by collision-induced dissociation (CID), higher energy collision dissociation (HCD), or electron-transfer dissociation (ETD) (Figure 1) [24–26]. The choice of fragmentation method is ultimately dependent on the type of modifications present, including PTMs and isotope tags for quantification, and sample analysis routinely involves the use of several different fragmentation methods to obtain complimentary MS2 spectra.
Figure 1.

Peptide bonds cleaved by MS2 fragmentation. A: Collison-induced dissociations fragment the amide bond, producing b- and y-ions. Also targeted are many PTMs (not shown). B: Electron-transfer dissociation targets the amino-α-carbon bond, producing c- and z-ions, and leaving PTMs intact.
3. Structural and Kinetic Investigations of Thiol Proteins by MS
This section describes various applications of MS (primarily ESI-TOF MS but others as well) to investigate the chemistry and kinetics underlying protein thiol oxoform formation, reactivity, and repair using intact thiol-containing proteins as model systems. Also discussed are MS methods for the identification of structural changes induced by thiol oxidative modifications in proteins. We use the term “oxoform” to denote thiol modifications resulting in an increase in sulfur oxidation state relative to the cysteine thiol (−2). Additionally, discussed modifications will be abbreviated in the format Cys-SR, where “S” is the cysteine sulfur and “R” is the added moiety.
General Considerations and Model Systems for Investigations of Protein Thiol Chemistry
Studies of intact proteins using ESI-MS provide critical and detailed information, but have several essential technical requirements though not all specific to studies of protein cysteine. First, on average, a minimum of 20–40 μg of a pure protein is needed for acceptable signal to noise. This value changes according to the mass and unique ionization efficiency of each protein, but is substantially more material than is required for Western blot (< 1 μg of pure protein). Nevertheless a greater level of chemical information may be obtained by MS compared with Western blot or by separating the protein by native or denaturing gel analysis. As stated previously, samples must be devoid of salts and detergents, requiring that the protein(s) or reaction mixture is able to withstand volatile buffers and/or the slightly acidic pH (4–5) used to facilitate ionization in positive ion mode. For quantification purposes, one must account for the ionization efficiencies of all species formed along the reaction time course, so that a change in the abundance of a particular ion may be related to its relative concentration in solution. Such standardizations are performed by analyzing known amounts of each relevant species in its pure form, when possible, or by fitting reaction data to theoretical kinetic models. An important caveat to consider is the possibility of gas phase reactions occurring during MS analysis, such as those known for cysteine sulfenic acid/sulfenamide equilibria and oxidation of methionine residues, among others [27, 28]. These technical requirements have not precluded ESI-MS from being a powerful tool in identifying new protein species and measuring the rates of protein reactions.
The numerous products of Cys oxidation can be classified based on their relative thermodynamic stabilities. The less stable forms, including sulfenic acid (Cys-SOH) and nitrosothiol (Cys-SNO), are reactive toward a variety of electron rich molecules but are also consumed by ambient elements like oxygen (Cys-SOH) and light (Cys-SNO). Semi-stable forms, including disulfides and thiosulfinates, are unstable toward reductants and some nucleophiles, but otherwise long-lived. The most thermodynamically stable Cys oxidation products include sulfinic and sulfonic acids, proven irreversible except in the specific case of the reduction of peroxiredoxin sulfinic acids by sulfiredoxin. The reductive reversibility of the less stable sulfur oxoforms implies roles as redox switches capable of impacting cell signaling by activating and deactivating enzyme pathways. However, the direct analysis of these is challenged by their short lifetimes in wild type proteins and overall limitations in obtaining significant amounts of pure eukaryotic proteins. For these reasons, a number of model protein systems have emerged in recent decades to provide means for the analysis of thiol oxoforms by MS and other methods. We will briefly discuss these along with some specific examples of our work using such models and methods.
To observe the Cys thiols in the less stable oxoforms, a few common strategies are employed. First is the use of a wild type protein and a derivatizing agent selective for a particular Cys-derived species. This type of approach was initially used by Allison to identify the formation of cysteine sulfenic acid in GAPDH via reaction of the nascent Cys-SOH with dimedone [29]. Other wild type proteins used in this manner are papain and the human and bovine serum albumins. Papain, a cysteine protease from papaya, contains 8 Cys in the mature form, though 6 are found in disulfide linkages. The active site Cys25 thiolate forms Cys-SOH in the presence of H2O2 [30] and the perthiol (Cys-SSH, also called persulfide) in response to H2S [31], and has thus been used to probe reactivity and kinetics of these species [32, 33]. Serum albumin (69 kDa), the principal antioxidant protein in plasma, has only one of 35 Cys residues present as a free thiol. This Cys was shown to be redox-sensitive in both the human (HSA) and bovine (BSA) proteins by oxidation to the corresponding sulfenic acids, S-glutathionylated (Cys-SSG) products [34, 35], and in the case of BSA, the persulfide Cys-SSH [35].
In contrast to the wild type eukaryotic proteins, which contain many Cys residues and other non-cysteine PTMs, a number of groups have developed mutant proteins, often from prokaryotes or lower eukaryotes that offer the simplicity of decreased Cys content, few or no modifications, and lower molecular weight. Mutations, including those of resolving Cys or all Cys residues except for the peroxidatic site, provide a method for generating rather quantitatively a particular oxoform and probing its reactivity. For example, an active site cysteine of the methionine sulfoxide reductases (Msr, 23 kDa) forms Cys-SOH en route to generating reduced methionine [36, 37]. Cysteine to serine mutations in free Msr (fRMsr, which reduces the free Met-(R)-sulfoxide) from E. coli, produce the single Cys118-containing Cys84Ser/Cys94Ser mutant, utilized to measure Cys-SOH capture by chemical probes [32]. Additionally, cysteine mutants of the transcription factor OxyR (34 kDa, E. coli), the peroxiredoxin AhpC (21 kDa, S. typhimurium), and glutathione peroxidase (Gpx, yeast, 18 kDa), leaving only the peroxidatic cysteines (Cys208Ser OxyR; Cys165Ser or Cys165Ala AhpC; Cys82Ser Gpx), have been used as model systems to probe the reactivities of Cys-SOH [32, 38–40], Cys-SNO [39–42], Cys-SO2H [39, 40], Cys-SSH [33], and other forms. Representative ESI-TOF mass spectra of several cysteine oxoforms of wt and mutant AhpC are shown in Figure 2.
Figure 2.

Representative ESI-TOF mass spectra for wild type and mutant AhpC from S. typhimurium. A: Wild type AhpC is observed as the monomer (20,616 amu) and the homodimer (41,230 amu). B: The Cys165Ser AhpC mutant, devoid of a resolving Cys, allows for the facile preparation of numerous oxoforms. R-SN refers to the cyclic sulfenamide Cys-SN formed from the intramolecular amine-mediated dehydration of Cys-SOH in the gas phase and/or in solution. Unpublished Data.
Overview of Methods for Investigation of Enzyme Kinetics by MS
Kinetic studies of the reactions of intact proteins by ESI-MS require a means of quenching the ongoing reaction at specified time points. Chemical quenching via the addition of a destabilizer like an acid, base, or chaotropic reagent halts the reaction but may introduce the need for a sample purification step before MS analysis. An alternative approach to reaction quenching involves passage through size-exclusion resins, providing the simultaneous removal of small molecule components and buffer salts, exchanging the protein solution into a volatile buffer amenable to ionization and MS (Figure 3A). For reactions occurring sufficiently fast to require the analysis of time points below 30 seconds, a number of time-resolved methods (TR-MS) have been developed using ESI desolvation as a means of quenching reactions. In this approach, two syringes, driven by the same pump, are connected by a mixing tee to a common fused silica capillary carrying the reaction solution to the ESI source (Figure 3B). One syringe is loaded with the protein of interest and the other with a substrate. The two solutions are mixed together in the tee at variable speeds to initiate the reaction, often high enough to ensure turbulent mixing, though some methods have been described using laminar flow [43–45]. The volume of tubing from the mixing tee to the ion source and the flow velocity allow for the determination of reaction time before the mixture is desolvated and quenched; variations in flow rate alter the reaction time, providing for the analysis of the reaction mixture at numerous time points and as short as 7 milliseconds [46]. Pulsed flow, stopped flow, and continuous flow approaches of TR-MS have been applied to studies of protein conformation and structure [47–49], investigations of enzyme mechanisms and the identification of intermediates [46, 50–54], and other kinetic studies [55, 56]. Recently, desorption ESI (DESI) has been utilized as an ionization source for TR-MS, providing an improved level of time resolution [57]. The DESI source samples from a jet liquid stream flowing at a greater velocity than is compatible with ESI sources and desolvates more rapidly than ESI, allowing for the analysis of earlier and more frequent time points than was previously possible [57, 58]. A number of these methods have been applied to investigate the mechanisms and kinetics of proteins relying on thiol chemistry and a few case studies are reviewed next.
Figure 3.

Discontinuous and continuous methods for kinetic investigations using intact proteins. A: Discontinuous approaches involve quenching reaction aliquots using conditions of low pH or a size exclusion resin followed by MS analysis of each time point. B: Continuous time-resolved kinetic analyses require the direct infusion using dual syringes with reaction occurring between the mixing tee and the ion source (called the aging loop). Step-wise alterations in flow velocity allow for the observation of multiple reaction time points in a single run. The methods allow for simultaneous monitoring of all covalent and in some cases non-covalent species present at each reaction time point.
MS to investigate the reactivities of thiol oxoforms
A long-standing interest in the field of thiol-based redox biology is the development of methods that enable the quantitative measurement of cysteine thiol modification by physiologically relevant molecules (e.g., H2O2, GSH, NO, HNO, etc.). Though several of these modified species are measured directly by MS or other methods, traditionally, chemical probes have been employed to target selectively and with high reactivity various cysteine oxoforms. An example of these is the use of 7-chloro-4-nitrobenzo-2-oxa-1,3-diazole (NBD chloride) to derivatize protein sulfenic acids for detection by UV-vis spectroscopy [59]. While this reagent is not selective for Cys-SOH modification, its reaction with Cys-SOH produces a signal that is unique to this Cys oxoform. In recent years our group and others have employed model thiol proteins, such as those discussed and referenced earlier in this section, to aid in the development of chemical probes with selectivity against specific thiol oxoforms, to quantify reactivity, and to troubleshoot workflows for labeling, enrichment and detection by MS of various thiol modified sub-proteomes generated by protein S-sulfenylation (Cys-SOH), S-sulfhydration (Cys-SSH), S-nitrosylation (Cys-SNO), and S-acylation (e.g., S-palmitate) in cells.
For example, in a recent publication we questioned whether the widely used thiol-blocking reagents iodoacetamide (IAM), N-ethylmaleimide (NEM) and methyl methanethiosulfonate (MMTS) react with protein Cys-SOH, and if so, what the consequences of such reactions would be on the ability to detect Cys-SOH in the broader context of the redox proteome using chemical probes or indirect tag-switch approaches. First, a kinetic profile was established by reacting Cys165Ser AhpC-SOH with each of the three electrophiles. Using an ESI-TOF-based intact protein analysis, we detected and quantified the kinetics of covalent adduct formation (termed Cys-SOR) formed from the reactions of AhpC-SOH with each of the three electrophiles, revealing the following rate profile: MMTS>NEM>IAM. Unanticipated reaction products were also identified by MS analysis, such as a Cys to dehydroalanine (DHA) conversion in the case of MMTS (Figure 4) [39]. In addition, we further employed ESI-TOF MS to probe the stability of the Cys-SOR species under reducing conditions (i.e., in the presence of DTT, TCEP, or ascorbate), a critical step in tag-switch approaches for redox proteomics studies. As shown in Figure 4, the reduction of Cys-SOR to Cys-SH was followed by ESI-TOF MS, revealing that the recovery of Cys165Ser AhpC-SH was dependent on both the blocking reagent and on the reductant used. The overall effects of this newly detailed chemistry on the outcome of experiments depend on the cysteine species of interest in each particular experiment. For example, general redox proteomics approaches focused on quantifying the global ratio of reduced and oxidized cysteine will likely be unaffected as a reductant step is utilized after labeling. In contrast, studies aimed at labeling Cys-SOH, such as with dimedone and related reagents, will be hampered by the cross-reactivity of thiol-blocking reagents in competition with the Cys-SOH-targeted probe. In our studies, MMTS reacted most rapidly with AhpC-SOH but the corresponding adduct was also the most sensitive to reduction. Certainly the relative stabilities of such adducts are anticipated to be protein- and microenvironment-dependent as well. These findings raised concerns about indirect methods of detection for protein thiol oxoforms and pointed to the need for continued development of probes that chemically target specific thiol modifications.
Figure 4.

Mutant AhpC-SOH readily captures electrophiles iodoacetamide (IAM), N-ethylmaleimide (NEM), and methyl methanethiosulfonate (MMTS) widely used in thiol blocking to yield covalent Cys-SOR adducts (exact connectivity unknown). Subsequent treatment with reductant (thiols, phosphines, ascorbate) converts Cys-SOR to Cys-SH at varying degrees. [DHA: dehydroalanine.] Adapted from Reisz et al. [39]
Similar MS-based approaches were employed to investigate the reaction of protein Cys-SOH with various chemical probes and to test the selectivity of the probes towards the Cys-SOH relative to their reaction with Cys-SH, Cys-SNO, and intermolecular disulfides. These chemical probes include dimedone and its derivatives [29, 60–62], S-ethyl cyclopentadiones [63], acyclic β-ketoesters [64], and most recently bicyclononynes [40].
MS analysis of protein sulfenylation in intact proteins
Sulfenic acids (Cys-SOH) are the initial products arising from the oxidation of Cys residues in proteins under physiological or stress conditions. The analysis of Cys-SOH formation is readily accessible using ESI-MS by monitoring the Δ mass of 16 Da relative to the parent protein. In most cases, formation of a Cys-SOH on the model proteins discussed earlier (e.g., by treatment with H2O2) is accompanied by higher oxidized products, including sulfinic (Cys-SO2H, +32 Da) and/or sulfonic acids (Cys-SO3H, +48 Da) derived either from the oxidative stimulus, during sample processing [65], or in the gas phase during MS acquisition [66]. In addition, we and others have observed in the MS analysis of Cys-SOH-containing intact proteins a signal corresponding to the cyclic sulfenamide (Cys-SN) product, a Cys-SOH dehydration product that is known for redox-sensitive proteins like PTP1B (structure shown in Figure 5) [67, 68]. In our hands, the ratios of Cys-SN to Cys-SOH signals observed by intact protein MS vary widely among different proteins [32, 39, 69]. Without clearly discerning whether the Cys-SN is formed in solution or in the gas phase, typically these species are considered together as Cys-SOH. It may be possible to distinguish solution versus gas phase formation of Cys-SN for a given protein by following an experimental design where 18O-labeled hydrogen peroxide is used to generate Cys-S18OH (Figure 5) and by taking advantage of the newly discovered reaction of Cys-SOH with iodoacetamide. The understanding of the functional roles of protein sulfenamides is still in its infancy, but has been described as a possible protective mechanism against Cys hyperoxidation (e.g., Prxs) [69] or as a circuit loop to sustain signaling. Using ESI MS, the biochemical impacts of sulfenylation have been assigned to a variety of targeted proteins [38, 65, 70–72] and have led to both gain [73] and loss of function [74–76].
Figure 5.

Hydration-dehydration equilibrium relating sulfenic acid and sulfenamide. 18O-Labeling of the sulfenamide leads to the formation of the 18O-labeled Cys-SOR species. SOH/SN equilibrium in the solution phase will decrease 18O incorporation in favor of the more abundant 16O.
MS analysis of protein S-sulfhydration in intact proteins
The term protein S-sulfhydration was recently introduced to define the PTM occurring on Cys residues upon their interactions with the gasotransmitter hydrogen sulfide (H2S) (e.g., persulfide and perthiol). However, prior to the description of this newly defined PTM, persulfides were known as key intermediates in the catalytic cycles of various enzymes, including cysteine desulfurase and sulfurtransferase, and also as ligands in Fe-S clusters [77–80]. Direct analysis of the persulfide modification using MS has been attempted with several intact proteins [33]. Protein persulfides are characterized using MS by the appearance of a signal of Δ mass +32 Da relative to the parent thiol. Despite the significant mass shift, control experiments are required to avoid misidentification due to the fact that other thiol PTMs, such as cysteine sulfinic acid (Cys-SO2H), also give a net Δ mass of +32 Da (see accurate masses in Table 1). Even with the wide use of high resolution instrumentation, distinguishing these two modifications on intact proteins remains a challenge as, for example, a mass accuracy of < 1 ppm would be needed to distinguish between these in a 20 kDa protein. Control experiments in this case could include 1) treatment with reducing agents (e.g., DTT or TCEP) to obtain the reduced thiol form of the analyzed protein and/or 2) reaction with thiol-blocking reagents such as IAM or NEM [33, 78, 79]. Both are critical as they allow for specific distinction of persulfides against sulfinic acid, noting that the latter cannot be reduced by DTT and is also inert to IAM and NEM treatment [39]. In selected cases and under certain conditions, persulfide formation is accompanied by protein polysulfinylation (Cys-S-(S)n-SH), resulting in higher spectral noise and decreased spectral quality. For example, oxidation of cysteine sulfurtransferase SufE by SufS-S-SH persulfide yields species with +32, +64, and +96 Da mass adducts, corresponding to the incorporation of one, two, and three sulfurs on the parent protein [78]. Though such polysulfides can obscure the signals of other species present, they may also serve as molecular fingerprints of reactive sulfur species as opposed to purely ROS-mediated oxidation to generate Cys-SO2H. Interestingly, polysulfides and persulfides are thought to coexist biologically and serve as signal carriers of H2S [81]. To this end, it is evident that the analysis of protein sulfhydration using MS is possible; however, the elimination of discrepancies in product identification requires: 1) the use of high-resolution MS instruments and/or inclusion of control experiments; 2) the exclusion of oxidants to maintain the polysulfide sulfane sulfur in the reduced state.
MS analysis of protein S-nitrosylation in intact proteins (also called protein S-nitrosation)
S-Nitrosothiols (Cys-SNO) are formed from the radical-involved reaction of sulfhydryl groups (Cys-SH) and nitric oxide (NO), resulting in a Δ mass of +29. An additional pathway by which protein Cys-SNO species are produced involves the transfer of a nitrosyl group from other protein SNOs or more commonly from low molecular weight SNOs, including free Cys-SNO, S-nitrosoglutathione (GSNO), and the recently discovered HSNO, to a Cys residue of a target protein in a process termed transnitrosation (or transnitrosylation) [82, 83]. The SNO moiety is particularly unstable for MS analysis and may be cleaved during ionization. Decomposition of the SNO functionality produces the parent sulfhydryl radical precursor and NO. Despite the intrinsic instability of S-nitrosothiols, in the past two decades many protein SNOs have been detected using ESI. For instance, the hydrolase enzyme p21ras was analyzed via ESI/triple-quadrupole, though the acquired spectrum suffered from a poor signal-to-noise ratio [84]. More recently, nitrosylated Trx1 was observed by ESI MS after careful optimization of the solvent composition and ESI ionization parameters [85]. Preparation and MS analysis of two model S-nitrosylated Trx1 peptides near neutral pH (6.8) showed reduced MS-induced denitrosylation in the presence of metal chelators such as EDTA or neocuproine. However, tuning of both cone voltage and collision energy was critical to minimize unwanted fragmentation of the Cys-SNO. Since these findings, the inclusion of chelator additives to the buffer has become common for sample preparation and MS analysis of protein nitrosylation. Other factors that challenge the analysis of intact S-nitrosylated proteins by MS are 1) the presence of disulfide-modified proteins formed during preparation of protein SNOs, which may obscure quantification of the SNO signal; 2) spatial transnitrosylation of adjacent Cys residues, which can limit the accurate identification of the “targeted” nitrosylation site. The latter may be overcome by introducing thiol-blocking reagents to alkylate free sulfhydryls after nitrosylation as discussed in Section 4. To this end, it is evident that careful optimization of experimental parameters are needed to fully exploit MS for the analysis of S-nitrosylated proteins, providing a fundamental and complementary technology to assay functional roles of Cys-SNO sites of pure proteins [84, 86, 87].
MS analysis of protein S-thiolation in intact proteins
Protein S-thiolation describes a PTM in which protein cysteines are bound to low molecular weight thiols via disulfide linkage. Mechanistically, S-thiolation originates from both enzymatic and non-enzymatic pathways, typically involving an oxidized, electrophilic cysteine (e.g., Cys-SSR, Cys-SOH, Cys-SNO) and a reduced, nucleophilic thiol, though radical reactions of thiyl and glutathionyl radicals also lead to S-thiolation. S-thiolation occurs as part of normal physiology and signaling in many organisms; however, this PTM is primarily observed in response to stress as a mechanism for preventing irreversible and irreparable protein oxidation of proteins, and in other instances, in the regulation of cell signaling [88]. In vivo, common S-thiolation products are the glutathionyl (Cys-SSG) and cysteinyl (Cys-SSCys) derivatives, and to a lesser extent, the homocysteinyl (Hcy) product [89, 90]. The high abundance of intracellular GSH, ranging from low mM to 10 mM, renders S-glutathionylation the predominant S-thiolation species [91]. Methods utilized to identify targets of glutathionylation on intact proteins include the metabolic incorporation of radiolabeled GSH (35S), chemically induced disulfide formation with biotinylated GSSG, and detection with antibodies against the S-glutathionyl moiety [92]. In recent years, other non-radioactive methods have been developed using the chemoenzymatic formation and incorporation of non-native/tagged-glutathione substrates to identify glutathionylation targets (e.g., biotin-labeled S-glutathionyl spermidine [93] and S-glutathionyl azidoalanine [94]. These methods and others have allowed the identification of over 2200 glutathionylation targets providing insights into structural motifs, protein domain and gene ontology associated with S-glutathionylation [95]. Though these methods are very useful to identify and study targets of S-glutathionylation, other S-thiolation PTMs are less investigated.
A general analysis of intact protein S-thiolation, including S-cysteinylation, S-homocysteinylation and S-glutathionylation, can be achieved using MS. Each modification gives rise to a characteristic mass amenable for simple identification of pure S-thiolated proteins even with low resolution MS. The characteristic Δ mass signals for S-cysteinylation, S-homocysteinylation, and S-glutathionylation from the parent protein are + 119, +143, and + 305 Da, respectively. To identify the type of S-thiolation, a control experiment may be performed in which the sample is treated with DTT followed by MS analysis to monitor the loss of mass corresponding to either Cys, Hcy, and GSH [96, 97]. Recent advances in MS approaches, like top-down proteomics, allow for the analysis of S-thiolation in intact single proteins and simple mixtures and have been recently demonstrated with both protein S-glutathionylation and S-cysteinylation [98, 99]. Recent reports have highlighted several considerations to be taken when analyzing S-thiolation via top-down proteomics, for example, handling protein samples anaerobically and utilizing thiol scavengers [100].
MS analysis of protein S-acylation in intact proteins
S-Acylation of protein cysteine residues is the covalent incorporation of a fatty acid chain on the sulfhydryl group through a thioester linkage, catalyzed by acyltransferase enzymes. A common example of S-acylation on proteins is S-palmitoylation, which is the attachment of a 16-carbon saturated fatty acid chain (palmitic acid), though other fatty acids can be incorporated as well, such as stearic and the unsaturated oleic acid. Assays that enable the detection of intact S-acylated proteins by MS are limited; however, several have provided useful information in the development of robust methods for the global analysis of the S-acylated proteome. These studies revealed that S-acylation can result in multiple fatty acid patterns, meaning that a protein may be modified at a given Cys residue by different fatty acids and also that multiple Cys residues within the same protein could harbor the same fatty acid or a different set of fatty acids [101]. In addition, fatty acid acylation has also been observed at serine and the protein N-terminus [102–104]. The analysis of acylated intact proteins by MS, typically using MALDI-TOF [44], is often coupled with deacylation of the sample of interest to determine the composition, content of the fatty acid (type and %), and amino acid/fatty acid chain connectivity (whether O-, N-, or S- linked). Deacylation is typically achieved via alkaline methanolysis [105, 106], aminolysis via hydroxylamine [107, 108], or hydrogenolysis [109] (Figure 6). Using deacylation conditions, the composition and content of the fatty acid adduct may be revealed, typically in conjunction with fatty acid identification using gas chromatography-MS (GC-MS). Aminolysis is widely used because of the chemoselectivity of hydroxylamine for the thioester over other types of amino acid/fatty acid chain connectivities, however, a recent report indicates that hydrogenolysis provides more reproducible and accurate fatty acid quantification [109].
Figure 6.

Protein deacylation strategies: A. alkaline methanolysis; B. aminolysis; C. hydrogenation [FA = formic acid]
MS to investigate the kinetics of 2-cysteine peroxiredoxin hyperoxidation and repair by sulfiredoxin
Peroxiredoxins (Prx) are highly abundant antioxidant enzymes whose active site Cys cycles through numerous thiol oxidation states during the reduction of H2O2 to water. Many organisms have Prx enzymes, from bacteria to humans, and though their catalytic mechanisms are quite similar, they differ widely in their propensity to form an active site Cys-SO2H, which inactivates the peroxidase. The kinetics of Prx oxidation, the structural factors underlying Prx hyperoxidation, and the sulfiredoxin (Srx)-catalyzed repair of yeast and mammalian Prx enzymes are under investigation using numerous methods including MS [11, 12]. The Srx repair mechanism is Mg2+ and ATP dependent, first involving the formation of a Prx sulfinic phosphoryl ester, which is captured by the Srx active site thiol to form the mixed Prx-Srx thiosulfinate. In the presence of a thiol-based reductant, the thiosulfinate collapses to release Prx-SOH and regenerate active Srx [110] (Figure 7).
Figure 7.

Catalytic cycle of 2-Cys peroxiredoxins and ESI-TOF mass spectra revealing the hyperoxidation sensitivity differences between the human Prx2 and Prx3 enzymes in the presence of H2O2. [Prx: peroxiredoxin; Trx: thioredoxin; TrxR: thioredoxin reductase] Adapted from Haynes et al. [69].
MS has been applied in several different experimental setups to investigate the kinetics of Prx hyperoxidation and repair by Srx. As an example, the first application of online TR-MS for investigations of thiol biochemistry was recently reported by our group in a study of the differential resistance of human Prx 2 and 3 to hyperoxidation/inactivation [69]. Prxs 2 and 3 readily detoxify H2O2 to water through a 2-Cys active site in which the peroxidatic Cys (Cp) first adds to H2O2 to form the Cys-SOH (Figure 7). Local unfolding allows for Cys-SOH capture by the resolving Cys (Cr) of another monomer, leading to the formation of a disulfide bond-linked Prx homodimer. However, with each turnover, a small fraction of Prx in the Cys-SOH state does not unfold and undergoes further reaction with H2O2 to produce the inactive Cys-SO2H (hyperoxidized) species. Prior to this study, it was well-documented that human Prx2 (cytoplasmic) was more susceptible to hyperoxidation than Prx3 (mitochondrial), though the molecular mechanisms underlying the differential resistance were unknown [111]. To investigate the structural factors conferring the intrinsic resistance of Prx3 to inactivation, the formation and reactivity of the protein Cys-SOH species were monitored in the native protein state using TR-MS. Previously, the repair of Cys-SO2H in Prx2 by Srx was monitored at short time points using rapid chemical quench followed by MS [110]. Though rapid chemical quench provides low ms time points, it requires strongly acidic or otherwise harsh quenching methods that often interfere with the detection of unstable intermediates. In the continuous flow TR-MS experiments, equimolar solutions of the Prx and H2O2 (100 μM), both in volatile ammonium-based buffers (bicarbonate or acetate), were loaded into separate syringes then mixed at variable flow rates. Reaction monitoring by TR-MS in the low second time range allowed for the first direct observation of the Cys-SOH for Prx2 (as a minor species), which was consumed by ~6 s. Under these conditions, the corresponding Cys-SOH of Prx3 was not observed, though the oxidized dimer was present, indicating a strong propensity for disulfide formation and a very short lifetime of the sulfenic acid. Additional aspects of this study uncovered key residues near the C-terminal region that impact the susceptibilities for hyperoxidation, however, only with TR-MS was a detailed comparative analysis of Cys-SOH chemistry in the two Prx enzymes possible.
Also reported at this time was a separate study of human Prx2 and Prx3 oxidation kinetics using both intact protein LC/MS and gel electrophoresis. Each Prx enzyme was incubated with varying amounts of substrate H2O2, then quenched by the addition of catalase, at which time reduced Prx Cys-SH were blocked with NEM [112]. Because the Prx SS homodimer ionizes less efficiently than monomeric species, resulting solutions were reduced with DTT to reduce dimeric species (both SS and SS+SO2H, where one Cp is a disulfide partner and the other Cp is a sulfinic acid) to the monomers Prx-SH and Prx-SO2H. Mass spectra obtained for the reduced samples were compared to those of the non-reduced samples to determine the overall ratios of these species for a given H2O2 concentration. These and other studies therein allowed for the determination of rate constants for Prx hyperoxidation and SS dimer formation for both Prx2 and Prx3. The rate constants for the reactions of the peroxidatic thiol (k = 2 × 107 M−1 s−1) and the resulting sulfenic acid (k = 1.2 × 104 M−1 s−1) with H2O2 are relatively equal for Prx2 and Prx3; the drastic difference lies in the relative rate constants for homodimer formation, 2 s−1 for Prx2 and 20 s−1 for Prx3, strongly suggesting that the more rapid rate of dimerization decreases the likelihood for Prx3 hyperoxidation and inactivation [112].
Thus, despite the different experimental and MS approaches used in these studies, the findings were consistent, illustrating the great versatility and advantages of utilizing MS in investigating enzyme kinetics in general and thiol-based kinetics in particular. In the context of Prx biochemistry, MS approaches were also employed to compare the sensitivities of Prx enzymes toward inactivation by peroxide [113], to identify Cys PTMs in mouse Prx6 [114], and to characterize nitration at Tyr193 as an activator of Prx2 activity and protector against hyperoxidation [115].
MS analyses to identify protein conformational changes induced by oxidation
Classical structural biology approaches to studying protein conformational changes resulting from oxidation or other chemical modifications involve the use of x-ray crystallography and nuclear magnetic resonance spectroscopy (NMR). Though these approaches provide unequivocal structural details and are widely used today, the application of MS as a tool for investigating protein structure has gained increasing attention for its relative ease of use and very small sample amount requirement. Studies of protein structure using MS use a diverse set of experimental approaches, though in this section our discussion will be limited to ion mobility MS (IMS-MS), hydrogen-deuterium exchange (HDX) and chemical cross-linking, all in the context of thiol modifications and structural changes imparted by these modifications in proteins.
Ion mobility MS (IMS-MS), as discussed previously, provides a convenient means of separating ESI-generated protein ions on the basis of their collision cross-sectional area (CCS). Changes in protein tertiary structure, such as those resulting from intramolecular disulfide bond formation following treatment with an oxidant, are detected as a result of their modified drift time in the ion mobility tube relative to the untreated species (Figure 8A). IM separation prior to MS acquisition provides CCS values for ions that may be compared to databases of characteristic CCS values to predict changes in protein scaffold, and equally importantly, provides a means of analytical separation of more complex samples, ensuring a more thorough measurement of m/z for relevant species. Using this approach, cysteine residues involved in disulfide bonds were measured by IMS-MS in IgG2 monoclonal antibodies, which are known to form higher order protein complexes [116]. In this study, the use of IMS-MS along with targeted Cys to Ser mutations elucidated the particular disulfide bonding patterns underlying the molecular behavior of IgG2 and provide a means for studying other antibodies, including those used as drug therapies [116]. In addition to intact proteins, IMS-MS has been successfully utilized to map disulfide bonds in peptides. A significant challenge in studying intrapeptide disulfides is the propensity for disulfide scrambling, particularly among peptides with multiple intramolecular disulfide linkages. An alternative approach relied on the cleavage of peptide disulfides and oxidation of disulfide sulfurs to the cysteine sulfonic acid (Cys-SO3H) using m-chloroperbenzoic acid (mCPBA) [117]. This innovation prevented disulfide shuffling on the model peptides and was easily coupled to MS2 sequencing (CID) of the peptides and the modified cysteine sites.
Figure 8.

Overview of strategies for investigating protein conformational changes by mass spectrometry. A: General scheme for the hydrogen-deuterium exchange (HDX) technique used to uncover flexible and/or solvent-exposed regions of protein tertiary and quaternary structure. B: Chemical cross-linking as a valuable means for probing protein conformation in native and perturbed states. In this example, nearby cysteine residues are cross-linked by a homobifunctional maleimide (Mal-Ph-Mal), forming a covalent cross-link stable toward proteolysis and MS ionization and fragmentation. Both intramolecular and intermolecular crosslinking events may occur depending on the unique structure of the protein or protein complex investigated. C: IMS-MS enables separation of protein conformers based on the protein tertiary structure.
Studies of protein tertiary and quaternary structure are also possible with ESI or MALDI MS. Since higher order protein structure is a function of many parameters, including covalent and non-covalent interactions, both inter- and intramolecular, determinations of structure often require milder sample and ionization conditions that do not disrupt structural attributes prior to data acquisition. Protein complexes are especially well suited for ESI and have been analyzed in a number of studies [see [118] for a review on this topic]. Additionally, MS analysis of intact proteins has proven useful in determining the binding stoichiometries of proteins and their cofactors or substrates, though the precise molecular locations where the interactions occur are not readily determined using such methods [119].
To understand dynamic changes in protein structure that do not result in mass (m/z) differences, the hydrogen-deuterium exchange (HDX) method, known for 2-dimensional NMR-based structural studies, was applied to MS. In HDX experiments, proteins in their native states are immersed in a solution of D2O for a controlled time during which exchangeable (relatively acidic) protons in solvent-accessible regions will be replaced with deuterons, inducing a mass change (Figure 8B). Depending on the individual study, the HDX experiments may performed using continuous labeling in which protein aliquots are removed, quenched, and analyzed by MS at different time points after addition of D2O, or using pulse labeling. In pulse labeling, the protein is incubated with D2O for a very short period of time (10 s) after inducing a perturbation in protein structure (e.g., ligand binding) [120]. Sites experiencing the greatest degree of exchange include protons on amide nitrogens and on heteroatoms of amino acid side chains (-OH, -NH2/3, -SH, etc), particularly those in regions of the protein that are at the surface along with those in unstructured or flexible regions. After allowing for sufficient deuterium (2H) incorporation, the exchange is quenched by a rapid reduction in pH (2–3) and temperature (0 °C), denaturing the protein and drastically slowing the exchange. This introduction of acid allows most side chain heteroatom deuterons to exchange back to protons, leaving the amide deuterons, which increase the mass by 1 Da per substitution [119]. These dynamic changes in structure as labeled by HDX are uncovered by the analysis of the intact protein or subsequent to digestion with an acid-stable protease like pepsin.
In the context of Cys active site enzymes, HDX-MS has been utilized to investigate the structural dynamics of peroxiredoxin AhpC from Salmonella typhimurium since the propensity for oxidative inactivation of all Prx enzymes is highly dependent on the structural environment surrounding the active site Cys [121]. In addition, HDX provided a means for analyzing the conformational aspects of monoclonal antibodies before and after drug conjugation, revealing the presence of interchain disulfide bonds that help to stabilize the antibody-drug conjugates [122]. The effects of S-nitrosylation of human glutathione transferase P1-1 at two Cys sites were analyzed using HDX, which revealed that modification of Cys101 is conformationally stabilizing, but dual nitrosylation of Cys47 and Cys101 results in a disruption of structural integrity at the active site and a corresponding loss of enzymatic activity [123]. The wide utility of HDX in studying many types of thiol-based proteins, including those involved in the machinery for Fe-S cluster formation [124], tumor suppressor proteins [125], and studies of biotherapeutics like IgGs [126], illustrate the undeniable importance of this technique in the field of MS-based structural proteomics.
Chemical labeling is a technique applied to investigate residues close in 3-dimensional space, including those involved in non-covalent interactions, using MS. Experimentally, proteins, including those in complexes, are reacted with a reagent that covalently links residues, then digested using a protease of choice. MS analysis of linked peptides provides sequence information of the two peptides linked along with the residues attached (Figure 8C). The length of linker reagents is tailored to gain information about residues at particular distances in the folded protein. Typically, cross-linking reagents are homobifunctional, and involve maleimides to target Cys residues [127] or N-hydroxysuccinimide (NHS) esters to target Lys residues. Lysine residues are more often targeted by researchers using cross-linking approaches for their higher frequency in the proteome relative to cysteine (6% for Lys; <2% for Cys) [128]. The library of available cross-linking reagents has recently become increasingly complex and versatile, including reagents that are equipped with biotin for enrichment of cross-linked peptides [129, 130] along with those that have cleavable sites to either decrease sample complexity prior to MS analysis or to cleave during CID fragmentation [131, 132]. The MS analysis of cross-linked peptides is complicated by their increased m/z, less predictable fragmentation pattern, and potential fragmentation of the linker itself, requiring levels of MS optimization along with advances in the chemical linkers themselves. A recent report details a development in which CID is used in parallel with electron-capture dissociation (ECD): CID cleaves the linker at a predictable site to generate reporter ions, while ECD does not induce linker cleavage and instead provides peptide sequence information [131]. In addition, disulfide-targeted cross-linking reagents have been developed which cleave only in negative ion mode, allowing for complimentary analyses on a single sample [133].
Aside from disulfide mapping, cross-linking studies have relied on the inherent nucleophilicity of cysteine to form adducts with heterobifunctional linkers. Carcinogen furan, a component of smoke, is activated in vivo by the cytochromes P450 to cis-2-butene-1,4-dial (BDA), which readily links Lys and redox-sensitive Cys in proteins, providing a unique signature for proteins or tissues exposed to furan [134]. Similarly, the formation of Cys-guanine-N7 (protein-DNA) conjugates in response to exposure of 1,3-butadiene, a component of vehicle exhaust and cigarette smoke, was identified by MS in over 150 proteins in human fibrosarcoma cells [135]. Lastly, HDX and cross-linking were recently employed in tandem to investigate tertiary structure in the non-cysteine-containing lysostaphin from Staphylococcus simulans [136]. First, HDX-MS was used to identify putative domain interfaces established by non-covalent interactions, then cysteine residues were engineered into the structure at positions predicted to be sufficiently close to promote disulfide bond formation [136]. The successful formation of disulfide bonds, as judged by MS, provided key evidence of the proximity of the two regions in 3-dimensional space, illustrating that studies of protein structure by MS are achievable without amino acid labeling using reactive compounds.
4. Analysis of Thiol Chemistry in Complex Systems Using MS
Though advances in MS technologies and methods have allowed for the direct observation of S-moieties in single intact proteins, the lability of many thiol modifications often hinders the detection of modification sites using digestion and MS/MS analytical methods, though examples of direct identification of various oxoforms by MS/MS exist (e.g., [137]). Global reduction strategies accompanied by differential alkylation provide a useful and robust means for quantifying the extent of protein thiol reversible oxidation. One such method, OxMRM, uses NEM (d0) to label Cys-SH in cell lysates followed by TCEP treatment and labeling of nascent thiols with an isotopically labeled (d5) NEM analog [138]. The differentially labeled peptides are quantified using multiple reaction monitoring (MRM), a quadrupole-based MS quantification method where precursor isolation and fragmentation is followed by the detection of MS2 ions. MRM methods are advantageous for the short times required for acquisition of the MS2 spectrum along with the low limit of quantification afforded by a targeted analysis [138]. Here we will discuss experimental strategies to identify oxidized cysteines in proteins and the residues of modification by MS and MS/MS analysis using selective reduction/switch methods, selective chemical probes, and enrichment strategies.
Detection of oxidized species using selective reduction and tag switch techniques
The reactivity profiles of Cys thiols in proteins was recognized early on as an asset in the development of chemical tools for quantitative proteomics studies by MS leading to development of ICAT described in a previous section. For example, the biotin-switch technique (BST), built on ICAT principles, is a multistep sequence in which first, protein thiols are blocked by an electrophile to generate stable and inert conjugates. Secondly, the thiol PTM of interest is converted back to reduced thiol, which is then captured by an electrophile carrying a detectable handle. Nascent labeled species are then quantified using imaging, Western blot, MS or MS/MS analysis. Most often utilized for thiol tagging is biotin-HPDP, a disulfide-containing reagent with a biotin moiety allowing for avidin-based enrichment. The accuracy of the BST and all selective reduction/switch procedures relies profoundly on the chemoselectivity and yield of each workflow step. For example, unreacted thiols remaining after the initial blocking step react efficiently with the electrophile in the subsequent step. Besides, several electrophiles often used as thiol-blocking reagents in reduction/switch protocols, including IAM, MMTS, and NEM, have shown cross-reactivity with protein Cys-SOH [39]. As discussed above this reactivity could affect result interpretation regardless on the oxoform targeted in the assay.
The chemoselective reduction step allows for a tailoring of the technique for the detection and quantification of a particular cysteine oxoform of interest. For example, ascorbate is typically used to reduce Cys-SNO in proteins [139] while arsenite allows for the selective reduction of Cys-SOH [140], DTT reduction releases disulfide, glutathionylated, and sulfhydrated sites [141, 142], and aminolysis via hydroxylamine exposes acylated sites [143]. Though numerous concerns have been raised regarding the chemoselectivity and putative copper-dependence of the ascorbate reduction for identifying Cys-SNO species [144–147], the reduction/switch methods have evolved, providing streamlined workflows and a focus on residue-level resolution of protein S-nitrosylation. A resin-assisted capture (RAC) version of the BST, called SNO-RAC, utilizes a thiol-reactive resin to capture protein thiols generated by ascorbate reduction of SNOs. RAC and SNO-RAC methods evolved from the concept of QCET (quantitative cysteine enrichment technique), originally applied for the peptide level quantification of Cys thiols using 18O/16O labeling and cysteinyl peptide enrichment [148]. Resin-SNO conjugation allows for the improved retention of formerly nitrosylated proteins and aids in the removal of unreacted ascorbate and unlabeled proteins and peptides. Furthermore, SNO-RAC is compatible with on-resin digestion and the various approaches for MS-based relative quantification (e.g., SILAC, iTRAQ, TMT) [149–151]. Recently, the resin-assisted capture strategy was expanded to the analysis of S-palmitoylated proteins [152]. For the global identification of Cys-SNO residues in proteins, the SNOSID (SNO site identification) method has been developed based on the BST approach, leading to the identification of 68 SNO sites on 56 proteins in GSNO-treated rat cerebellum lysates [153]. In the SNOSID workflow, the BST with biotin-HPDP labeling is followed by proteolytic digestion and a subsequent streptavidin-based enrichment of labeled peptides, facilitating the identification of peptide sequences containing Cys residues sensitive to nitrosylation [153–155]. SNOSID is also compatible with strategies for peptide quantification; methods coupling SNOSID with SILAC and TMT have been described recently [156, 157]. Though the abovementioned BST approaches have enabled the identification and quantification of a wide range of S-nitrosylated proteins in both cell and tissue lysates, the potential pitfalls regarding incomplete blocking of free thiols by IAM, NEM, and MMTS should be considered, as well as the cross-reactivity of these reagents with Cys-SOH. The latter generates a covalent product found to be partially reducible in switch conditions used to identify Cys-SNO, including in the presence of 1 mM ascorbate/Cu(cat), illustrating the possibility of overestimation of nitrosylated Cys sites and proteins.
Selective reduction/switch approaches have also been utilized for the detection of S-glutathionylated proteins by using the enzymatic activity of recombinant glutaredoxins (Grx) to chemoselectively reduce glutathionylated cysteine residues to the corresponding thiols, which are then tagged for imaging or enrichment purposes. Both E. coli and human Grx enzymes have been utilized in this role, which has successfully uncovered S-glutathionylated proteins even in cells and tissue sections [158–161]. Important experimental considerations include performing the assay in the absence of denaturants, unlike the majority of reduction/switch workflows, to preserve the activity of Grx. Additionally, as active Grx is regenerated through the activity of the glutathione reductant system, other necessary reaction components include GSH, GSSG reductase, and NADPH.
Detection of reactive thiols and oxidized species using chemical probes and enrichment strategies
A summary of chemical probes for the detection of modified Cys is included in Table 2.
Table 2.
Chemical probes for the detection of reversibly oxidized cysteine
| Cys-modification | Probe | Structure | Product | References |
|---|---|---|---|---|
|
| ||||
|
Sulfenic acid |
1,3-dicarbonyl core
|
Cyclic R1= H, biotin, azide or alkynyl side chains R2, R3= H, CH3, CD3 |

|
Poole et al. 2007 [60] Klomsiri et al. 2010 [32] Truong et al. 2011 [62] Seo et al. 2011 [174] |
 R= biotin side chain |

|
Qian et al. 2011 [63] | ||
|
| ||||
Acyclic
|

|
Qian et al. 2012 [64] | ||
|
| ||||
| arylboronic acid |

|

|
Liu et al. 2013 [178] | |
|
| ||||
| bicyclononyne |

|

|
Poole et al. 2014 [40] | |
|
| ||||
|
Persulfide |
Methylsulfonyl benzothiazole |
 R1= CH3 or CH2CO2H |

|
Zhang et al. 2014 [35] |
|
| ||||
|
S-Nitrosothiol |
phenylmercury |

|

|
Doulias et al. 2010 [186] |
|
| ||||
| triarylphosphines |

|
|
Zhang et al. 2010 [190] | |

|

|
Bechtold et al. 2010 [42] | ||
|
| ||||
| benzenesulfinic acid |

|

|
Reeves et al. 2013 [195] | |
|
| ||||
|
S-acylation |
metabolic incorporation |

|

|
Hang et al. 2007 [200] Yount et al. 2010 [203] |
Chemical probes for the detection of reduced protein thiols (Cys-SH)
In light of the available strategies for detecting global changes in protein Cys oxidation ([138] and others), a number of studies have investigated how oxidants differentially modify the Cys proteome under a given set of cellular conditions. Though outcomes are affected by a number of factors, including the identity of the oxidant, oxidant concentration, and cellular redox balance between oxidants and antioxidant systems, critical factors are the relative susceptibility of Cys residues to modification and the relative stability of a particular oxoform formed on different residues. Differential Cys nucleophilicity has been probed using the isoTOP-ABPP (isotopic tandem orthogonal proteolysis-activity-based protein profiling) approach, where protein Cys sites are tagged with an alkyne-containing derivative of iodoacetamide later coupled to a biotinylated azide using click chemistry [162]. Isotope labeling of the azide click partner allows for relative quantification of labeled Cys sites in various samples. The premise of this approach is that high (mM range) concentrations of the IAM-based electrophile label protein Cys to provide information of abundance; low (μM) concentrations, at a sub-stoichiometric level, instead capture the most nucleophilic Cys thiols, termed hyper-reactive [162]. Monitoring labeling at various concentrations of IA-probe revealed a small subset of Cys sites (~10% of the Cys proteome) that were equally labeled across all concentrations of IA-probe tested, inferring a hyper-reactive site. Subsequent functional analysis found that more than one-third of residues in the hyper-reactive set are known active site nucleophiles or disulfide partners, illustrating the use of this strategy in identifying functional Cys sites and predicting their function in uncharacterized or less-studied proteins [162].
Endogenous Cys-reactive molecules (and/or their derivatives) are often utilized to experimentally uncover the proteins and the specific Cys sites targeted by modification. A biotinylated ethyl ester of GSH (BioGEE) was designed for cell permeability and enrichment strategies, providing a means to uncover protein Cys sites targeted by S-glutathionylation [163]. Similarly, treatment of human plasma and cells with alkynyl derivatives of reactive endogenous electrophiles like 4-hydroxynonenal (HNE, Figure 9) was coupled with click chemistry, enrichment, and photorelease to identify proteins and cellular pathways differentially targeted by endogenous protein alkylation [164, 165].
Figure 9.

Structures of selected thiol-targeted electrophiles
Additional approaches for investigating the cellular pool of reduced Cys-SH involve labeling of thiols with synthetic derivatives of iodoacetamide, maleimides, or vinyl pyridine that contain biotin (for enrichment) or fluorophores (for cellular imaging) (Figure 9). One such example is the tagging of Cys-SH-containing tryptic peptides with a biotin-containing iodoacetyl probe IBB (N-(2-(2-(2-(2-(3-(1-hydroxy-2-oxo-2-phenylethyl)phenoxy)acetamido)ethoxy)-ethoxy)ethyl)-5-(2-oxohexahydro-1H-thieno[3,4-d]imdazol-4-yl)pentanamide), followed by enrichment and mild cleavage (benzoin moiety cleaved upon treatment with 50 mM ammonium bicarbonate at r.t.) [166]. In addition, because of selectivity shortfalls with IAM and NEM, several groups have recently developed new thiol-blocking electrophiles that offer distinct mechanisms from IAM and NEM [167, 168]. Nucleophilic aromatic substitution has been employed to irreversibly and selectively block thiols (over other amino acids) using methylsulfonyl benzothiazole (MSBT) in nearly quantitative yields in under thirty minutes [169]. The MSBT probe is also reactive with S-sulfhydrated Cys (Cys-SSH) and is discussed in more detail in that context. In a recent report, a new set of heteroaromatic substrates tethered to a methylsulfonyl leaving group such as phenyltetrazole and phenyloxadiazole revealed much faster reaction kinetics [170]. In contrast to S-maleimide adducts, the resulting S-heteroaromatic products (including S-benzothiazole product) showed significant stability under basic conditions and in the presence of GSH [170]. 3-Arylpropiolonitriles (APN) are electron-deficient acetylene compounds that serve as Michael acceptors for Cys-SH [171]. The 3-phenyl analog showed kinetic superiority to IAM, vinyl pyridine, and other moieties, and conjugation to TMPP (tris(2,4,6-trimethoxyphenyl)phosphonium) provided improved LC and MS properties, most notably conferring an increased charge to labeled Cys-containing peptides relative to their corresponding unlabeled partners [171].
Chemical probes for the detection of protein sulfenylation (Cys-SOH)
Sulfenylated Cys sites possess both electrophilic and nucleophilic character, providing a diverse reactivity profile that has led to various methods of detection. Early work revealed that dimedone and alkene functionalities react with Cys-SOH, illustrating their electrophilic character in the presence of carbon nucleophiles [29]. Departing from this seminal work, cyclic 1,3-diketones (dimedone and S-ethyl-cyclopentanedione), alkyl acetoacetate (methyl acetoacetate), and 1,3-cyclic diamides (dimethylbarbituric acid) have been studied to capture Cys-SOH in proteins. In addition, there are instances where sulfenic acid acts as potent nucleophile, reacting with alkylating agents and Michael acceptors [39]. Nonetheless, the majority of studies regarding the detection and identification of sulfenylated sites use cyclic or acyclic 1,3-dicarbonyl reagents due to their high chemoselectivity towards sulfenic acids over other electrophilic PTMs such as persulfides, S-nitrosothiols and mixed disulfides [33, 35]. In this context, dicarbonyl reagents containing biotin and fluorescent tags are available for immunoblotting and imaging analysis [60, 63, 172].
Specific applications of these probes in chemical biology have shown the capability to assign sites of modification and ascribe functional roles of redox-sensitive Cys when used in conjunction with MS [73, 75, 173]. Quantitative proteomic analyses are possible using relative quantification (SILAC, TMT, etc) along with sulfenylated proteome labeling with any of the currently available probes. Alternatively, isotopic labeling of the probe itself provides another avenue for relative redox quantification. A series of dimedone-based light and heavy isotopically coded probes are currently available to quantify redox changes after stimuli, including the thiol-reactive iododimedone (I-dimedone)/d6-dimedone and the Cys-SOH-reactive DAz-2/d6-DAz-2, which were used to quantify the redox status of single proteins [62, 174]. The majority of known Cys-SOH probes are tagged with biotin to enable the enrichment of labeled proteins or contain a terminal azide or alkyne for attachment via click chemistry of the complementary alkyne or azide biotin or fluorophore. Early studies have found that cleavage of the biotin moiety, which suppresses peptide ionization, significantly improved MS and MS/MS spectral quality [175]. In line with these findings, our group disclosed the use of an acetoacetate ester equipped with an alkyne moiety (referred to as alkyne-β-ketoester) to label protein Cys-SOH followed by enrichment via click chemistry with biotinylated azides [64]. The biotin affinity tag was readily removed using mild cleavable conditions such as hydroxylamine (NH2OH) to generate an isoxazolone with improved MS properties relative to dimedone. In a recent report on the influences of various 1,3-dicarbonyl probes also revealed the suppression of ionization induced by dimethylbarbituric acid, though in this study, dimedone and cyclopentane-1,3-dione exhibited less of an effect [176]. Collectively, the loss of sensitivity for the detection of enriched peptides using MS is likely due to the presence of poorly ionizing, readily fragmenting biotin.
Despite the vast amount of knowledge regarding the reactivity and selectivity of 1,3-dicarbonyl probes accumulated throughout the years, thus far the applications of these probes for global MS analyses are limited [61, 173]. There is, however, one very recent report in which isotopically labeled dimedone-based probes DYn-2 (light) and DYn-2-d6 (heavy) were utilized in conjunction with MS to identify and quantify several hundreds of protein Cys-SOH sites within the cellular proteome [177]. This constitutes a significant step forward towards characterization of the thiol redox proteome, though it is difficult at this point to determine the factors contributing to the difference in labeling under H2O2 or growth factor stimulation. For example, the quantitative analysis was performed by incubating the cells with H2O2 or epidermal growth factor for 5–10 min followed by 1–2 h incubation with 5 mM DYn-2. Previous studies have established that these levels of dimedone can be cytotoxic to cells by inducing a strong increase in intracellular H2O2 (likely through inactivation of peroxidases) [64]. Thus the quantification of protein Cys-SOH following this procedure may reflect the effects of the probe addition to cells more than the modifications associated with redox signaling induced by EGF. Indeed, the progress in this area has been until recently limited by the kinetic features of 1,3-dicarbonyl probes, hampering the quantitative labeling of sulfenic acid sites, along with steric effects surrounding redox-sensitive Cys and/or the intrinsic instability of a given Cys-SOH. In addition, the uncertainty of whether the protein Cys-SN is present in solution or is formed only in the gas phase during the MS analysis raises the question of whether the 1,3-dicarbonyl probes are also reactive with this species and implicates this dynamic equilibrium as potentially influencing the kinetic profile of Cys-SOH probes. In early work, cyclopenta-1,3-dione was found to react with a small molecule surrogate of the cyclic sulfenamide, suggesting that perhaps this can occur with protein sulfenamides as well [71]. The finding of IAM reactivity with Cys-SOH opens a possible avenue to address this question and an approach is described in Figure 5. These challenges have prompted investigations of other reactive partners for Cys-SOH such as aryl boronic acid substrates [178] and the recently reported strained bicyclononynes, which covalently trap Cys-SOH by mechanisms distinct from the 1,3-dicarbonyls [40]. The latter has revealed reaction rates with protein Cys-SOH >300 times faster than dimedone allowing for effective trapping of Cys-SOH using micromolar amounts of probe and much shorter incubation times. Furthermore, the corresponding sulfoxide adduct is strongly amenable to MS detection on both intact proteins by ESI-TOF MS and on labeled peptides by MS2. In particular, this most recent series of reagents show great promise for future quantitative analysis of protein S-sulfenylation by alleviating at least some of the current caveats of 1,3-dicarbonyl probes. In an ideal workflow the probes would rapidly react to quantitatively consume the Cys-SOH instantaneously at the time of lysis before these moieties convert into other oxidized species (i.e., Cys-SOH probes should effectively compete with disulfide bond formation). Addition of probes to cells should be avoided, as protein modification by the probes would alter protein activity and lead to shifts in signaling and metabolic fluxes independent of the physiological or pathological stimuli being investigated.
Chemical agents and strategies for the analysis of protein persulfides (Cys-SSH) by MS
Chemical strategies for the global analysis of protein persulfides are being developed at a rapid pace, but there is a limited number of reagents and methods that have been thoroughly validated in the field. Currently available methods are primarily challenged by the similar reactivity of Cys-SSH compared to the sulfhydryl group (Cys-SH) and persulfide (Cys-SSH), which has prompted the utilization of blocking reagents [33, 142]. Therefore, it is not surprising that various BST- and switch-derived methods have been developed [141, 142]. At the same time, the notion of activating the S-SH moiety (for subsequent labeling) and simultaneously blocking the sulfhydryl group has also been explored [35]. S-Benzothiazole disulfide adducts derived from the reaction of Cys-SSH and methylsulfonylbenzothiazole (MSBT), were found to activate the sulfur atom of the Cys counterpart to undergo nucleophilic substitution reaction with methyl or biotin-tagged α-cyanoacetate (MCA), generating a thioether-linked protein in a tag-switch assay (Figure 10) [169]. Using a biotinylated α-cyanoacetate probe, this strategy enables the detection of protein Cys-SSH in cells by fluorescence imaging and/or immunoblotting. Recently, the method was evaluated for the global analysis of protein persulfides in the A549 cell line, establishing its effectiveness for use in proteomics studies [81]. Of great interest is the fact that this method can also detect polysulfide-modified proteins, suggesting that persulfides and polysulfides coexist in cells, and inferring that both PTMs may serve as signaling effector species. Given the potential of the tag-switch assay for analyzing the persulfide/polysulfide proteome, control experiments are needed to subtract any discrepancies in the event that the α-cyanoacetate probe reacts with endogenous protein sulfenic acids.
Figure 10.
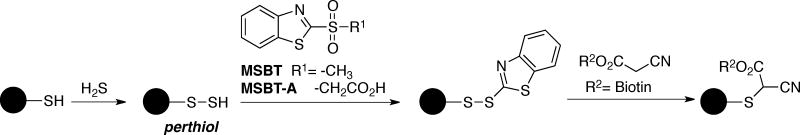
Tag-switch method for protein sulfhydration
Chemical agents and strategies for the analysis of protein S-nitrosylation (Cys-SNO)
The instability of S-nitrosothiols toward light, heat, and in non-physiological pH requires chemical derivatization for the global analysis of Cys-SNO-containing proteins and peptides. The approaches for nitrosothiol bioconjugation often rely on the electrophilicity and/or Lewis basicity of the moiety. As with sulfenic acids, the low physiological abundance of nitrosothiols makes the use of enrichment methods necessary prior to MS analysis. Described next are several transition metal and nucleophile reagents with demonstrated success for the enrichment and detection of nitrosothiols in proteins and peptides.
Gold Nanoparticles
S-Nitrosothiols react readily with metal centers, particularly those from groups 8, 10 and 11 of the periodic table (e.g., Cu(II), Fe(III), Hg(II), Au(III)) [82, 179–182]. This reactivity has been exploited in the development of detection and quantification methods for both low molecular weight and proteins S-nitrosothiols. A gold nanoparticle-(AuNP)-based method provides a means for the enrichment of Cys-SNO-containing peptides after digestion [183]. The workflow of this method consists of 1) thiol blocking; 2) proteolysis; 3) binding to AuNP and enrichment; 4) DTT treatment to release AuNP-bound peptides; 5) identification of denitrosylated peptides by MS. The effectiveness of this strategy was shown with S-nitrosylated peptides of protein disulfide isomerase and cysteine-based protein tyrosine phosphatase hYVH1 [183]. However, AuNP also exerted high affinity toward thioethers, including those derived from thiol blocking with IAM, methionine residues, and glutathionylated cysteine peptides. The method was modified to distinguish Cys-SNO-containing peptides from other Cys modifications by differentially tagging denitrosylated peptides with NEM much like biotin-HPDP labeling in the BST assay. Nonetheless, the feasibility of this modified method in complex biological samples needs to be addressed.
Phenylmercury resin and biotin
The chemical reduction of Cys-SNO using mercury salts was the landmark of the Saville assay, one of the earliest detection methods for nitrosylated cysteine [184]. On the basis of this method, an approach was later developed that utilized phenylmercury tethered to an agarose resin or biotin to enrich S-nitrosylated proteins, both pure proteins [185] and in more complex samples like cells and tissues [186]. The workflow of this method consists of four main steps: 1) thiol blocking using MMTS; 2) conjugation of Cys-SNO proteins with phenylmercury; 3) proteolysis; 4) release of bound peptides from Hg(II) using performic acid. The final step produces a cysteine sulfonic acid, which serves as the MS-fingerprint for site identification. In recent studies the selectivity of the organomercurial compound for Cys-SNO against other endogenously or exogenously produced Cys-modified proteins was addressed using GAPDH as a model. In addition to the fully reduced form, modified GAPDH species tested included GAPDH-S-NEM (derived from the thiol-blocking step), S-glutathionylated, and hyperoxidized protein (e.g., GAPDH-SO2H and GAPDH-SO3H). Each form of GAPDH showed homologous modification at active site Cys245 [185]. Only the S-nitrosylated peptide containing Cys245 was released from phenylmercury upon treatment with reductant β-mercaptoethanol, and no other modifications yielded a detectable signal for this peptide, establishing the selectivity of this method toward S-nitrosylated proteins. However, negative controls are essential in this approach to subtract any false positive signals generated from inefficient thiol blocking. Nonetheless, this approach has enabled the detection of over one thousand Cys-SNO sites in various tissues in mice including liver, heart, kidney, lung and others with high confidence [187].
Triarylphosphine-based bioorthogonal reactions
Increased desire to exclude the use of thiol-blocking reagents and to simplify data analysis has triggered the development of chemical probes selective for S-nitrosothiols. Organophosphine compounds equipped with electrophilic traps were found to mediate chemical ligations of S-nitrosothiols and have been exploited as a detection tool for small molecule and protein SNOs [188–191] and for the quantification of intracellular GSNO [192]. In addition, the use of fully aqueous soluble, sterically hindered organophosphine tris(4,6-dimethyl-3-sulfonatophenyl)phosphine trisodium salt hydrate (TXPTS), that does not possess an electrophilic trap, was also demonstrated to react with S-nitrosothiols, yielding a unique covalent adduct [42]. The use of Cys165Ser AhpC-SNO, generated via transnitrosylation by Cys-SNO, revealed that TXPTS reacted with the nitrosothiol to provide a covalent adduct proposed as S-alkylthiophosphonium by ESI-TOF MS and complementary NMR-based approaches (31P NMR, 1H/31P COSY, 1H/13C HMQC) [42]. This adduct was found to be stable under aqueous conditions, and in addition, control experiments indicated that TXPTS is inert toward disulfide cleavage (using the Cys165Ser AhpC-S-S-Cys mixed disulfide) at 42 hours of reaction time. Despite the potential of TXPTS as labeling tool for protein Cys-SNO using MS, this phosphine has not yet been investigated with more complex samples such as cell lysates. Recently, the electrophilicities of BSA-SNO and GSNO have been exploited for their detection by derivatization with benzenesulfinic acid (PhSO2H) to generate a benzenethiosulfonate covalent adduct, a well-known reaction for low molecular weight S-nitrosothiols [193–195]. This reactivity provides an exciting new avenue for Cys-SNO detection, but its utility in cell lysate mixtures may be hindered by susceptibility of thiosulfonates toward reduction by cellular reductants such as thiols.
Chemical agents and strategies for the analysis of protein S-acylation (Cys-S(C=O)R) by MS
Metabolic incorporation of radiolabeled fatty acids was the initial chemical and analytic tool to identify physiological targets of S-acylation [196]. With the rapid dissemination that azido-(-N3)- and alkynyl-containing bioactive molecules are metabolically incorporated into macromolecules and amenable for bioorthogonal reactions, the use of radiolabeled molecules has become less attractive [197]. In light of these studies, various groups have developed “tagged” fatty acids, both azido- and alkynyl-modified, to allow metabolic incorporation and endogenous S-acylation of proteins [198, 199]. Both the Staudinger ligation and click reactions have been effective for the labeling and detection of S-palmitoylated proteins [198–202]. A large-scale proteomic analysis using the click chemistry approach has been exploited, resulting in the identification of over one hundred S-palmitoylated proteins and sites [203]. Despite the unbiased enrichment of proteins and identification of S-palmitoylated cysteine sites using this approach, in some cases the total S-acylated sites and proteins can be underestimated, for instance, if switch-like control experiments (thiol blocking/deacylation/thiol labeling) are not performed to account for thioester cleavage during MS analysis [204].
5. Concluding remarks
Redox reactions and associated reactive oxygen and nitrogen species (ROS/RNS) are key drivers of growth and survival in biological systems, and widely acknowledged today as integral regulatory components in signaling, metabolism, and epigenetics [205]. While the controlled localized production of ROS/RNS is critical to normal physiology and health, the disruption of redox homeostasis through the acute or chronic accumulation of these reactive oxidants leads to numerous pathologies in humans including cancer, cardiovascular diseases, diabetes, and others. Thus, investigation of the molecular mechanisms linking redox balance to disease has been the focus of numerous studies in the scientific community spanning virtually all areas of research. The modification of protein thiols by ROS/RNS has been a major component of this large research endeavor with over 10,000 PubMed references on the topic of thiol oxidation alone. While the current technologies and methods to study the chemical behavior and kinetics of specific thiol modifications in isolated proteins are adequate, pinpointing these changes with high selectivity in complex systems is still a great chemical and instrumentation challenge due to the low stoichiometry of modification. As highlighted in this review, qualitative and quantitative analysis of Cys modification in proteins is complicated by the overlapping reactivity profiles of several Cys-derived species along with a limited number of reliable chemical tools available for some thiol oxoforms. Thus careful consideration of the analytical limitations for detecting cysteine PTMs and the potential for chemical cross-reactivity among the species and their probes is highly critical to this field of investigation. Development of selective and highly reactive chemical reagents capable of parallel tracking of specific ROS/RNS species and their protein cysteine targets in real time continues to be a significant challenge in the field of protein thiol chemistry. Unlike phosphorylation and other PTMs, thiol modifications in proteins are highly dynamic events and one can have multiple oxidative PTMs at the same thiol site in the population of protein molecules (e.g., Cys-SNO, Cys-SOH, Cys-SSG), as well as further conversion of initial oxidized species over time (e.g., Cys-SOH to Cys-SN or Cys-SSCys) or through reactions with other molecules within cells. Since in most cases these reactions are rapid, not only selectivity but also the kinetic parameters of chemical probes for these species are critically important. While targeted and broad scale MS-based time course studies of specific oxidative PTMs have been published using tag-switch methods or highly selective reagents, the difficulty comes with interpreting the results. For example, what can we conclude when there is an increase or decrease in a given oxidative PTM? Typically this is presented as having more or less of the oxidized PTM at a given time point. However, one cannot exclude the possibility that more labeling at longer time points is due not to an increase in the oxidized species but to the kinetics of the probe reaction with the oxidative PTM or to the continuous cycling through the respective, potentially transient species. This is particularly an issue for slower reacting probes and especially when the probes are added to cells in time course studies if the incubation time with the probe is not strictly controlled. Similarly, it is difficult to interpret a decrease in a particular oxidative PTM when this is monitored alone and not in the context of other PTMs. Is the decrease due to reduction back to the -SH state or is it due to conversion into other oxidative PTMs (e.g., Cys-SOH conversion to Cys-SO2/3, Cys-SSCys, Cys-SN, Cys-SSG). For now, these questions are being addressed on a case-by-case basis but given the accomplishments thus far, we will have in the near future a set of chemical probes for each of the multiple oxidative PTMs (and the reduced SH) to enable parallel monitoring using MS-based technologies. Equally and critically important would be the integration of oxidative PTMs in computational analysis of cellular networks. Most of the current efforts on modeling cellular signaling and metabolism rely on protein expression and phosphorylation as the main parameters driving the pathway activity. However, direct oxidation of proteins has been shown over the last decades to profoundly impact the enzyme activity and subcellular localization of proteins. Yet, the timing and amplitude of oxidative modifications and their effects on regulation of cellular networks are rarely considered in systems biology studies. An important requisite to enable such studies is the creation of a comprehensive thiol redox database with annotated sites of modification, nature of modification, consequence of modification on protein activity and the biological context where the respective modification was observed. Compartmentalization of redox signaling and metabolism is another area of investigation that continues to evolve and needs to be considered in both experimental and computational studies. Unlike other PTMs, oxidative PTMs are expected to be highly dependent on the subcellular localization of both the enzymes involved in ROS/RNS metabolism and their protein targets and are also influenced by the redox potential of cellular compartments and that of the extracellular microenvironment.
Thus, many challenging tasks lie ahead and though these may seem daunting, the field of thiol-based signaling and metabolism is poised to make these significant contributions by combining improved chemical tools with omics technologies such as MS and advanced computational methods. The hard work and innovation brought about by many groups whose work we cited in this review and many others has profoundly impacted and will continue to advance this critically important field of research with broad implications in biology and medicine.
Acknowledgments
Financial support was provided by the National Cancer Institute of the National Institutes of Health under award numbers R01 CA136810 and R33 CA177461 (C.M.F.), and TRADONC fellowship T32 CA113267 (N.O.D.B.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting galley proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Thomson JJL., VIII On the masses of the ions in gases at low pressures. Philos Mag Series 5. 1899;48:547–567. [Google Scholar]
- 2.Karas M, Hillenkamp F. Laser desorption ionization of proteins with molecular masses exceeding 10,000 Daltons. Anal Chem. 1988;60:2299–2301. doi: 10.1021/ac00171a028. [DOI] [PubMed] [Google Scholar]
- 3.Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Electrospray ionization for mass spectrometry of large biomolecules. Science. 1989;246:64–71. doi: 10.1126/science.2675315. [DOI] [PubMed] [Google Scholar]
- 4.Ganem B, Li YT, Henion JD. Detection of noncovalent receptor-ligand complexes by mass spectrometry. J Am Chem Soc. 1991;113:6294–6296. [Google Scholar]
- 5.Siuzdak G. Probing viruses with mass spectrometry. J Mass Spectrom. 1998;33:203–211. doi: 10.1002/(SICI)1096-9888(199803)33:3<203::AID-JMS653>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 6.Henzel WJ, Billeci TM, Stults JT, Wong SC, Grimley C, Watanabe C. Identifying proteins from two-dimensional gels by molecular mass searching of peptide fragments in protein sequence databases. Proc Natl Acad Sci U S A. 1993;90:5011–5015. doi: 10.1073/pnas.90.11.5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol. 1999;17:994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 8.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 9.Thompson A, Schafer J, Kuhn K, Kienle S, Schwarz J, Schmidt G, Neumann T, Johnstone R, Mohammed AK, Hamon C. Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal Chem. 2003;75:1895–1904. doi: 10.1021/ac0262560. [DOI] [PubMed] [Google Scholar]
- 10.Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, Purkayastha S, Juhasz P, Martin S, Bartlet-Jones M, He F, Jacobson A, Pappin DJ. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics. 2004;3:1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 11.Biteau B, Labarre J, Toledano MB. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature. 2003;425:980–984. doi: 10.1038/nature02075. [DOI] [PubMed] [Google Scholar]
- 12.Chang TS, Jeong W, Woo HA, Lee SM, Park S, Rhee SG. Characterization of mammalian sulfiredoxin and its reactivation of hyperoxidized peroxiredoxin through reduction of cysteine sulfinic acid in the active site to cysteine. J Biol Chem. 2004;279:50994–51001. doi: 10.1074/jbc.M409482200. [DOI] [PubMed] [Google Scholar]
- 13.Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 14.Gama MR, Collins CH, Bottoli CB. Nano-liquid chromatography in pharmaceutical and biomedical research. J Chromatogr Sci. 2013;51:694–703. doi: 10.1093/chromsci/bmt023. [DOI] [PubMed] [Google Scholar]
- 15.Boersema PJ, Mohammed S, Heck AJ. Hydrophilic interaction liquid chromatography (HILIC) in proteomics. Anal Bioanal Chem. 2008;391:151–159. doi: 10.1007/s00216-008-1865-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Horvatovich P, Hoekman B, Govorukhina N, Bischoff R. Multidimensional chromatography coupled to mass spectrometry in analysing complex proteomics samples. J Sep Sci. 2010;33:1421–1437. doi: 10.1002/jssc.201000050. [DOI] [PubMed] [Google Scholar]
- 17.Kanu AB, Dwivedi P, Tam M, Matz L, Hill HH., Jr Ion mobility-mass spectrometry. J Mass Spectrom. 2008;43:1–22. doi: 10.1002/jms.1383. [DOI] [PubMed] [Google Scholar]
- 18.Uetrecht C, Rose RJ, van Duijn E, Lorenzen K, Heck AJ. Ion mobility mass spectrometry of proteins and protein assemblies. Chem Soc Rev. 2010;39:1633–1655. doi: 10.1039/b914002f. [DOI] [PubMed] [Google Scholar]
- 19.Zhong Y, Hyung S-J, Ruotolo BT. Ion mobility-mass spectrometry for structural proteomics. Expert Rev Proteomics. 2012;9:47–58. doi: 10.1586/epr.11.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Caprioli RM, Farmer TB, Gile J. Molecular imaging of biological samples: Localization of peptides and proteins using MALDI-TOF MS. Anal Chem. 1997;69:4751–4760. doi: 10.1021/ac970888i. [DOI] [PubMed] [Google Scholar]
- 21.Gessel MM, Norris JL, Caprioli RM. MALDI imaging mass spectrometry: Spatial molecular analysis to enable a new age of discovery. J Proteomics. 2014;107c:71–82. doi: 10.1016/j.jprot.2014.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ho CS, Lam CW, Chan MH, Cheung RC, Law LK, Lit LC, Ng KF, Suen MW, Tai HL. Electrospray ionisation mass spectrometry: principles and clinical applications. Clin Biochem Rev. 2003;24:3–12. [PMC free article] [PubMed] [Google Scholar]
- 23.Hu Q, Noll RJ, Li H, Makarov A, Hardman M, Graham Cooks R. The Orbitrap: a new mass spectrometer. J Mass Spectrom. 2005;40:430–443. doi: 10.1002/jms.856. [DOI] [PubMed] [Google Scholar]
- 24.Olsen JV, Macek B, Lange O, Makarov A, Horning S, Mann M. Higher-energy C-trap dissociation for peptide modification analysis. Nat Methods. 2007;4:709–712. doi: 10.1038/nmeth1060. [DOI] [PubMed] [Google Scholar]
- 25.Syka JEP, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc Natl Acad Sci U S A. 2004;101:9528–9533. doi: 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mikesh LM, Ueberheide B, Chi A, Coon JJ, Syka JE, Shabanowitz J, Hunt DF. The utility of ETD mass spectrometry in proteomic analysis. Biochim Biophys Acta. 2006;12:30. doi: 10.1016/j.bbapap.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen M, Cook KD. Oxidation artifacts in the electrospray mass spectrometry of Aβ Peptide. Anal Chem. 2007;79:2031–2036. doi: 10.1021/ac061743r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zang L, Carlage T, Murphy D, Frenkel R, Bryngelson P, Madsen M, Lyubarskaya Y. Residual metals cause variability in methionine oxidation measurements in protein pharmaceuticals using LC-UV/MS peptide mapping. J Chromatogr B Analyt Technol Biomed Life Sci. 2012;896:71–76. doi: 10.1016/j.jchromb.2012.03.016. [DOI] [PubMed] [Google Scholar]
- 29.Benitez LV, Allison WS. The inactivation of the acyl phosphatase activity catalyzed by the sulfenic acid form of glyceraldehyde 3-phosphate dehydrogenase by dimedone and olefins. J Biol Chem. 1974;249:6234–6243. [PubMed] [Google Scholar]
- 30.Lin WS, Armstrong DA, Gaucher GM. Formation and repair of papain sulfenic acid. Can J Biochem. 1975;53:298–307. doi: 10.1139/o75-042. [DOI] [PubMed] [Google Scholar]
- 31.Francoleon NE, Carrington SJ, Fukuto JM. The reaction of H2S with oxidized thiols: Generation of persulfides and implications to H2S biology. Arch Biochem Biophys. 2011;516:146–153. doi: 10.1016/j.abb.2011.09.015. [DOI] [PubMed] [Google Scholar]
- 32.Klomsiri C, Nelson KJ, Bechtold E, Soito L, Johnson LC, Lowther WT, Ryu S-E, King SB, Furdui CM, Poole LB. Chapter 3 - Use of Dimedone-Based Chemical Probes for Sulfenic Acid Detection: Evaluation of Conditions Affecting Probe Incorporation into Redox-Sensitive Proteins. In: Enrique C, Lester P, editors. Methods in Enzymology. Academic Press; 2010. pp. 77–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pan J, Carroll KS. Persulfide reactivity in the detection of protein S-sulfhydration. ACS Chem Biol. 2013;8:1110–1116. doi: 10.1021/cb4001052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carballal S, Radi R, Kirk MC, Barnes S, Freeman BA, Alvarez B. Sulfenic acid formation in human serum albumin by hydrogen peroxide and peroxynitrite. Biochemistry. 2003;42:9906–9914. doi: 10.1021/bi027434m. [DOI] [PubMed] [Google Scholar]
- 35.Zhang D, Macinkovic I, Devarie-Baez NO, Pan J, Park C-M, Carroll KS, Filipovic MR, Xian M. Detection of protein S-sulfhydration by a tag-switch technique. Angew Chem Int Ed Engl. 2014;53:575–581. doi: 10.1002/anie.201305876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boschi-Muller S, Azza S, Sanglier-Cianferani S, Talfournier F, Van Dorsselear A, Branlant G. A sulfenic acid enzyme intermediate is involved in the catalytic mechanism of peptide methionine sulfoxide reductase from Escherichia coli. J Biol Chem. 2000;275:35908–35913. doi: 10.1074/jbc.M006137200. [DOI] [PubMed] [Google Scholar]
- 37.Lin Z, Johnson LC, Weissbach H, Brot N, Lively MO, Lowther WT. Free methionine-(R)-sulfoxide reductase from Escherichia coli reveals a new GAF domain function. Proc Natl Acad Sci U S A. 2007;104:9597–9602. doi: 10.1073/pnas.0703774104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ellis HR, Poole LB. Roles for the two cysteine residues of AhpC in catalysis of peroxide reduction by alkyl hydroperoxide reductase from Salmonella typhimurium. Biochemistry. 1997;36:13349–13356. doi: 10.1021/bi9713658. [DOI] [PubMed] [Google Scholar]
- 39.Reisz JA, Bechtold E, King SB, Poole LB, Furdui CM. Thiol-blocking electrophiles interfere with labeling and detection of protein sulfenic acids. FEBS J. 2013;280:6150–6161. doi: 10.1111/febs.12535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Poole TH, Reisz JA, Zhao W, Poole LB, Furdui CM, King SB. Strained cycloalkynes as new protein sulfenic acid traps. J Am Chem Soc. 2014;136:6167–6170. doi: 10.1021/ja500364r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim SO, Merchant K, Nudelman R, Beyer WF, Jr, Keng T, DeAngelo J, Hausladen A, Stamler JS. OxyR: a molecular code for redox-related signaling. Cell. 2002;109:383–396. doi: 10.1016/s0092-8674(02)00723-7. [DOI] [PubMed] [Google Scholar]
- 42.Bechtold E, Reisz JA, Klomsiri C, Tsang AW, Wright MW, Poole LB, Furdui CM, King SB. Water-soluble triarylphosphines as biomarkers for protein S-nitrosation. ACS Chem Biol. 2010;5: 405–414. doi: 10.1021/cb900302u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wilson DJ, Konermann L. A capillary mixer with adjustable reaction chamber volume for millisecond time-resolved studies by electrospray mass spectrometry. Anal Chem. 2003;75:6408–6414. doi: 10.1021/ac0346757. [DOI] [PubMed] [Google Scholar]
- 44.Wilson DJ, Konermann L. Mechanistic studies on enzymatic reactions by electrospray ionization MS using a capillary mixer with adjustable reaction chamber volume for time-resolved measurements. Anal Chem. 2004;76:2537–2543. doi: 10.1021/ac0355348. [DOI] [PubMed] [Google Scholar]
- 45.Wilson DJ, Konermann L. Ultrarapid desalting of protein solutions for electrospray mass spectrometry in a microchannel laminar flow device. Anal Chem. 2005;77:6887–6894. doi: 10.1021/ac050902o. [DOI] [PubMed] [Google Scholar]
- 46.Li ZL, Sau AK, Shen SD, Whitehouse C, Baasov T, Anderson KS. A snapshot of enzyme catalysis using electrospray ionization mass spectrometry. J Am Chem Soc. 2003;125:9938–9939. doi: 10.1021/ja0354768. [DOI] [PubMed] [Google Scholar]
- 47.Konermann L, Pan JX, Wilson DJ. Protein folding mechanisms studied by time-resolved electrospray mass spectrometry. Biotechniques. 2006;40:135–141. doi: 10.2144/06402TE01. [DOI] [PubMed] [Google Scholar]
- 48.Rob T, Liuni P, Gill PK, Zhu SL, Balachandran N, Berti PJ, Wilson DJ. Measuring dynamics in weakly structured regions of proteins using microfluidics-enabled subsecond H/D exchange mass spectrometry. Anal Chem. 2012;84:3771–3779. doi: 10.1021/ac300365u. [DOI] [PubMed] [Google Scholar]
- 49.Liuni P, Jeganathan A, Wilson DJ. Conformer selection and intensified dynamics during catalytic turnover in chymotrypsin. Angew Chem Int Ed Engl. 2012;51:9666–9669. doi: 10.1002/anie.201204903. [DOI] [PubMed] [Google Scholar]
- 50.Paiva AA, Tilton RF, Jr, Crooks GP, Huang LQ, Anderson KS. Detection and identification of transient enzyme intermediates using rapid mixing, pulsed-flow electrospray mass spectrometry. Biochemistry. 1997;36:15472–15476. doi: 10.1021/bi971883i. [DOI] [PubMed] [Google Scholar]
- 51.Zechel DL, Konermann L, Withers SG, Douglas DJ. Pre-steady state kinetic analysis of an enzymatic reaction monitored by time-resolved electrospray ionization mass spectrometry. Biochemistry. 1998;37:7664–7669. doi: 10.1021/bi980445o. [DOI] [PubMed] [Google Scholar]
- 52.Li ZL, Sau AK, Furdui CM, Anderson KS. Probing the role of tightly bound phosphoenolpyruvate in Escherichia coli 3-deoxy-D-manno-octulosonate 8-phosphate synthase catalysis using quantitative time-resolved electrospray ionization mass spectrometry in the millisecond time range. Analytical Biochemistry. 2005;343:35–47. doi: 10.1016/j.ab.2005.04.021. [DOI] [PubMed] [Google Scholar]
- 53.Roberts A, Furdui C, Anderson KS. Observation of a chemically labile, noncovalent enzyme intermediate in the reaction of metal-dependent Aquifex pyrophilus KDO8PS by time-resolved mass spectrometry. Rapid Commun Mass Spectrom. 2010;24:1919–1924. doi: 10.1002/rcm.4594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Clarke DJ, Stokes AA, Langridge-Smith P, Mackay CL. Online quench-flow electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry for elucidating kinetic and chemical enzymatic reaction mechanisms. Anal Chem. 2010;82:1897–1904. doi: 10.1021/ac9026302. [DOI] [PubMed] [Google Scholar]
- 55.Konermann L, Collings BA, Douglas DJ. Cytochrome c folding kinetics studied by time-resolved electrospray ionization mass spectrometry. Biochemistry. 1997;36:5554–5559. doi: 10.1021/bi970046d. [DOI] [PubMed] [Google Scholar]
- 56.Kolakowski BM, Simmons DA, Konermann L. Stopped-flow electrospray ionization mass spectrometry: A new method for studying chemical reaction kinetics in solution. Rapid Commun Mass Spectrom. 2000;14:772–776. doi: 10.1002/(SICI)1097-0231(20000515)14:9<772::AID-RCM942>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 57.Miao Z, Chen H, Liu P, Liu Y. Development of submillisecond time-resolved mass spectrometry using desorption electrospray ionization. Anal Chem. 2011;83:3994–3997. doi: 10.1021/ac200842e. [DOI] [PubMed] [Google Scholar]
- 58.Perry RH, Splendore M, Chien A, Davis NK, Zare RN. Detecting reaction intermediates in liquids on the millisecond time scale using desorption electrospray ionization. Angew Chem Int Ed Engl. 2011;50:250–254. doi: 10.1002/anie.201004861. [DOI] [PubMed] [Google Scholar]
- 59.Poole LB. Measurement of protein sulfenic acid content. Curr Protoc Toxicol. 2008;17 doi: 10.1002/0471140856.tx1702s38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Poole LB, Klomsiri C, Knaggs SA, Furdui CM, Nelson KJ, Thomas MJ, Fetrow JS, Daniel LW, King SB. Fluorescent and affinity-based tools to detect cysteine sulfenic acid formation in proteins. Bioconjug Chem. 2007;18:2004–2017. doi: 10.1021/bc700257a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Leonard SE, Reddie KG, Carroll KS. Mining the thiol proteome for sulfenic acid modifications reveals new targets for oxidation in cells. ACS Chem Biol. 2009;4:783–799. doi: 10.1021/cb900105q. [DOI] [PubMed] [Google Scholar]
- 62.Truong TH, Garcia FJ, Seo YH, Carroll KS. Isotope-coded chemical reporter and acid-cleavable affinity reagents for monitoring protein sulfenic acids. Bioorg Med Chem Lett. 2011;21:5015–5020. doi: 10.1016/j.bmcl.2011.04.115. [DOI] [PubMed] [Google Scholar]
- 63.Qian J, Klomsiri C, Wright MW, King SB, Tsang AW, Poole LB, Furdui CM. Simple synthesis of 1,3-cyclopentanedione derived probes for labeling sulfenic acid proteins. Chem Commun (Camb) 2011;47:9203–9205. doi: 10.1039/c1cc12127h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Qian J, Wani R, Klomsiri C, Poole LB, Tsang AW, Furdui CM. A simple and effective strategy for labeling cysteine sulfenic acid in proteins by utilization of β-ketoesters as cleavable probes. Chem Commun (Camb) 2012;48:4091–4093. doi: 10.1039/c2cc17868k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kim JS, Raines RT. A misfolded but active dimer of bovine seminal ribonuclease. Eur J Biochem. 1994;224:109–114. doi: 10.1111/j.1432-1033.1994.tb20001.x. [DOI] [PubMed] [Google Scholar]
- 66.Steen H, Mann M. Similarity between condensed phase and gas phase chemistry: fragmentation of peptides containing oxidized cysteine residues and its implications for proteomics. J Am Soc Mass Spectrom. 2001;12:228–232. doi: 10.1016/S1044-0305(00)00219-1. [DOI] [PubMed] [Google Scholar]
- 67.Salmeen A, Andersen JN, Myers MP, Meng TC, Hinks JA, Tonks NK, Barford D. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature. 2003;423:769–773. doi: 10.1038/nature01680. [DOI] [PubMed] [Google Scholar]
- 68.van Montfort RL, Congreve M, Tisi D, Carr R, Jhoti H. Oxidation state of the active-site cysteine in protein tyrosine phosphatase 1B. Nature. 2003;423:773–777. doi: 10.1038/nature01681. [DOI] [PubMed] [Google Scholar]
- 69.Haynes AC, Qian J, Reisz JA, Furdui CM, Lowther WT. Molecular basis for the resistance of human mitochondrial 2-Cys peroxiredoxin 3 to hyperoxidation. J Biol Chem. 2013;288:29714–29723. doi: 10.1074/jbc.M113.473470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fuangthong M, Helmann JD. The OhrR repressor senses organic hydroperoxides by reversible formation of a cysteine-sulfenic acid derivative. Proc Natl Acad Sci U S A. 2002;99:6690–6695. doi: 10.1073/pnas.102483199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shiau TP, Erlanson DA, Gordon EM. Selective reduction of peptide isothiazolidin-3-ones. Org Lett. 2006;8:5697–5699. doi: 10.1021/ol062077j. [DOI] [PubMed] [Google Scholar]
- 72.Regazzoni L, Panusa A, Yeum KJ, Carini M, Aldini G. Hemoglobin glutathionylation can occur through cysteine sulfenic acid intermediate: electrospray ionization LTQ-Orbitrap hybrid mass spectrometry studies. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877:3456–3461. doi: 10.1016/j.jchromb.2009.05.020. [DOI] [PubMed] [Google Scholar]
- 73.Paulsen CE, Truong TH, Garcia FJ, Homann A, Gupta V, Leonard SE, Carroll KS. Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nat Chem Biol. 2011;8:57–64. doi: 10.1038/nchembio.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Denu JM, Tanner KG. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: Evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry. 1998;37:5633–5642. doi: 10.1021/bi973035t. [DOI] [PubMed] [Google Scholar]
- 75.Wani R, Qian J, Yin L, Bechtold E, King SB, Poole LB, Paek E, Tsang AW, Furdui CM. Isoform-specific regulation of Akt by PDGF-induced reactive oxygen species. Proc Natl Acad Sci U S A. 2011;108:10550–10555. doi: 10.1073/pnas.1011665108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wu J, Cheng Z, Reddie K, Carroll K, Hammad LA, Karty JA, Bauer CE. RegB kinase activity is repressed by oxidative formation of cysteine sulfenic acid. J Biol Chem. 2013;288:4755–4762. doi: 10.1074/jbc.M112.413492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zal F, Leize E, Lallier FH, Toulmond A, Van Dorsselaer A, Childress JJ. S-Sulfohemoglobin and disulfide exchange: the mechanisms of sulfide binding by Riftia pachyptila hemoglobins. Proc Natl Acad Sci U S A. 1998;95:8997–9002. doi: 10.1073/pnas.95.15.8997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ollagnier-de-Choudens S, Lascoux D, Loiseau L, Barras F, Forest E, Fontecave M. Mechanistic studies of the SufS-SufE cysteine desulfurase: evidence for sulfur transfer from SufS to SufE. FEBS Lett. 2003;555:263–267. doi: 10.1016/s0014-5793(03)01244-4. [DOI] [PubMed] [Google Scholar]
- 79.Matthies A, Nimtz M, Leimkuhler S. Molybdenum cofactor biosynthesis in humans: identification of a persulfide group in the rhodanese-like domain of MOCS3 by mass spectrometry. Biochemistry. 2005;44:7912–7920. doi: 10.1021/bi0503448. [DOI] [PubMed] [Google Scholar]
- 80.Sawyer EB, Stephens E, Ferguson SJ, Allen JW, Barker PD. Aberrant attachment of heme to cytochrome by the Ccm system results in a cysteine persulfide linkage. J Am Chem Soc. 2010;132:4974–4975. doi: 10.1021/ja908241v. [DOI] [PubMed] [Google Scholar]
- 81.Ida T, Sawa T, Ihara H, Tsuchiya Y, Watanabe Y, Kumagai Y, Suematsu M, Motohashi H, Fujii S, Matsunaga T, Yamamoto M, Ono K, Devarie-Baez NO, Xian M, Fukuto JM, Akaike T. Reactive cysteine persulfides and S-polythiolation regulate oxidative stress and redox signaling. Proc Natl Acad Sci U S A. 2014;14:14. doi: 10.1073/pnas.1321232111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Singh RJ, Hogg N, Joseph J, Kalyanaraman B. Mechanism of nitric oxide release from S-nitrosothiols. J Biol Chem. 1996;271:18596–18603. doi: 10.1074/jbc.271.31.18596. [DOI] [PubMed] [Google Scholar]
- 83.Filipovic MR, Miljkovic J, Nauser T, Royzen M, Klos K, Shubina T, Koppenol WH, Lippard SJ, Ivanovic-Burmazovic I. Chemical characterization of the smallest S-nitrosothiol, HSNO; cellular cross-talk of H2S and S-nitrosothiols. J Am Chem Soc. 2012;134:12016–12027. doi: 10.1021/ja3009693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lander HM, Ogiste JS, Pearce SF, Levi R, Novogrodsky A. Nitric oxide-stimulated guanine nucleotide exchange on p21ras. J Biol Chem. 1995;270:7017–7020. doi: 10.1074/jbc.270.13.7017. [DOI] [PubMed] [Google Scholar]
- 85.Wang Y, Liu T, Wu C, Li H. A strategy for direct identification of protein S-nitrosylation sites by quadrupole time-of-flight mass spectrometry. J Am Soc Mass Spectrom. 2008;19:1353–1360. doi: 10.1016/j.jasms.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ferranti P, Malorni A, Mamone G, Sannolo N, Marino G. Characterisation of S-nitrosohaemoglobin by mass spectrometry. FEBS Lett. 1997;400:19–24. doi: 10.1016/s0014-5793(96)01258-6. [DOI] [PubMed] [Google Scholar]
- 87.Knipp M, Braun O, Gehrig PM, Sack R, Vasak M. Zn(II)-free dimethylargininase-1 (DDAH-1) is inhibited upon specific Cys-S-nitrosylation. J Biol Chem. 2003;278:3410–3416. doi: 10.1074/jbc.M209088200. [DOI] [PubMed] [Google Scholar]
- 88.Gallogly MM, Mieyal JJ. Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Curr Opin Pharmacol. 2007;7:381–391. doi: 10.1016/j.coph.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 89.Priora R, Coppo L, Salzano S, Di Simplicio P, Ghezzi P. Measurement of mixed disulfides including glutathionylated proteins. Methods Enzymol. 2010;473:149–159. doi: 10.1016/S0076-6879(10)73007-X. [DOI] [PubMed] [Google Scholar]
- 90.Jakubowski H. Homocysteine in Protein Structure/Function and Human Disease. Springer; Vienna: 2013. S-Homocysteinylated Proteins; pp. 121–135. [Google Scholar]
- 91.Wu G, Fang YZ, Yang S, Lupton JR, Turner ND. Glutathione metabolism and its implications for health. J Nutr. 2004;134:489–492. doi: 10.1093/jn/134.3.489. [DOI] [PubMed] [Google Scholar]
- 92.Gao XH, Bedhomme M, Veyel D, Zaffagnini M, Lemaire SD. Methods for analysis of protein glutathionylation and their application to photosynthetic organisms. Mol Plant. 2009;2:218–235. doi: 10.1093/mp/ssn072. [DOI] [PubMed] [Google Scholar]
- 93.Chiang BY, Chou CC, Hsieh FT, Gao S, Lin JC, Lin SH, Chen TC, Khoo KH, Lin CH. In vivo tagging and characterization of S-glutathionylated proteins by a chemoenzymatic method. Angew Chem Int Ed Engl. 2012;51:5871–5875. doi: 10.1002/anie.201200321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Samarasinghe KT, Munkanatta Godage DN, VanHecke GC, Ahn YH. Metabolic synthesis of clickable glutathione for chemoselective detection of glutathionylation. J Am Chem Soc. 2014;136:11566–11569. doi: 10.1021/ja503946q. [DOI] [PubMed] [Google Scholar]
- 95.Chen YJ, Lu CT, Lee TY. dbGSH: a database of S-glutathionylation. Bioinformatics. 2014;30:2386–2388. doi: 10.1093/bioinformatics/btu301. [DOI] [PubMed] [Google Scholar]
- 96.Liu P, Tarnowski MA, O’Mara BW, Wu W, Zhang H, Tamura JK, Ackerman MS, Tao L, Grace MJ, Russell RJ. Characterization of S-thiolation on secreted proteins from E. coli by mass spectrometry. Rapid Commun Mass Spectrom. 2009;23:3343–3349. doi: 10.1002/rcm.4247. [DOI] [PubMed] [Google Scholar]
- 97.Cabras T, Manconi B, Iavarone F, Fanali C, Nemolato S, Fiorita A, Scarano E, Passali GC, Manni A, Cordaro M, Paludetti G, Faa G, Messana I, Castagnola M. RP-HPLC-ESI-MS evidenced that salivary cystatin B is detectable in adult human whole saliva mostly as S-modified derivatives: S-Glutathionyl, S-cysteinyl and S-S 2-mer. J Proteomics. 2012;75:908–913. doi: 10.1016/j.jprot.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 98.Ansong C, Wu S, Meng D, Liu X, Brewer HM, Deatherage Kaiser BL, Nakayasu ES, Cort JR, Pevzner P, Smith RD, Heffron F, Adkins JN, Pasa-Tolic L. Top-down proteomics reveals a unique protein S-thiolation switch in Salmonella typhimurium in response to infection-like conditions. Proc Natl Acad Sci U S A. 2013;110:10153–10158. doi: 10.1073/pnas.1221210110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Iavarone F, Cabras T, Pisano E, Sanna MT, Nemolato S, Vento G, Tirone C, Romagnoli C, Cordaro M, Fanos V, Faa G, Messana I, Castagnola M. Top-down HPLC-ESI-MS detection of S-glutathionylated and S-cysteinylated derivatives of cystatin B and its 1-53 and 54-98 fragments in whole saliva of human preterm newborns. J Proteome Res. 2013;12:917–926. doi: 10.1021/pr300960f. [DOI] [PubMed] [Google Scholar]
- 100.Auclair JR, Salisbury JP, Johnson JL, Petsko GA, Ringe D, Bosco DA, Agar NY, Santagata S, Durham HD, Agar JN. Artifacts to avoid while taking advantage of top-down mass spectrometry based detection of protein S-thiolation. PROTEOMICS. 2014;14:1152–1157. doi: 10.1002/pmic.201300450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Liang X, Lu Y, Neubert TA, Resh MD. Mass spectrometric analysis of GAP-43/neuromodulin reveals the presence of a variety of fatty acylated species. J Biol Chem. 2002;277:33032–33040. doi: 10.1074/jbc.M204607200. [DOI] [PubMed] [Google Scholar]
- 102.Pepinsky RB, Zeng C, Wen D, Rayhorn P, Baker DP, Williams KP, Bixler SA, Ambrose CM, Garber EA, Miatkowski K, Taylor FR, Wang EA, Galdes A. Identification of a palmitic acid-modified form of human Sonic hedgehog. J Biol Chem. 1998;273:14037–14045. doi: 10.1074/jbc.273.22.14037. [DOI] [PubMed] [Google Scholar]
- 103.Takada R, Satomi Y, Kurata T, Ueno N, Norioka S, Kondoh H, Takao T, Takada S. Monounsaturated fatty acid modification of Wnt protein: its role in Wnt secretion. Dev Cell. 2006;11:791–801. doi: 10.1016/j.devcel.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 104.Linder ME, Deschenes RJ. Palmitoylation: policing protein stability and traffic. Nat Rev Mol Cell Biol. 2007;8:74–84. doi: 10.1038/nrm2084. [DOI] [PubMed] [Google Scholar]
- 105.Messier AM, Bizzozero OA. Conserved fatty acid composition of proteolipid protein during brain development and in myelin subfractions. Neurochem Res. 2000;25:449–455. doi: 10.1023/a:1007551723752. [DOI] [PubMed] [Google Scholar]
- 106.Bizzozero OA, Malkoski SP, Mobarak C, Bixler HA, Evans JE. Mass-spectrometric analysis of myelin proteolipids reveals new features of this family of palmitoylated membrane proteins. J Neurochem. 2002;81:636–645. doi: 10.1046/j.1471-4159.2002.00852.x. [DOI] [PubMed] [Google Scholar]
- 107.Bernstein LS, Linder ME, Hepler JR. Analysis of RGS protein palmitoylation. Methods Mol Biol. 2004;237:195–204. doi: 10.1385/1-59259-430-1:195. [DOI] [PubMed] [Google Scholar]
- 108.Drisdel RC, Alexander JK, Sayeed A, Green WN. Assays of protein palmitoylation. Methods. 2006;40:127–134. doi: 10.1016/j.ymeth.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 109.Sorek N, Yalovsky S. Analysis of protein S-acylation by gas chromatography-coupled mass spectrometry using purified proteins. Nat Protoc. 2010;5:834–840. doi: 10.1038/nprot.2010.33. [DOI] [PubMed] [Google Scholar]
- 110.Jonsson TJ, Tsang AW, Lowther WT, Furdui CM. Identification of intact protein thiosulfinate intermediate in the reduction of cysteine sulfinic acid in peroxiredoxin by human sulfiredoxin. J Biol Chem. 2008;283:22890–22894. doi: 10.1074/jbc.C800124200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cox AG, Pearson AG, Pullar JM, Jonsson TJ, Lowther WT, Winterbourn CC, Hampton MB. Mitochondrial peroxiredoxin 3 is more resilient to hyperoxidation than cytoplasmic peroxiredoxins. Biochem J. 2009;421:51–58. doi: 10.1042/BJ20090242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Peskin AV, Dickerhof N, Poynton RA, Paton LN, Pace PE, Hampton MB, Winterbourn CC. Hyperoxidation of peroxiredoxins 2 and 3: rate constants for the reactions of the sulfenic acid of the peroxidatic cysteine. J Biol Chem. 2013;288:14170–14177. doi: 10.1074/jbc.M113.460881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Nelson KJ, Parsonage D, Karplus PA, Poole LB. Evaluating peroxiredoxin sensitivity toward inactivation by peroxide substrates. Methods Enzymol. 2013;527:21–40. doi: 10.1016/B978-0-12-405882-8.00002-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Jeong J, Kim Y, Kyung Seong J, Lee KJ. Comprehensive identification of novel post-translational modifications in cellular peroxiredoxin 6. PROTEOMICS. 2012;12:1452–1462. doi: 10.1002/pmic.201100558. [DOI] [PubMed] [Google Scholar]
- 115.Randall LM, Manta B, Hugo M, Gil M, Batthyany C, Trujillo M, Poole LB, Denicola A. Nitration transforms a sensitive peroxiredoxin 2 into a more active and robust peroxidase. J Biol Chem. 2014;9:9. doi: 10.1074/jbc.M113.539213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bagal D, Valliere-Douglass JF, Balland A, Schnier PD. Resolving disulfide structural isoforms of IgG2 monoclonal antibodies by ion mobility mass spectrometry. Anal Chem. 2010;82:6751–6755. doi: 10.1021/ac1013139. [DOI] [PubMed] [Google Scholar]
- 117.Echterbille J, Quinton L, Gilles N, De Pauw E. Ion mobility mass spectrometry as a potential tool to assign disulfide bonds arrangements in peptides with multiple disulfide bridges. Anal Chem. 2013;85:4405–4413. doi: 10.1021/ac303686w. [DOI] [PubMed] [Google Scholar]
- 118.Heck AJ, Van Den Heuvel RH. Investigation of intact protein complexes by mass spectrometry. Mass Spectrom Rev. 2004;23:368–389. doi: 10.1002/mas.10081. [DOI] [PubMed] [Google Scholar]
- 119.Kaltashov IA, Bobst CE, Abzalimov RR. Mass spectrometry-based methods to study protein architecture and dynamics. Protein Sci. 2013;22:530–544. doi: 10.1002/pro.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wales TE, Engen JR. Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom Rev. 2006;25:158–170. doi: 10.1002/mas.20064. [DOI] [PubMed] [Google Scholar]
- 121.Nirudodhi S, Parsonage D, Karplus PA, Poole LB, Maier CS. Conformational studies of the robust 2-Cys peroxiredoxin Salmonella typhimurium AhpC by solution phase hydrogen/deuterium (H/D) exchange monitored by electrospray ionization mass spectrometry. Int J Mass Spectrom. 2011;302:93–100. doi: 10.1016/j.ijms.2010.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pan LY, Salas-Solano O, Valliere-Douglass JF. Conformation and dynamics of interchain cysteine-linked antibody-drug conjugates as revealed by hydrogen/deuterium exchange mass spectrometry. Anal Chem. 2014;86:2657–2664. doi: 10.1021/ac404003q. [DOI] [PubMed] [Google Scholar]
- 123.Balchin D, Stoychev SH, Dirr HW. S-Nitrosation destabilizes glutathione transferase P1-1. Biochemistry. 2013;52:9394–9402. doi: 10.1021/bi401414c. [DOI] [PubMed] [Google Scholar]
- 124.Singh H, Dai Y, Outten FW, Busenlehner LS. Escherichia coli SufE sulfur transfer protein modulates the SufS cysteine desulfurase through allosteric conformational dynamics. J Biol Chem. 2013;288:36189–36200. doi: 10.1074/jbc.M113.525709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kim MS, Jeong J, Shin DH, Lee KJ. Structure of Nm23-H1 under oxidative conditions. Acta Crystallogr D Biol Crystallogr. 2013;69:669–680. doi: 10.1107/S0907444913001194. [DOI] [PubMed] [Google Scholar]
- 126.Zhou S, Mozziconacci O, Kerwin BA, Schoneich C. The photolysis of disulfide bonds in IgG1 and IgG2 leads to selective intramolecular hydrogen transfer reactions of cysteine thiyl radicals, probed by covalent H/D exchange and RPLC-MS/MS analysis. Pharm Res. 2013;30:1291–1299. doi: 10.1007/s11095-012-0968-1. [DOI] [PubMed] [Google Scholar]
- 127.Taylor AM, Zhu Q, Casey JR. Cysteine-directed cross-linking localizes regions of the human erythrocyte anion-exchange protein (AE1) relative to the dimeric interface. Biochem J. 2001;359:661–668. doi: 10.1042/0264-6021:3590661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Chen F, Nielsen S, Zenobi R. Understanding chemical reactivity for homo- and heterobifunctional protein cross-linking agents. J Mass Spectrom. 2013;48:807–812. doi: 10.1002/jms.3224. [DOI] [PubMed] [Google Scholar]
- 129.Rinner O, Seebacher J, Walzthoeni T, Mueller LN, Beck M, Schmidt A, Mueller M, Aebersold R. Identification of cross-linked peptides from large sequence databases. Nat Methods. 2008;5: 315–318. doi: 10.1038/nmeth.1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Leitner A, Reischl R, Walzthoeni T, Herzog F, Bohn S, Forster F, Aebersold R. Expanding the chemical cross-linking toolbox by the use of multiple proteases and enrichment by size exclusion chromatography. Mol Cell Proteomics. 2012;11:27. doi: 10.1074/mcp.M111.014126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Chowdhury SM, Munske GR, Tang X, Bruce JE. Collisionally activated dissociation and electron capture dissociation of several mass spectrometry-identifiable chemical cross-linkers. Anal Chem. 2006;78:8183–8193. doi: 10.1021/ac060789h. [DOI] [PubMed] [Google Scholar]
- 132.Kao A, Chiu CL, Vellucci D, Yang Y, Patel VR, Guan S, Randall A, Baldi P, Rychnovsky SD, Huang L. Development of a novel cross-linking strategy for fast and accurate identification of cross-linked peptides of protein complexes. Mol Cell Proteomics. 2011;10:24. doi: 10.1074/mcp.M110.002212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Calabrese AN, Wang T, Bowie JH, Pukala TL. Negative ion fragmentations of disulfide-containing cross-linking reagents are competitive with aspartic acid side-chain-induced cleavages. Rapid Commun Mass Spectrom. 2013;27:238–248. doi: 10.1002/rcm.6445. [DOI] [PubMed] [Google Scholar]
- 134.Lu D, Peterson LA. Identification of furan metabolites derived from cysteine-cis-2-butene-1,4-dial-lysine cross-links. Chem Res Toxicol. 2010;23:142–151. doi: 10.1021/tx9003215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gherezghiher TB, Ming X, Villalta PW, Campbell C, Tretyakova NY. 1,2,3,4-Diepoxybutane-induced DNA–protein cross-linking in human fibrosarcoma (HT1080) cells. J Proteome Res. 2013;12:2151–2164. doi: 10.1021/pr3011974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lu H-R, Gu M-G, Huang Q, Huang J-J, Lu W-Y, Lu H, Huang Q-S. Hydrogen/deuterium exchange mass spectrometry and site-directed disulfide cross-linking suggest an important dynamic interface between the two lysostaphin domains. Antimicrob Agents Chemother. 2013;57:1872–1881. doi: 10.1128/AAC.02348-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Jeong J, Jung Y, Na S, Lee E, Kim MS, Choi S, Shin DH, Paek E, Lee HY, Lee KJ. Novel oxidative modifications in redox-active cysteine residues. Mol Cell Proteomics. 2011;10:10. doi: 10.1074/mcp.M110.000513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Held JM, Danielson SR, Behring JB, Atsriku C, Britton DJ, Puckett RL, Schilling B, Campisi J, Benz CC, Gibson BW. Targeted quantitation of site-specific cysteine oxidation in endogenous proteins using a differential alkylation and multiple reaction monitoring mass spectrometry approach. Mol Cell Proteomics. 2010;9:1400–1410. doi: 10.1074/mcp.M900643-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat Cell Biol. 2001;3:193–197. doi: 10.1038/35055104. [DOI] [PubMed] [Google Scholar]
- 140.Saurin AT, Neubert H, Brennan JP, Eaton P. Widespread sulfenic acid formation in tissues in response to hydrogen peroxide. Proc Natl Acad Sci U S A. 2004;101:17982–17987. doi: 10.1073/pnas.0404762101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Krishnan N, Fu C, Pappin DJ, Tonks NK. H2S-Induced sulfhydration of the phosphatase PTP1B and its role in the endoplasmic reticulum stress response. Sci Signal. 2011;4:2002329. doi: 10.1126/scisignal.2002329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Mustafa AK, Gadalla MM, Sen N, Kim S, Mu W, Gazi SK, Barrow RK, Yang G, Wang R, Snyder SH. H2S signals through protein S-sulfhydration. Sci Signal. 2009;2:2000464. doi: 10.1126/scisignal.2000464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wan J, Roth AF, Bailey AO, Davis NG. Palmitoylated proteins: purification and identification. Nat Protoc. 2007;2:1573–1584. doi: 10.1038/nprot.2007.225. [DOI] [PubMed] [Google Scholar]
- 144.Wang X, Kettenhofen NJ, Shiva S, Hogg N, Gladwin MT. Copper dependence of the biotin switch assay: modified assay for measuring cellular and blood nitrosated proteins. Free Radic Biol Med. 2008;44:1362–1372. doi: 10.1016/j.freeradbiomed.2007.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Landino LM, Koumas MT, Mason CE, Alston JA. Ascorbic acid reduction of microtubule protein disulfides and its relevance to protein S-nitrosylation assays. Biochem Biophys Res Commun. 2006;340:347–352. doi: 10.1016/j.bbrc.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 146.Huang B, Chen C. An ascorbate-dependent artifact that interferes with the interpretation of the biotin switch assay. Free Radic Biol Med. 2006;41:562–567. doi: 10.1016/j.freeradbiomed.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 147.Giustarini D, Dalle-Donne I, Colombo R, Milzani A, Rossi R. Is ascorbate able to reduce disulfide bridges? A cautionary note. Nitric Oxide. 2008;19:252–258. doi: 10.1016/j.niox.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 148.Liu T, Qian WJ, Strittmatter EF, Camp DG, 2nd, Anderson GA, Thrall BD, Smith RD. High-throughput comparative proteome analysis using a quantitative cysteinyl-peptide enrichment technology. Anal Chem. 2004;76:5345–5353. doi: 10.1021/ac049485q. [DOI] [PubMed] [Google Scholar]
- 149.Forrester MT, Thompson JW, Foster MW, Nogueira L, Moseley MA, Stamler JS. Proteomic analysis of S-nitrosylation and denitrosylation by resin-assisted capture. Nat Biotechnol. 2009;27:557–559. doi: 10.1038/nbt.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lu XM, Lu M, Tompkins RG, Fischman AJ. Site-specific detection of S-nitrosylated PKBα/Akt1 from rat soleus muscle using CapLC-Q-TOF(micro) mass spectrometry. J Mass Spectrom. 2005;40:1140–1148. doi: 10.1002/jms.885. [DOI] [PubMed] [Google Scholar]
- 151.Thompson JW, Forrester MT, Moseley MA, Foster MW. Solid-phase capture for the detection and relative quantification of S-nitrosoproteins by mass spectrometry. Methods. 2013;62:130–137. doi: 10.1016/j.ymeth.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Forrester MT, Hess DT, Thompson JW, Hultman R, Moseley MA, Stamler JS, Casey PJ. Site-specific analysis of protein S-acylation by resin-assisted capture. J Lipid Res. 2011;52:393–398. doi: 10.1194/jlr.D011106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Hao G, Derakhshan B, Shi L, Campagne F, Gross SS. SNOSID, a proteomic method for identification of cysteine S-nitrosylation sites in complex protein mixtures. Proc Natl Acad Sci U S A. 2006;103:1012–1017. doi: 10.1073/pnas.0508412103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Derakhshan B, Wille PC, Gross SS. Unbiased identification of cysteine S-nitrosylation sites on proteins. Nat Protocols. 2007;2:1685–1691. doi: 10.1038/nprot.2007.210. [DOI] [PubMed] [Google Scholar]
- 155.Greco TM, Hodara R, Parastatidis I, Heijnen HFG, Dennehy MK, Liebler DC, Ischiropoulos H. Identification of S-nitrosylation motifs by site-specific mapping of the S-nitrosocysteine proteome in human vascular smooth muscle cells. Proc Natl Acad Sci U S A. 2006;103:7420–7425. doi: 10.1073/pnas.0600729103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Torta F, Bachi A. Quantitative analysis of S-nitrosylated proteins. Methods Mol Biol. 2012;893:405–416. doi: 10.1007/978-1-61779-885-6_25. [DOI] [PubMed] [Google Scholar]
- 157.Murray CI, Uhrigshardt H, O’Meally RN, Cole RN, Van Eyk JE. Identification and quantification of S-nitrosylation by cysteine reactive tandem mass tag switch assay. Mol Cell Proteomics. 2012;11:29. doi: 10.1074/mcp.M111.013441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Lind C, Gerdes R, Hamnell Y, Schuppe-Koistinen I, von Lowenhielm HB, Holmgren A, Cotgreave IA. Identification of S-glutathionylated cellular proteins during oxidative stress and constitutive metabolism by affinity purification and proteomic analysis. Arch Biochem Biophys. 2002;406:229–240. doi: 10.1016/s0003-9861(02)00468-x. [DOI] [PubMed] [Google Scholar]
- 159.Aesif SW, Janssen-Heininger YM, Reynaert NL. Protocols for the detection of S-glutathionylated and S-nitrosylated proteins in situ. Methods Enzymol. 2010;474:289–296. doi: 10.1016/S0076-6879(10)74017-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Su D, Gaffrey MJ, Guo J, Hatchell KE, Chu RK, Clauss TRW, Aldrich JT, Wu S, Purvine S, Camp DG, Smith RD, Thrall BD, Qian W-J. Proteomic identification and quantification of S-glutathionylation in mouse macrophages using resin-assisted enrichment and isobaric labeling. Free Radic Biol Med. 2014;67:460–470. doi: 10.1016/j.freeradbiomed.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Guo J, Gaffrey MJ, Su D, Liu T, Camp DG, II, Smith RD, Qian W-J. Resin-assisted enrichment of thiols as a general strategy for proteomic profiling of cysteine-based reversible modifications. Nat Protocols. 2014;9:64–75. doi: 10.1038/nprot.2013.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Weerapana E, Wang C, Simon GM, Richter F, Khare S, Dillon MB, Bachovchin DA, Mowen K, Baker D, Cravatt BF. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature. 2010;468:790–795. doi: 10.1038/nature09472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Sullivan DM, Levine RL, Finkel T. Detection and affinity purification of oxidant-sensitive proteins using biotinylated glutathione ethyl ester. Methods Enzymol. 2002;353:101–113. doi: 10.1016/s0076-6879(02)53040-8. [DOI] [PubMed] [Google Scholar]
- 164.Kim H-YH, Tallman KA, Liebler DC, Porter NA. An azido-biotin reagent for use in the isolation of protein adducts of lipid-derived electrophiles by streptavidin catch and photorelease. Mol Cell Proteomics. 2009;8:2080–2089. doi: 10.1074/mcp.M900121-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Codreanu SG, Ullery JC, Zhu J, Tallman KA, Beavers WN, Porter NA, Marnett LJ, Zhang B, Liebler DC. Alkylation damage by lipid electrophiles targets functional protein systems. Mol Cell Proteomics. 2014;13:849–859. doi: 10.1074/mcp.M113.032953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Lin D, Li J, Slebos RJC, Liebler DC. Cysteinyl peptide capture for shotgun proteomics: Global assessment of chemoselective fractionation. J Proteome Res. 2010;9:5461–5472. doi: 10.1021/pr1007015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Ying J, Clavreul N, Sethuraman M, Adachi T, Cohen RA. Thiol oxidation in signaling and response to stress: detection and quantification of physiological and pathophysiological thiol modifications. Free Radic Biol Med. 2007;43:1099–1108. doi: 10.1016/j.freeradbiomed.2007.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Yang Z, Attygalle AB. LC/MS characterization of undesired products formed during iodoacetamide derivatization of sulfhydryl groups of peptides. J Mass Spectrom. 2007;42:233–243. doi: 10.1002/jms.1157. [DOI] [PubMed] [Google Scholar]
- 169.Zhang D, Devarie-Baez NO, Li Q, Lancaster JR, Jr, Xian M. Methylsulfonyl benzothiazole (MSBT): A selective protein thiol blocking reagent. Org Lett. 2012;14:3396–3399. doi: 10.1021/ol301370s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Toda N, Asano S, Barbas CF., 3rd Rapid, stable, chemoselective labeling of thiols with Julia-Kocienski-like reagents: A serum-stable alternative to maleimide-based protein conjugation. Angew Chem Int Ed Engl. 2013;52:12592–12596. doi: 10.1002/anie.201306241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Koniev O, Leriche G, Nothisen M, Remy J-S, Strub J-M, Schaeffer-Reiss C, Van Dorsselaer A, Baati R, Wagner A. Selective irreversible chemical tagging of cysteine with 3-arylpropiolonitriles. Bioconjug Chem. 2014;25:202–206. doi: 10.1021/bc400469d. [DOI] [PubMed] [Google Scholar]
- 172.Nelson KJ, Klomsiri C, Codreanu SG, Soito L, Liebler DC, Rogers LC, Daniel LW, Poole LB. Use of dimedone-based chemical probes for sulfenic acid detection methods to visualize and identify labeled proteins. Methods Enzymol. 2010;473:95–115. doi: 10.1016/S0076-6879(10)73004-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Charles RL, Schroder E, May G, Free P, Gaffney PR, Wait R, Begum S, Heads RJ, Eaton P. Protein sulfenation as a redox sensor: Proteomics studies using a novel biotinylated dimedone analogue. Mol Cell Proteomics. 2007;6:1473–1484. doi: 10.1074/mcp.M700065-MCP200. [DOI] [PubMed] [Google Scholar]
- 174.Seo YH, Carroll KS. Quantification of protein sulfenic acid modifications using isotope-coded dimedone and iododimedone. Angew Chem Int Ed Engl. 2011;50:1342–1345. doi: 10.1002/anie.201007175. [DOI] [PubMed] [Google Scholar]
- 175.Yi EC, Li XJ, Cooke K, Lee H, Raught B, Page A, Aneliunas V, Hieter P, Goodlett DR, Aebersold R. Increased quantitative proteome coverage with 13C/12C-based, acid-cleavable isotope-coded affinity tag reagent and modified data acquisition scheme. PROTEOMICS. 2005;5:380–387. doi: 10.1002/pmic.200400970. [DOI] [PubMed] [Google Scholar]
- 176.Martinez-Acedo P, Gupta V, Carroll KS. Proteomic analysis of peptides tagged with dimedone and related probes. J Mass Spectrom. 2014;49:257–265. doi: 10.1002/jms.3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Yang J, Gupta V, Carroll KS, Liebler DC. Site-specific mapping and quantification of protein S-sulphenylation in cells. Nat Commun. 2014;5:4776. doi: 10.1038/ncomms5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Liu CT, Benkovic SJ. Capturing a sulfenic acid with arylboronic acids and benzoxaborole. J Am Chem Soc. 2013;135:14544–14547. doi: 10.1021/ja407628a. [DOI] [PubMed] [Google Scholar]
- 179.Cook JA, Kim SY, Teague D, Krishna MC, Pacelli R, Mitchell JB, Vodovotz Y, Nims RW, Christodoulou D, Miles AM, Grisham MB, Wink DA. Convenient colorimetric and fluorometric assays for S-nitrosothiols. Analytical Biochemistry. 1996;238:150–158. doi: 10.1006/abio.1996.0268. [DOI] [PubMed] [Google Scholar]
- 180.Vanin AF, Papina AA, Serezhenkov VA, Koppenol WH. The mechanisms of S-nitrosothiol decomposition catalyzed by iron. Nitric Oxide. 2004;10:60–73. doi: 10.1016/j.niox.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 181.Jia HY, Liu Y, Zhang XJ, Han L, Du LB, Tian Q, Xu YC. Potential oxidative stress of gold nanoparticles by induced-NO releasing in serum. J Am Chem Soc. 2009;131:40–41. doi: 10.1021/ja808033w. [DOI] [PubMed] [Google Scholar]
- 182.Zhang S, Celebi-Olcum N, Melzer MM, Houk KN, Warren TH. Copper(I) nitrosyls from reaction of copper(II) thiolates with S-nitrosothiols: mechanism of NO release from RSNOs at Cu. J Am Chem Soc. 2013;135:16746–16749. doi: 10.1021/ja406476y. [DOI] [PubMed] [Google Scholar]
- 183.Faccenda A, Bonham CA, Vacratsis PO, Zhang X, Mutus B. Gold nanoparticle enrichment method for identifying S-nitrosylation and S-glutathionylation sites in proteins. J Am Chem Soc. 2010;132:11392–11394. doi: 10.1021/ja103591v. [DOI] [PubMed] [Google Scholar]
- 184.Saville B. A scheme for the colorimetric determination of microgram amounts of thiols. Analyst. 1958;83:670–672. [Google Scholar]
- 185.Doulias PT, Tenopoulou M, Raju K, Spruce LA, Seeholzer SH, Ischiropoulos H. Site specific identification of endogenous S-nitrosocysteine proteomes. J Proteomics. 2013;92:195–203. doi: 10.1016/j.jprot.2013.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Doulias PT, Greene JL, Greco TM, Tenopoulou M, Seeholzer SH, Dunbrack RL, Ischiropoulos H. Structural profiling of endogenous S-nitrosocysteine residues reveals unique features that accommodate diverse mechanisms for protein S-nitrosylation. Proc Natl Acad Sci U S A. 2010;107:16958–16963. doi: 10.1073/pnas.1008036107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Doulias PT, Tenopoulou M, Greene JL, Raju K, Ischiropoulos H. Nitric oxide regulates mitochondrial fatty acid metabolism through reversible protein S-nitrosylation. Sci Signal. 2013;6:2003252. doi: 10.1126/scisignal.2003252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Wang H, Xian M. Fast reductive ligation of S-nitrosothiols. Angew Chem Int Ed Engl. 2008;47:6598–6601. doi: 10.1002/anie.200801654. [DOI] [PubMed] [Google Scholar]
- 189.Zhang J, Wang H, Xian M. An unexpected Bis-ligation of S-nitrosothiols. J Am Chem Soc. 2009;131:3854–3855. doi: 10.1021/ja900370y. [DOI] [PubMed] [Google Scholar]
- 190.Zhang J, Li S, Zhang D, Wang H, Whorton AR, Xian M. Reductive ligation mediated one-step disulfide formation of S-nitrosothiols. Org Lett. 2010;12:4208–4211. doi: 10.1021/ol101863s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Zhang D, Chen W, Miao Z, Ye Y, Zhao Y, King SB, Xian M. A reductive ligation based fluorescent probe for S-nitrosothiols. Chem Commun (Camb) 2014;50:4806–4809. doi: 10.1039/c4cc01288g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Seneviratne U, Godoy LC, Wishnok JS, Wogan GN, Tannenbaum SR. Mechanism-based triarylphosphine-ester probes for capture of endogenous RSNOs. J Am Chem Soc. 2013;135:7693–7704. doi: 10.1021/ja401565w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Hart TW. Some observations concerning the S-nitroso and S-phenylsulphonyl derivatives of L-cysteine and glutathione. Tetrahedron Lett. 1985;26:2013–2016. [Google Scholar]
- 194.Hart TW, Vine MB, Walden NR. Triolsulphonate derivatives of amino acids. Tetrahedron Lett. 1985;26:3879–3882. [Google Scholar]
- 195.Reeves BD, Hilmer JK, Mellmann L, Hartzheim M, Poffenberger K, Johnson K, Joshi N, Singel DJ, Grieco PA. Selective trapping of SNO-BSA and GSNO by benzenesulfinic acid sodium salt: mechanistic study of thiosulphonate formation and feasibility as a protein S-nitrosothiol detection strategy. Tetrahedron Lett. 2013;54:011. doi: 10.1016/j.tetlet.2013.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Resh MD. Use of analogs and inhibitors to study the functional significance of protein palmitoylation. Methods. 2006;40:191–197. doi: 10.1016/j.ymeth.2006.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Saxon E, Bertozzi CR. Cell surface engineering by a modified Staudinger reaction. Science. 2000;287:2007–2010. doi: 10.1126/science.287.5460.2007. [DOI] [PubMed] [Google Scholar]
- 198.Hannoush RN, Sun J. The chemical toolbox for monitoring protein fatty acylation and prenylation. Nat Chem Biol. 2010;6:498–506. doi: 10.1038/nchembio.388. [DOI] [PubMed] [Google Scholar]
- 199.Hang HC, Wilson JP, Charron G. Bioorthogonal chemical reporters for analyzing protein lipidation and lipid trafficking. Acc Chem Res. 2011;44:699–708. doi: 10.1021/ar200063v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Hang HC, Geutjes EJ, Grotenbreg G, Pollington AM, Bijlmakers MJ, Ploegh HL. Chemical probes for the rapid detection of fatty acylated proteins in mammalian cells. J Am Chem Soc. 2007;129:2744–2745. doi: 10.1021/ja0685001. [DOI] [PubMed] [Google Scholar]
- 201.Kostiuk MA, Corvi MM, Keller BO, Plummer G, Prescher JA, Hangauer MJ, Bertozzi CR, Rajaiah G, Falck JR, Berthiaume LG. Identification of palmitoylated mitochondrial proteins using a bio-orthogonal azido-palmitate analogue. FASEB J. 2008;22:721–732. doi: 10.1096/fj.07-9199com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Charron G, Zhang MM, Yount JS, Wilson J, Raghavan AS, Shamir E, Hang HC. Robust fluorescent detection of protein fatty-acylation with chemical reporters. J Am Chem Soc. 2009;131:4967–4975. doi: 10.1021/ja810122f. [DOI] [PubMed] [Google Scholar]
- 203.Yount JS, Moltedo B, Yang YY, Charron G, Moran TM, Lopez CB, Hang HC. Palmitoylome profiling reveals S-palmitoylation-dependent antiviral activity of IFITM3. Nat Chem Biol. 2010;6:610–614. doi: 10.1038/nchembio.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Ji Y, Leymarie N, Haeussler DJ, Bachschmid MM, Costello CE, Lin C. Direct detection of S-palmitoylation by mass spectrometry. Anal Chem. 2013;85:11952–11959. doi: 10.1021/ac402850s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Finkel T. Signal transduction by reactive oxygen species. J Cell Biol. 2011;194:7–15. doi: 10.1083/jcb.201102095. [DOI] [PMC free article] [PubMed] [Google Scholar]