

Abstract
Background
The native ability of Clostridium thermocellum to rapidly consume cellulose and produce ethanol makes it a leading candidate for a consolidated bioprocessing (CBP) biofuel production strategy. C. thermocellum also synthesizes lactate, formate, acetate, H2, and amino acids that compete with ethanol production for carbon and electrons. Elimination of H2 production could redirect carbon flux towards ethanol production by making more electrons available for acetyl coenzyme A reduction to ethanol.
Results
H2 production in C. thermocellum is encoded by four hydrogenases. Rather than delete each individually, we targeted hydrogenase maturase gene hydG, involved in converting the three [FeFe] hydrogenase apoenzymes into holoenzymes. Further deletion of the [NiFe] hydrogenase (ech) resulted in a mutant that functionally lacks all four hydrogenases. H2 production in ∆hydG∆ech was undetectable, and the ethanol yield nearly doubled to 64% of the maximum theoretical yield. Genomic analysis of ∆hydG revealed a mutation in adhE, resulting in a strain with both NADH- and NADPH-dependent alcohol dehydrogenase activities. While this same adhE mutation was found in ethanol-tolerant C. thermocellum strain E50C, ∆hydG and ∆hydG∆ech are not more ethanol tolerant than the wild type, illustrating the complicated interactions between redox balancing and ethanol tolerance in C. thermocellum.
Conclusions
The dramatic increase in ethanol production suggests that targeting protein post-translational modification is a promising new approach for simultaneous inactivation of multiple enzymes.
Electronic supplementary material
The online version of this article (doi:10.1186/s13068-015-0204-4) contains supplementary material, which is available to authorized users.
Keywords: Cellulosic ethanol, Clostridium thermocellum, Hydrogenase maturation, Metabolic engineering
Background
A sustainable future will likely be dependent on large-scale production of fuels, chemicals, and products from renewable resources. One of the most promising approaches to this end is microbially catalyzed conversion of lignocellulosic biomass [1]. Abundant plant biomass resources are available that have the potential to be used as feedstocks [2], but economical bioconversion of plant material into fuel has been elusive [3]. Microbes may ultimately play a central role in the conversion of biomass to fuels and chemicals. While current technologies for biomass fermentation to fuels tend to rely on added cellulolytic enzymes to solubilize hemicellulose and cellulose prior to fermentation, enzyme production represents a significant cost and hinders economic production of biofuels [4]. An alternative approach is to utilize one or more microorganisms to ferment plant cell walls to fuels in one step without added enzymes; this process is called consolidated bioprocessing (CBP). However, no known naturally occurring microbe is capable of robust, high yield, and high titer fuel production from lignocellulose. Thus, genetic modification will be required to create an organism with all of the desired properties for bioconversion of lignocellulosic biomass to fuels.
Clostridium thermocellum, a thermophilic, cellulolytic member of the Firmicutes phylum, is a potential platform to engineer into a CBP organism due to its native ability to efficiently solubilize cellulose and produce ethanol as a fermentation product. However, wild-type C. thermocellum is limited by its low ethanol yield and titer, producing acetate, lactate, H2, formate, and free amino acids as additional fermentation products [5], and native strains tolerate low levels of ethanol [6,7]. Recently, methodologies for genetic manipulation have been developed for C. thermocellum [8-12], raising the possibility that it can be engineered to economically produce fuels from cellulosic substrates. While these tools are still laborious to use, they have now been successfully used to improve industrially important properties in C. thermocellum. For instance, ethanol tolerance has been correlated with mutations in the bifunctional acetaldehyde/alcohol dehydrogenase, adhE, in three independent strains of C. thermocellum [6,13], and heterologous expression of a mutant adhE gene in an otherwise wild-type strain conferred ethanol tolerance [13]. Metabolic flux to major end products has also been blocked via gene deletion, including production of acetic acid [11], lactic acid in an adhE mutant [14], and acetic and lactic acid simultaneously [8], increasing ethanol yield. C. thermocellum does not encode a pyruvate kinase and is instead thought to divert significant flux from phosphoenolpyruvate → oxaloacetate → malate → pyruvate in what is called a malate shunt, which would generate NADPH while oxidizing NADH to NAD+, possibly creating a redox imbalance [15-17]. Heterologous expression of pyruvate kinase and disruption of the malate shunt in C. thermocellum also substantially improved flux towards ethanol [16], but further increases in ethanol yield are still needed.
While acetic and lactic acids are the primary soluble fermentation products competing with ethanol synthesis, H2 production is a major sink for electrons that could otherwise be directed toward ethanol production. By using protons as an electron sink rather than glycolytic intermediates, acetyl coenzyme A (acetyl-CoA) becomes available for production of acetate and ATP. While this provides more usable energy for the cell, it decreases the pool of electrons available for reduction of acetyl-CoA.
H2 production is catalyzed by a class of enzymes called hydrogenases, which broadly fall into three primary categories based on the metals in the active site: [Fe] hydrogenases, which are thus far only found in methanogens, [FeFe] hydrogenases, and [NiFe] hydrogenases. C. thermocellum encodes three putative [FeFe] hydrogenases and one ferredoxin-dependent [NiFe] energy-converting hydrogenase (Ech) (Figure 1A, [18,19]). Hydrogenase active sites are complex organometallic catalysts that require a dedicated enzymatic system for post-translational assembly. [FeFe] hydrogenases utilize a single system for active site assembly, consisting of the maturases HydE, HydF, and HydG (Figure 1B, reviewed in [20]). HydF acts as a scaffold upon which the binuclear Fe active site is assembled. HydE likely produces the ligand that bridges the two active site Fe molecules, while HydG cleaves tyrosine to generate the -CN and -CO ligands on the active site Fe molecules. Each is required for [FeFe] hydrogenase activity, and thus represents a novel target for simultaneous inactivation of multiple hydrogenases. We hypothesized that targeting electron flux to H2 can be a fruitful approach to increasing flux to ethanol. Therefore, we targeted inactivation of hydG as part of a strategy to eliminate H2 as a fermentation product and redirect metabolic flux toward ethanol.
Figure 1.
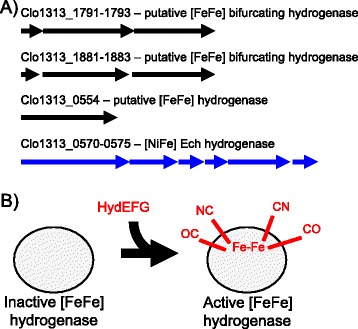
C. thermocellum hydrogenases and hydrogenase maturation. A) Chromosomal loci of C. thermocellum encoding three [FeFe] hydrogenases (in black) and one [NiFe] hydrogenase (in blue). B) Together, HydE, HydF, and HydG assemble the active site of [FeFe] hydrogenases to make active enzymes.
Results
Hydrogenase maturase deletion simplifies elimination of H2 as a fermentation product
To redirect electrons away from H2 and toward ethanol, we deleted the [FeFe] hydrogenase maturase gene hydG to prevent conversion of the hydrogenase apoenzymes into holoenzymes. Because HydEFG is only involved in maturation of the [FeFe] hydrogenases, we further deleted the genes encoding the [NiFe] Ech hydrogenase (Additional file 1). Deletion of hydG dramatically decreased H2 production, with a 15-fold reduction (Figure 2). Further deletion of ech, resulting in strain ΔhydGΔech, completely eliminated detectable H2 production and verified that deleting hydG simultaneously eliminated all [FeFe] hydrogenase activity.
Figure 2.
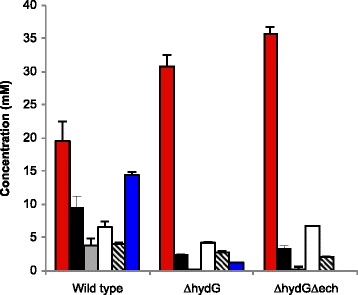
Fermentation products of C. thermocellum . The wild-type, ∆hydG, and ∆hydG∆ech strains grown on minimal medium with 5 g/L cellobiose. Red bar, ethanol; black bar, acetate; gray bar, lactate; white bar, formate; diagonal line bar, total amino acids; blue bar, H2. H2 concentration is reported as mmol gaseous H2 per L liquid medium to facilitate comparison to soluble products. Error bars represent one standard deviation.
In anaerobic batch fermentation, the ΔhydG mutant, which was later found to also contain a point mutation in the bifunctional acetaldehyde/alcohol dehydrogenase adhE (Clo1313_1798) (see below), produced 63% more ethanol than the parent strain (Figure 2). It decreased acetate production by 74%, lactate production was nearly eliminated, formate production decreased by 34% compared to the wild type, and total secreted amino acid levels decreased as well. Further deletion of ech in the ∆hydG background completely eliminated H2 production, while ethanol production increased by 90% compared to the wild type and 16% relative to ∆hydG. This represents an overall ethanol yield from cellobiose of 64% of the theoretical yield. The maximum optical density was 12% lower in ∆hydG (OD600 = 0.70) relative to the wild type (OD600 = 0.80), and the growth rate was also slower (wild type = 0.26 h−1, ∆hydG = 0.12 h−1). The growth yield and rate of ΔhydGΔech were between these values, with a maximum OD600 = 0.76 and a growth rate of 0.22 h−1 (Figure 3).
Figure 3.
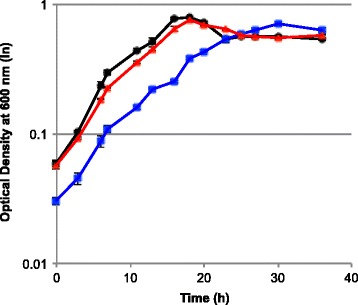
Growth profile of C. thermocellum strains on minimal medium. Symbols: Black circles, wild-type C. thermocellum; blue squares , C. thermocellum ∆hydG; red triangles , C. thermocellum ∆hydG∆ech. Error bars represent one standard deviation.
As C. thermocellum is known to divert a significant flux toward production of secreted amino acids, we further examined the abundance of individual amino acids in the supernatant. Among the other secreted amino acids, L-valine was produced in significant amounts in all the strains (Figure 4) as previously observed [21], but was 38% lower in ΔhydGΔech. Interestingly, the wild-type strain removed about 1.6 mM of 2.4 mM cysteine in the medium, while C. thermocellum ΔhydG and ΔhydGΔech did not consume any cysteine from the medium (Figure 4).
Figure 4.
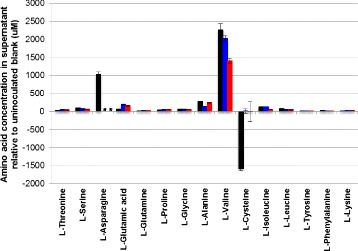
Concentration of amino acids produced by wild-type, ΔhydG, and ∆hydG∆ech strains. *, asparagine quantification for two samples was prevented by interference. Error bars represent one standard deviation. Black bars, wild-type C. thermocellum; blue bars, C. thermocellum ∆hydG; red bars, C. thermocellum ∆hydG∆ech.
Mutation in adhE altered cofactor specificity but not ethanol tolerance
During strain validation, a point mutation in the bifunctional acetaldehyde/alcohol dehydrogenase adhE (Clo1313_1798) was discovered in C. thermocellum ∆hydG, converting aspartate 494 into glycine (D494G). This exact same mutation was identified in the ethanol-tolerant C. thermocellum mutant E50C [6], though the effect of this mutation has not been validated enzymatically or genetically. A different mutant AdhE from the ethanol-tolerant strain C. thermocellum adhE* (EA) containing two mutations (P704L and H734R) was found to alter coenzyme specificity, eliminating NADH-dependent acetaldehyde reduction but allowing NADPH-dependent acetaldehyde reduction [13]. We therefore tested the effect of the D494G mutation on ADH cofactor specificity in C. thermocellum ∆hydG. While the wild-type ADH specific activity was almost exclusively NADH-dependent (7.03 ± 1.06 μmol/min/mg protein with NADH; 0.10 ± 0.08 μmol/min/mg protein with NADPH), ADH specific activity was high with each cofactor in C. thermocellum ∆hydG (6.31 ± 1.27 μmol/min/mg protein with NADH; 5.89 ± 0.89 μmol/min/mg protein with NADPH). As a control, the specific activity of ADH from the C. thermocellum adhE* (EA) strain containing the P704L and H734R mutations was tested, resulting in 0.006 ± 0.007 μmol/min/mg protein NADH-dependent activity and 0.25 ± 0.005 μmol/min/mg protein NADPH-dependent activity, consistent with previous results [13]. Because previously identified adhE mutations in C. thermocellum, including D494G, correlated with an ethanol tolerance phenotype [6,13], we examined the effect of added ethanol on the growth of C. thermocellum ∆hydG. In rich (Additional file 2, left) and minimal media (Additional file 2, right), ∆hydG and ∆hydG∆ech did not display an ethanol-tolerant phenotype.
Discussion
Genetic manipulation is possible in an increasing number of Firmicutes, and tremendous progress in these systems has been made in recent years. However, depending on the gene and bacterium, the effort to delete a single gene can be time-consuming and a barrier to advanced metabolic engineering approaches. Here, we identified a commonality between multiple desired enzymatic targets and designed a strategy to target them simultaneously. Targeting post-translational modification machinery might be a generally useful approach to streamline metabolic engineering under certain circumstances. Particularly in H2-producing organisms such as Clostridium cellulolyticum (three predicted [NiFe] hydrogenases; four predicted [FeFe] hydrogenases), Clostridium acetobutylicum (two predicted [FeFe] hydrogenases), and Clostridium beijerinckii (six predicted [FeFe] hydrogenases) [18], this same approach of targeting hydrogenase maturation to divert flux away from H2 + acetate or butyrate is quite promising. It may also enable fundamental studies of H2 metabolism in organisms such as sulfate-reducing bacteria [22-24].
Altering electron flux via elimination of H2 production had a dramatic effect on carbon flux in C. thermocellum (Figure 5). Previous C. thermocellum metabolic engineering efforts primarily focused on altering carbon flux directly; deletion of phosphotransacetylase (pta) nearly eliminated acetate as a fermentation product but had little impact on ethanol yield [11]. Similarly, deletion of pta and lactate dehydrogenase together only increased ethanol yield slightly to 27% of the theoretical yield, but strain evolution allowed for increased conversion up to 59% of the theoretical yield [8]. Heterologous expression of a pyruvate kinase and blockage of the malate shunt via deletion of malic enzyme resulted in an ethanol yield of 47% of the theoretical yield [16]. Instead of targeting carbon flux directly, we constrained electron flux by both blocking [FeFe] hydrogenase activity and deleting the Ech hydrogenase. Combined with the spontaneous mutation in adhE, this dramatically reduced production of acetate and eliminated H2, resulting in the highest yet achieved ethanol yield in C. thermocellum, 64% of the theoretical maximum without yet evolving the strain for improved performance. The impact of the adhE mutation on product yields in these strains is currently unclear, but the evolved strain E50C [6,7] contains the exact same point mutation, and the ethanol yield is comparable to that of the wild type. While ΔhydG grew slowly, likely due to difficulty balancing redox reactions, it attained a similar maximum OD to that of the wild type (Figure 3). The metabolic load of producing hydrogenase apoproteins could contribute to the diminished growth rate, especially if they are overproduced in response to the ΔhydG mutation, but the hydrogenases were expressed in ΔhydG and ΔhydGΔech at the same level as in the wild type (CM Wilson, unpublished data), suggesting that metabolic load does not play a significant role in the growth defect. Further deletion of ech allowed for a growth rate more similar to that of the wild type while also redirecting more flux to ethanol. This demonstrates that C. thermocellum is capable of sufficiently rerouting metabolism to accommodate the lack of H2 production.
Figure 5.
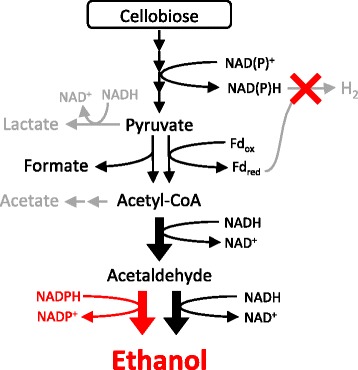
Overview of metabolic changes in C. thermocellum ΔhydGΔech . When hydrogenases were inactivated (red X), flux to ethanol increased. Further, the alcohol dehydrogenase mutated, allowing use of NADPH as a cofactor for ethanol production (red pathway). Simultaneously, fluxes to H2, lactate, and acetate decreased or were eliminated (gray pathways).
Production of acetic acid plus CO2 is obligately coupled to production of a more reduced compound, either H2 or formate in C. thermocellum, in order to balance redox reactions. Thus, decreasing or eliminating H2 production without altering acetate production would result in a redox imbalance, and decreasing flux through this pathway could help alleviate this imbalance. Therefore, the decrease in acetate production seen here is expected upon the elimination of H2 production. While elimination of the [FeFe] hydrogenases alone increased ethanol yield substantially, the additional removal of the Ech hydrogenase was needed to further increase yield, clearly demonstrating the importance of Ech in this system. This result might be unexpected, because previous proteomics studies showed a lack of Ech protein in the proteome of related strain C. thermocellum ATCC27405 [25]. The cause of this discrepancy might well be the difficulty of detecting membrane proteins during proteomics studies, differences between C. thermocellum strains ATCC27405 and DSM1313, or differences in physiology due to the deletion of hydG in our study.
The hydrogenase mutants also stopped synthesizing lactate via lactate dehydrogenase (ldh). In C. thermocellum Ldh is known to be allosterically activated by fructose-1,6-bisphosphate (FbP) [26], presenting an alternate mechanism by which lactate production could have been eliminated. Perhaps the hydrogenase deletion strains have low levels of FbP, preventing Ldh from being active. Further, FbP-activated Ldh enzymes from other anaerobic thermophiles are also regulated by other metabolites, such as inhibition by NADPH in Thermoanaerobacter ethanolicus [27] and inhibition by pyrophosphate and activation by ATP in Caldicellulosiruptor saccharolyticus [28]. While these potential allosteric regulators have not been tested on the C. thermocellum Ldh, deletion of the hydrogenases would likely result in an accumulation of reduced ferredoxin via pyruvate: ferredoxin oxidoreductase (PFOR). This reduced ferredoxin plus NADH could then be used as substrates by NfnAB (Clo1313_1848 - 1849) to transfer electrons to NADPH [29], potentially resulting in low levels of NADH and an overabundance of NADPH, leading to inhibition of Ldh. With decreased Ldh activity and an AdhE capable of using both NADH and NADPH, the flux to lactate will decrease and flux to ethanol would then naturally increase to regenerate the cofactors.
Previous characterization of an ethanol-tolerant mutant of C. thermocellum revealed that it contained, among many other mutations, two point mutations in adhE, resulting in amino acid changes P704L and H734R. This mutation was reconstructed in an otherwise wild-type strain, resulting in altered alcohol dehydrogenase (ADH) enzyme cofactor specificity from NADH-dependent to NADPH-dependent and increased ethanol tolerance [13]. This suggested that an unknown redox imbalance occurs as the ethanol concentration increases. Additional evolved ethanol-tolerant C. thermocellum strains E50C and E50A also acquired independent mutations in adhE, resulting in amino acid changes to D494G and G553R, respectively [6]. However, the effect of the latter two mutations in an otherwise wild-type strain has not yet been determined. Here, the hydG mutant independently acquired the exact same D494G mutation as strain E50C. Unlike the previous P704L-H734R mutant, NADH-dependent activity was not eliminated. Instead, the hydG deletion strain contained high levels of both NADH- and NADPH-dependent ADH activities. Interestingly, ΔhydG was not ethanol tolerant, illustrating the complexity of the C. thermocellum ethanol tolerance phenotype. While it is possible that the D494G mutation does not contribute to the ethanol tolerance phenotype of strain E50C, a more likely explanation is that the additional redox imbalance(s) caused by deletion of hydG prevent the D494G mutation from rebalancing metabolism. Regardless, this gain of function mutation that allows use of either NADH or NADPH for reduction of acetaldehyde to ethanol will be useful for further metabolic engineering efforts by minimizing NADH/NADPH redox imbalances. Indeed, C. thermocellum ΔhydGΔech, with its high ethanol yield, near wild-type growth rate, and ADH capable of utilizing both NADH and NADPH, is now an ideal platform for further engineering of C. thermocellum for production of cellulosic fuels and chemicals.
Conclusions
While Clostridium thermocellum degrades cellulose well, efforts to engineer this strain are laborious and time-consuming. In addition to making ethanol, C. thermocellum converts cellulosic to lactate, formate, acetate, H2, ethanol, amino acids, and other products that must be eliminated to achieve high yield. Here, we developed a methodology to simultaneously inactivate multiple enzymes by targeting a common post-translational modification system, simplifying strain construction by minimizing the necessary genetic modifications. We eliminated H2 production and successfully redirected electron and carbon flux towards desired product. This strain will serve as a platform for further engineering to economically produce fuels and chemicals from lignocellulosic biomass.
Materials and methods
Strains and culture conditions
Standard molecular methods were used for plasmid cloning [30,31]. Escherichia coli TOP10 and BL21 were grown in LB medium supplemented with 12 μg ml−1 chloramphenicol when appropriate. Clostridium thermocellum DSM1313 and mutant strains were grown in a modified DSM122 medium similar to that described by Tripathi et al. [11] supplemented with 5 μg ml−1 thiamphenicol (TM; Sigma-Aldrich, St. Louis, MO, USA), 50 μg ml−1 5-fluoro-2′-deoxyuridine (FUdR, Sigma-Aldrich, St. Louis, MO, USA), and 500 μg ml−1 8-azahypoxanthine (8AZH; Acros Organics, Pittsburgh, PA, USA) as needed and MTC minimal medium [32] prepared as described in [6]. The modified DSM122 medium composition was (L−1): 3 g sodium citrate tribasic dehydrate, 1.3 g ammonium sulfate, 1.43 g potassium phosphate monobasic, 1.8 g potassium phosphate dibasic trihydrate, 0.5 g cysteine-HCl, 10.5 g 3-morpholino-propane-1-sulfonic acid (MOPS), 6 g glycerol-2-phosphate disodium, 0.41 g sodium acetate, 5 g cellobiose, 4.5 g yeast extract, 0.13 g calcium chloride dehydrate, 2.6 g magnesium chloride hexahydrate, 0.0011 g ferrous sulfate heptahydrate, and 0.0001 g resazurin, adjusted to pH 7.0. The minimal medium consisted of (L−1): 2 g sodium citrate dehydrate, 1.25 g citric acid monohydrate, 1 g sodium sulfate, 1 g potassium phosphate dibasic trihydrate, 2.5 g sodium bicarbonate, 1.5 g ammonium chloride, 2 g urea, 1 g magnesium chloride hexahydrate, 0.2 g calcium chloride dehydrate, 0.1 g ferrous chloride tetrahydrate, 1 g L-cysteine hydrochloride monohydrate, 5 g cellobiose, 0.001 g resazurin, 5 g MOPS, 20 mg pyridoxamine dihydrochloride, 1 mg riboflavin, 1 mg nicotinamide, 0.5 mg DL-thioctic acid, 4 mg 4-amino benzoic acid, 4 mg D-biotin, 0.025 mg folic acid, 2 mg cyanocobalamin, 4 mg thiamine hydrochloride, 0.5 mg MnCl2.4H2O, 0.5 mg CoCl2.6H2O, 0.2 mg ZnSO4.7H2O, 0.05 mg CuSO4.5H2O, 0.05 mg HBO3, 0.05 mg Na2MoO4.2H2O, and 0.05 mg NiCl2.6H2O.
Plasmid and strain constructions
Strains were constructed as previously described [8-10] in strain C. thermocellum ∆hpt, which is a derivative of strain DSM1313 and is herein referred to as wild type. Plasmid pAMG278 (Additional file 3) was used to delete hydG (Clo1313_1571), resulting in strain C. thermocellum ∆hpt ∆hydG, hereafter referred to as C. thermocellum ∆hydG. In the C. thermocellum ∆hydG background, plasmid pAMG275 (Additional file 3) was used to delete the [NiFe] maturase and hydrogenase enzymes hypABFCDE (Clo1313_0564 to 0569) and echABCDEF (Clo1313_0570 to 0575), resulting in strain C. thermocellum ∆hpt ∆hydG ∆hyp-ech, hereafter referred to as C. thermocellum ∆hydG∆ech. All genetic manipulation was carried out using the modified DSM122 medium. Briefly, the plasmids were each isolated from a dcm− strain of E. coli (Guss et al., 2012) [9], transformed into C. thermocellum ∆hpt via electroporation, and plated in medium supplemented with TM. Colonies were picked into liquid medium with TM, grown at 51°C, and plated in medium supplemented with TM and FUdR. Colonies were single colony purified and picked into liquid medium with TM. These cultures were then subcultured without TM and plated with 8AZH to select for the final deletion, followed by single colony purification to obtain pure cultures [8-10]. Deletion strains were confirmed by PCR (Additional file 1; primer sequences in Additional file 4). Strain purity was further confirmed by 16S rRNA gene sequencing.
Fermentation conditions
The inoculum for batch fermentation was prepared by growing the mutants in MTC medium overnight at 55°C in an anaerobic chamber (COY Laboratory Products, Inc., Grass Lake, MI, USA). The fermentation was carbon limited and carried out in 27-ml Balch tubes with 10 ml of MTC media containing 5 g L−1 of cellobiose under a N2 headspace sealed with butyl rubber stoppers. The tubes were inoculated with 0.5% v/v culture and incubated at 55°C. The fermentation products were determined after 53 hours of growth. The final cellobiose concentration was usually less than 0.5 mM, suggesting that fermentation activity was complete. Fermentations were performed at least two times with three independent biological replicates each. Growth rate was calculated using the change in absorbance during mid-log phase, from an OD600 of approximately 0.1 to 0.3.
Ethanol tolerance
Ethanol tolerance was tested in Balch tubes containing minimal medium with 0, 1, 2, 3, 4, and 5% v/v added ethanol, inoculated with 0.5% of overnight grown culture, and incubated at 55°C. Growth was monitored by measuring the optical density at 600 nm on a Unico 1200 spectrophotometer (Unico, Dayton, NJ, USA). Growth experiments were performed at least two times with three independent biological replicates each.
Analytical methods
Fermentation products, including ethanol, acetate, lactate, and formate, were analyzed on a Breeze 2 High Performance Liquid Chromatograph system using an Aminex-HPX-87H column with a 5 mM sulfuric acid mobile phase. H2 was measured using an Agilent Technologies 6850 Series II Gas Chromatograph (Agilent, Santa Clara, CA, USA) using a thermal conductivity detector at 190°C with a N2 reference flow and a Carboxen 1010 PLOT (30.0 m × 530 μm I.D.; model Supelco 25467) column.
Enzyme assay of alcohol dehydrogenase (ADH)
50-ml cultures of C. thermocellum cells were grown anaerobically to OD600 = 0.3 and harvested by centrifugation at 2800 × g for 30 minutes at 4°C and stored at −80°C. Before the assay, the pellet was resuspended anaerobically in 0.5 ml Assay Buffer (0.1 M Tris–HCl, 0.1 mM Fe2+, 0.1 mM DTT, pH 7.5). The cells were lysed with 2 μl Ready-Lyse Lysozyme from Epicentre Technologies (Madison, WI, USA), and 1 μl DNase I from New England Biolabs (Ipswich, MA, USA) was added to reduce viscosity. The resulting solution was centrifuged at 17,000 × g for 10 minutes at room temperature, and the supernatant was used as cell-free extracts for enzyme assays. For the ADH (acetaldehyde reduction) reactions, the anaerobic reaction mixture contained 0.25 mM NAD(P)H, 18 mM acetaldehyde, 1 to 10 μl cell-free extract, and 800 μl of Assay Buffer. Decrease in absorbance at 340 nm caused by NAD(P)H oxidation was monitored by an Agilent 8453 UV–vis spectrophotometer with Peltier controlled heating set at 55°C inside an anaerobic chamber. Protein concentration was determined by using the Bradford method [33].
Amino acid analysis
Amino acid analysis was carried out by AminoAcids.com (St. Paul, MN, USA). Samples were prepared for analysis by appropriate dilution with a deproteinizing solution (13.5% w/v 5-sulfosalicylic acid hydrate), L-2-amino-3-guanidinopropionic acid hydrochloride, and eluent lithium buffer. The mixture was vortexed, micro-centrifuged, and filtered through a 0.2-μm filter. Amino acid analysis was performed on a Hitachi Model L-8900 HPLC with 10-cm cation exchange columns, four sequential lithium-based eluents (Hitachi and Pickering Laboratories), and lithium hydroxide for column regeneration. Absorbance was measured at 440 and 570 nm following post-column color development by ninhydrin reagent at 135°C. Data are an average of three independent biological replicates. AminoAcids.com reported that an unknown peak overlapped with the asparagine peak, preventing quantification in both the ΔhydG and ΔhydGΔech samples. As the ninhydrin method detects primary and secondary amines, the interfering peak presumably represents an as yet unidentified amino compound.
Identification of mutation in alcohol dehydrogenase gene in ∆hydG strain
The adhE region was PCR amplified as 982-bp product using primers XD520 and XD521 (Additional file 3). The primer region was 70 bp away from the D494G mutation [6], which was sequenced and confirmed with primer XD560 (Additional file 4).
Acknowledgements
This work was supported by the BioEnergy Science Center, U.S. DOE Bioenergy Research Center supported by the Office of Biological and Environmental Research in the DOE Office of Science. Oak Ridge National Laboratory is managed by UT-Battelle, LLC, for the U.S. DOE under contract DE-AC05-00OR22725. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Disclaimer
This manuscript has been authored by UT-Battelle, LLC, under Contract No. DE-AC05-00OR22725 with the U.S. Department of Energy. The United States Government retains and the publisher, by accepting the article for publication, acknowledges that the United States Government retains a non-exclusive, paid-up, irrevocable, world-wide license to publish or reproduce the published form of this manuscript, or allow others to do so, for United States Government purposes.
Abbreviations
- ADH
alcohol dehydrogenase
- 8AZH
8-azahypoxanthine
- CBP
consolidated bioprocessing
- FUdR
5-fluoro-2′-deoxyuridine
- MOPS
3-morpholino-propane-1-sulfonic acid
- TM
thiamphenicol
Additional files
Confirmation of C. thermocellum deletion mutants. A) Three primer sets were used for confirmation of the hydG deletion. Primer set (P1) and (P2) amplified 863-bp and 729-bp fragments of the hydG, respectively, which are present in the wild type but absent in C. thermocellum ∆hydG. Primer set (P3) amplifies the 3,500-bp region of the wild-type locus, while amplification from ∆hydG mutant results in a 2,100-bp fragment. B) Similar primer sets (P4, P5, and P6) are used to confirm deletion of ech in the C. thermocellum ∆hydG background. P4 and P5 amplify 701-bp and 721-bp regions of ech, respectively, which are present in the ∆hydG and wild-type strains but absent in the ∆hydG∆ech strain. Primer set (P6) would amplify a 12,000-bp region of the wild-type locus and a 2,000-bp fragment in the ∆hydG∆ech strain. C) PCR confirmation of deletion of hydG and ech. Lane m, DNA ladder with molecular weights noted (in kilobases); lane a, ∆hydG∆ech template; lane b, ∆hydG template; lane c, C. thermocellum wild-type template; lane d, no template PCR control.
Maximum optical density (OD) attained by wild-type and mutant strains of C. thermocellum. (Left) rich and (right) minimal medium supplemented with 0 to 5% (v/v) added ethanol. Symbols: Open black squares, C. thermocellum wild type; closed gray squares, ethanol-tolerant control C. thermocellum adhE*(EA); closed blue triangles, C. thermocellum ∆hydG; and open red triangles, C. thermocellum ∆hydG∆ech. Error bars represent one standard deviation.
Annotated plasmid sequences of pAMG275 and pAMG278.
List of primers used in this study.
Footnotes
Competing interests
LRL is a co-founder and employee of Mascoma Corporation, at which DGO was formerly employed. Mascoma has a financial interest in biomass conversion processes.
Authors’ contributions
RB participated in the design of the study, participated in strain construction, characterized growth and fermentation, and drafted the manuscript. TZ carried out the enzyme assays. DGO participated in the design of the study. LRL helped conceive of the study and participated in its design. AMG helped conceive of the study, participated in its design and coordination, participated in strain construction, and drafted the manuscript. All authors read and approved the final manuscript.
Contributor Information
Ranjita Biswas, Email: ranjitabiswas@gmail.com.
Tianyong Zheng, Email: tianyong.zheng.gr@dartmouth.edu.
Daniel G Olson, Email: dan268@gmail.com.
Lee R Lynd, Email: lee.r.lynd@dartmouth.edu.
Adam M Guss, Email: gussam@ornl.gov.
References
- 1.Lynd LR, Weimer PJ, van Zyl WH, Pretorius IS. Microbial cellulose utilization: fundamentals and biotechnology. Microbiol Mol Biol Rev. 2002;66:506–77. doi: 10.1128/MMBR.66.3.506-577.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.US Department of Energy . US billion-ton update: biomass supply for a bioenergy and bioproducts industry. Oak Ridge, TN: Oak Ridge National Laboratory; 2011. [Google Scholar]
- 3.Blumer-Schuette SE, Brown SD, Sander KB, Bayer EA, Kataeva I, Zurawski JV, et al. Thermophilic lignocellulose deconstruction. FEMS Microbiol Rev. 2014;38:393–448. doi: 10.1111/1574-6976.12044. [DOI] [PubMed] [Google Scholar]
- 4.Lynd LR, Laser MS, Bransby D, Dale BE, Davison B, Hamilton R, et al. How biotech can transform biofuels. Nat Biotechnol. 2008;26:169–72. doi: 10.1038/nbt0208-169. [DOI] [PubMed] [Google Scholar]
- 5.Ellis LD, Holwerda EK, Hogsett D, Rogers S, Shao X, Tschaplinski T, et al. Closing the carbon balance for fermentation by Clostridium thermocellum (ATCC 27405) Bioresource Technol. 2012;103:293–9. doi: 10.1016/j.biortech.2011.09.128. [DOI] [PubMed] [Google Scholar]
- 6.Shao X, Raman B, Zhu M, Mielenz JR, Brown SD, Guss AM, et al. Mutant selection and phenotypic and genetic characterization of ethanol-tolerant strains of Clostridium thermocellum. Appl Microbiol Biotechnol. 2011;92:641–52. doi: 10.1007/s00253-011-3492-z. [DOI] [PubMed] [Google Scholar]
- 7.Williams TI, Combs JC, Lynn BC, Strobel HJ. Proteomic profile changes in membranes of ethanol-tolerant Clostridium thermocellum. Appl Microbiol Biotechnol. 2007;74:422–32. doi: 10.1007/s00253-006-0689-7. [DOI] [PubMed] [Google Scholar]
- 8.Argyros DA, Tripathi SA, Barrett TF, Rogers SR, Feinberg LF, Olson DG, et al. High ethanol titers from cellulose using metabolically engineered thermophilic, anaerobic microbes. Appl Environ Microbiol. 2011;77:8288–94. doi: 10.1128/AEM.00646-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guss AM, Olson DG, Caiazza NC, Lynd LR. Dcm methylation is detrimental to plasmid transformation in Clostridium thermocellum. Biotechnol Biofuels. 2012;5:30. doi: 10.1186/1754-6834-5-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Olson DG, Lynd LR. Transformation of Clostridium thermocellum by electroporation. Methods Enzymol. 2012;510:317–30. doi: 10.1016/B978-0-12-415931-0.00017-3. [DOI] [PubMed] [Google Scholar]
- 11.Tripathi SA, Olson DG, Argyros DA, Miller BB, Barrett TF, Murphy DM, et al. Development of pyrF-based genetic system for targeted gene deletion in Clostridium thermocellum and creation of a pta mutant. Appl Environ Microbiol. 2010;76:6591–9. doi: 10.1128/AEM.01484-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mohr G, Hong W, Zhang J, Cui GZ, Yang Y, Cui Q, et al. A targetron system for gene targeting in thermophiles and its application in Clostridium thermocellum. PLoS One. 2013;8:e69032. doi: 10.1371/journal.pone.0069032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown SD, Guss AM, Karpinets TV, Parks JM, Smolin N, Yang S, et al. Mutant alcohol dehydrogenase leads to improved ethanol tolerance in Clostridium thermocellum. Proc Natl Acad Sci U S A. 2011;108:13752–7. doi: 10.1073/pnas.1102444108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Biswas R, Prabhu S, Lynd LR, Guss AM. Increase in ethanol yield via elimination of lactate production in an ethanol-tolerant mutant of Clostridium thermocellum. PLoS One. 2014;9:e86389. doi: 10.1371/journal.pone.0086389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rydzak T, McQueen PD, Krokhin OV, Spicer V, Ezzati P, Dwivedi RC, et al. Proteomic analysis of Clostridium thermocellum core metabolism: relative protein expression profiles and growth phase-dependent changes in protein expression. BMC Microbiol. 2012;12:214. doi: 10.1186/1471-2180-12-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deng Y, Olson DG, Zhou J, Herring CD, Joe Shaw A, Lynd LR. Redirecting carbon flux through exogenous pyruvate kinase to achieve high ethanol yields in Clostridium thermocellum. Metab Eng. 2013;15:151–8. doi: 10.1016/j.ymben.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 17.Raman B, McKeown CK, Rodriguez M, Jr, Brown SD, Mielenz JR. Transcriptomic analysis of Clostridium thermocellum ATCC 27405 cellulose fermentation. BMC Microbiol. 2011;11:134. doi: 10.1186/1471-2180-11-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Calusinska M, Happe T, Joris B, Wilmotte A. The surprising diversity of clostridial hydrogenases: a comparative genomic perspective. Microbiol-Sgm. 2010;156:1575–88. doi: 10.1099/mic.0.032771-0. [DOI] [PubMed] [Google Scholar]
- 19.Feinberg L, Foden J, Barrett T, Davenport KW, Bruce D, Detter C, et al. Complete genome sequence of the cellulolytic thermophile Clostridium thermocellum DSM1313. J Bacteriol. 2011;193:2906–7. doi: 10.1128/JB.00322-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mulder DW, Shepard EM, Meuser JE, Joshi N, King PW, Posewitz MC, et al. Insights into [FeFe]-hydrogenase structure, mechanism, and maturation. Structure. 2011;19:1038–52. doi: 10.1016/j.str.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 21.van der Veen D, Lo J, Brown SD, Johnson CM, Tschaplinski TJ, Martin M, et al. Characterization of Clostridium thermocellum strains with disrupted fermentation end-product pathways. J Ind Microbiol Biotechnol. 2013;40:725–34. doi: 10.1007/s10295-013-1275-5. [DOI] [PubMed] [Google Scholar]
- 22.Caffrey SM, Park HS, Voordouw JK, He Z, Zhou J, Voordouw G. Function of periplasmic hydrogenases in the sulfate-reducing bacterium Desulfovibrio vulgaris Hildenborough. J Bacteriol. 2007;189:6159–67. doi: 10.1128/JB.00747-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Casalot L, De Luca G, Dermoun Z, Rousset M, de Philip P. Evidence for a fourth hydrogenase in Desulfovibrio fructosovorans. J Bacteriol. 2002;184:853–6. doi: 10.1128/JB.184.3.853-856.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Casalot L, Valette O, De Luca G, Dermoun Z, Rousset M, de Philip P. Construction and physiological studies of hydrogenase depleted mutants of Desulfovibrio fructosovorans. FEMS Microbiol Lett. 2002;214:107–12. doi: 10.1111/j.1574-6968.2002.tb11332.x. [DOI] [PubMed] [Google Scholar]
- 25.Rydzak T, Grigoryan M, Cunningham ZJ, Krokhin OV, Ezzati P, Cicek N, et al. Insights into electron flux through manipulation of fermentation conditions and assessment of protein expression profiles in Clostridium thermocellum. Appl Microbiol Biotechnol. 2014;98:6497–510. doi: 10.1007/s00253-014-5798-0. [DOI] [PubMed] [Google Scholar]
- 26.Ozkan M, Yilmaz EI, Lynd LR, Ozcengiz G. Cloning and expression of the Clostridium thermocellum L-lactate dehydrogenase gene in Escherichia coli and enzyme characterization. Can J Microbiol. 2004;50:845–51. doi: 10.1139/w04-071. [DOI] [PubMed] [Google Scholar]
- 27.Bryant FO. Characterization of the fructose 1,6-bisphosphate-activated, L(+)-lactate dehydrogenase from Thermoanaerobacter ethanolicus. J Enzyme Inhib. 1991;5:235–48. doi: 10.3109/14756369109080062. [DOI] [PubMed] [Google Scholar]
- 28.Willquist K, van Niel EW. Lactate formation in Caldicellulosiruptor saccharolyticus is regulated by the energy carriers pyrophosphate and ATP. Metab Eng. 2010;12:282–90. doi: 10.1016/j.ymben.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 29.Wang S, Huang H, Moll J, Thauer RK. NADP+ reduction with reduced ferredoxin and NADP+ reduction with NADH are coupled via an electron-bifurcating enzyme complex in Clostridium kluyveri. J Bacteriol. 2010;192:5115–23. doi: 10.1128/JB.00612-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maniatis T, Fritsch EF, Sambrook J. Molecular cloning: a laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1982. [Google Scholar]
- 31.Shanks RMQ, Kadouri DE, MacEachran DP, O’Toole GA. New yeast recombineering tools for bacteria. Plasmid. 2009;62:88–97. doi: 10.1016/j.plasmid.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hogsett D. Cellulose hydrolysis and fermentation by Clostridium thermocellum for the production of ethanol [PhD thesis] Hanover, NH: Dartmouth College, Thayer School of Engineering; 1995. [Google Scholar]
- 33.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]