

The surface properties and dynamics of PKD-like domains from ColG and ColH differ.
Keywords: polycystic kidney disease-like domains, ColG, ColH, Clostridium histolyticum
Abstract
Clostridium histolyticum collagenases ColG and ColH are segmental enzymes that are thought to be activated by Ca2+-triggered domain reorientation to cause extensive tissue destruction. The collagenases consist of a collagenase module (s1), a variable number of polycystic kidney disease-like (PKD-like) domains (s2a and s2b in ColH and s2 in ColG) and a variable number of collagen-binding domains (s3 in ColH and s3a and s3b in ColG). The X-ray crystal structures of Ca2+-bound holo s2b (1.4 Å resolution, R = 15.0%, R free = 19.1%) and holo s2a (1.9 Å resolution, R = 16.3%, R free = 20.7%), as well as of Ca2+-free apo s2a (1.8 Å resolution, R = 20.7%, R free = 27.2%) and two new forms of N-terminally truncated apo s2 (1.4 Å resolution, R = 16.9%, R free = 21.2%; 1.6 Å resolution, R = 16.2%, R free = 19.2%), are reported. The structurally similar PKD-like domains resemble the V-set Ig fold. In addition to a conserved β-bulge, the PKD-like domains feature a second bulge that also changes the allegiance of the subsequent β-strand. This β-bulge and the genesis of a Ca2+ pocket in the archaeal PKD-like domain suggest a close kinship between bacterial and archaeal PKD-like domains. Different surface properties and indications of different dynamics suggest unique roles for the PKD-like domains in ColG and in ColH. Surface aromatic residues found on ColH s2a-s2b, but not on ColG s2, may provide the weak interaction in the biphasic collagen-binding mode previously found in s2b-s3. B-factor analyses suggest that in the presence of Ca2+ the midsection of s2 becomes more flexible but the midsections of s2a and s2b stay rigid. The different surface properties and dynamics of the domains suggest that the PKD-like domains of M9B bacterial collagenase can be grouped into either a ColG subset or a ColH subset. The conserved properties of PKD-like domains in ColG and in ColH include Ca2+ binding. Conserved residues not only interact with Ca2+, but also position the Ca2+-interacting water molecule. Ca2+ aligns the N-terminal linker approximately parallel to the major axis of the domain. Ca2+ binding also increases stability against heat and guanidine hydrochloride, and may improve the longevity in the extracellular matrix. The results of this study will further assist in developing collagen-targeting vehicles for various signal molecules.
1. Introduction
Clostridium histolyticum collagenases are causative agents of gas gangrene. The two classes of collagenase, ColG and ColH, differ in domain structures (s1, s2, s3a and s3b for ColG and s1, s2a, s2b and s3 for ColH; Matsushita et al., 2001 ▶; Fig. 1 ▶) and work synergistically to degrade collagen fibers (Breite et al., 2011 ▶). The s1 collagenase module belongs to metallopeptidase subfamily M9B. The amino-acid sequences of s2, s2a and s2b resemble the polycystic kidney disease domain (PKD) that was first identified in the polycystic kidney disease protein PKD1 (The International Polycystic Kidney Disease Consortium, 1995 ▶). The C-terminal domains s3a, s3b and s3 are collagen-binding homologues that are a subclass of bacterial pre-peptidase C-terminal domains (PPC superfamily; Wilson et al., 2003 ▶; Philominathan, Koide et al., 2009 ▶; Philominathan et al., 2012 ▶; Bauer et al., 2013 ▶). The collagen-binding segment composed of the PKD-like domain and collagen-binding domain (CBD) is not necessary to degrade gelatin (denatured, non-triple-helical collagen) and acid-solubilized collagen. However, this segment is necessary to degrade insoluble collagen fibers.
Figure 1.

Domain map of collagenases ColG and ColH from C. histolyticum. The pre-pro-peptide (grey hatching) is cleaved from the mature enzyme and indicated by sequence numbering N1–N110 (ColG) and N1–N40 (ColH). The collagenase module is composed of an activator subdomain (olive) and peptidase subdomain (dark olive) that is accompanied by a helper subdomain. The PKD-like domain(s) (yellow for ColG; cyan and green for ColH) connect the collagenase module to the C-terminal CBD(s) (red for ColG; salmon for ColH) that are responsible for collagen binding.
Understanding the interaction of Ca2+ is significant owing to its role in regulating both stability and enzyme activity in the extracellular matrix (ECM; Philominathan, Matsushita et al., 2009 ▶; Bauer et al., 2013 ▶; Ohbayashi et al., 2013 ▶). Full-length ColH has been shown to undergo Ca2+-dependent structural changes as demonstrated using SAXS and limited proteolysis (Ohbayashi et al., 2013 ▶). In ColG, Ca2+ triggers the linker between s3a and s3b to undergo secondary-structure transformation from an α-helix to a β-strand to increase collagen affinity (Wilson et al., 2003 ▶; Sides et al., 2012 ▶). Similar to the N-terminal linker structure of s3b (Philominathan, Matsushita et al., 2009 ▶), the N-terminal linker structure of the PKD-like domain is also thought to be Ca2+-dependent, and thus high-resolution structures of both apo and holo states of the PKD-like domains are needed in order to elucidate their activation mechanism. Thus far, crystallographic methods have been used to describe apo s1 from ColG (Eckhard et al., 2011 ▶), the holo peptidase subdomains of ColH (Eckhard et al., 2013 ▶), apo and holo s2 (Eckhard et al., 2011 ▶, 2013 ▶), apo and holo s3b of ColG (Wilson et al., 2003 ▶) and holo s3 of ColH (Bauer et al., 2013 ▶). In the apo s2 structure, however, the conserved Pro688 near the Ca2+-binding site was mutated to Thr. As a side note, we use the amino-acid sequence numbering for the mature enzymes. The numbering for s2b and s2a accounts for the cleavage of a 40-amino-acid pre-pro-peptide present in ColH, while the numbering for s2 accounts for the cleavage of a 110-amino-acid pre-pro-peptide present in ColG. In this paper, we describe crystal structures of ColG and, for the first time, ColH PKD-like domains. Thermal and chemical stability differences upon Ca2+ binding for the PKD-like domains are also reported.
The collagenolytic mechanism differs between mammalian matrix metalloproteinases (MMPs) and bacterial collagenases (Adhikari et al., 2012 ▶; Duarte et al., 2014 ▶). Unlike bacterial collagenases, MMPs are sequence-specific and are proposed to actively unwind the triple helix (Bertini et al., 2012 ▶; Nagase & Fushimi, 2008 ▶; Welgus et al., 1981a ▶,b ▶). Meanwhile, each domain in bacterial collagenase is believed to play a unique role in collagenolysis (Fields, 2013 ▶). The C-terminal CBD unidirectionally binds to under-twisted sites in the triple-helical collagenous peptide (Philominathan et al., 2012 ▶; Bauer et al., 2013 ▶). The CBD does not unwind mini-collagen, and hence targeting under-twisted regions of tropocollagen may circumvent the energy barrier required for unwinding the helix. Various roles have been proposed for the PKD-like domains. The PKD-like domain has been shown to swell, but not unwind, collagen fibrils (Wang et al., 2010 ▶). Clostridial PKD-like domains do not bind tightly to collagen fibrils (Matsushita et al., 1998 ▶, 2001 ▶), although s2b has been shown to enhance the ability of s3 to bind to the collagen fiber. S2b-s3 binding is biphasic; the initial low affinity (K d = 2.11 × 10−6 M) leads to higher affinity (K d = 3.39 × 10−7 M) (Matsushita et al., 1998 ▶). The N-terminal collagenase module, s1, has a two-domain architecture that disbands the collagen microfibril into monomeric triple helices and then cleaves the exposed peptide bond preceding the Gly residue (Eckhard et al., 2011 ▶, 2013 ▶). Crystallographic packing analysis of s2 suggested a side-by-side assembly of s1, s2, s3a and s3b that matches the width of the collagen microfibril (Eckhard et al., 2011 ▶; Eckhard & Brandstetter, 2011 ▶). The proposed holo ColG structure is compact; s2 helps to align the active site of s1 with the binding clefts of s3a and s3b. In contrast, the solution envelope of ColH resembles a tadpole (Ohbayashi et al., 2013 ▶), and thus the role of the PKD-like domains in ColH could differ from that in ColG. The work presented here provides a structural framework to better decipher the role of the PKD-like domain.
Despite their detrimental role in bacterial infection, bacterial collagenases and their collagen-binding segments have been investigated for therapeutic applications. A cocktail of C. histolyticum ColG and ColH is used in both nonsurgical treatment of Dupuytren’s contracture (Hurst et al., 2009 ▶) and the isolation of pancreatic islets (McCarthy et al., 2011 ▶; Fujio et al., 2013 ▶). Other applications are in preclinical stages (Duarte et al., 2014 ▶). Moreover, fusion proteins consisting of growth factors, cytokines or hormones and the collagen-binding segment s2b-s3 are non-diffusing and long-lasting at wound sites (Nishi et al., 1998 ▶; Akimoto et al., 2013 ▶; Uchida et al., 2013 ▶; Saito et al., 2013 ▶), and hence the binding segments are being developed as drug-delivery vehicles. Therapeutic strategies based on these results are proposed to enhance efficacy by minimizing the quantity of signal molecules necessary and reducing side effects. In contrast, bone distribution of the fusion protein of parathyroid hormone with s3 only (PTH-s3) was efficacious in increasing bone mineral density in osteoporotic models, although fusion proteins of PTH-s2b-s3 demonstrated little efficacy (Ponnapakkam et al., 2012 ▶, 2013 ▶; Ponnapakkam, Katikaneni, Miller et al., 2011 ▶; Ponnapakkam, Katikaneni, Nichols et al., 2011 ▶). When applied to skin, PTH-s3 was efficacious in hair-follicle regeneration in alopecia models (Katikaneni et al., 2012 ▶, 2014 ▶). This study of PKD-like domains is necessary for commercialization and optimization efforts for various drug candidates.
2. Methods
2.1. Expression and purification of s2a, s2b and s2
Expression and purification of each PKD-like domain as a glutathione S-transferase (GST)-fusion protein was achieved using previously described methods (Matsushita et al., 2001 ▶).
2.2. 15N-HSQC NMR characterization of apo s2
Although stably folded and monodisperse in solution, s2 with its N-terminal linker did not crystallize. 15N-enriched apo s2 was produced to measure the dynamics of the protein using NMR. 15N-enriched s2 was prepared as described in Philominathan et al. (2008 ▶). NMR experiments were performed at 298 ± 0.5 K on a Bruker 700 MHz spectrometer equipped with a cryoprobe. The concentration of the protein used was 0.1 mM in 50 mM Tris–HCl pH 7.5. In the HSQC spectra, 13 residues could not be identified owing to line broadening (Supplementary Fig. S1). Using the homology-modeled s2 (based on PDB entry 2c4x; Najmudin et al., 2006 ▶), we reasoned that the unobserved HSQC peaks corresponded to a highly dynamic N-terminus that hindered crystallization. Guided by the solution data, 13 residues were truncated from the N-terminus. The truncated s2 crystallized readily.
2.3. Crystal structure determination of PKD-like domains
Initial conditions suitable to grow crystals of apo s2a and holo s2b and two crystal forms of apo s2 were identified by the sitting-drop method using a high-throughput screen (Hampton Research Crystal Screen HT). Subsequent crystallization trials using the initial conditions were carried out using the hanging-drop method. Apo s2a (at a concentration of 30.5 mg ml−1) was crystallized from 3 M ammonium sulfate, 0.1 M MES pH 4.5, 15%(w/v) PEG 4000 at 290 K, whereas holo s2a (at a concentration of 4.8 mg ml−1) was crystallized from 30%(w/v) PEG 4000, 0.2 M MgCl2, 0.1 M Tris–HCl pH 8.5 at 17°C. These crystals were subsequently soaked in 15%(w/v) PEG 4000, 0.2 M MgCl2, 50 mM CaCl2 , 0.1 M Tris–HCl pH 8.5 before initial unit-cell characterization and data collection. Meanwhile, holo s2b (at a concentration of 13.7 mg ml−1) was crystallized from 35%(w/v) PEG 5000, 0.2 M ammonium sulfate, 0.1 M MES pH 6.5 at 277 K. Both crystal forms of s2 (12.0 mg ml−1) were grown from 41–49% 2-methyl-2,4-pentanediol (MPD), 100 mM bis-tris pH 5.5, 0.1–0.3 M ammonium acetate at 277 K. The in-house X-ray diffraction facility (Rigaku MicroMax-007, Osmic Blue confocal mirrors, Saturn CCD detector) was used for initial characterization of each PKD-like domain crystal, and in the case of the s2a crystals was also used for data collection at 113 K. s2b and s2 crystals were cryocooled and subsequently stored in liquid nitrogen until data collection. Diffraction data were collected at 109 K on the 19-ID beamline of the Advanced Photon Source at Argonne National Laboratory, USA.
Each s2a data set was indexed and scaled using d*TREK (Pflugrath, 1999 ▶), whereas each s2b and s2 data set was indexed and scaled using HKL-3000 (Minor et al., 2006 ▶). In each case, a data set truncated to 3 Å resolution was used for molecular replacement using Phaser (McCoy et al., 2007 ▶). The PKD-like domain from the carbohydrate-binding module (PDB entry 2c4x) was used as the search model during the structure determination of s2. s2 form I was subsequently used as the search model during structure determination of s2a and s2b. Four molecules were found in the asymmetric unit of the apo s2a crystals, while eight molecules were found in the asymmetric unit of the holo s2a crystals. Meanwhile, two molecules were found in the asymmetric unit of the holo s2b crystals and each form of the apo s2 crystals.
The subsequent structure determination for each model was accomplished using an iterative process of manual adjustments aided by the use of MIFit (McRee, 1999 ▶) and refinement using REFMAC (Murshudov et al., 2011 ▶). During manual adjustments, ARP/wARP (Perrakis et al., 1999 ▶) was used to place water molecules. R free was lowered for the s2 form I, apo s2a and holo s2a models by utilizing Babinet’s principle for bulk-solvent scaling. In each s2a model, R free was also lowered by applying TLS and tight NCS restraints. The s2a models were refined with isotropic B factors, whereas the s2 form I and s2b models were refined with anisotropic B factors. PARVATI (Merritt, 2012 ▶) calculated the anisotropy of s2b and s2 to be 0.5 ± 0.1 and 0.4 ± 0.1, respectively. Isotropic B factors would result in an anisotropy of 1.0. Each model exhibited excellent geometry as analyzed by MolProbity (Chen et al., 2010 ▶). Full data-collection and refinement statistics are summarized in Table 1 ▶ for s2a, s2b and s2 form I and in Supplementary Table S1 for s2 form II. Alternate conformations are detailed in Supplementary Table S2.
Table 1. Data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| Holo s2a | Apo s2a | Holo s2b | Apo s2 form I | |
|---|---|---|---|---|
| Data collection | ||||
| X-ray wavelength () | 1.54 | 1.54 | 0.97937 | 0.919 |
| Space group | C2 | P61 | P21 | P21 |
| Unit-cell parameters | ||||
| a () | 102.3 | 88.3 | 49.38 | 25.00 |
| b () | 87.6 | 88.3 | 38.87 | 71.76 |
| c () | 104.4 | 123.6 | 54.66 | 47.89 |
| () | 90.0 | 90.0 | 90.0 | 90.0 |
| () | 116.0 | 90.0 | 98.43 | 95.56 |
| () | 90.0 | 120.0 | 90.00 | 90.00 |
| Resolution () | 93.81.9 (1.981.91) | 76.51.8 (1.811.76) | 22.191.42 (1.441.42) | 47.671.40 (1.441.40) |
| No. of reflections | 224470 | 252508 | 137190 | 124611 |
| Multiplicity | 3.6 (3.1) | 4.7 (2.1) | 3.5 (2.9) | 3.3 (2.4) |
| Completeness (%) | 96.4 (93.8) | 99.4 (95.7) | 96.7 (85.8) | 92.5 (83.7) |
| I/(I) | 13.8 (3.0) | 11.5 (2.3) | 33.0 (3.1) | 21.3 (2.0) |
| R meas † (%) | 5.4 (31.0) | 7.5 (37.4) | 4.6 (38.5) | 8.1 (46.9) |
| Refinement | ||||
| Unique reflections | 58720 | 50932 | 35993 | 28786 |
| R cryst ‡ (%) | 16.3 (25.5) | 20.7 (26.8) | 15.0 (25.4) | 16.9 (28.3) |
| R free § (%) | 20.7 (29.7) | 27.2 (32.0) | 19.1 (33.2) | 21.2 (35.6) |
| Average B factor (2) | ||||
| Chain A, main chain | 18.9 | 17.6 | 9.6 | 12.5 |
| Chain A, side chains | 23.3 | 21.1 | 13.7 | 16.5 |
| Chain B, main chain | 18.9 | 17.9 | 8.3 | 12.5 |
| Chain B, side chains | 23.5 | 21.4 | 12.2 | 16.8 |
| Chain C, main chain | 18.9 | 18.9 | N/A | N/A |
| Chain C, side chains | 23.6 | 22.0 | N/A | N/A |
| Chain D, main chain | 18.7 | 19.3 | N/A | N/A |
| Chain D, side chains | 23.1 | 22.3 | N/A | N/A |
| Chain E, main chain | 19.0 | N/A | N/A | N/A |
| Chain E, side chains | 23.5 | N/A | N/A | N/A |
| Chain F, main chain | 18.7 | N/A | N/A | N/A |
| Chain F, side chains | 23.0 | N/A | N/A | N/A |
| Chain G, main chain | 18.8 | N/A | N/A | N/A |
| Chain G, side chains | 23.1 | N/A | N/A | N/A |
| Chain H, main chain | 19.0 | N/A | N/A | N/A |
| Chain H, side chains | 23.5 | N/A | N/A | N/A |
| Solvent | 46.1 | 42.2 | 25.6 | 31.1 |
| Ramachandran statistics | ||||
| Favored (%) | 98.5 | 99.4 | 98.9 | 99.4 |
| Additionally allowed (%) | 1.5 | 0.6 | 1.1 | 0.6 |
| Outliers (%) | 0 | 0 | 0 | 0 |
R
meas = 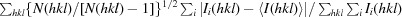 .
.
R
cryst = 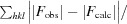
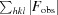 for the 95% of reflection data used for refinement.
for the 95% of reflection data used for refinement.
R
free =
for the 5% of reflection data excluded from refinement.
2.4. Equilibrium denaturation of PKD-like domains measured using fluorescence spectroscopy
PKD-like domains share similar topology, and unfolding of each domain was monitored by the change in intrinsic fluorescence of a conserved Trp residue. All experiments were carried out at room temperature using a Hitachi F-2500 fluorimeter with excitation and emission bandwidths at 2.5 and 10 nm, respectively. The excitation wavelength used was 280 nm, and fluorescence emissions were monitored between 300 and 450 nm. For s2b, s2a and s2, λmax for the folded state occurred at 325, 328 and 327 nm, respectively. For each domain, λmax for the denatured state occurred at 350 nm. The ratio of intensity at 350 nm versus the intensity at the native-state λmax was used to track the unfolding process. During thermal denaturation trials, the temperature of the protein solution was maintained with a Neslab RTE-110 circulating water bath (Thermo Scientific, Newington, New Hampshire, USA). In the thermal denaturation trials for s2b and s2, the protein concentration was 3 µM. In the chemical denaturation trials, the protein concentration was 1.5 µM. In the case of s2a, the protein concentration for both thermal and chemical denaturation was 5 µM. Each holo PKD-like domain was supplemented with 2 mM CaCl2, while each apo PKD-like domain was supplemented with 2 mM EDTA. In all cases, the protein was diluted in 10 mM Tris–HCl pH 7.5, 100 mM NaCl. When heat was used as the denaturant, each domain was exposed to temperatures that linearly increased from 280.5 to 363 K in 2.5 K increments. When guanidine hydrochloride (Gu–HCl) was used as the denaturant, each domain was exposed to concentrations of denaturant that increased linearly from 0.0 to 5.8 M in 0.2 M increments. ΔG HOH, C M and m values were calculated as described previously (Philominathan, Matsushita et al., 2009 ▶; Bauer et al., 2013 ▶).
3. Results and discussion
The X-ray crystal structures of Ca2+-bound holo s2a (space group C2), Ca2+-free apo s2a (space group P61), Ca2+-bound holo s2b (space group P21) and two new forms of N-terminally truncated wild-type apo s2 (space groups P21 and P212121) are reported for the first time. Of the novel s2a and s2b structures, that of s2b was solved at higher resolution and therefore is described in the most detail. New insights into s2 are subsequently reported.
3.1. Overall structure descriptions of apo and holo s2a
In the following discussion, holo s2a will be described first (Fig. 2 ▶ a). The eight holo s2a molecules are virtually identical (average r.m.s.d. of 0.2 ± 0.1 Å). Here, the molecules spiral along the crystallographic (1, 0, 1) axis. Along this axis, molecule pairs A and G, B and E, C and F, and D and H are related by NCS translation that results in an off-origin peak that is 63.9% of the origin peak in the Patterson map.
Figure 2.

Structural comparison of holo s2a (a), apo s2a (b), holo s2b (c) and apo s2 (d). Hydrogen bonds that stabilize β-bulges are highlighted. This figure was prepared using PyMOL (Schrödinger).
Similar to the molecules in the holo s2a crystal, the four apo s2a molecules are similar (average r.m.s.d. of 0.5 ± 0.2 Å), with molecules C and D being the most similar (r.m.s.d. = 0.2 Å) and molecules A and D being the most divergent (r.m.s.d. = 0.8 Å). Molecules A and B, as well as molecules C and D, are related by a noncrystallographic twofold. Temperature factors for each structure are relatively high (Table 1 ▶), possibly as a consequence of the high solvent content in the crystal (61.8%).
The holo and apo structures resemble each other (r.m.s.d. = 0.6 ± 0.2 Å). As expected, the most notable difference between the structures occurs near the N-terminus, where Ca2+ reorients the interacting residues Asn685 and Ser686. While neither structure could be refined using anisotropic B factors, comparison of B factors revealed that with the exception of the N-terminal residues, no significant change in B factors occurred upon Ca2+ binding.
Unlike s2b or s2, s2a truncates β-strand A through an approximately 126° rotation of the ψ bond of Ile692. To accommodate the change, Tyr696 packs with Phe706 and is involved in a Δ4 Tyr corner, in which the side-chain hydroxyl group hydrogen-bonds to the backbone four residues prior to it (Hemmingsen et al., 1994 ▶). Interestingly, the Tyr corner also stabilizes the nonprolyl cis-peptide bond between Gly694 and Thr695 of s2a that forms the bulge that realigns β-strand A′ to interact with β-strand G (Fig. 2 ▶ a and 2 ▶ b). A second bulge between β-strands B and B′ is stabilized by a hydrogen-bonding network that features a monodentate interaction between Asn735 and Ser708.
3.2. Overall structure description of holo s2b
Similar to holo s2a, the two NCS-related holo s2b structures are virtually identical (r.m.s.d. of 0.4 Å). In the structures, one Ca2+ was found near the N-terminal linker (Fig. 2 ▶ c). Each holo s2b structure begins at residue 766 and the last residue is 860. The PKD-like domain resembles a V-set Ig fold that lacks two strands (Omit β-strand C′ of the Ig fold corresponds to β-strand D of the PKD-like domain fold). β-strands B, D, and E in the PKD-like domain structures are shorter than the corresponding β-strands of a prototypical V-set Ig fold (PDB entry 1bre; Schormann et al., 1995 ▶), while strands F and G in the PKD-like domains are longer than the corresponding β-strands in the V-set Ig fold. In the PKD-like domain structures, β-strands A, B, B′ and E form one sheet, while strands A′, C, D, F and G form the opposing sheet. β-strands A′ and G form a parallel β-sheet, while the remaining strands form antiparallel β-sheets. Meanwhile, β-strand B forms a sheet with β-strand E, with the exception of Tyr796, which is aligned with β-strand A. Given the β-sheet sandwich fold, the PKD-like domain has been predicted to resemble the CBD (Yeats et al., 2003 ▶), although structural alignment of holo s2b with holo s3 (PDB entry 3jqw; Bauer et al., 2013 ▶) suggests little similarity.
Two prominent features are the conserved bulges in the domain that interrupt β-strands A and B and help to form a ridge along the ABE face (Fig. 2 ▶). The first bulge occurs when Pro784 breaks β-strand A and pushes the subsequent Lys785 outwards, which in turn leads to an approximately 127° angle between β-strands A and A′ that also changes the allegiance of β-strand A′ to β-strand G. The second conserved bulge is introduced by Lys798 and Gly799. This bulge removes the backbone hydrogen-bonding partner of Tyr780 in β-strand A. To help stabilize the bulge, the side-chain hydroxyl group of the conserved Thr800 hydrogen-bonds to the backbone amide of Tyr780. To further stabilize the bulge and to position the hydroxyl group of Thr800, the carbonyl O atom of Gly797 hydrogen-bonds to the side-chain hydroxyl group and the main-chain amide of Thr800. The bulge is also stabilized by the conserved Asn825 hydrogen-bonding to the amides of Gly797 and Lys798 (Fig. 2 ▶ c). The second bulge helps to form a prominent ridge. Surface-exposed aromatic residues are found on both sides of the ridge and are discussed later.
Temperature factors for both NCS-related structures are low (the average B factor for molecule A is 11.7 Å2 and that for molecule B is 9.9 Å2). Anisotropic B-factor analyses using the Anisotropic Network Model (ANM) web server (Eyal et al., 2006 ▶) showed that the main chain is mostly isotropic and that potential correlated movement occurs exclusively at the N-terminal linker. The calcium coordination for both holo s2b and holo s2a is described in detail in a later section.
3.3. Structure descriptions of apo s2
Despite being solved in two crystal forms, the crystal structures of forms I and II of apo s2 from ColG are similar. Each asymmetric unit contains two noncrystallographic twofold symmetry-related molecules. All four molecules are virtually identical to each other (r.m.s.d. of <0.5 Å). Each structure begins at residue 685, although the first two residues (Gly-Ser) are remnants from GST-tag cleavage. The last residue is either 770 or 771. The NCS-related molecules are stabilized by antiparallel-type intermolecular interactions between β-strands A. Comparison of our apo s2 structures with the previously solved apo s2 structure (PDB entry 2y72; Eckhard et al., 2011 ▶), in which the conserved Pro688 is mutated, showed that the N-terminal mutation pushes the N-terminus out by 3 Å at the Cα atom of Ala687. Furthermore, while the residues that make up the previously described bulges are conserved, the interaction pattern differs slightly from the pattern found in s2a and s2b. Here, the hydroxyl group of Ser707 mediates interaction between the conserved Asn735 and the backbone amides of Gly708 and Lys709 (Fig. 2 ▶ d).
3.4. Structure-based sequence comparisons of PKD-like domains
Sequence comparison of divergent PKD-like domains revealed conserved residues that are essential for the overall fold and for Ca2+ chelation. The residues involved in Ca2+ chelation will be discussed in the next section. Peptidase M9 family members are all thought to be collagenases and consist of subfamilies A (Vibrio) and B (Bacillus and Clostridium). M9A enzymes lack CBDs, and consequently may utilize different approaches to collagenolysis. Therefore, this section will discuss M9B-derived PKD-like domains exclusively, and will utilize s2b numbering. The PKD-like domains found in M9B enzymes share two conserved clusters of residues (Fig. 3 ▶). The first conserved stretch, 802DxDGx IxxYxWDFGDG817, contains β-strand C (the underlined residues form the β-strand). This stretch is conserved for its Ca2+-binding and architectural importance. The first two Asp residues chelate Ca2+. The invariant Tyr810 is accommodated by the second β-bulge. Asp813 is sometimes replaced by an acidic Glu residue. The side chain of Glu may easily fulfill the role of the side chain of Asp813, which terminates β-strand C and stabilizes the subsequent sharp turn by hydrogen-bonding to the amide of Gly815. Phe814 stacks against the conserved His828 so that the imidazole ring can also form a salt bridge with the conserved Asp816. Gly817 allows Asp816 to also stabilize the turn by hydrogen-bonding to the amide of Ser818. Although a sharp turn follows β-strand C in all PKD-like domains, the type of turn is different. In s2b, the insertion of Asp819 results in the region adopting an ω-loop (i→i + 10). Both s2a and s2 lack Asp819 in this stretch, and subsequently each is involved in an α-turn (i→i + 4) that forms a β-hairpin. The second conserved stretch, 825NPx HxY xxx GxYxVxLxVxD xxG847, forms β-strands E and F. Tyr830 and Tyr836 stack against each other to stabilize the interactions between the sheets. Tyr836 further stabilizes the sheets by forming a Δ4 Tyr corner. Asp844 is responsible for one of the axial interactions with Ca2+. The β-strands also wrap around the conserved turn and β-strand C to form the most unique feature of the PKD-like domain. Gly847 allows β-strands F and G to be separated by a β-turn (i→i + 3).
Figure 3.
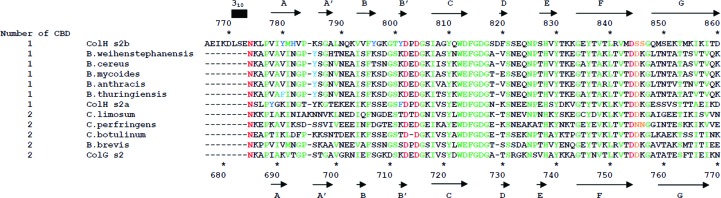
Structure-based sequence alignment of PKD-like domains from M9B. Residues responsible for Ca2+ binding and for positioning the Ca2+-interacting water, architecturally critical residues and surface aromatic residues are shown in red, orange, green and blue, respectively. Sequence numbering and secondary-structure positions for s2b are shown at the top of the figure. Secondary-structure positions for the s2 structure are similar, although the 310-helix is absent. Sequence numbering for s2a and s2, as well as secondary-structure positions for s2a, are shown at the bottom of the figure. Sequence alignment was aided by the use of ClustalW2 (Thompson et al., 1994 ▶).
3.5. Ca2+ chelation in s2a and s2b
The Ca2+ coordinations in s2a, s2b and s2 are virtually identical to each other. Since holo s2 has been described, this section describes the binding sites in s2a and s2b in detail. One calcium-binding site with a pentagonal bipyramidal geometry was identified near the N-terminus in each domain (Fig. 4 ▶). The pentagonal base is composed of OD1 of Asn685, the main-chain carbonyl of Ser686, OD1 and OD2 of Asp713 and a water molecule, while the axial positions are filled by OD1 of Asp715 and OD2 of Asp754. The Ca—O bond distances and planar deviations amongst the O atoms responsible for the pentagonal base (Table 2 ▶) are similar to the values found in clostridial CBDs (Bauer et al., 2013 ▶). The Ca2+ coordination geometry in PKD-like domains has been described as octahedral (Najmudin et al., 2006 ▶; Eckhard & Brandstetter, 2011 ▶), although our results demonstrate that a water is involved in forming a pentagonal base. The coordinating water molecule is positioned by OD1 of Asp755 (Fig. 4 ▶ a). The calcium coordination in s2 (PDB entry 4aqo; Eckhard et al., 2013 ▶) resembles that in s2a. Both the water and the calcium ion are ordered (B factor < 8.1 Å2). Based on the sequence alignment (Fig. 3 ▶) of PKD-like domains, residues contributing side-chain interactions with calcium are strictly conserved. s2b chelates with a Ca2+ ion very similarly, except for the residues that position the water molecule (Fig. 4 ▶ b): s2b utilizes OG from both Ser845 and Ser846 in lieu of Asp755 in s2a.
Figure 4.

Ca2+ coordination in s2a (a) and s2b (b). Seven O atoms from five residues and one water molecule coordinate to Ca2+ in a pentagonal bipyramidal geometry. Pentagonal base interactions are indicated using brown dashes, while axial interactions are indicated using yellow dashes. Residue-to water interactions are indicated with blue dashes. Either one aspartate (s2a) or adjacent serines (s2b) are responsible for positioning the water molecule along the pentagonal base. This figure was prepared using PyMOL (Schrödinger).
Table 2. CaO bond distances and planar deviations for holo s2b and holo s2a.
| Residue | Atom | Distance () | Planar deviation () | |
|---|---|---|---|---|
| s2b | ||||
| Molecule A | HOH | O | 2.49 | 0.09 |
| Asn774 | OD1 | 2.39 | 0.20 | |
| Lys775 | O | 2.41 | 0.27 | |
| Asp802 | OD1 | 2.42 | 0.38 | |
| Asp802 | OD2 | 2.44 | 0.22 | |
| Asp804: axial | OD1 | 2.34 | N/A | |
| Asp844: axial | OD2 | 2.42 | N/A | |
| Molecule B | HOH | O | 2.40 | 0.14 |
| Asn774 | OD1 | 2.37 | 0.24 | |
| Lys775 | O | 2.38 | 0.28 | |
| Asp802 | OD1 | 2.46 | 0.38 | |
| Asp802 | OD2 | 2.49 | 0.19 | |
| Asp804: axial | OD1 | 2.34 | N/A | |
| Asp844: axial | OD2 | 2.39 | N/A | |
| s2a | ||||
| Average of molecules A H | HOH | O | 2.53 (5) | 0.03 (2) |
| Asn685 | OD1 | 2.52 (6) | 0.10 (3) | |
| Ser686 | O | 2.43 (6) | 0.14 (3) | |
| Asp713 | OD1 | 2.51 (3) | 0.15 (3) | |
| Asp713 | OD2 | 2.50 (6) | 0.09 (4) | |
| Asp715: axial | OD1 | 2.42 (3) | NA | |
| Asp754: axial | OD2 | 2.40 (5) | NA | |
3.6. Ca2+-induced structural change
Ca2+ chelation appears to align the N-terminal linker approximately parallel to the major axis of the domain (Supplementary Fig. S2). In s2b, Ca2+ chelation by Asn774 and Lys775 could stabilize a 310-helix (residues 770–774) that aligns with the cylinder axis. In s2 and s2a, the N-terminal residues are positioned so that the N-terminal linker could also be positioned parallel to the major axis of the domain. The residues prior to Asn685 cannot be observed in the electron density, and consequently the secondary structure of the region remains ambiguous. Structural comparison of the Ca2+-binding site between the apo and holo PKD-like domains revealed that Ca2+ has a varied influence on the loop between β-strands B′ and C. The proline positioned between the aspartates equivalent to Asp802 and Asp804 restricts the loop flexibility so that minimal change occurs between the apo and holo states. However, in s2, which does not have a proline positioned between Asp713 and Asp715, the Ca2+ interaction moves Asp715 2 Å from the binding site (Supplementary Fig. 2 ▶ c).
Overall, the average B factors of s2b and s2 are low and the average B factors of apo and holo s2a are similar (Table 1 ▶). Despite different symmetry interactions, the B factor per-residue trend of 18 PKD-like domains (eight holo s2a molecules, four apo s2a molecules, two holo s2b molecules and four apo s2 molecules) are very similar to each other (Supplementary Figs. S3 ▶ a–S3d). Holo s2, however, does not follow this trend (Supplementary Fig. S3e). Comparison of holo and apo structures revealed a stark contrast between s2a and s2 in the influence of Ca2+ (Figs. 5 ▶ a and 5 ▶ c). Decreases in the Cα B factor in holo s2 are found in three stretches (Gly698–Ile704, Gly708–Tyr721 and His738–Thr761) that are immediately preceded by stretches in which the B factor is higher (Lys691–Thr697, Glu705–Ser707 and Gly733–Val737). In the holo s2 structure the mid-section became more flexible, although both terminal regions became more rigid. Differences in crystal packing could account for the reversal in dynamics, although it is possible that the crystal packing is a consequence of dynamic changes. Both termini of holo s2 are pinned down by symmetry-related molecules that could suppress their dynamics, while the mid-section of the barrel lacks the intermolecular interactions observed in the apo state. In apo s2, intermolecular antiparallel β-sheet interactions involving β-strand A could suppress dynamics of the region.
Figure 5.

Cα B-factor changes upon Ca2+ binding for s2a (a) and s2 (b). The Cα B factors of holo s2b (c) are also shown. This figure was prepared using PyMOL (Schrödinger).
In contrast, comparison of the holo s2a and apo s2a structures revealed that Ca2+ did not increase the B factor of the mid-section (Fig. 5 ▶ a). In the s2a structures the termini are the most flexible. As mentioned, the B-factor trends in all holo and apo s2a structures are similar despite the difference in crystal packing. Cα B factors for the mid-section of holo s2b are low (Fig. 5 ▶ b) and suggest that s2b behaves similarly to s2a. Overall, the starkly contrasting dynamics between ColG-derived and ColH-derived PKD-like domains suggest diverging roles during collagenolysis.
3.7. Ca2+-induced stability gain of PKD-like domains
The apo states of s2a, s2b and s2 are thermally stable proteins (T m of ∼373 K), but they gain further stability in the presence of Ca2+. The fluorescence of Trp812 in s2b was monitored, while in s2a and s2 the fluorescence of Trp723 was monitored. During fluorescence-monitored thermal denaturation, none of the PKD-like domains fully unfolded in the holo state (Figs. 6 ▶ a, 6 ▶ b and 6 ▶ c). Such hyperthermostability has also been observed for the holo states of s3 (Bauer et al., 2013 ▶) and s3b (Philominathan, Matsushita et al., 2009 ▶). The stability of both PKD-like domains and CBD may allow prolonged collagenolytic activity in the ECM. Heat is thought to denature proteins by disrupting electrostatic interactions. As such, the conserved hydrogen-bonding network around the bulges may play a strong role in the overall stability of the domains, while the Ca2+–O interactions may contribute to increased stability in the holo state.
Figure 6.

Results of fluorescence-measured equilibrium denaturation of (a, d) s2a, (b, e) s2b and (c, f) s2 in their apo (open circles) and holo (closed circles) forms.
PKD-like domains consist of a conserved (shown in green in Fig. 3 ▶) well packed core, and are likewise stable against Gu–HCl denaturation. Here, denaturation occurs through a cooperative transition from the folded state to unfolded states (Figs. 6d, 6e and 6f). In contrast to heat, Gu–HCl is thought to denature proteins predominately by disrupting hydrophobic interactions (Monera et al., 1994 ▶). Of the three domains, s2b is the most stable (Table 3 ▶). The difference in ΔG H2O between the apo and holo states (ΔΔG H2O) is approximately the same for all PKD-like domains. In addition to reorienting the N-terminal linker, the proposed Ca2+-induced helical base of the N-terminal linker may have a partial capping effect that shields the core against Gu–HCl. It is also well documented that metalloproteins are more stable in the presence of their metal ligand (Kellis et al., 1991 ▶).
Table 3. Stability parameters for GuHCl denaturation of PKD-like domains.
| Holo s2b | Apo s2b | Holo s2a | Apo s2a | Holo s2 | Apo s2 | |
|---|---|---|---|---|---|---|
| G (kcalmol1) | 9.8 | 6.9 | 7.8 | 4.9 | 6.0 | 4.3 |
| m (kcalmol1 M 1) | 3.1 | 2.6 | 3.6 | 3.1 | 2.4 | 2.7 |
| C M (M) | 3.2 | 2.6 | 2.2 | 1.6 | 2.5 | 1.6 |
In the clostridial collagen-binding domain, Ca2+-induced stability could be partially accounted for by a reduction in void volume and an increase in hydrogen bonds (Philominathan, Matsushita et al., 2009 ▶). Analysis of the void volume of PKD-like domains using CASTp (Dundas et al., 2006 ▶) revealed that a common cavity located near the N-terminus shrinks. The common cavity located near the C-terminus of the holo and apo pairs of both s2a and s2 curiously remains essentially unchanged upon Ca2+ binding. Furthermore, Ca2+ binding does not lead to a significant change in hydrogen-bond totals in any of the domains (Supplementary Table S3 ▶). For therapeutic applications, the in vitro stability of s2b and s3 may explain the prolonged activity of growth factors and signal molecules when fused to s2b-s3 in vivo.
3.8. Surface characteristics of ColG and ColH PKD-like domains
s2a and s2b, unlike s2, contain two and four surface aromatic residues, respectively, that are located on the ABE face (Figs. 7 ▶ a and 7 ▶ b). Interestingly, these residues are also located along the previously mentioned ridge. These residues could be involved in collagen binding given that aromatic residues are found at the hotspot of the collagen-binding pocket in CBD. In s3b, mutations of Tyr970, Ty994 and Tyr996 to Ala greatly reduced binding to collagenous peptide as monitored by surface plasmon resonance (Wilson et al., 2003 ▶). NMR studies also showed that these aromatic residues are involved in collagen binding (Philominathan, Koide et al., 2009 ▶). A structure-based sequence alignment of PKD-like domains (Fig. 3 ▶) suggests that the PKD-like domain of collagenases consisting of only one CBD will likely contain surface aromatic residues. Conversely, the PKD-like domain of collagenases consisting of multiple CBDs, such as s2, appears to have no surface aromatic residues. Collagenases from B. brevis, C. botulinum and C perfringens contain multiple CBDs. Their respective PKD-like domains lack aromatic residues and hence may not directly interact with collagen.
Figure 7.

Surface aromatic residues in s2a (a) and s2b (b). The boxed regions correspond to residues Ala766–Asp770, which are observed in both the s2a and s2b structures and were used to help assemble the full holo ColH structure (c). This structure is assembled from the crystal structures of the peptidase domains of s1, s2a, s2b and s3, as well as the homology-modeled activator domain of s1. Homology modeling was accomplished using SWISS-MODEL (Biasini et al., 2014 ▶). Surface-exposed aromatic residues of the peptidase domain of s1 and s2a-s2b as well as the conserved collagen-interacting aromatic residues of s3 are shown in yellow. Ca2+ is shown as orange spheres. This figure was prepared using PyMOL (Schrödinger).
A putative structure of holo ColH can be built from the homology-modeled activator domain of s1 and helical linker (residues 7–301 based on PDB entry 2y50; Eckhard et al., 2011 ▶) and the crystal structures of the peptidase domain (residues 302–681; PDB entry 4ar1; Eckhard et al., 2013 ▶), s2a (residues 685–770), s2b (residues 766–860) and s3 (residues 861–981; PDB entry 3jqw; Bauer et al., 2013 ▶). The overall dimensions (length 133 Å, height 36 Å, width 88 Å) match the tadpole shape observed in the SAXS envelope of holo ColH (Ohbayashi et al., 2013 ▶). In the model, the five-residue overlap between the s2a and s2b structures was superimposed (r.m.s.d. = 1.0 ± 0.1 Å) to assist with formation of the s2a-s2b segment. The aromatic residues mentioned are found on one side of s2a-s2b (Fig. 7 ▶ c). In this model, the surface aromatic residues on s2a-s2b may either span across multiple tropocollagen molecules on the surface of the fibril or bind along one tropocollagen molecule when the binding surface of s3 is docked onto the collagen fibril surface. The interactions may serve to prevent the collagen-binding segment from diffusing away after the s3–collagen interaction is transiently broken. Likewise, the domains may provide loose contacts with the collagen fibril that allow the enzyme to scan the fibril surface for optimal regions for tight CBD interaction. In these roles, the PKD-like domain strengthens collagen avidity so that only one CBD is required for collagen binding. The zinc ion involved in activation of a water molecule is approximately 115 Å away from Tyr962 found in the collagen-binding pocket of s3. In this model, the PKD-like domains may also be critical in positioning the catalytic domain with respect to CBD.
3.9. Potential role of PKD-like domains in synergistic collagenolysis
The apparent differences between the ColG-derived PKD-like domain and the ColH-derived PKD-like domains may aid synergistic collagenolysis. The putative holo ColH structure and the structure-based insights into the PKD-like domains allow us to begin to speculate on how ColG and ColH work together to degrade collagen. Currently, it is not known whether any of the clostridial PKD-like domains swell collagen fibers. Both s3 and s3b share a common preference for under-twisted regions of collagen (Bauer et al., 2013 ▶), although ColG and ColH initially cleave different sites in collagen (French et al., 1992 ▶). When digesting the insoluble fiber, ColH is the workhorse (Breite et al., 2011 ▶). The higher collagen affinity observed for s2b-s3 may increase further on the addition of s2a. The increased affinity could hold ColH close to the collagen fibril so that it can slide along the fibril and find vulnerable regions. Meanwhile, holo ColG has been propsed to adopt a compact structure in which the domains of the collagen-bonding segment are aligned by intermolecular β-sheet-type hydrogen-bond interactions (Eckhard et al., 2011 ▶). The tandem CBDs of ColG may allow the enzyme to anchor itself to the most vulnerable region of the fibril. In this context, the spring-like dynamics of s2 may allow it to swell the fibril. The swelled fibril would then expose the interior of the fibril and expose new sites for ColH collagenolysis.
3.10. PKD evolution
Human PKD1 (PDB entry 1b4r; Bycroft et al., 1999 ▶) and PKD-like domains from archaea and bacteria share a high degree of structural similarity that suggests that the fold has laterally transferred across the kingdoms. As expected, s2a, s2b and s2 resemble the C. thermocellum endoglucanase PKD-like domain (PDB entry 2c4x; Najmudin et al., 2006 ▶) more closely than either the archaeal surface protein PKD-like domain (PDB entry 1l0q; Jing et al., 2002 ▶) or human PKD1. While the bulge between β-strands A and A′ appears to be well conserved only in bacterial PKD-like domains, the bulge between β-strands B and B′ is also conserved in the archaeal PKD-like domain but is not conserved in PKD1. Correspondingly, residues Thr800 and Asn825, which are critical for stabilizing this bulge, are conserved in the archaeal PKD-like domain. Oddly, only Thr800 is conserved in the endoglucanase PKD-like domain. Normally, surface interactions are not well conserved, but surprisingly the salt bridge formed between Asp816 and His828 in s2b is found in the archaeal PKD-like domain. Structurally equivalent residues in the endoglucanase PKD-like domain, Asp47 and Tyr60, utilize hydrogen-bonding between OD1 and OD2 of Asp and OH of Tyr in lieu of the salt bridge. In the domains, these interactions serve to stack the β-sheets together and stabilize the region where β-strand D of the Ig fold is deleted. The interaction is not found between the equivalent Asp and His residues in the NMR structure of human PKD1, although it should be noted that the NMR structure is derived from main-chain NOE restraints and therefore the side-chain orientations are not experimentally obtained. Thus, these residues may also assist in the interaction of β-strands C and E. Within the core, Trp812 and Phe795 are conserved throughout the three kingdoms. Our PKD-like domain structures suggest that this Phe residue strengthens the interactions of β-strands B and C through packing with the strictly conserved Trp and Phe in strand D. The residue further supports the barrel architecture through hydrophobic packing with the C-terminal region of the barrel.
Comparison of bacterial holo PKD-like domains with either archaeal or mammalian PKDs suggests that the Ca2+-binding site in bacteria evolved from archaea. In addition to the overall structural similarity, five of the seven O atoms that coordinate to Ca2+ are present in the archaeal PKD-like domain. The archaeal domain lacks the initial asparagine residue and one of the axial aspartate residues required for Ca2+ binding. The Ca2+-interacting residues Asn774 and Lys775 of s2b are replaced by Pro302 and Val303 in archaea. In addition to removing an O atom responsible for the pentagonal base, Pro appears to constrain Val303 O to the position occupied by Ca2+ (Fig. 8 ▶). The archeal PKD-like domain possesses a bidentate Asp802 equivalent. However, the loop is significantly shortened compared with the bacterial domains, which consequently removes the Asp804 equivalent. It should be noted that the water-positioning residues in s2b appear to be conserved in archaea (Ser845 is conserved, while Ser846 is replaced by asparagine). The mammalian PKD, meanwhile, lacks all of the residues that interact with Ca2+.
Figure 8.

Proposed evolution of the Ca2+-binding pocket in bacterial PKD-like domains (holo s2b is shown on the left) from the archaeal PKD-like domain (PDB entry 1loq, shown on the right). This figure was prepared using PyMOL (Schrödinger).
Comparison of clostridial PKD-like domain structures with V-set kappa light-chain Bence–Jones protein (PDB entry 1bre; Schormann et al., 1995 ▶) as well as with archaeal PKD-like domains and human PKD suggests that the PKD1 domain fold in eukaryotes descended from the simpler Ig fold and may then have spread to archaea. From archaea, the fold spread laterally to bacteria. Characteristics of the V-set Ig fold that are shared with the PKD-like domain fold are the following. (i) The tertiary structure consists of a two-faced β-sheet architecture made up from a well packed hydrophobic core. (ii) β-Strand A is broken by a conserved bulge that changes the allegiance of the subsequent β-strand A′. (iii) β-Strand C contains the conserved tryptophan, and along with β-strand C′ (β-strand D of the PKD-like domain fold) forms a β-hairpin connected by an approximately i→i + 8 ω-loop. (iv) The turn leading into β-strand F is stabilized by a Δ4 Tyr corner.
4. Conclusion
Comparison of crystal structures of ColG s2 with crystal structures of ColH s2a and s2b suggests that despite common tertiary folds, PKD-like domains can be grouped into two subsets. The subset containing ColH-derived domains exhibits exposed aromatic residues and is found in M9B collagenases with a single CBD. The surface aromatic residues could be involved in secondary interactions that allow weak collagen binding. In contrast, the subset containing s2 is likely to be different; the lack of surface aromatic residues on s2 suggests that the domain is less directly involved in interactions with collagen. Overall, this subset is found in M9B collagenases with multiple CBDs. The unique differences in dynamics and surface characteristics between s2a-s2b and s2 may aid in synergistic collagenolysis.
Meanwhile, the N-terminal linker structure of a PKD-like domain is described for the first time in the holo s2b structure and suggests that Ca2+ repositions the linker along the barrel axis. The helical structure of the linker upon Ca2+ binding may shorten the distance between s2b and s2a, and may help to account for the previously described proteinase resistance (Ohbayashi et al., 2013 ▶). Lastly, our stability data show that the domains are extremely stable in the presence of physiological Ca2+. Structural and stability data are critical for the development of PKD-like domains as part of the site-directed delivery of signal molecules such as growth factors and cytokines.
Supplementary Material
PDB reference: ColG PKD domain 2, 4jrw
PDB reference: 4tn9
PDB reference: ColH PKD domain 2a, apo, 4u6t
PDB reference: in the presence of calcium, 4u7k
PDB reference: ColH PKD domain 2b, in the presence of calcium, 4jgu
Supporting Information.. DOI: 10.1107/S1399004714027722/mn5081sup1.pdf
Acknowledgments
This work has been supported by the National Institutes of Health (GM103429 and GM103450), Arkansas Biosciences Institute, a Grant-in Aid for Scientific Research (C) from the Japan Society for the Promotion of Science and Kagawa University Project Research Fund 2005–2006. SG was supported by a NSF summer fellowship. RB, KT and EL were supported by the Honors College of UA. RB was also supported by a Student Undergraduate Research Fellowship from Arkansas Department of Higher Education and a Doctoral Academy Fellowship. We thank the staff at XSD-12ID and at SBC-19ID and BM at the Advanced Photon Source, which is supported by the Department of Energy.
References
- Adhikari, A. S., Glassey, E. & Dunn, A. R. (2012). J. Am. Chem. Soc. 134, 13259–13265. [DOI] [PMC free article] [PubMed]
- Akimoto, M., Takeda, A., Matsushita, O., Inoue, J., Sakamoto, K., Hattori, M., Kounoike, N. & Uchinuma, E. (2013). Plast. Reconstr. Surg. 131, 717–725. [DOI] [PubMed]
- Bauer, R., Wilson, J. J., Philominathan, S. T., Davis, D., Matsushita, O. & Sakon, J. (2013). J. Bacteriol. 195, 318–327. [DOI] [PMC free article] [PubMed]
- Bertini, I., Fragai, M., Luchinat, C., Melikian, M., Toccafondi, M., Lauer, J. L. & Fields, G. B. (2012). J. Am. Chem. Soc. 134, 2100–2110. [DOI] [PMC free article] [PubMed]
- Biasini, M., Bienert, S., Waterhouse, A., Arnold, K., Studer, G., Schmidt, T., Kiefer, F., Cassarino, T. G., Bertoni, M., Bordoli, L. & Schwede, T. (2014). Nucleic Acids Res. 42, W252–W258. [DOI] [PMC free article] [PubMed]
- Breite, A. G., McCarthy, R. C. & Dwulet, F. E. (2011). Transplant. Proc. 43, 3171–3175. [DOI] [PubMed]
- Bycroft, M., Bateman, A., Clarke, J., Hamill, S. J., Sandford, R., Thomas, R. L. & Chothia, C. (1999). EMBO J. 18, 297–305. [DOI] [PMC free article] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Duarte, A. S., Correia, A. & Esteves, A. C. (2014). Crit. Rev. Microbiol., 10.3109/1040841X.2014.904270. [DOI] [PubMed]
- Dundas, J., Ouyang, Z., Tseng, J., Binkowski, A., Turpaz, Y. & Liang, J. (2006). Nucleic Acids Res. 34, W116–W118. [DOI] [PMC free article] [PubMed]
- Eckhard, U. & Brandstetter, H. (2011). Biol. Chem. 392, 1039–1045. [DOI] [PubMed]
- Eckhard, U., Schönauer, E. & Brandstetter, H. (2013). J. Biol. Chem. 288, 20184–20194. [DOI] [PMC free article] [PubMed]
- Eckhard, U., Schönauer, E., Nüss, D. & Brandstetter, H. (2011). Nature Struct. Mol. Biol. 18, 1109–1114. [DOI] [PMC free article] [PubMed]
- Eyal, E., Yang, L.-W. & Bahar, I. (2006). Bioinformatics, 22, 2619–2627. [DOI] [PubMed]
- Fields, G. B. (2013). J. Biol. Chem. 288, 8785–8793. [DOI] [PMC free article] [PubMed]
- French, M. F., Bhown, A. & Van Wart, H. E. (1992). J. Protein Chem. 11, 83–97. [DOI] [PubMed]
- Fujio, A., Murayama, K., Yamagata, Y., Watanabe, K., Imura, T., Inagaki, A., Ohbayashi, N., Shima, H., Sekiguchi, S., Fujimori, K., Igarashi, K., Ohuchi, N., Satomi, S. & Goto, M. (2013). Transplantation, 96, S141–S142.
- Hemmingsen, J. M., Gernert, K. M., Richardson, J. S. & Richardson, D. C. (1994). Protein Sci. 3, 1927–1937. [DOI] [PMC free article] [PubMed]
- Hurst, L. C., Badalamente, M. A., Hentz, V. R., Hotchkiss, R. N., Kaplan, F. T., Meals, R. A., Smith, T. M. & Rodzvilla, J. (2009). N. Engl. J. Med. 361, 968–979. [DOI] [PubMed]
- Jing, H., Takagi, J., Liu, J.-H., Lindgren, S., Zhang, R.-G., Joachimiak, A., Wang, J.-H. & Springer, T. A. (2002). Structure, 10, 1453–1464. [DOI] [PubMed]
- Katikaneni, R., Ponnapakkam, T., Matsushita, O., Sakon, J. & Gensure, R. (2014). Anticancer Drugs, 25, 30–38. [DOI] [PMC free article] [PubMed]
- Katikaneni, R., Ponnapakkam, T., Suda, H., Miyata, S., Sakon, J., Matsushita, O. & Gensure, R. C. (2012). Int. J. Cancer, 131, E813–E821. [DOI] [PMC free article] [PubMed]
- Kellis, J. T. Jr, Todd, R. J. & Arnold, F. H. (1991). Nature Biotechnol. 9, 994–995. [DOI] [PubMed]
- Matsushita, O., Jung, C.-M., Minami, J., Katayama, S., Nishi, N. & Okabe, A. (1998). J. Biol. Chem. 273, 3643–3648. [DOI] [PubMed]
- Matsushita, O., Koide, T., Kobayashi, R., Nagata, K. & Okabe, A. (2001). J. Biol. Chem. 276, 8761–8770. [DOI] [PubMed]
- McCarthy, R. C., Breite, A. G., Green, M. L. & Dwulet, F. E. (2011). Transplantation, 91, 137–145. [DOI] [PMC free article] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- McRee, D. E. (1999). Practical Protein Crystallography, 2nd ed. San Diego: Academic Press.
- Merritt, E. A. (2012). Acta Cryst. D68, 468–477. [DOI] [PMC free article] [PubMed]
- Minor, W., Cymborowski, M., Otwinowski, Z. & Chruszcz, M. (2006). Acta Cryst. D62, 859–866. [DOI] [PubMed]
- Monera, O. D., Kay, C. M. & Hodges, R. S. (1994). Protein Sci. 3, 1984–1991. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Nagase, H. & Fushimi, K. (2008). Connect. Tissue Res. 49, 169–174. [DOI] [PubMed]
- Najmudin, S., Guerreiro, C. I. P. D., Carvalho, A. L., Prates, J. A. M., Correia, M. A. S., Alves, V. D., Ferreira, L. M., Romao, M. J., Gilbert, H. J., Bolam, D. N. & Fontes, C. M. G. A. (2006). J. Biol. Chem. 281, 8815–8828. [DOI] [PubMed]
- Nishi, N., Matsushita, O., Yuube, K., Miyanaka, H., Okabe, A. & Wada, F. (1998). Proc. Natl Acad. Sci. USA, 95, 7018–7023. [DOI] [PMC free article] [PubMed]
- Ohbayashi, N., Matsumoto, T., Shima, H., Goto, M., Watanabe, K., Yamano, A., Katoh, Y., Igarashi, K., Yamagata, Y. & Murayama, K. (2013). Biophys. J. 104, 1538–1545. [DOI] [PMC free article] [PubMed]
- Perrakis, A., Morris, R. & Lamzin, V. S. (1999). Nature Struct. Biol. 6, 458–463. [DOI] [PubMed]
- Pflugrath, J. W. (1999). Acta Cryst. D55, 1718–1725. [DOI] [PubMed]
- Philominathan, S. T., Koide, T., Hamada, K., Yasui, H., Seifert, S., Matsushita, O. & Sakon, J. (2009). J. Biol. Chem. 284, 10868–10876. [DOI] [PMC free article] [PubMed]
- Philominathan, S. T., Koide, T., Matsushita, O. & Sakon, J. (2012). Protein Sci. 21, 1554–1565. [DOI] [PMC free article] [PubMed]
- Philominathan, S. T., Matsushita, O., Gensure, R. & Sakon, J. (2009). FEBS J. 276, 3589–3601. [DOI] [PMC free article] [PubMed]
- Philominathan, S. T., Matsushita, O., Jordan, J. B. & Sakon, J. (2008). Biomol. NMR Assign. 2, 127–129. [DOI] [PMC free article] [PubMed]
- Ponnapakkam, T., Katikaneni, R., Miller, E., Ponnapakkam, A., Hirofumi, S., Miyata, S., Suva, L. J., Sakon, J., Matsushita, O. & Gensure, R. C. (2011). Calcif. Tissue Int. 88, 511–520. [DOI] [PubMed]
- Ponnapakkam, T., Katikaneni, R., Nichols, T., Tobin, G., Sakon, J., Matsushita, O. & Gensure, R. C. (2011). J. Endocrinol. Invest. 34, e392–e397. [DOI] [PubMed]
- Ponnapakkam, T., Katikaneni, R., Sakon, J., Stratford, R. & Gensure, R. C. (2013). Drug Discov. Today, 19, 204–208 [DOI] [PMC free article] [PubMed]
- Ponnapakkam, T., Katikaneni, R., Suda, H., Miyata, S., Matsushita, O., Sakon, J. & Gensure, R. C. (2012). Calcif. Tissue Int. 91, 196–203. [DOI] [PMC free article] [PubMed]
- Saito, W., Uchida, K., Ueno, M., Matsushita, O., Inoue, G., Nishi, N., Ogura, T., Hattori, S., Fujimaki, H., Tanaka, K. & Takaso, M. (2013). J. Biomed. Mater. Res. A, 102, 3049–3055 [DOI] [PubMed]
- Schormann, N., Murrell, J. R., Liepnieks, J. J. & Benson, M. D. (1995). Proc. Natl Acad. Sci. USA, 92, 9490–9494. [DOI] [PMC free article] [PubMed]
- Sides, C. R., Liyanage, R., Lay, J. O. Jr, Philominathan, S. T., Matsushita, O. & Sakon, J. (2012). J. Am. Soc. Mass Spectrom. 23, 505–519. [DOI] [PMC free article] [PubMed]
- The International Polycystic Kidney Disease Consortium (1995). Cell, 81, 289–298. [DOI] [PubMed]
- Thompson, J. D., Higgins, D. G. & Gibson, T. J. (1994). Nucleic Acids Res. 22, 4673–4680. [DOI] [PMC free article] [PubMed]
- Uchida, K., Matsushita, O., Naruse, K., Mima, T., Nishi, N., Hattori, S., Ogura, T., Inoue, G., Tanaka, K. & Takaso, M. (2013). J. Biomed. Mater. Res. A, 102, 1737–1743. [DOI] [PMC free article] [PubMed]
- Wang, Y.-K., Zhao, G.-Y., Li, Y., Chen, X.-L., Xie, B.-B., Su, H.-N., Lv, Y.-H., He, H.-L., Liu, H., Hu, J., Zhou, B.-C. & Zhang, Y.-Z. (2010). J. Biol. Chem. 285, 14285–14291. [DOI] [PMC free article] [PubMed]
- Welgus, H. G., Jeffrey, J. J. & Eisen, A. Z. (1981a). J. Biol. Chem. 256, 9511–9515. [PubMed]
- Welgus, H. G., Jeffrey, J. J. & Eisen, A. Z. (1981b). J. Biol. Chem. 256, 9516–9521. [PubMed]
- Wilson, J. J., Matsushita, O., Okabe, A. & Sakon, J. (2003). EMBO J. 22, 1743–1752. [DOI] [PMC free article] [PubMed]
- Yeats, C., Bentley, S. & Bateman, A. (2003). BMC Microbiol. 3, 3. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: ColG PKD domain 2, 4jrw
PDB reference: 4tn9
PDB reference: ColH PKD domain 2a, apo, 4u6t
PDB reference: in the presence of calcium, 4u7k
PDB reference: ColH PKD domain 2b, in the presence of calcium, 4jgu
Supporting Information.. DOI: 10.1107/S1399004714027722/mn5081sup1.pdf