

Abstract
Fetal alcohol syndrome (FAS), presenting with a constellation of neuro-/psychological, craniofacial and cardiac abnormalities, occurs frequently in offspring of women who consume alcohol during pregnancy, with a prevalence of 1–3 per 1000 livebirths. The present study was designed to test the hypothesis that alcohol alters global DNA methylation, and modulates expression of the DNA methyltransferases (DNMTs) and various methyl CpG-binding proteins. Murine embryonic fibroblasts (MEFs), utilized as an in vitro embryonic model system, demonstrated ~5% reduction in global DNA methylation following exposure to 200 mM ethanol. In addition, ethanol induced degradation of DNA methyltransferases (DNMT-1, DNMT-3a, and DNMT-3b), as well as the methyl CpG-binding proteins (MeCP-2, MBD-2 and MBD-3), in MEF cells by the proteasomal pathway. Such degradation could be completely rescued by pretreatment of MEF cells with the proteasomal inhibitor, MG-132. These data support a potential epigenetic molecular mechanism underlying the pathogenesis of FAS during mammalian development.
Keywords: Fetal alcohol syndrome, Alcohol, Mouse, Embryo, Fibroblast, Proteasome, Epigenetic, DNA methylation
1. Introduction
Maternal alcohol use during pregnancy, a significant public health problem, affects an estimated 1% liveborn infants in the United States [1]. Exposure to alcohol during gestation has been associated with a variety of adverse pregnancy outcomes including spontaneous abortion, stillbirth, prematurity, pre- and postnatal growth retardation, and a spectrum of congenital malformations and neurological defects [2,3]. The most severe manifestation of maternal alcohol use in offspring is fetal alcohol syndrome (FAS), a spectrum of congenital anomalies characterized by craniofacial and cardiac dysmorphologies, growth retardation, brain damage and other physical abnormalities that occur with increased frequency in offspring of women who consume alcohol during pregnancy. FAS is thought to occur in 30–50% of the offspring of alcoholic women with an estimated incidence in the United States of 1–3 cases per 1000 livebirths and an international incidence of approximately 1.9 cases per 1000 livebirths [4].
The multisystem abnormalities resulting from in utero alcohol exposure were first described by Lemoine et al. [5] and later described as “fetal alcohol syndrome” (FAS) by Jones and Smith [6], and Jones et al. [7]. In the United States, studies document that up to 30% of women consume alcohol at some point during their pregnancy and significant evidence exists to support the fact that alcohol consumption during pregnancy can be harmful to the developing fetus [8].
A typical constellation of abnormalities, including growth retardation, developmental delay and mental deficiency, is observed in children with FAS. Moreover, multiple cranial and facial abnormalities are also characteristic of this syndrome [9]. Despite extensive study (for reviews, see [10–13]), the cellular and molecular mechanisms underlying the developmental toxicity of alcohol are still poorly defined. Recent studies imply that, apart from genetic and environmental factors, epigenetic mechanisms, known to regulate cell proliferation, differentiation, and migration during embryonic development (for reviews, see [14–17]), may be associated with the etiology of a range of developmental abnormalities, including FAS [18–20]. Recent studies have reported that prenatal exposure to alcohol results in alterations in DNA methylation associated with fetal genetic and phenotypic changes [21–25]. Acute ethanol administration to pregnant mice (from GD 9.0 to 11.0), resulted in hypomethylation of fetal DNA [26]. Diminishing Dnmt3b mRNA levels have been detected in the sperm of alcohol-exposed male rats [27], and findings from a range of studies highlight a crucial role for DNA methylation in neural cell lineage differentiation and early neurogenesis [15,28]. Using a mouse model for fetal alcohol spectrum disorders (FASD), alcohol exposure during early neurulation resulted in aberrant changes in DNA methylation patterns with associated changes in gene expression, cell cycle regulation, and neural development [29].
Mammalian DNA methylation, occurring exclusively at cytosine residues within CpG dinucleotides, is catalyzed by a family of active DNA methyl transferases (DNMTs) that includes DNMT-1, -3a, -3b. The “maintenance” methyltransferase, DNMT-1, exclusively associated with the methylation of hemimethylated DNA, ensures clonal transmission of lineage-specific DNA methylation patterns from maternal to daughter cells during cell division, whereas, the de novo methyltransferases, DNMT-3a and DNMT-3b, display a level of target specificity and diverse temporal activity [30–32]. The importance of the three active DNMTs (DNMT-1, -3a, and -3b) during embryogenesis, is well documented. Disruption/mutation of the Dnmt1 gene results in developmental anomalies and embryonic lethality [33,34]. Targeted disruption of the Dnmt3b gene also leads to embryonic lethality [31], whereas Dnmt3a null mice die in utero or shortly after birth [31,35]. Mutations in the human DNMT3B gene cause the immunodeficiency-centromeric instability-facial anomalies (ICF) syndrome, characterized by hypomethylation of pericentromeric repeats [36]. Altered neuronal differentiation and maturation in DNMT3B-deficient human embryonic stem cells also has been reported recently [37].
Methyl-CpG-binding domain (MBD) proteins, characterized by the presence of a methyl CpG-binding domain, act as “interpreters” of the DNA methylation mark, and function as key regulators of several epigenetic processes. Of the five known MBD proteins, four (MeCP-2 and MBDs 1–3) generally operate as transcriptional repressors, whereas MBD-4 is involved in DNA repair (for reviews, see [38,39]). MBD proteins are thought to govern normal embryogenesis by a range of mechanisms, many of which are still unknown [38,40]. Notably, mice harboring a Mecp2 deletion exhibit a phenotype markedly similar to that presented in individuals with the symptoms of Rett Syndrome, a neurological disorder in human caused by MECP2 mutation [41,42]. In addition, MBD-3 is essential for development as Mbd3-null mutations are embryonically lethal [43].
The consequences of in utero exposure to ethanol on the expression of DNMTs and MBDs are unknown. As the developing craniofacial complex, which is targeted by in utero alcohol exposure, is contributed to by neural stem cells, neural crest cells, and mesoderm-derived mesenchyme [44], each of these cell types in culture represents a biologically relevant experimental model system for molecular mechanistic studies relating to the cellular/molecular basis of FAS. Consequently, in the present study, the outcomes of ethanol exposure on the expression of various DNMTs and MBDs were investigated in an in vitro embryonic model, murine embryonic fibroblasts cells. Fetal embryonic fibroblasts are derived from primitive mesenchyme and hence are of mesodermal origin. They maintain a degree of pluripotency and can differentiate into other cell types including chondrocytes, osteocytes, adipocytes and smooth muscle cells [45]. Interestingly, findings from this study reveal that ethanol exposure resulted in global DNA hypomethylation, and for the first time, document significant proteasomal degradation of all three DNA methyltransferases (DNMT-1, DNMT-3a, and DNMT-3b) and the methyl CpG-binding proteins, MeCP-2, MBD-2 and MBD-3. Moreover, such ethanol-induced degradation could be completely prevented via pretreatment of the cells with the proteasome inhibitor, MG-132.
2. Materials and methods
2.1. Cell culture
Mouse embryonic fibroblasts (MEFs; NIH/3T3) were obtained from American Type Culture Collection (ATCC; Rockville, MD, USA) at passage 56. These cells were originally derived from embryonic day (E) 13.5 mouse embryos (strain 129/Sv-C57BL/6). Cells were grown at 37 °C in an atmosphere of 90% air/10% CO2, and maintained in Dulbecco’s modified Eagle medium (DMEM) containing high glucose and supplemented with 10% fetal bovine serum (FBS), 4 mM L-glutamine, and antibiotics (100 units/ml penicillin G, 100 mg/ml streptomycin sulfate and 0.25 mg/ml amphotericin B) (Invitrogen – Life Technologies; Grand Island, NY, USA). Prior to treatment of the cells, growth medium was replaced every 48 h. Cells were utilized for experimental purposes at passage numbers 60–62, when the cells were actively proliferating.
2.2. Global DNA methylation assay
MEF cells were plated in 60 mm tissue culture dishes (Nalge Nunc International; Rochester, NY, USA) at an initial density of 1.5 × 104 cells/dish. Cells were grown to confluence in the growth medium (as described under Section 2.1), washed with phosphate-buffered saline (PBS) and treated with either vehicle (PBS) or ethanol at a final concentration of 25 or 200 mM for 24 h. Culture dishes (both treated and control) were sealed with parafilm to prevent evaporation of ethanol and incubated at 37 °C. After 24 h, the growth medium was replaced with fresh medium containing vehicle or appropriate concentrations of ethanol, again sealed with parafilm, and the cell cultures were incubated for an additional 24 h. In addition, other MEF cell cultures, identically grown and maintained (for a total duration of 48 h), were treated only with 10 μM 5-azacytidine (a DNA methylation inhibitor used as a negative control) (Sigma; St. Louis, MO, USA) for 24 h.
Genomic DNA was extracted from vehicle, ethanol- or azacytidine-treated MEFs using the Qiagen DNeasy Blood & Tissue Kit according to the manufacturer’s instructions (Qiagen; Valencia, CA, USA). Global DNA methylation was assessed using the Methylamp Global DNA Methylation Quantification Ultra Kit (Epigentek; Brooklyn, NY, USA) according to the manufacturer’s protocol. In this assay, DNA was added to a strip well coated with a specific methylated-DNA binder. The methylated fraction of DNA is recognized by an anti-5-methylcytosine (5mCyt) antibody, and then quantified through an ELISA-like reaction, assessing optical densities (ODs) at 450 nm, using a spectrophotometric plate reader (VICTOR™ X3 Multilabel Plate Reader, Perkin Elmer; Waltham, MA, USA).
2.3. Methyl-cytosine immunostaining
MEF cells grown on 4-well Lab-Tek® chamber slides (Nalge Nunc International) were treated with either 25 mM or 200 mM ethanol, PBS (vehicle control), or 10 μM 5-azacytidine, as described under Section 2.2. Control and treated cells were washed in PBS, fixed for 5mCyt staining in ice-cold methanol for 10 min at −20 °C, treated with 2 N HCl for 20 min at 37 °C and then with boric buffer to neutralize the acid for 10 min at 37 °C. Subsequently, cells were washed in PBS, permeabilized in PBS/0.1% Triton X-100, and blocked with PBS/0.1% TritonX-100/0.5% BSA/10% FBS at 37 °C for 30 min to minimize nonspecific staining. Cells were then incubated overnight with a 1:200 dilution of the anti-5-methylcytosine rabbit polyclonal antibody (Megabase Research; Omaha, NE, USA). Following repeated washings with PBS to remove the primary antibody, cells were incubated with anti-rabbit IgG conjugated to Alexa Fluor 594 (1:200 dilution; Molecular Probes – Life Technologies) for 60 min at 37 °C, and then washed again with PBS. Cellular DNA was counterstained with 300 nM 4′-6′-diamidino-2-phenylindole (DAPI) (Molecular Probes – Life Technologies) and mounted under Fluoromount-G (Molecular Probes – Life Technologies) to preserve fluorescence. Cells were visualized and photographed with a Nikon Eclipse TE 2000-U microscope equipped with epifluorescence optics.
2.4. TaqMan® quantitative real-time PCR (RT-PCR)
Total RNA (from control or ethanol-treated MEF cells) was isolated using the RNeasy Protect Mini Kit (Qiagen) following the manufacturer’s recommendations. The quality and quantity of extracted total RNAs were assessed using the Agilent 2100 Bioanalyzer (Agilent Technologies; Santa Clara, CA, USA) and spectro-photometric UV absorbance at 260/280 nm. Total RNA was treated with DNase I in the presence of RNase OUT (Invitrogen – Life Technologies) to remove contaminating DNA before cDNA synthesis. cDNA was synthesized from total RNA using random hexamer primers and Superscript II reverse transcriptase (Invitrogen – Life Technologies). Real-time PCR analysis was performed on a TaqMan® ABI Prism 7000 Sequence Detection system (Applied Biosystems; Foster City, CA, USA). Primers and their corresponding fluorescence probes (Assays-on-Demand) were purchased from Applied Biosystems. For each gene analyzed, both forward and reverse primers were used at a concentration of 900 nM and the final fluorescent probe concentration was 200 nM. The PCR reaction was performed in a total volume of 25 μl containing 0.2 mM each of dATP, dCTP, and dGTP, 0.4 mM dUTP, 0.625 unit of Amplitaq Gold (Applied Biosystems, UK) and 2 μl (8.0 ng) of cDNA template. Cycling parameters were: 50 °C for 2 min for probe and primer activation, 95 °C for 10 min for denaturation of DNA strands, 40 cycles of denaturation at 95 °C for 15 s, and primer extension at 60 °C for 1 min. For each reaction, a parallel reaction lacking template served as a negative control. Raw data were acquired and processed with ABI Sequence Detector System software, version 1.0 (Applied Biosystems). mRNA amounts for each gene were normalized to 18S ribosomal RNA (rRNA) present in each sample.
2.5. Preparation of nuclear extracts and Western blotting
MEF cells were treated and maintained for 48 h as described under Section 2.2. Nuclear-enriched protein extracts were prepared from 25 mM- or 200 mM ethanol- or vehicle (PBS)-treated MEF cells using the NE-PER kit (Pierce; Rock-ford, IL, USA) following the manufacturer’s recommendations. Steady state levels of various DNMT and MBD proteins were determined by Western (immuno) blotting as previously described [46]. Protein was separated (15 μg/lane) by SDS-PAGE followed by electrophoretic transfer to PVDF membranes. To visualize proteins and ensure the efficiency of transfer, gels were stained with Coomassie blue [47] and membranes with 0.1% fast green. Blots were blocked by incubation in 5% non-fat dry milk in TBST buffer (50 mM Tris, pH 7.6; 150 mM NaCl; 0.1%Tween-20) for 1 h at room temperature. Antibodies (see below) were prepared by dilution in blocking solution. Blots were incubated with primary antibody overnight at 4 °C with gentle shaking, washed extensively and then incubated for 45 min at room temperature with the appropriate horseradish peroxidase-conjugated secondary antibody. Immune complexes were detected on film using the ECL-Plus™ chemiluminescent detection system (GE Healthcare Life Sciences; Pittsburgh, PA, USA) according to the manufacturer’s instructions. Molecular weights of proteins on each gel were estimated by reference to a Magic Mark™ (20–200 kDa) protein ladder (Invitrogen – Life Technologies). A methodological control, wherein buffer was utilized in place of primary antibody, was utilized to distinguish between specific immunoreactive bands and nonspecific bands due to interaction of the secondary antibody with endogenous proteins. Immunoblots of control and MEF cell extracts were replicated with similar results utilizing a minimum of three complete and independent sets of cell samples. Commercially available primary antibodies utilized included DNMT-1 (D63A6), DNMT-3a (D23G1), and MeCP-2 (D4F3) rabbit monoclonal antibodies (catalog #5032, #3598, and #3456, respectively) and DNMT-3b and MBD-3 rabbit polyclonal antibodies (catalog #2161 and #3896, respectively) (all from Cell Signaling; Danvers, MA, USA), MBD-2 goat polyclonal antibody (catalog #3598; Epigentek; Brooklyn, NY, USA) and mouse monoclonal anti-actin antibody (catalog #sc-8432; Santa Cruz Biotechnology; Santa Cruz, CA, USA). Secondary antibodies included horseradish peroxidase conjugated goat anti-rabbit IgG and donkey anti-goat IgG (catalog #sc-2004 and #sc-2020, respectively, Santa Cruz) and rabbit anti-mouse IgG1 (catalog #61-0120; Zymed Laboratories; San Francisco, CA, USA). To confirm equal loading of proteins and conduct normalization of immunoblotted samples during densitometric analysis, blots were stripped and reprobed for β-actin, a housekeeping (control) protein.
2.6. Proteasome inhibitor studies
MEF cells, grown to confluence in 60 mm tissue culture dishes (Nalge Nunc International), were pre-treated with either vehicle (DMSO) or 1-, 3- or 5-μM of the proteasome inhibitor MG-132 (Sigma) for 3 h. The drug was removed, and fresh medium containing DMSO (vehicle), or ethanol (25 mM- or 200 mM) was added, and the cells maintained for 48 h as described under Section 2.2. A separate set of control experiments, wherein the cells were treated with only 5-μM MG-132 (for 3 h), was also performed. Nuclear protein extracts were then prepared and examined by Western analysis as described in the preceding paragraph.
2.7. DNMT-1 ELISA
Nuclear extracts from 48-h vehicle-, or ethanol- (alone or after 3-h MG-132 pre-treatment) treated MEF cells, were assayed for DNMT-1 protein using the Epiquik DNMT-1 assay kit (Epigentek). A standard curve was generated utilizing protein standards of known concentrations (20, 10, 5 and 2 ng). The amount of DNMT-1 protein was estimated as: DNMT protein (ng/ml) = (sample OD − blank OD/standard slope) × sample dilution, following the manufacturer’s instructions.
2.8. Densitometric analysis of Western blots
Densitometric analyses of DNMT-1, -3a, -3b, MeCP-2, MBD-2, MBD-3 and β-actin protein bands were performed with Image J (version 1.38) software [48]. Film-detected chemiluminescent signals from immunoblotting were scanned and analyzed by densitometry. Intensities of the β-actin bands were recorded and used as an internal control to correct for differences in sample loading. Densitometric data for each protein band of interest were normalized to that of β-actin in that lane by subtracting the intensity value for the specific protein band from the corresponding intensity value for the β-actin band for each sample.
2.9. Statistical analyses
Statistical analyses were performed using SAS v9.3 (SAS Institute Inc.; Cary, NC, USA). Effects of ethanol on (1) percent global DNA methylation, (2) normalized protein band intensities (relative to steady state levels of proteins), (3) normalized Ct values (ΔCt ) from qRT-PCR for gene expression and (4) normalized DNMT-1 protein levels (relative to steady state level of DNMT-1 protein), were all analyzed using one-way ANOVA models. For the global methylation experiments, control samples were compared to samples treated with 25 mM EtOH, 200 mM EtOH, and 10 μM Azacytidine. Experiments #2 (protein band intensity) and #3 (Ct values) only involved comparisons between the EtOH treated and control samples. For the ELISA of DNMT-1 protein levels in MEF cells, control samples were compared to samples treated with 25 mM EtOH, 200 mM EtOH, and 5 μM MG-132 plus 200 mM EtOH. Post hoc comparisons to vehicle (control) were presented with Dunnett’s adjustment for multiple comparisons. Means and standard deviations from three independent experiments, followed by differences or fold-changes relative to control samples were presented with simultaneous 95% confidence limits and statistical significance after multiple comparison adjustment. p-Values of <0.05 were considered significant.
3. Results
3.1. Effect of ethanol on MEF cell global DNA methylation
Ethanol (200 mM) significantly decreased global DNA methylation in MEF cells by ~5.8% (Table 1). While treatment with 25 mM ethanol also resulted in a decrease in global DNA methylation, the effect was not statistically significant. The apparent magnitude of the ethanol-induced decrease in global DNA methylation, although small, is likely biologically relevant in view of the finding that 5-azacytidine – an extremely potent DNA methylation inhibitor –elicited a ~21% decrease in global DNA methylation in these cells (Table 1).
Table 1.
Effect of ethanol on global DNA methylation in MEF cells.
| Sample typea | % global DNA methylationb,c | % change (vs. control) |
|---|---|---|
| Control | 1.41 ± 0.13 | – |
| 25 mM EtOH | 1.39 ± 0.12 | 1.0% decrease |
| 200 mM EtOH | 1.33 ± 0.12* | 5.8% decrease |
| 10 μM azacytidine | 1.10 ± 0.11*** | 21.6% decrease |
MEF cells were treated with vehicle (control) or ethanol (25 or 200 mM) for 48 h, or 10 μM azacytidine as described in Section 2.
Global DNA methylation was quantified using the Methylamp Global DNA Methylation Quantification Ultra Kit as described in Section 2.
Data represent mean ± standard deviation, obtained from three independent experiments.
P < 0.05.
P < 0.001.
3.2. Immunostaining of 5-methyl-cytosine in ethanol-treated MEF cells
MEF cells treated with either 200 mM ethanol or 10 μM 5-azacytidine, demonstrated markedly reduced levels of 5-methyl-cytosine immunostaining when compared to that seen in cells treated with vehicle alone (Fig. 1). Treatment of MEF cells with 25 mM ethanol (48 h) did not result in any detectable change in immunostaining for 5-methyl-cytosine (data not shown).
Fig. 1.

Immunostaining of methylated DNA in murine embryonic fibroblasts (MEF) cells treated with (a) vehicle (b) 200 mM ethanol and (c) 10 μM 5-Azacytidine (positive control). MEF cells were fixed, permeabilized, and incubated with anti-5-methyl-cytosine antibody, followed by incubation with an Alexa Fluor 594-conjugated secondary antibody (lower row of panels). Upper row of panels represent brightfield images of cells, while middle row of panels depict DAPI staining of same cells. The last column of panels labeled as (d) represents a methodological “control” wherein primary antibody was omitted from the immunostaining procedure. Results from one out of three independent sets of experiments are shown in the figure.
3.3. Expression of DNA methyl transferases and methyl CpG/CpG domain binding proteins in nuclear extracts of MEF cells
MEF cells were examined for the expression of DNMT-1, -3a, -3b, MeCP-2, MBD-2 and MBD-3 by immunoblotting using either monoclonal or polyclonal antibodies as described under Section 2.5 in Section 2. Multiple (3) bands in the molecular weight range of 200–115 kDa and 140–110 kDa were detected for DNMT-1 and DNMT-3a, respectively, whereas a single major 98 kDa band was detected for DNMT-3b, on immunoblots of nuclear extracts of MEF cells treated for 48 h with vehicle or 25 mM ethanol (Fig. 2a–c). Likewise, immunoblotting of nuclear extracts of MEF cells treated for 48 h with vehicle- or 25 mM ethanol, detected multiple (3) bands in the molecular weight range of 34–28 kDa for MBD-3 and a single major band at 49 kDa and 75 kDa for MBD-2 or MECP-2, respectively. Notably, treatment of MEF cells with 200 mM ethanol for 48 h, resulted in considerable degradation of all six proteins as evidenced by the absence of the major bands and/or appearance of lower molecular weight bands (Fig. 2d–f). Densitometric analysis of film-detected chemiluminescent signals confirmed the changes in individual protein steady state levels noted above (Table 2). The presence of multiple bands for DNMT-1, DNMT-3a and MBD-3 may be explained by the existence of varied known isoforms and/or modified forms (e.g., phosphorylated) of these proteins. To confirm equal loading of proteins and normalize samples during densitometric analysis of immunoblots, Western blots were stripped and reprobed for β-actin, a housekeeping protein. As shown in Fig. 2g, the abundance of β-actin in the three lanes of the blot was similar. These results indicate that all three DNA methyl transferases (DNMT-1, -3a and 3b) and the three methyl CpG-binding proteins (MeCP-2, MBD-2 and -3) are expressed in MEF cells and that treatment (48 h) with 200 mM ethanol results in extensive degradation of these proteins in the nucleus. In contrast, the expression/integrity of β-actin, the housekeeping protein, remained unaffected following 48 h treatment of MEF cells with 200 mM ethanol (Fig. 2g).
Fig. 2.

Immunoblots demonstrating steady-state levels of DNMT-1 (a), DNMT-3a (b), DNMT-3b (c), MeCP-2 (d), MBD-2 (e) and MBD-3 (f) proteins in nuclear extracts derived from murine embryonic fibroblasts following treatment (48 h) with either 25 mM or 200 mM ethanol or vehicle control (PBS). Equal amounts of protein (15 μg) were resolved by SDS-PAGE on 12% polyacrylamide bis–tris gels, transferred to PVDF membranes, probed with specific antibodies and immunoreactive species detected by chemiluminescence, as detailed in Section 2. Molecular weights of the marker proteins are indicated to the left of each panel. The lowermost panel (g) depicts one representative immunoblot of the normalization control, β-actin. Each immunoblot is representative of no less than three independent blots from three unique sets of extracts from control and ethanol treated MEF cells.
Table 2.
Densitometric analysis of DNMT and methyl CpG/CpG domain binding protein immunoblots of MEF cell nuclear extracts.
| Proteina | Treatment | Band intensitiesb | Difference in band intensities (vs. vehicle)b (95% confidence limits) |
|---|---|---|---|
| DNMT-1 | Vehicle | 51.95 ± 2.00 | |
| 25 mM EtOH | 80.30 ± 6.51 | +28.35 (19.16, 37.54)*** | |
| 200 mM EtOH | 0.00 ± 0.00 | −51.95 (−61.14, −42.76)*** | |
| DNMT-3a | Vehicle | 53.88 ± 0.83 | |
| 25 mM EtOH | 82.21 ± 3.71 | +28.33 (22.51, 34.14)*** | |
| 200 mM EtOH | 33.21 ± 2.03 | −20.67 (−26.49, −14.86)*** | |
| DNMT-3b | Vehicle | 67.56 ± 1.31 | |
| 25 mM EtOH | 77.54 ± 6.34 | +9.97 (1.23, 18.72)* | |
| 200 mM EtOH | 9.96 ± 0.07 | −57.61 (−66.35, −48.87)*** | |
| MeCP-2 | Vehicle | 667.33 ± 76.00 | |
| 25 mM EtOH | 675.33 ± 69.14 | +8.00 (−148.46,164.46) | |
| 200 mM EtOH | 369.98 ± 53.72 | −297.35 (−453.81, −140.89)** | |
| MBD-2 | Vehicle | 43.44 ± 32.68 | |
| 25 mM EtOH | 78.47 ± 58.08 | +35.03 (−54.95, 125.01) | |
| 200 mM EtOH | 2.64 ± 2.12 | −40.80 (−130.78, 49.18) | |
| MBD-3 | Vehicle | 122.38 ± 19.68 | |
| 25 mM EtOH | 100.00 ± 24.25 | −22.38 (−64.55, 19.79) | |
| 200 mM EtOH | 4.73 ± 0.95 | −117.66 (−159.82, −75.49)*** |
Steady state levels of DNMT-1, DNMT-3a, DNMT-3b, MeCP-2, MBD-2 and MBD-3 proteins were determined by immunoblotting (Fig. 4) of nuclear extracts of MEF cells treated (48 h) with either vehicle (PBS), 25 mM ethanol or 200 mM ethanol.
The relative levels of DNMT-1, DNMT-3a, DNMT-3b, MeCP-2, MBD-2 and MBD-3 on immunoblots (Fig. 4) were analyzed by densitometry using the Image J software, as described in Section 2. Densitometric analysis of protein steady state levels was conducted on no less than three independent blots of nuclear extracts of MEF cells treated with either vehicle, 25 mM ethanol or 200 mM ethanol. β-Actin was used as an internal control for sample normalization. The data are presented as the mean ± standard deviation from three independent experiments. Differences from vehicle (+) indicates higher band intensity in ethanol treated samples, whereas (−) indicates lower band intensity in ethanol treated samples.
P < 0.05.
P < 0.01.
P < 0.001.
3.4. Differential effect of ethanol on the expression of genes encoding DNMT and methyl CpG/CpG domain binding proteins in MEF cells
Total RNA extracted from MEF cells treated (for 48 h) with either vehicle, 25 mM ethanol, or 200 mM ethanol was analyzed by TaqMan® RT-PCR. Comparison of Ct values [49] for genes encoding the three DNA methyltransferases, DNMT-1, -3a and -3b as well as those encoding the methyl CpG-binding proteins, MeCP-2, MBD-2 and -3, demonstrated a divergent effect of ethanol on their mRNA expression levels. Expression of Dnmt-1 was significantly decreased (>2.0-fold) following treatment of MEF cells with 200 mM ethanol but was unchanged following treatment with 25 mM ethanol (Table 3). Expression of Dnmt-3b was significantly up-regulated (1.5-fold) following treatment with the lower concentration (25 mM) of ethanol only, while treatment of MEF cells with both 25- and 200 mM ethanol resulted in stimulation of Dnmt-3a mRNA expression (1.35- and 1.25-fold, respectively) (Table 3). Expression of the gene encoding the methyl-CpG-binding protein, MeCP-2 demonstrated 1.28- and 1.25-fold upregulation, whereas that encoding MBD-2 displayed 1.40- and 1.50-fold down-regulation subsequent to treatment of MEF cells with 25- and 200 mM ethanol, respectively (Table 3). Mbd3 expression was diminished by 1.20-fold following treatment of MEF cells with 200 mM ethanol but remained unaltered when cells were exposed to 25 mM ethanol (Table 3). Mean Ct value corresponding to each gene was representative of no less than three separate assays of unique RNA extracts prepared from cultures of ethanol- or vehicle-treated MEF cells.
Table 3.
Effect of ethanol on the expression of genes encoding various DNMT and Methyl CpG/CpG domain binding proteins in MEF cells.
| Gene | Type of culturea | ΔCtb,c | Fold changed (2−ΔΔCt ) (95% confidence limits) |
|---|---|---|---|
| DNMT-1 | Control | 10.80 ± 0.01 | |
| 25 mM EtOH | 10.84 ± 0.06 | −1.03 (−1.44, 1.07) | |
| 200 mM EtOH | 11.86 ± 0.08 | −2.08 (−2.27, −1.89)*** | |
| DNMT-3a | Control | 14.08 ± 0.00 | |
| 25 mM EtOH | 13.65 ± 0.01 | 1.35 (1.32, 1.38)*** | |
| 200 mM EtOH | 13.54 ± 0.02 | 1.45 (1.42, 1.48)*** | |
| DNMT-3b | Control | 14.09 ± 0.01 | |
| 25 mM EtOH | 13.52 ± 0.02 | 1.48 (1.45, 1.51)*** | |
| 200 mM EtOH | 13.99 ± 0.01 | 1.07 (1.05, 1.10)*** | |
| MeCP-2 | Control | 12.37 ± 0.09 | |
| 25 mM EtOH | 12.02 ± 0.06 | 1.27 (1.15, 1.42)** | |
| 200 mM EtOH | 12.05 ± 0.03 | 1.26 (1.13, 1.40)** | |
| MBD-2 | Control | 10.54 ± 0.02 | |
| 25 mM EtOH | 10.99 ± 0.00 | −1.37 (−1.39, −1.35)*** | |
| 200 mM EtOH | 11.10 ± 0.00 | −1.47 (−1.49, −1.45)*** | |
| MBD-3 | Control | 11.67 ± 0.04 | |
| 25 mM EtOH | 11.56 ± 0.06 | 1.08 (1.01, 1.15)* | |
| 200 mM EtOH | 11.93 ± 0.01 | −1.20 (−1.28, −1.12)*** |
cDNA samples were prepared from control, 25 mM- and 200 mM Ethanol treated MEF cells and subjected to TaqMan® quantitative real-time PCR (RT-PCR) for each target gene. Analyses were performed in triplicate using data from three independent experiments.
Ct values represent the number of cycles during the exponential phase of amplification necessary to reach a predetermined threshold level of PCR product as measured by fluorescence. The more template present at the start of a reaction, the fewer the cycles required to synthesize enough fluorescent product to be recorded as statistically above background. All data were normalized to the amplification signal from the housekeeping gene, 18S rRNA. The ΔCt values represent these normalized signals, ΔCt = Ctsample − Ct18S rRNA. Data presented represent mean ΔCt ± standard deviation for three replicates.
Negative methodological control reactions, which lacked reverse transcriptase, did not amplify any detectable product.
Fold-change (FC) values were determined according to the relationship: FC = 2−ΔΔCt, where ΔΔCt is the difference in ΔCt values between 25 mM and 200 mM EtOH-MEF samples vs. Control-MEF samples [49]. Statistical analysis comparing the three cultures (control, 25 mM and 200 mM EtOH) was done with one-way ANOVA of the ΔCt values and adjustment for multiple comparisons using Dunnett’s method. 95% confidence intervals for the FC were calculating by taking the appropriate transformation of the 95% confidence limits for the estimated difference in ΔCt values. A negative fold-change value indicates down regulation of gene expression relative to control samples and a positive fold-change value indicates up regulation.
P < 0.05.
P < 0.01.
P < 0.001.
3.5. Ethanol reduces cellular levels of DNA methyltransferases and methyl CpG/CpG domain binding proteins in MEF cells via the proteasome pathway
Treatment of MEF cells with 200 mM ethanol resulted in the appearance of lower molecular weight proteins detected by immunoblotting for DNMT-1, DNMT-3a, MeCP-2 and MBD-3 proteins, most likely due to proteomic degradation of these proteins. Pretreatment with increasing doses of the proteasome inhibitor MG-132 (1–5 μM) led to a dose-dependent disappearance of lower molecular weight bands in immunoblots for DNMT-1, DNMT-3a, MeCP-2 and MBD-3 (Fig. 3a, b, d and f). Treatment of MEF cells with only MG-132 (5 μM) did not result in any degradation, or change in expression of the aforementioned proteins (data not shown).
Fig. 3.

Immunoblots demonstrating steady-state levels of DNMT-1 (a), DNMT-3a (b), DNMT-3b (c), MeCP-2 (d), MBD-2 (e) and MBD-3 (f) proteins in nuclear extracts derived from murine embryonic fibroblasts that were pre-treated (3 h) with either DMSO (vehicle/control) or 1-, 3- or 5 μM of the proteasomal inhibitor, MG-132, followed by a 48-h treatment with either 200 mM ethanol or vehicle control (PBS), as detailed in Section 2. Equal amounts of protein (15 μg) were resolved by SDS-PAGE on 12% polyacrylamide bis–tris gels, transferred to PVDF membranes, probed with specific antibodies and immunoreactive species detected by chemiluminescence as detailed in Section 2. Molecular weights of the marker proteins are indicated to the left of each panel. The lowermost panel (g) depicts one representative immunoblot of the normalization control, β-actin. Each immunoblot is representative of no less than three independent blots from three unique sets of extracts from control, inhibitor and ethanol treated MEF cells. C: pre-treatment with DMSO + treatment with PBS; E: pre-treatment with DMSO + treatment with 200 mM Ethanol; 1 + E: pre-treatment with 1 μM MG-132 + treatment with 200 mM Ethanol; 3 + E: pre-treatment with 3 μM MG-132 + treatment with 200 mM Ethanol; 5 + E: pre-treatment with 5 μM MG-132 + treatment with 200 mM Ethanol.
3.6. Determination of DNMT-1 protein levels in MEF cells
Analysis of the DNMT-1 protein levels using an ELISA-based method revealed a significant reduction (>70%) following treatment of MEF cells with 200 mM ethanol (Fig. 4). Treatment of MEF cells with 25 mM EtOH did not result in any notable alteration in the amount of DNMT-1 protein. Pre-treatment of MEF cells with 5 μM of the proteasomal inhibitor MG-132 resulted in total prevention of ethanol-induced DNMT-1 protein degradation – further substantiating the involvement of the proteasome pathway in ethanol-induced degradation of DNA methyltransferases and methyl CpG/CpG domain binding proteins (Fig. 3).
Fig. 4.
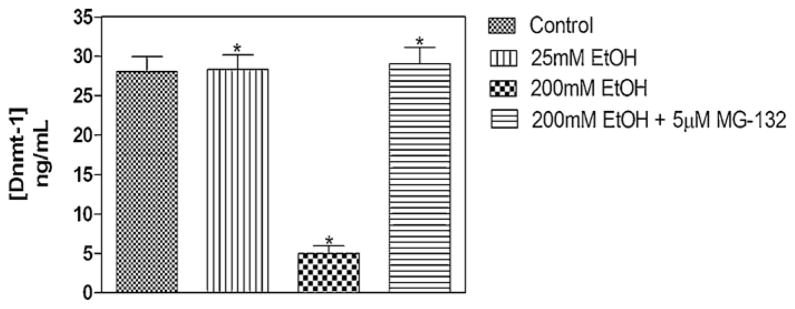
ELISA of DNMT-1 protein levels in MEF cells. Nuclear extracts from 48-h vehicle-, and ethanol- (alone or after 3-h MG-132 pre-treatment) treated MEFs, were assayed for DNMT-1 protein using the Epiquik DNMT-1 assay kit (Epigentek). A standard curve was generated utilizing protein standards of known concentrations (2, 5, 10 and 20 ng). The amount of DNMT-1 protein was estimated as: DNMT protein (ng/ml) = (sample OD − blank OD/standard slope) × sample dilution, following the manufacturer’s instructions. Treatment of MEF cells with 25 mM EtOH did not result in any notable alteration in the amount of DNMT-1 protein (*P < 0.05). Treatment of MEF cells with 200 mM ethanol revealed a significant reduction in DNMT-1 protein level (*P < 0.05, one-way analysis of variance [ANOVA]). Pre-treatment of MEF cells with 5 μM of the proteasomal inhibitor MG-132 resulted in total prevention of ethanol-induced DNMT-1 protein degradation (*P < 0.05, one-way ANOVA).
4. Discussion
Epigenetic control of gene transcription, via dynamic modification of DNA, plays a central role in the regulation of gene expression. Such DNA modification – brought about in a tissue- and time-specific manner – appears to be critically important for normal embryonic development. Environmental factors such as exposure to alcohol, cigarette smoke, numerous drugs and chemicals, microbial infection, and lack of nutrients (e.g., trace elements and folate) have been implicated in the etiology of numerous developmental anomalies [21,24,50–53]. A potential molecular mechanism underlying many of these environmental insults is aberrant methylation of susceptible genes which results in the altered expression of genes indispensable for normal embryogenesis.
The role of ethanol in the emerging field of “alcohol metabolism and epigenetic effects on tissue” is becoming increasingly apparent. Normal development of various embryonic tissues is adversely affected by in utero exposure to demethylating agents as well as by ethanol, suggesting that the developing embryo is a potential target of ethanol-mediated alterations in DNA methylation [54–56]. A number of recent studies have convincingly documented the teratogenic effect of ethanol on the epigenome of the developing embryo. Kaminen-Ahola et al. [21], utilizing a murine model of gestational ethanol exposure, reported changes in the expression of an epigenetically-sensitive allele, Agouti viable yellow (Avy), in offspring, thus demonstrating that ethanol can modulate the adult phenotype by altering the epigenome of the early embryo. These authors also observed postnatal growth restriction and craniofacial dysmorphology reminiscent of fetal alcohol syndrome, in congenic a/a siblings of the Avy mice. Investigating the effect of alcohol exposure during early embryonic neurulation on genome-wide DNA methylation and gene expression, Liu et al. [29] reported alterations in the methylation of imprinted genes, as well as genes governing cell cycle, growth, apoptosis, and olfaction. These authors also reported changes in the methylation of several genes (Wbscr 1, Wbscr 22, Ptpn11, Nipbl) involved in three developmental syndromes (e.g., Williams-, Noonan-, and Brachman-Delange syndrome) that share common phenotypic traits with FAS. Furthermore, MeDIP-chip analysis also identified alcohol-induced changes in methylation levels in genes associated with other developmental syndromes such as Angelmann (UBe3a), Bartter (Bsnd), Cleft palate (Pdgfra, snai1), and Hurler (Idua). In addition, it was recently shown by Zhou et al. [57], that alcohol exposure retarded neurogenesis and the migration of neural stem cells (NSCs) and a genome-wide diversification of DNA methylation – resulting in hyper- and hypomethylation of many moderately methylated genes implicated in neural development, neurotransmission, and olfaction. Collectively, these studies support the notion that in utero alcohol exposure can epigenetically modify genes crucial in mediating normal ontogenesis and developmental anomalies associated with FAS and FASD.
During mammalian embryogenesis – when the developing embryo undergoes genomic reprogramming–substantial alterations in global DNA methylation levels occur. Several earlier studies documented that exposure to ethanol, a potential teratogen, can result in adverse developmental outcomes by adversely affecting the embryonic epigenome [21,29,58]. In the present study, ethanol exposure significantly altered global DNA methylation, reduced 5-methyl-cytosine immunostaining, and diminished the activity and expression of DNMT-1 both at mRNA and protein levels in MEF cells. Since epigenetic modifications, such as DNA methylation, are often used to regulate cell/tissue- and stage-specific gene expression (for a review, see [59]), unique temporospatial epigenetic signatures underlie the entirety of embryogenesis. Hence, it is somewhat difficult to unilaterally compare the epigenetic modifications resulting from alcohol exposure in different cell types. Moreover, it is not surprising that alcohol has been found to have disparate effects on global and gene specific DNA methylation in varied cells/tissues. Nevertheless, some experimental comparisons are noteworthy.
Findings in the present study are consistent with a study demonstrating that maternal ethanol consumption decreased the activity of DNMT in fetal mice resulting in DNA hypomethylation [26]. They are, however, in contrast with another study which indicated that ethanol exposure resulted in an increased DNMT activity as well as hypermethylation and decreased expression of cell cycle genes in neural stem cells (NSCs) [60]. Despite this observation, and consistent with our data, the Dnmt-1 transcript was down-regulated by alcohol in both neural stem cells [60] and rat sperm [61]. Numerous studies have reported the effects of ethanol exposure on gene expression [29,62–69]. Intriguingly, in the present study, ethanol stimulated the expression of the genes encoding the “de novo” methyltransferases (DNMT-3a and -3b), but not the “maintenance” methyltransferase (DNMT-1). This differential effect is consistent with previous reports [27]. Ethanol has a similar differential effect on the expression of genes encoding the methyl CpG-binding proteins – upregulating Mecp2, but downregulating Mbd2 and Mbd3 in MEF cells. This represents the first report of the effect(s) of ethanol exposure on the expression (either at the level of mRNA or protein) of MeCP-2, MBD-2 and MBD-3. However, a recent study reported that alcohol exposure significantly reduced 5-MeC staining and MBD-1 expression both in the dorsal and ventral neural tube, and also led to major developmental delay [58]. The further demonstration that direct inhibition of DNA methylation by 5-azacytidine (5-AZA) resulted in similar growth retardation – reinforces the notion that alcohol can affect embryogenesis through an epigenetic pathway [58]. Collectively, these data support the notion that alcohol can impact the cellular DNA methylation machinery via modulating expression of the DNMTs and the MBDs, and that the resultant effects vary depending on its concentration and the target tissue and/or cell type.
To further investigate the underlying mechanism of ethanol-induced modulation of expression of the DNA methyltransferases (DNMT-1, -3a and -3b) and methyl CpG-binding proteins (MeCP-2, MBD-2 and -3), protein levels were determined following ethanol exposure of MEF cells. Interestingly, ethanol exposure resulted in substantial proteasome-mediated degradation of all three DNMTs and methyl CpG-binding proteins. While inconsistent with the observed down-regulation of the proteasome pathway when ethanol-responsive KEGG pathways were examined [63], our data are consistent with alcohol-induced alteration of proteins with roles in the ubiquitin-proteasome pathways [70]. In our study, treatment with the proteasome inhibitor, MG-132 prevented degradation of all the DNMT and MBD proteins. This strongly suggests that toxicant (alcohol) modulation of the DNA methylation machinery (DNA methyltransferases and methyl CpG binding proteins) is mediated through the ubiquitin/proteasomal degradation pathway. Such modulation can be included in the wide range of cellular substrates and critical biological processes that are controlled by the ubiquitin proteasome system [71].
Interestingly, in the current study, ethanol stimulated the expression of transcripts encoding DNMT-3a, -3b and MeCP-2, but diminished their expression at the protein level. A similar contrasting effect of ethanol on the expression of Dnmt-1 mRNA and protein in NSCs and fetal brain has been reported [60]. While little is known about the interaction between ethanol exposure and proteasome activity in the embryo [70,71], several studies have documented alterations in proteasome function as one of several multifactorial and detrimental changes in hepatocellular function subsequent to ethanol exposure sufficient to result in alcoholic liver disease (ALD) [72–74]. Chronic ethanol exposure has been shown to decrease the activity of the proteasome [72–75]. Ethanol-induced alterations in proteasome activity in hepatocytes contributes to impaired detoxification activity and sensitizes the hepatocyte to necrotic and apoptotic cell death (for a review, see [76]). In the present study, however, a totally contrasting effect of ethanol on proteasome function was observed in MEF cells. As discussed in the preceding section, ethanol stimulated proteasome function in MEF cells and induced proteasomal degradation of all three DNMTs and methyl CpG-binding proteins within the nucleus. Thus, one of several potential mechanisms underlying ethanol’s teratogenic effect on the embryo could be proteasome-mediated degradation of crucial effectors/regulators of DNA methylation (such as DNMT-1, -3a, -3b and MeCP-2, MBD-2 and -3). In conclusion, the present study is the first to document that ethanol reduces expression of proteins crucial for regulating DNA methylation in an ubiquitin-proteasome-dependent manner. Experimental evidence from the current study elucidates a potential novel molecular mechanism underlying the pathogenesis of FAS, through which ethanol may modulate methylation of DNA via alteration of DNMTS and methyl CpG-binding proteins, within cells of the developing embryo.
Acknowledgments
The authors thank Mrs. Savi Appana and Dr. Guy Brock for help with bioinformatics. This research was supported in part by NIH grant P20 RR017702 from the COBRE Program of the National Center for Research Resources and the NIGMS, NIH grants AA13205, HD053509, DE018215, and a grant from the Kentucky Science and Engineering Foundation grant KSEF-1420-RDE-010.
Footnotes
Conflict of interest statement
The authors declare that there are no conflicts of interest.
References
- 1.May PA, Gossage JP. Estimating the prevalence of fetal alcohol syndrome. A summary Alcohol Research & Health. 2001;25:159–67. [PMC free article] [PubMed] [Google Scholar]
- 2.Kuehn D, Aros S, Cassorla F, Avaria M, Unanue N, Henriquez C, et al. A prospective cohort study of the prevalence of growth, facial, and central nervous system abnormalities in children with heavy prenatal alcohol exposure. Alcoholism, Clinical and Experimental Research. 2012;36:1811–9. doi: 10.1111/j.1530-0277.2012.01794.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lipinski RJ, Hammond P, O’Leary-Moore SK, Ament JJ, Pecevich SJ, Jiang Y, et al. Ethanol-induced face-brain dysmorphology patterns are correlative and exposure-stage dependent. PLoS ONE. 2012;7:e43067. doi: 10.1371/journal.pone.0043067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sampson PD, Streissguth AP, Bookstein FL, Little RE, Clarren SK, Dehaene P, et al. Incidence of fetal alcohol syndrome and prevalence of alcohol-related neurodevelopmental disorder. Teratology. 1997;56:317–26. doi: 10.1002/(SICI)1096-9926(199711)56:5<317::AID-TERA5>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 5.Lemoine P, Harrousseau H, Borteyru J, Menuet J. Les enfants de parents alcooliques: Anomalies observées à propos de 127 cas. Quest Medical. 1968;25:476–82. [Google Scholar]
- 6.Jones KL, Smith DW. Recognition of the fetal alcohol syndrome in early infancy. Lancet. 1973;302:999–1001. doi: 10.1016/s0140-6736(73)91092-1. [DOI] [PubMed] [Google Scholar]
- 7.Jones KL, Smith DW, Ulleland CN, Streissguth P. Pattern of malformation in offspring of chronic alcoholic mothers. Lancet. 1973;1:1267–71. doi: 10.1016/s0140-6736(73)91291-9. [DOI] [PubMed] [Google Scholar]
- 8.Floyd RL, O’Connor MJ, Sokol RJ, Bertrand J, Cordero JF. Recognition and prevention of fetal alcohol syndrome. Obstetrics and Gynecology. 2005;106:1059–64. doi: 10.1097/01.AOG.0000181822.91205.6f. [DOI] [PubMed] [Google Scholar]
- 9.Klingenberg CP, Wetherill L, Rogers J, Moore E, Ward R, Autti-Ramo I, et al. Prenatal alcohol exposure alters the patterns of facial asymmetry. Alcohol. 2010;44:649–57. doi: 10.1016/j.alcohol.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haycock PC. Fetal alcohol spectrum disorders: the epigenetic perspective. Biology of Reproduction. 2009;81:607–17. doi: 10.1095/biolreprod.108.074690. [DOI] [PubMed] [Google Scholar]
- 11.Moonat S, Starkman BG, Sakharkar A, Pandey SC. Neuroscience of alcoholism: molecular and cellular mechanisms. Cellular and Molecular Life Sciences. 2010;67:73–88. doi: 10.1007/s00018-009-0135-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pandey SC, Ugale R, Zhang H, Tang L, Prakash A. Brain chromatin remodeling: a novel mechanism of alcoholism. Journal of Neuroscience. 2008;28:3729–37. doi: 10.1523/JNEUROSCI.5731-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shukla SD, Velazquez J, French SW, Lu SC, Ticku MK, Zakhari S. Emerging role of epigenetics in the actions of alcohol. Alcoholism, Clinical and Experimental Research. 2008;32:1525–34. doi: 10.1111/j.1530-0277.2008.00729.x. [DOI] [PubMed] [Google Scholar]
- 14.Kiefer JC. Epigenetics in development. Developmental Dynamics. 2007;236:1144–56. doi: 10.1002/dvdy.21094. [DOI] [PubMed] [Google Scholar]
- 15.MacDonald JL, Roskams AJ. Epigenetic regulation of nervous system development by DNA methylation and histone deacetylation. Progress in Neurobiology. 2009;88:170–83. doi: 10.1016/j.pneurobio.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 16.Meshorer E. Chromatin in embryonic stem cell neuronal differentiation. Histology and Histopathology. 2007;22:311–9. doi: 10.14670/HH-22.311. [DOI] [PubMed] [Google Scholar]
- 17.Mohammad HP, Baylin SB. Linking cell signaling and the epigenetic machinery. Nature Biotechnology. 2010;28:1033–8. doi: 10.1038/nbt1010-1033. [DOI] [PubMed] [Google Scholar]
- 18.Lalande M, Calciano MA. Molecular epigenetics of Angelman syndrome. Cellular and Molecular Life Sciences. 2007;64:947–60. doi: 10.1007/s00018-007-6460-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shahbazian MD, Zoghbi HY. Rett syndrome and MeCP2: linking epigenetics and neuronal function. American Journal of Human Genetics. 2002;71:1259–72. doi: 10.1086/345360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ueda Y, Okano M, Williams C, Chen T, Georgopoulos K, Li E. Roles for Dnmt3b in mammalian development: a mouse model for the ICF syndrome. Development. 2006;133:1183–92. doi: 10.1242/dev.02293. [DOI] [PubMed] [Google Scholar]
- 21.Kaminen-Ahola N, Ahola A, Maga M, Mallitt KA, Fahey P, Cox TC, et al. Maternal ethanol consumption alters the epigenotype and the phenotype of offspring in a mouse model. PLoS Genetics. 2010;6:e1000811. doi: 10.1371/journal.pgen.1000811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miranda RC, Pietrzykowski AZ, Tang Y, Sathyan P, Mayfield D, Keshavarzian A, et al. MicroRNAs: master regulators of ethanol abuse and toxicity. Alcoholism, Clinical and Experimental Research. 2010;34:575–87. doi: 10.1111/j.1530-0277.2009.01126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oberlander TF, Weinberg J, Papsdorf M, Grunau R, Misri S, Devlin AM. Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (NR3C1) and infant cortisol stress responses. Epigenetics. 2008;3:97–106. doi: 10.4161/epi.3.2.6034. [DOI] [PubMed] [Google Scholar]
- 24.Ouko LA, Shantikumar K, Knezovich J, Haycock P, Schnugh DJ, Ramsay M. Effect of alcohol consumption on CpG methylation in the differentially methylated regions of H19 and IG-DMR in male gametes: implications for fetal alcohol spectrum disorders. Alcoholism, Clinical and Experimental Research. 2009;33:1615–27. doi: 10.1111/j.1530-0277.2009.00993.x. [DOI] [PubMed] [Google Scholar]
- 25.Qiang M, Denny A, Chen J, Ticku MK, Yan B, Henderson G. The site specific demethylation in the 5′-regulatory area of NMDA receptor 2B subunit gene associated with CIE-induced up-regulation of transcription. PLoS ONE. 2010;5:e8798. doi: 10.1371/journal.pone.0008798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garro AJ, McBeth DL, Lima V, Lieber CS. Ethanol consumption inhibits fetal DNA methylation in mice: implications for the fetal alcohol syndrome. Alcoholism, Clinical and Experimental Research. 1991;15:395–8. doi: 10.1111/j.1530-0277.1991.tb00536.x. [DOI] [PubMed] [Google Scholar]
- 27.Bonsch D, Lenz B, Fiszer R, Frieling H, Kornhuber J, Bleich S. Lowered DNA methyltransferase (DNMT-3b) mRNA expression is associated with genomic DNA hypermethylation in patients with chronic alcoholism. Journal of Neural Transmission. 2006;113:1299–304. doi: 10.1007/s00702-005-0413-2. [DOI] [PubMed] [Google Scholar]
- 28.Feng J, Fouse S, Fan G. Epigenetic regulation of neural gene expression and neuronal function. Pediatric Research. 2007;61:58R–63R. doi: 10.1203/pdr.0b013e3180457635. [DOI] [PubMed] [Google Scholar]
- 29.Liu Y, Balaraman Y, Wang G, Nephew KP, Zhou FC. Alcohol exposure alters DNA methylation profiles in mouse embryos at early neurulation. Epigenetics. 2009;4:500–11. doi: 10.4161/epi.4.7.9925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hermann A, Goyal R, Jeltsch A. The Dnmt1 DNA-(cytosine-C5)-methyltransferase methylates DNA processively with high preference for hemimethylated target sites. Journal of Biological Chemistry. 2004;279:48350–9. doi: 10.1074/jbc.M403427200. [DOI] [PubMed] [Google Scholar]
- 31.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–57. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 32.Smith SS, Kaplan BE, Sowers LC, Newman EM. Mechanism of human methyl-directed DNA methyltransferase and the fidelity of cytosine methylation. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:4744–8. doi: 10.1073/pnas.89.10.4744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Howell CY, Bestor TH, Ding F, Latham KE, Mertineit C, Trasler JM, et al. Genomic imprinting disrupted by a maternal effect mutation in the Dnmt1 gene. Cell. 2001;104:829–38. doi: 10.1016/s0092-8674(01)00280-x. [DOI] [PubMed] [Google Scholar]
- 34.Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–26. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- 35.Kaneda M, Okano M, Hata K, Sado T, Tsujimoto N, Li E, et al. Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature. 2004;429:900–3. doi: 10.1038/nature02633. [DOI] [PubMed] [Google Scholar]
- 36.Hansen RS, Wijmenga C, Luo P, Stanek AM, Canfield TK, Weemaes CM, et al. The DNMT3B DNA methyltransferase gene is mutated in the ICF immunodeficiency syndrome. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:14412–7. doi: 10.1073/pnas.96.25.14412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martins-Taylor K, Schroeder DI, LaSalle JM, Lalande M, Xu RH. Role of DNMT3B in the regulation of early neural and neural crest specifiers. Epigenetics. 2012;7:71–82. doi: 10.4161/epi.7.1.18750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bogdanovic O, Veenstra GJ. DNA methylation and methyl-CpG binding proteins: developmental requirements and function. Chromosoma. 2009;118:549–65. doi: 10.1007/s00412-009-0221-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Defossez PA, Stancheva I. Biological functions of methyl-CpG-binding proteins. Progress in Molecular Biology and Translational Science. 2011;101:377–98. doi: 10.1016/B978-0-12-387685-0.00012-3. [DOI] [PubMed] [Google Scholar]
- 40.Ruddock-D’Cruz NT, Xue J, Wilson KJ, Heffernan C, Prashadkumar S, Cooney MA, et al. Dynamic changes in the localization of five members of the methyl binding domain (MBD) gene family during murine and bovine preimplantation embryo development. Molecular Reproduction and Development. 2008;75:48–59. doi: 10.1002/mrd.20712. [DOI] [PubMed] [Google Scholar]
- 41.Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nature Genetics. 1999;23:185–8. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 42.Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nature Genetics. 2001;27:322–6. doi: 10.1038/85899. [DOI] [PubMed] [Google Scholar]
- 43.Kaji K, Nichols J, Hendrich B. Mbd3, a component of the NuRD co-repressor complex, is required for development of pluripotent cells. Development. 2007;134:1123–32. doi: 10.1242/dev.02802. [DOI] [PubMed] [Google Scholar]
- 44.Greene RM, Pisano MM. Palate morphogenesis: current understanding and future directions. Birth Defects Research C Embryo Today. 2010;90:133–54. doi: 10.1002/bdrc.20180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lodish H, Berk A, Kaiser CA, Krieger M, Bretscher A, Ploegh H, et al. Molecular cell biology. 7. New York: W.H. Freeman and Co; 2012. Culturing, visualizing and perturbing cell. [Google Scholar]
- 46.Mukhopadhyay P, Webb CL, Warner DR, Greene RM, Pisano MM. BMP signaling dynamics in embryonic orofacial tissue. Journal of Cellular Physiology. 2008;216:771–9. doi: 10.1002/jcp.21455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sasse J, Gallagher SR. Staining proteins in gels. Current Protocols in Molecular Biology. 2009;Chapter 10:10.6.1–27. doi: 10.1002/0471142727.mb1006s85. [DOI] [PubMed] [Google Scholar]
- 48.Abramoff MD, Magelhaes PJ, Ram SJ. Image processing with Image J. Biophotons International. 2004;11:36–42. [Google Scholar]
- 49.Gibson UE, Heid CA, Williams PM. A novel method for real time quantitative RT-PCR. Genome Research. 1996;6:995–1001. doi: 10.1101/gr.6.10.995. [DOI] [PubMed] [Google Scholar]
- 50.Boyles AL, Billups AV, Deak KL, Siegel DG, Mehltretter L, Slifer SH, et al. Neural tube defects and folate pathway genes: family-based association tests of gene–gene and gene–environment interactions. Environmental Health Perspectives. 2006;114:1547–52. doi: 10.1289/ehp.9166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Knudsen TB, Kleinstreuer NC. Disruption of embryonic vascular development in predictive toxicology. Birth Defects Research C Embryo Today. 2011;93:312–23. doi: 10.1002/bdrc.20223. [DOI] [PubMed] [Google Scholar]
- 52.Obican SG, Finnell RH, Mills JL, Shaw GM, Scialli AR. Folic acid in early pregnancy: a public health success story. FASEB Journal. 2010;24:4167–74. doi: 10.1096/fj.10-165084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Suarez L, Ramadhani T, Felkner M, Canfield MA, Brender JD, Romitti PA, et al. Maternal smoking, passive tobacco smoke, and neural tube defects. Birth Defects Research Part A: Clinical and Molecular Teratology. 2011;91:29–33. doi: 10.1002/bdra.20743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kakutani T, Jeddeloh JA, Flowers SK, Munakata K, Richards EJ. Developmental abnormalities and epimutations associated with DNA hypomethylation mutations. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:12406–11. doi: 10.1073/pnas.93.22.12406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Martin CC, Laforest L, Akimenko MA, Ekker M. A role for DNA methylation in gas-trulation and somite patterning. Developmental Biology. 1999;206:189–205. doi: 10.1006/dbio.1998.9105. [DOI] [PubMed] [Google Scholar]
- 56.Matsuda M, Yasutomi M. Inhibition of cephalic neural tube closure by 5-azacytidine in neurulating rat embryos in vitro. Anatomy and Embryology (Berlin) 1992;185:217–23. doi: 10.1007/BF00211820. [DOI] [PubMed] [Google Scholar]
- 57.Zhou FC, Balaraman Y, Teng M, Liu Y, Singh RP, Nephew KP. Alcohol alters DNA methylation patterns and inhibits neural stem cell differentiation. Alcoholism, Clinical and Experimental Research. 2011;35:735–46. doi: 10.1111/j.1530-0277.2010.01391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhou FC, Chen Y, Love A. Cellular DNA methylation program during neurulation and its alteration by alcohol exposure. Birth Defects Research Part A: Clinical and Molecular Teratology. 2011;91:703–15. doi: 10.1002/bdra.20820. [DOI] [PubMed] [Google Scholar]
- 59.Geiman TM, Muegge K. DNA methylation in early development. Molecular Reproduction and Development. 2010;77:105–13. doi: 10.1002/mrd.21118. [DOI] [PubMed] [Google Scholar]
- 60.Hicks SD, Middleton FA, Miller MW. Ethanol-induced methylation of cell cycle genes in neural stem cells. Journal of Neurochemistry. 2010;114:1767–80. doi: 10.1111/j.1471-4159.2010.06886.x. [DOI] [PubMed] [Google Scholar]
- 61.Bielawski DM, Zaher FM, Svinarich DM, Abel EL. Paternal alcohol exposure affects sperm cytosine methyltransferase messenger RNA levels. Alcoholism, Clinical and Experimental Research. 2002;26:347–51. [PubMed] [Google Scholar]
- 62.Da Lee R, Rhee GS, An SM, Kim SS, Kwack SJ, Seok JH, et al. Differential gene profiles in developing embryo and fetus after in utero exposure to ethanol. Journal of Toxicology and Environmental Health Part A. 2004;67:2073–84. doi: 10.1080/15287390490515001. [DOI] [PubMed] [Google Scholar]
- 63.Green ML, Singh AV, Zhang Y, Nemeth KA, Sulik KK, Knudsen TB. Reprogramming of genetic networks during initiation of the fetal alcohol syndrome. Developmental Dynamics. 2007;236:613–31. doi: 10.1002/dvdy.21048. [DOI] [PubMed] [Google Scholar]
- 64.Grenett HE, Aikens ML, Tabengwa EM, Davis GC, Booyse FM. Ethanol down-regulates transcription of the PAI-1 gene in cultured human endothelial cells. Thrombosis Research. 2000;97:247–55. doi: 10.1016/s0049-3848(99)00172-3. [DOI] [PubMed] [Google Scholar]
- 65.Higashiyama D, Saitsu H, Komada M, Takigawa T, Ishibashi M, Shiota K. Sequential developmental changes in holoprosencephalic mouse embryos exposed to ethanol during the gastrulation period. Birth Defects Research Part A: Clinical and Molecular Teratology. 2007;79:513–23. doi: 10.1002/bdra.20367. [DOI] [PubMed] [Google Scholar]
- 66.Kumari M, Ticku MK. Ethanol and regulation of the NMDA receptor subunits in fetal cortical neurons. Journal of Neurochemistry. 1998;70:1467–73. doi: 10.1046/j.1471-4159.1998.70041467.x. [DOI] [PubMed] [Google Scholar]
- 67.Miller MW, Jacobs JS, Neg Yokoyama R. a nerve growth factor-stimulated gene expressed by fetal neocortical neurons that is downregulated by ethanol. Journal of Comparative Neurology. 2003;460:212–22. doi: 10.1002/cne.10651. [DOI] [PubMed] [Google Scholar]
- 68.Varodayan FP, Pignataro L, Harrison NL. Alcohol induces synaptotagmin 1 expression in neurons via activation of heat shock factor 1. Neuroscience. 2011;193:63–71. doi: 10.1016/j.neuroscience.2011.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang LH, Yang JY, Cui W, Shin YK, Wu CF. Involvement of promyelocytic leukemia protein in the ethanol-induced apoptosis in mouse embryo fibroblasts. Yakugaku Zasshi: Journal of the Pharmaceutical Society of Japan. 2008;128:1067–71. doi: 10.1248/yakushi.128.1067. [DOI] [PubMed] [Google Scholar]
- 70.Mason S, Anthony B, Lai X, Ringham HN, Wang M, Witzmann FA, et al. Ethanol exposure alters protein expression in a mouse model of fetal alcohol spectrum disorders. International Journal of Proteomics. 2012;2012:867141. doi: 10.1155/2012/867141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Myung J, Kim KB, Crews CM. The ubiquitin-proteasome pathway and proteasome inhibitors. Medicinal Research Reviews. 2001;21:245–73. doi: 10.1002/med.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bousquet-Dubouch MP, Nguen S, Bouyssie D, Burlet-Schiltz O, French SW, Monsarrat B, et al. Chronic ethanol feeding affects proteasome-interacting proteins. Proteomics. 2009;9:3609–22. doi: 10.1002/pmic.200800959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Donohue TM, Jr, Kharbanda KK, Casey CA, Nanji AA. Decreased proteasome activity is associated with increased severity of liver pathology and oxidative stress in experimental alcoholic liver disease. Alcoholism, Clinical and Experimental Research. 2004;28:1257–63. doi: 10.1097/01.alc.0000134233.89896.19. [DOI] [PubMed] [Google Scholar]
- 74.Thomes PG, Trambly CS, Thiele GM, Duryee MJ, Fox HS, Haorah J, et al. Protea-some activity and autophagosome content in liver are reciprocally regulated by ethanol treatment. Biochemical and Biophysical Research Communications. 2012;417:262–7. doi: 10.1016/j.bbrc.2011.11.097. [DOI] [PubMed] [Google Scholar]
- 75.Bardag-Gorce F, Li J, French BA, French SW. The effect of ethanol-induced CYP2E1 on proteasome activity: the role of 4-hydroxynonenal. Experimental and Molecular Pathology. 2005;78:109–15. doi: 10.1016/j.yexmp.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 76.Albano E. Oxidative mechanisms in the pathogenesis of alcoholic liver disease. Molecular Aspects of Medicine. 2008;29:9–16. doi: 10.1016/j.mam.2007.09.004. [DOI] [PubMed] [Google Scholar]