

Abstract
New agents are urgently needed for the therapeutic treatment of Staphylococcus aureus infections. In that regard, S. aureus RNase RnpA may represent a promising novel dual-function antimicrobial target that participates in two essential cellular processes, RNA degradation and tRNA maturation. Accordingly, we previously used a high-throughput screen to identify small-molecule inhibitors of the RNA-degrading activity of the enzyme and showed that the RnpA inhibitor RNPA1000 is an attractive antimicrobial development candidate. In this study, we used a series of in vitro and cellular assays to characterize a second RnpA inhibitor, RNPA2000, which was identified in our initial screening campaign and is structurally distinct from RNPA1000. In doing so, it was found that S. aureus RnpA does indeed participate in 5′-precursor tRNA processing, as was previously hypothesized. Further, we show that RNPA2000 is a bactericidal agent that inhibits both RnpA-associated RNA degradation and tRNA maturation activities both in vitro and within S. aureus. The compound appears to display specificity for RnpA, as it did not significantly affect the in vitro activities of unrelated bacterial or eukaryotic ribonucleases and did not display measurable human cytotoxicity. Finally, we show that RNPA2000 exhibits antimicrobial activity and inhibits tRNA processing in efflux-deficient Gram-negative pathogens. Taken together, these data support the targeting of RnpA for antimicrobial development purposes, establish that small-molecule inhibitors of both of the functions of the enzyme can be identified, and lend evidence that RnpA inhibitors may have broad-spectrum antimicrobial activities.
INTRODUCTION
Staphylococcus aureus is a major human pathogen that is becoming increasingly difficult to treat, primarily due to the emergence of antibiotic resistance. Indeed, the organism has developed resistance to every standard-of-care antibiotic available, including vancomycin, daptomycin, linezolid, and tigecycline, and it recently surpassed HIV/AIDS as an annual cause of death in the United States (1–5). The shrinking arsenal of effective therapeutics for the treatment of S. aureus infections necessitates novel antibiotic drug discovery programs to successfully combat this pathogen.
Bacterial mRNA degradation is an essential cellular process that has been well characterized in the Gram-negative model organism Escherichia coli, in which bulk cellular mRNA degradation is catalyzed by an RNA degradosome complex consisting of RNase E, enolase (Eno), polynucleotide phosphorylase (PNPase), and RNA helicase B (RhlB) (6–8). RNase E is an essential enzyme and the key component of the E. coli degradosome, catalyzing the initial rate-limiting endoribonucleolytic event during substrate decay and also serving as a scaffold for the assembly of other degradosome subunits (9–11). In addition to participating in mRNA degradation, RNase E catalyzes the maturation of regulatory and structured RNA species, such as tRNAs and rRNAs (12–17). Thus, RNase E may represent a promising antimicrobial target, as the corresponding inhibitors would affect cellular mRNA degradation, regulatory processes, and/or translation. Nonetheless, Gram-positive bacteria, such as S. aureus, do not encode an RNase E amino acid ortholog, which has hindered both the identification and targeting of the Gram-positive RNA turnover machinery for use in antimicrobial development.
We recently showed that S. aureus mRNA turnover is likely to be mediated by an RNA degradosome-like complex consisting of enolase, RNA helicase (CshA), RNase J1, RNase J2, RNase Y (also known as CvfA and YmdA), PNPase, phosphofructokinase (Pfk), and RnpA that is very similar to the recently described Bacillus subtilis RNA degradosome (18, 19). Further, we hypothesized that this complex represents an attractive antimicrobial development target for several reasons. First, at least five of the putative S. aureus degradosome complex subunits, RnpA, RNase J1, RNase J2, enolase, and Pfk, are thought to be required for viability and, ostensibly, represent antimicrobial targets (20). Second, the molecular components and mechanisms by which prokaryotic and eukaryotic cells catalyze mRNA decay fundamentally differ, providing an opportunity to develop agents that selectively inhibit the bacterial process (reviewed in reference 21). Third, small-molecule inhibitors of bacterial mRNA turnover would represent first-in-class agents that are likely to be structurally distinct from current antibiotic classes and less susceptible to inactivation by the currently encountered enzymatic antibiotic resistance mechanisms.
In considering which S. aureus RNA degradosome subunit may represent the most attractive antimicrobial target, it was recognized that in addition to participating in mRNA degradation, RnpA is also likely to participate in a second essential S. aureus biological process, tRNA maturation (22). Indeed, in both the Gram-negative and Gram-positive model organisms E. coli and B. subtilis, RnpA has been shown to interact with an RNA ribozyme, rnpB, to form RNase P ribonucleoprotein complexes that catalyze the removal of the 5′-leader sequences from precursor tRNAs (ptRNAs), thereby generating mature tRNA molecules needed for translation (22–27). Admittedly, to our knowledge, S. aureus RnpA has not been formally shown to confer RNase P activity. Nonetheless, RNase P function is thought to be conserved across bacterial species; thus, it seems very probable that RnpA is required for S. aureus RNase P activity. Consequently, small-molecule inhibitors of S. aureus RnpA may serve as novel dual-function antimicrobial agents that interfere with both the RNA degradation and tRNA processing activities of the organism. As a result, bacterial resistance to RnpA inhibitors would likely be slow to develop, because RnpA mutations that interfere with compound binding may be tolerated by one holoenzyme (i.e., RNA degradosome) but inactivate the second (i.e., RNase P). From these perspectives, we hypothesized that RnpA is a unique and promising antimicrobial target.
As a first step toward validating RnpA as an antimicrobial target, we previously performed a high-throughput screening campaign to identify inhibitors of S. aureus RnpA RNA-degrading activity (28). The screening results revealed three structurally distinct classes of inhibitors, RNPA1000, RNPA2000, and RNPA3000, which displayed antistaphylococcal activity but did not elicit significant mammalian cytotoxicity. The RnpA inhibitor RNPA1000 was found to exhibit antimicrobial properties toward all Gram-positive bacterial pathogens tested, and the antibacterial activity of the compound correlated with the inhibition of RnpA-associated RNA decay within bacterial cells (28). In addition, RNPA1000 was active against S. aureus biofilms with equal or greater efficacy compared to that of daptomycin, linezolid, and vancomycin, could be incorporated into biomedical materials, and protected mice in a lethal S. aureus model of infection, suggesting that RnpA inhibitors may have tremendous therapeutic potential (28, 29).
The goal of the current study was to characterize a second RnpA inhibitor, RNPA2000, which was identified in our initial screening campaign. The results revealed that like RNPA1000, RNPA2000 displays antimicrobial activities toward Gram-positive pathogens. However, distinct differences between the antimicrobial features of the two RnpA inhibitors were also observed. More specifically, while RNPA1000 is a bacteriostatic agent, RNPA2000 was found to be bactericidal toward the S. aureus strains tested. This prompted us to evaluate whether the two RnpA inhibitors displayed differing effects on the putative tRNA processing function(s) of the enzyme. Our data indicate that RnpA is indeed required for S. aureus RNase P-dependent 5′ processing of precursor tRNA molecules within the tRNA maturation pathway, as previously hypothesized, confirming the multifunctional activities of the enzyme in S. aureus. Further, we show that RNPA2000 inhibits both RnpA-mediated cellular RNA degradation and tRNA maturation activities within S. aureus cells, whereas RNPA1000 inhibits RnpA-mediated cellular RNA degradation only. We also found that RNPA2000 exhibits antimicrobial activity and inhibits cellular RNase P activity in efflux-deficient Gram-negative bacteria. Taken together, RNPA2000 appears to be a dual-function inhibitor of both known cellular functions of S. aureus RnpA, whereas in Gram-negative bacteria, the agent acts as an inhibitor of the tRNA processing function of the enzyme, suggesting that broad-spectrum RnpA inhibitors can be developed for the therapeutic intervention of both Gram-positive and Gram-negative bacterial species. The RnpA inhibitor RNPA2000 may represent an excellent starting antimicrobial scaffold for lead optimization.
MATERIALS AND METHODS
Protein purification.
S. aureus RnpA was purified, as described previously (28). Briefly, E. coli strain BL21(DE3) cells harboring plasmid pET-30 Ek/ligation-independent cloning (LIC) (Novagen, Madison, WI) containing a hexahistidine tag fused to the N terminus of the S. aureus RnpA coding region under the control of the plasmid's T7 promoter strain were grown in the presence of 0.5 mM isopropyl-β-d-1-thiogalactopyranoside (IPTG) for 4 h to induce protein production. The cells were collected by centrifugation at 2,830 × g for 20 min at 4°C and then suspended in 50 ml of buffer A (300 mM NaCl, 50 mM Na2HPO4 [pH 7.4]) containing a mini EDTA-free protease inhibitor tablet (Roche, Branford, CT) and 20 mM imidazole. The cells were mechanically ruptured by five passes at 14,000 lb/in2 through a French pressure cell press (SLM-Aminco, Pittsford, NY), and cell debris was removed by centrifugation at 17,000 × g and 4°C for 10 min. The supernatants were collected, filtered through a 0.2-μm syringe filter, and then loaded onto the DuoFlow Maximizer medium-pressure chromatography system (Bio-Rad, Hercules, CA) containing a 5-ml HisPur cobalt column (Thermo Fisher Scientific, Waltham, MA). Protein was eluted using an imidazole gradient (80 mM to 500 mM); the fractions were assessed for protein presence and purity on SDS-PAGE gels via Coomassie blue staining and Western blotting using anti-His antibody (Invitrogen, Carlsbad, CA), as previously described (28). S. aureus RNase J1 and RNase J2 were purified as described above, except that RNase J1 was cloned into pTrcHis2A (Invitrogen) under the control of the trc (trp-lac) promoter of the plasmid and contained a C-terminal hexahistidine tag.
In vitro transcription of RNA.
The RNA component of S. aureus RNase P (rnpB) and the RNA substrates for RNase activity assays (spa mRNA) and tRNA processing assays (mature and precursor tyrosine tRNAs [tRNATyr and ptRNATyr]) were generated in vitro. To do so, the DNA sequence corresponding to each transcript species was PCR amplified using S. aureus strain UAMS-1 chromosomal DNA as a template and the following oligonucleotide primer pairs (the forward primers contained an RNA polymerase T7 promoter sequence [underlined]): 5′-GATTACATAATACGACTCACTATAGGGTGATATTTCGGGTAATCGCTATA (forward) and 5′-ACTAGTAGTGATATTTCTATAAGCCATG (reverse) for rnpB, 5′-GATTACATAATACGACTCACTATAGGGGGAGGGGTAGCGAAGTGGC (forward) and 5′-TGGTGGAGGGGGGCAGATTC (reverse) for tRNATyr, 5′-GATTACATAATACGACTCACTATAGGGCACCATTTATGGAGGGGTAGCG (forward) and the same reverse primer as tRNATyr for ptRNATyr, and 5′-GATTACATAATACGACTCACTATAGGGTTATAGTTCGCGACGACGTCCAG (forward) and 5′-TTGAAAAAGAAAAACATTTATTCAATTCGTAAACTAGG (reverse) for spa.
The resulting PCR products were electrophoresed in an agarose gel, purified using the QIAquick nucleotide removal kit, according to the manufacturer's instructions (Qiagen, Gaithersburg, MD), and sequenced by ACGT, Inc. (Wheeling, IL) to ensure the integrity of each PCR product. Each PCR product was used as a template for in vitro transcription using the TranscriptAid T7 high-yield transcription kit, according to the manufacturer's recommendations (Fermentas, Burlington, Ontario, Canada). The resulting RNA was treated with DNase I for 30 min, repurified using the RNeasy minikit (Qiagen), and quantified using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific).
RNA degradation assays.
RnpA-mediated RNA degradation assays were performed as previously described (28). Briefly, either 1 μg of total S. aureus RNA or 1 pmol of in vitro-synthesized spa mRNA was incubated with 20 pmol of RnpA at 37°C for 15 to 30 min in reaction buffer (50 mM Tris-HCl [pH 8.0], 2 mM NaCl, 2 mM MgCl2) in the absence or presence of the indicated compound. The reactions were stopped by adding an equal volume of 2× RNA loading dye (95% formamide, 0.025% SDS, 0.025% bromophenol blue, 0.025% xylene cyanol FF, 0.5 mM EDTA), run on a denaturing 1.0% agarose–0.66 M formaldehyde gel, and stained with ethidium bromide. The RNA substrates and corresponding degradation products were visualized using a FluorChem 5500 system (Alpha Innotech, San Leandro, CA). The inhibitory effects of the test compounds were measured using the ImageJ densitometry software (National Institutes of Health, Bethesda, MD) to quantify the signal intensity of the RNA band(s) in the negative control (RNA alone), positive control (RnpA plus RNA plus dimethyl sulfoxide [DMSO]), and experimental samples (RnpA plus RNA plus test compound). The percent enzyme inhibition of the test compounds was calculated using the following equation: percent inhibition = [(experimental signal − positive control)/(negative-control signal − positive-control signal)] × 100.
RNase P ptRNA processing assay.
S. aureus RNase P activity assays were performed under the conditions previously used to measure B. subtilis RNase P-mediated cleavage of precursor tRNATyr 5′ leader in low-salt buffer (50 mM Tris-HCl [pH 8.0], 5 mM MgCl2) or in high-salt buffer (50 mM Tris-HCl [pH 8.0], 100 mM MgCl2, 800 mM NH4Cl) (27). For all the reactions, ptRNATyr, tRNATyr, or rnpB RNA species were first denatured by heating to 95°C for 3 min and then being slowly cooled to room temperature. RNase P was reconstituted by mixing an equimolar ratio of rnpB and RnpA for 15 min at 37°C. The precursor tRNA processing reactions (20 μl) were performed by mixing 1.25 pmol of RNase P (RnpA plus rnpB), RnpA, or rnpB with an equal volume of 2× low-salt buffer or 2× high-salt buffer and 10 pmol of ptRNATyr. The mixtures were incubated for 15 min at 37°C. The reactions were stopped by adding 20 μl of 2× RNA loading dye (95% formamide, 0.025% SDS, 0.025% bromophenol blue, 0.025% xylene cyanol FF, 0.5 mM EDTA), and 30 μl of each sample was electrophoresed in a 7 M urea–8% polyacrylamide gel and then stained with ethidium bromide (0.5 μg/ml). Where indicated, the reactions were repeated in the presence of the indicated amount of putative RnpA inhibitors or dimethyl sulfoxide (DMSO). A FluorChem 5500 imaging system was used to visualize the RNA, and the relative abundance of the mature tRNATyr band in the positive control (RNase P plus DMSO) or in samples containing the test compounds was measured using ImageJ densitometry software (NIH). The percent RNase P activity was then calculated using the following equation: (test compound tRNATyr signal/positive-control tRNATyr signal) × 100.
Antimicrobial susceptibility testing.
MIC testing was performed according to Clinical and Laboratory Standards Institute (CLSI) guidelines (30). Briefly, individual wells of a 96-well microtiter plate were inoculated with ∼1 × 105 CFU of the indicated organism, containing 2-fold increasing concentrations (0 to 256 μg ml−1) of the indicated antibiotic or putative RnpA inhibitor, and incubated at 37°C for 18 h in Mueller-Hinton broth. The assays were also performed in the presence of 2.5% bovine serum albumin. The MIC was defined as the lowest concentration of antibiotic at which there was no visible bacterial growth in the wells. Minimum bactericidal concentration testing was performed by enumerating the bacterial cells in each of the wells containing treatments at and above the MIC. The concentration of the test agent that resulted in 99.9% cell death of the starting inoculum was determined to be the minimum bactericidal concentration, as previously described (31, 32).
Fractional inhibitory concentration (FIC) testing was performed to determine the combined effects of the indicated antibiotic and the RnpA inhibitors RNPA1000 and RNPA2000, as previously described (33). To do so, individual wells of a 96-well microtiter plate were inoculated with 1 × 105 CFU of S. aureus UAMS-1 in Mueller-Hinton broth. Each row of the plate contained increasing concentrations of RNPA1000 or RNPA2000 (2-fold increments; 0×, 0.004×, 0.008×, 0.016×, 0.03×, 0.06×, 0.125×, 0.25×, 0.5×, 1×, 2×, or 4× the MIC), whereas each column contained increasing concentrations of the indicated antibiotic (2-fold increments; 0×, 0.06×, 0.125×, 0.25×, 0.5×, 1×, 2×, or 4× the MIC). The plates were incubated for 18 h at 37°C, and growth was detected by the unaided eye. The FIC was determined using the following formula: (MIC of drug A in combination/MIC of drug A alone) + (MIC of drug B in combination/MIC of drug B alone). A synergistic interaction was defined as an FIC of ≤0.5, no interaction as an FIC of 0.5 to 4, and an antagonistic interaction as an FIC of >4 (33).
The spontaneous resistance frequencies for rifampin, RNPA1000, and RNPA2000 were determined by growing S. aureus UAMS-1 to the exponential phase and concentrating to approximately 1 × 1012 CFU ml−1. The cells were plated on Mueller-Hinton agar to determine the starting inoculum, and the plates were supplemented with 2× the MIC of each RnpA inhibitor or 10 μg ml−1 rifampin. The resistance frequency was determined as the number of resistant colonies divided by the total CFU of the inoculum.
RNA isolation.
S. aureus UAMS-1 or E. coli tolC imp cells were grown to mid-exponential phase, treated as indicated, and then mixed with an equal volume of ice-cold acetone-ethanol (1:1 [vol/vol]) to inactivate the cellular RNases. The cell pellets were collected by centrifugation at 1,510 × g and 4°C for 15 min, resuspended in 500 μl of ice-cold TE buffer (10 mM Tris-HCl [pH 8.0], 1 mM EDTA), transferred to a FastPrep bead beater tube containing 0.1-mm silica spheres, and mechanically disrupted using a FastPrep-24 instrument (MP Biomedicals, Santa Ana, CA). The resulting cell debris was removed by centrifugation at 17,000 × g and 4°C for 15 min, and RNA molecules >200 nucleotides (nt) in length were purified using an RNeasy minikit, whereas RNA species of ≤200 nt were purified using the miRNeasy minikit, according to the manufacturer's recommendations (Qiagen, Gaithersburg, MD). The RNA quantity and quality were measured by spectrophotometry (optical density at 260 nm [OD260] of 1.0 at 40 μg ml−1) and by assessing rRNA integrity following electrophoresis in a 1.0% agarose–0.66 M formaldehyde agarose gel.
GeneChip analysis.
S. aureus Affymetrix GeneChips (Santa Clara, CA) were used to measure the cellular mRNA turnover properties of RNPA2000-treated and RnpA-depleted cells, as previously described (28, 34–36). For the RNA2000-treated cell studies, S. aureus UAMS-1 was grown in tryptic soy broth (TSB) to an optical density at 600 nm (OD600) of 0.25 and treated with either 0.5× the MIC of RNPA2000 or DMSO for 30 min; it was then treated with rifampin to arrest de novo RNA synthesis. RNA was isolated from the aliquots removed at 0, 5, and 15 min posttranscriptional arrest and processed as described below. To measure the RNA turnover profiles of the RnpA-depleted cells, the S. aureus vector control (RN4220 pML100, containing a chloramphenicol-selectable marker and anhydrotetracycline-inducible promoter) or the RnpA depletion strain (RN4220 pML100::rnpA-A.S., containing rnpA-directed antisense RNA under the control of an anhydrotetracycline promoter) were grown to early exponential phase (OD600, 0.25) in TSB, diluted to a concentration of approximately 1 × 105 CFU ml−1 in fresh TSB containing 60 ng ml−1 anhydrotetracycline and 5 μg ml−1 chloramphenicol, and grown for 8 h to induce antisense transcript expression; these conditions limit RnpA production but do not decrease RnpA levels to the point that viability is affected, as was described previously (28). Transcript synthesis was then inhibited by adding rifampin (200 μg ml−1), and RNA was isolated from the aliquots taken at 0, 5, and 15 min posttranscriptional arrest.
For microarray analysis, 10 μg of each total bacterial RNA sample was reverse transcribed using SuperScript II reverse transcriptase (Invitrogen), purified using the QIAquick PCR purification kit (Qiagen), and partially digested using DNase I (Ambion). Fragmented DNA was then 3′-biotinylated using the Enzo BioArray terminal labeling kit (Enzo Life Sciences, Farmingdale, NY), and 2 μg of labeled cDNA was hybridized to a commercially available Affymetrix S. aureus GeneChip and processed according to the manufacturer's recommendations for prokaryotic arrays (Affymetrix, Santa Clara, CA). The signal intensity values for each RNA species were normalized and averaged using the GeneSpring 7.2 software (Agilent Technologies, Redwood City, CA), and the half-life of each transcript was calculated as the posttranscriptional arrest time point at which the time 0 (t0) signal decreased by a factor of 2, as previously described (28, 34–37).
Cellular tRNATyr population measures.
Northern blotting was used to measure ptRNATyr and tRNATyr cellular pools within S. aureus or E. coli cells treated with the indicated agent. To do so, S. aureus UAMS-1 or E. coli strain AT100 (tolC imp) was grown in TSB or Mueller-Hinton broth (MHB), respectively, to mid-exponential phase (OD600, 0.25 to 0.4). The cultures were then treated with the indicated agent for 1 h, and RNA was isolated using the miRNeasy kit (Qiagen), as described above. For Northern blotting, 10 μg of total bacterial RNA was resolved on a 7 M urea–8% polyacrylamide gel and transferred overnight to an Amersham Hybond-N+ membrane (GE, Fairfield, CT) using a Bio-Rad wet tank transfer in 0.5× Tris-borate-EDTA (TBE) buffer at 20 V and 4°C overnight. The blots were UV-cross-linked, washed, and probed with digoxigenin (DIG)-labeled PCR products representing S. aureus tRNATyr or E. coli tRNATyr or tRNAVal; these were generated using the primers 5′GGAGGGGTAGCGAAGTGGCT and 5′TGGTGGAGGGGGGCAGATTC for S. aureus tRNATyr, 5′GGTGGGGTTCCCGAGCGGCC and 5′TGGTGGTGGGGGAAGGATTCGAACC for E. coli tRNATyr, or 5′GCGTTCATAGCTCAGTTGGTTAGAGC and 5′TGGTGCGTTCAATTGGACTCGAACC for E. coli tRNAVal, using the PCR DIG probe synthesis kit, according to the manufacturer's instructions (Roche). The blots were subsequently probed with anti-DIG antibody conjugated to alkaline phosphatase (Roche), and the disodium 3-(4-methoxyspiro[1,2-dioxetane-3,2′-(5′-chloro)tricyclodecan]-4-yl) phenyl phosphate (CSPD) chemiluminescent substrate (Roche) was added to visualize the tRNA species by exposing the blot to BioMax light film (Kodak, Rochester, NY).
Reverse transcription-quantitative PCR.
Following RNA isolation, 1 μg of total bacterial RNA was treated with 10 units of DNase I (Ambion) for 1 h at 37°C and repurified using either the RNeasy minikit (>200 nucleotides [nt]) or miRNeasy (<200 nt) kit. The iScript cDNA synthesis kit (Bio-Rad, Hercules, CA) was used to synthesize cDNA, which was subsequently amplified via quantitative PCR (qPCR) using the Bio-Rad iQ SYBR green supermix, according to the manufacturer's instructions.
Polycistronic S. aureus tRNAPhe,Thr,Tyr levels were measured using 5′-TTCAGTAGCTCAGTTGGTAGAGCAATG (forward) and 5′-TGGTGGAGGGGGGCAGATTC (reverse), compared to S. aureus 16S rRNA (primers 5′TAACCTACCTATAAGACTGGGATAA and 5′GCTTTCACATCAGACTTAAAAA) as an internal control (ΔΔCT method), and plotted as a fold change compared to the indicated control.
5′-rapid amplification of cDNA ends.
The 5′ ends of S. aureus cellular tRNATyr species were determined following mock or RNPA2000 treatment using the 5′-rapid amplification of cDNA ends (RACE) system, according to the manufacturer's recommendations (Invitrogen). To do so, S. aureus UAMS-1 was grown in TSB to an OD600 of 0.25 and treated with 0× or 2× the MIC of RNPA2000 for 1 h. RNA was isolated using an miRNeasy kit, as described above. First-strand cDNA synthesis was carried out using 500 ng of RNA from each sample and the tRNATyr reverse primer 5′TGGTGGAGGGGGGCAGATTC; the resulting cDNA was purified and 3′-end labeled using terminal deoxynucleotidyl transferase. The poly(C)-tailed cDNA was used as a template for PCR using the kit-supplied abridged anchor primer and the nested tRNATyr-specific primer 5′GGGCAGATTCGAACTGCCGAAC. The resulting 5′-RACE PCR products were gel purified from an agarose gel using the QIAquick gel extraction kit (Qiagen), cloned into pCRII-TOPO (Invitrogen, Carlsbad, CA), and sequenced by ACGT, Inc.
Human cytotoxicity measures.
Human HepG2 hepatocytes (provided by S. Carson, University of Nebraska Medical Center, Omaha, NE) were seeded in individual wells of a 96-well cell culture-treated microtiter plate at a concentration of 1 × 105 cells per well and incubated overnight in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum at 37°C with 5% carbon dioxide. The cells were then treated with mitomycin C (50 μg ml−1; positive control) or 0, 16, or 64 μg ml−1 RNPA2000 for 24 h. The MTT [3-(4,5-dimethylthiazolyl-2)-2, 5-diphenyltetrazolium bromide] cell proliferation assay kit (American Type Culture Collection, Manassas, VA) was used to assess viability, according to the manufacturer's instructions, by spectrophotometrically measuring the reduction of the yellow tetrazolium MTT to the purple formazan by metabolically active cells.
RESULTS
RNPA2000 is an antimicrobial compound that inhibits in vitro RnpA-mediated RNA degradation.
S. aureus RnpA is a RNase component of the RNA degradosome-like complex of the organism and represents a promising target in antimicrobial development (28, 38). In an earlier high-throughput screening campaign, we identified structurally distinct small-molecule inhibitors of RnpA-mediated RNA degradation and subsequently found that one of these compounds, RNPA1000, represents an attractive scaffold for antimicrobial development (28). The same screen also identified RNPA2000 (Fig. 1A), a compound that is structurally distinct from RNPA1000 and has not been characterized. A gel-based RNA degradation assay that measures the ability of S. aureus RnpA to catalyze the digestion of spa mRNA (28) was used to verify that RNPA2000 is a modest inhibitor of the activity of the enzyme RNase (apparent 50% inhibitory concentration [IC50], 125 μM) rather than a screening artifact (Fig. 1B). Moreover, RNPA2000 appeared to display specificity for RnpA, as the compound did not significantly inhibit the RNase activity of commercially available E. coli RNase III, bovine RNase A, or in-house-purified S. aureus RNase J1 or RNase J2 at the concentrations tested (0 to 1,000 μM; data not shown).
FIG 1.
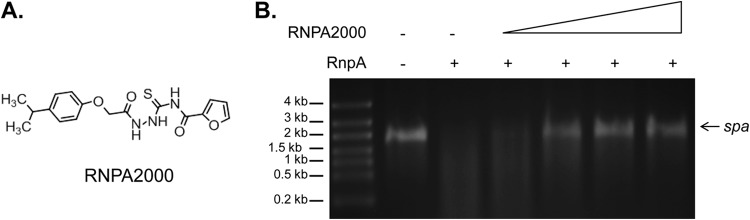
RNPA2000 inhibits S. aureus RnpA-mediated RNA degradation in vitro. (A) Structure of RNPA2000. (B) Mobility of spa mRNA (1 pmol) following incubation in the absence (−) or presence (+) of 20 pmol of RnpA with increasing concentrations of RNPA2000 (62.5, 125, 250, or 500 μM).
MIC testing was used to evaluate the antimicrobial activity of the compound toward a panel of Gram-positive and Gram-negative bacterial species. As shown in Table 1, RNPA2000 demonstrated moderate antimicrobial activity (MIC, 8 to 16 μg ml−1) toward all S. aureus strains tested in the absence or presence of serum albumin, including two well-characterized genotypically diverse methicillin-susceptible clinical isolates, methicillin-resistant S. aureus (MRSA), and vancomycin-resistant S. aureus (VRSA) clinical isolates. The compound also displayed antimicrobial activity (MIC, 4 to 16 μg ml−1) toward all other Gram-positive species tested, including Staphylococcus epidermidis, Streptococcus pyogenes, and Enterococcus faecium. Minimum bactericidal concentration testing indicated that RNPA2000 acts as a bactericidal agent against S. aureus, and human cell cytotoxicity studies revealed that the compound did not elicit toxicity following 24 h of exposure to 16× the MIC of the compound against S. aureus (256 μg ml−1; see Fig. S1 in the supplemental material). RNPA2000 did not exhibit antimicrobial activity (MIC, >256 μg ml−1) toward any of the Gram-negative species evaluated (Table 1). In that regard, we previously predicted that the lack of activity of S. aureus RnpA inhibitors toward Gram-negative species might be attributable to the low sequence conservation (∼33% amino acid identity) of the S. aureus enzyme within these species (28, 29).
TABLE 1.
Antimicrobial properties of RNPA2000
| Organism | Strain | MIC (μg ml−1) | Reference or sourcea |
|---|---|---|---|
| Staphylococcus aureus | UAMS-1 | 16 | 55 |
| Newman | 8 | 56 | |
| LAC | 16 | 57 | |
| Mu50 | 8 | 58 | |
| VRSA-1 | 8 | 28 | |
| VRSA-2 | 8 | 28 | |
| Staphylococcus epidermidis | ATCC 14990 | 4 | ATCC |
| Enterococcus faecium | 824-05 | 32 | 59 |
| Enterococcus faecium | ATCC 19634 | 4 | ATCC |
| Streptococcus pyogenes | 8255 | 16 | 28 |
| Acinetobacter baumannii | 98-37-09 | >256 | 60 |
| Pseudomonas aeruginosa | PA01 | >256 | 61 |
| Klebsiella pneumoniae | CKP4 | >256 | 62 |
| Escherichia coli | TOP10 | >256 | Invitrogen |
ATCC, American Type Culture Collection.
Taken together, RNPA2000 appears to resemble RNPA1000 in being an in vitro RnpA RNase inhibitor that displays antimicrobial activity toward Gram-positive bacteria and little or no toxicity toward human cells. However, the RnpA inhibitor RNPA1000 is a bacteriostatic agent (28), whereas RNPA2000 was found to exhibit bactericidal activity, suggesting that the two compounds display differing cellular potencies and/or affect different S. aureus cellular processes.
S. aureus RnpA participates in tRNA processing.
In addition to participating in cellular RNA turnover, S. aureus RnpA is hypothesized to be a component of the RNase P holoenzyme of the organism, which is thought to be a ubiquitous bacterial ribonucleoprotein complex composed of RnpA and the ribozyme rnpB that catalyzes the removal of precursor tRNA (ptRNA) 5′-leader sequences (23). This process is thought to generate the mature tRNAs needed for translation. While S. aureus RnpA has been modeled to interact with rnpB, and S. aureus rnpB can functionally complement RNase P activity in B. subtilis, to our knowledge, no studies have formally evaluated whether RnpA is indeed required for S. aureus RNase P function (22, 39). Nonetheless, we considered the possibility that the differing antimicrobial properties of RNPA1000 (bacteriostatic) and RNPA2000 (bactericidal) might be attributable to the corresponding differences in their ability to inhibit RnpA-mediated S. aureus RNase P precursor tRNA processing activity.
Accordingly, we initially set out to determine whether S. aureus RnpA together with rnpB does indeed confer functional RNase P activity in vitro. To do so, the experimental procedures used were those to characterize the in vitro properties of B. subtilis RNase P (27), in which we measured the ability of the S. aureus ribonucleoprotein complex to catalyze the removal of a 5′-leader sequence from S. aureus precursor tRNATyr (ptRNATyr) to yield the mature tRNATyr species. As shown in Fig. 2A, under low-salt buffer conditions (50 mM Tris-HCl [pH 8.0], 5 mM MgCl2), 1.25 pmol of reconstituted RNase P (1.25 pmol of RnpA plus 1.25 pmol of rnpB RNA) efficiently catalyzed the removal of a 10-nucleotide 5′-leader sequence from in vitro-synthesized ptRNATyr substrates to yield lower-molecular-weight tRNATyr molecules, whereas neither rnpB (Fig. 2B) nor RnpA (Fig. 2C) alone was able to process ptRNATyr. Notably, under high-salt conditions, B. subtilis rnpB is capable of catalyzing ptRNA processing in the absence of RnpA; the same was found to be true for S. aureus rnpB. As shown in Fig. 2D, under high-salt buffer conditions (50 mM Tris-HCl [pH 8.0], 100 mM MgCl2, 800 mM NH4Cl), S. aureus rnpB catalyzed ptRNATyr processing, whereas RnpA alone did not (data not shown). Taken together, these results suggest that S. aureus RnpA together with rnpB is capable of forming functional RNase P activity in vitro and that the ribonucleoprotein complex behaves similarly to other characterized bacterial RNase P enzymes; both RnpA and rnpB are required to catalyze 5′-ptRNA processing under physiologically relevant salt conditions, whereas rnpB alone modulates ptRNA processing under high-salt conditions (27).
FIG 2.
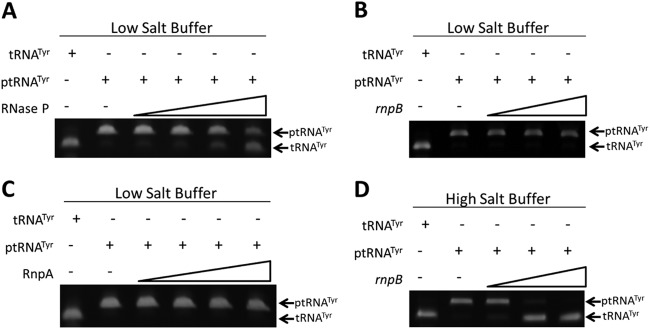
S. aureus ribonucleoprotein complex RNase P processes precursor tRNATyr in vitro. Gel mobility of precursor tRNA (ptRNATyr) following incubation with increasing concentrations of reconstituted S. aureus RNase P (RnpA plus rnpB) (A), rnpB alone (B), or RnpA alone (C) under low-salt buffer conditions. (D) Mobility of ptRNATyr in the presence of rnpB alone under high-salt buffer conditions. The mobilities of mock-treated ptRNATyr and mature tRNATyr are also shown.
Inhibitors of RnpA-mediated RNA degradation also inhibit RnpA-mediated precursor tRNA processing.
The above results support the hypothesis that RnpA may represent a unique antimicrobial target that participates in two essential cellular processes, and it might also explain the differential antimicrobial effects of RNPA1000 (bacteriostatic) and RNPA2000 (bactericidal); while both agents appear to inhibit RnpA-mediated RNA degradation, they may display differing effects on the precursor tRNA processing activity(ies) of the enzyme. As an initial test of that possibility, RNPA1000 and RNPA2000 were introduced into the RNase P processing assay and assessed for their ability to inhibit tRNA maturation. The results revealed that both RNPA1000 (Fig. 3A; apparent IC50, 175 μM) and RNPA2000 (Fig. 3B; apparent IC50, 130 μM) prevented RnpA-dependent RNase P processing of ptRNATyr under physiological salt conditions, whereas antibiotics affecting other cellular processes, such as mupirocin, linezolid, erythromycin, and vancomycin, did not (see Fig. S2 in the supplemental material). To distinguish whether the RNase P inhibitory effects of RNPA1000 and/or RNPA2000 were RnpA dependent as opposed to rnpB dependent, the assays were repeated using rnpB alone under high-salt conditions. As shown in Fig. S3A and B in the supplemental material, neither compound inhibited rnpB-mediated tRNA processing in high-salt buffer, suggesting that the RNase P inhibitory effects of each agent are RnpA dependent.
FIG 3.
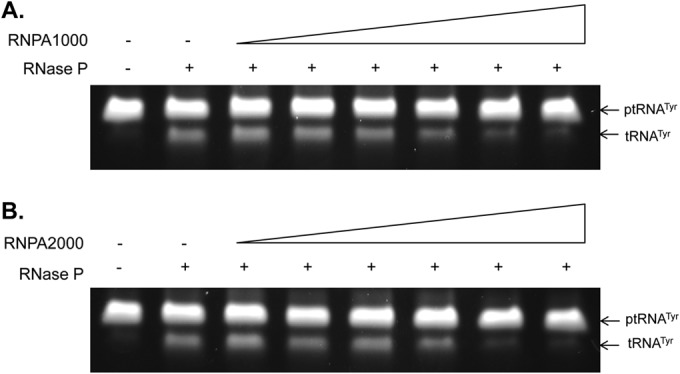
RNPA1000 and RNPA2000 inhibit RNase P-mediated ptRNATyr processing in vitro. RNase P activity assays measuring the ability of the enzyme to catalyze conversion of ptRNATyr to tRNATyr in the absence or presence of increasing concentrations (0, 31.25, 62.5, 125, 250, and 500 μM) of RNPA1000 (A) and RNPA2000 (B).
Collectively, these data indicate that the inhibitors of RnpA-mediated RNA degradation also inhibit RnpA-mediated RNase P tRNA processing in vitro and may represent novel dual-function antimicrobial compounds. Further, while overt differences between the in vitro inhibitory activities of RNPA1000 and RNPA2000 were not observed that might explain their differential antimicrobial activities, it was recognized that the complexity/physiology of the cell vastly differs from what can be modeled under in vitro conditions. Thus, the compounds may exhibit differing RnpA inhibitory properties within bacterial cells. In that regard, we previously showed that the antimicrobial properties of RNPA1000 correlate with the inhibition of cellular RnpA-associated mRNA degradation, but we did not previously evaluate whether the compound also inhibits RnpA-dependent tRNA processing within S. aureus cells (28). Likewise, it is unknown whether the antimicrobial activity of RNPA2000 correlates with the inhibition of RnpA-mediated RNA degradation and/or tRNA processing activity(ies). Accordingly, we set out to determine whether the antimicrobial effects of RNPA2000 were indeed RnpA dependent, define whether the agent inhibits cellular RnpA-mediated RNA degradation and/or tRNA processing, and compare those results to the cellular effects of RNPA1000.
S. aureus RNPA2000 susceptibility is dependent on cellular RnpA levels.
The conditional expression of antisense RNA fragments that are complementary to a given mRNA target has been proven to be a powerful tool to characterize bacterial cells that have been depleted of their cognate target. Indeed, such an antisense RNA approach has defined the repertoire of S. aureus enzymes, including RnpA, that are essential for survival under laboratory culture conditions (40). More recently, antisense RNA target knockdown strains have been used to define the cellular mechanism of action of antimicrobial agents, the premise being that cells depleted of a given target display hypersusceptibility to compounds that inhibit the depleted enzyme but not agents that target other bacterial processes (41). Similarly, we previously used an antisense rnpA mRNA expression strain to create depleted RnpA cells that are hypersusceptible to the RnpA inhibitor RNPA1000, verifying that the enzyme is a cellular target of RNPA1000 (28). Using the same approach, we evaluated whether RnpA is likely to be a cellular target of RNPA2000.
As shown in Fig. 4A (left), the S. aureus RnpA depletion strain, which contains a plasmid-borne antisense rnpA fragment that is complementary to 230 bp upstream and 81 bp into the coding region of rnpA mRNA, under the control of an anhydrotetracycline (ATc)-inducible promoter, displayed a severe growth defect when dilution plated on agar plates under high-level antisense RNA induction conditions (10 ng ml−1 ATc) in comparison to vector (pML100)-containing cells; this supports earlier studies establishing the essentiality of the protein. Further, plating the RnpA depletion strain on low-level anhydrotetracycline plates (5 ng ml−1 ATc) maintained the viability of the strain (Fig. 4A, right), despite mildly reducing the cellular concentration of the protein, as measured by Western blotting (see Fig. S4 in the supplemental material). As shown in Fig. 4B, under low-level antisense RNA expression conditions, the RnpA-depleted strain exhibited increased susceptibilities to the known RnpA inhibitor RNPA1000 and RNPA2000 in comparison to that with the vector-containing control cells. Conversely, the RnpA-depleted cells were not hypersusceptible to antibiotics known to affect other cellular targets, including vancomycin, rifampin, mupirocin, daptomycin, or ciprofloxacin, ensuring that RnpA-depleted conditions do not elicit nonspecific antimicrobial hypersusceptibility (see Fig. S5 in the supplemental material). Taken together, these results suggest that the antibacterial activity of RNPA2000 correlates with reduced cellular RnpA levels, raising the possibility that the compound may be inhibiting RnpA-mediated cellular RNA degradation, tRNA processing, or both biological processes.
FIG 4.
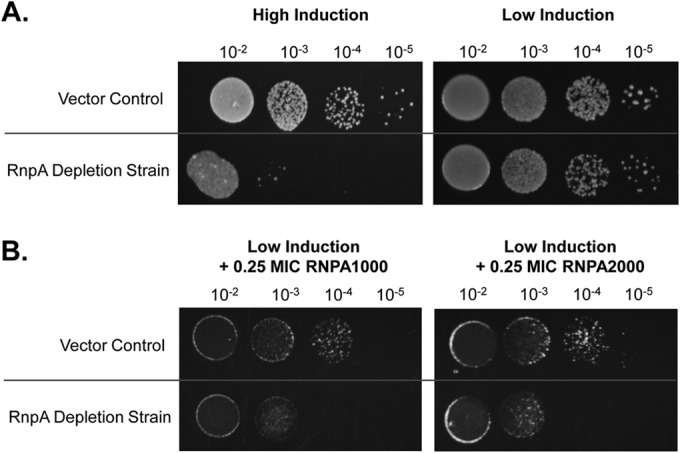
Antisense rnpA RNA levels influence S. aureus viability and susceptibility to RnpA inhibitors. (A) Serially diluted S. aureus vector (pML100) or isogenic RnpA depletion (pML100::A.S.-rnpA; rnpA mRNA-directed antisense molecule) cells plated on tryptic soy agar (TSA) containing high induction of the antisense transcript (10 ng ml−1 anhydrotetracycline [aTC], left) or under low-induction conditions (5 ng ml−1 ATc, right). (B) As in panel A, except strains were plated under low-induction conditions (5 ng ml−1 ATc) supplemented with 0.25× the MIC RNPA1000 (left) or RNPA2000 (right).
RNPA2000 inhibits S. aureus RNA turnover.
Microarrays have been used as an efficient means to measure the global mRNA degradation properties of bacterial species and bacteria in response to exogenous stressors (reviewed in reference 42). In these studies, the strains of interest are typically grown, de novo transcript synthesis is arrested by adding the RNA polymerase inhibitor rifampin, and the transcript titers of mRNA species are measured at T0 and various posttranscriptional arrest time points to monitor the degradation properties of RNA species as a function of time. Using this approach, we previously showed that global cellular RNA degradation is dramatically reduced in RnpA-depleted cells and closely resembles that of wild-type cells treated with 0.5× the MIC of RNPA1000, indicating that the compound inhibits the cellular RnpA-mediated RNA degradation function (28). The same approach was used to determine whether RNPA2000 also affects the cellular RNA degradation properties of S. aureus. To do so, S. aureus UAMS-1 was treated with 0.5× the MIC of RNPA2000 for 30 min, and Affymetrix GeneChips were used compare the global mRNA turnover properties of the treated and mock-treated cells (Table 2).
TABLE 2.
S. aureus RNA degradation properties
| Strain | Relevant condition | % with half-life of all mRNA species of: |
|
|---|---|---|---|
| ≤5 min | >5 min | ||
| UAMS-1 | Mock treatment | 88.9 | 11.1 |
| UAMS-1 | RNPA2000 | 3.6 | 96.4 |
| RN4220 pML100 (vector) | Mock treatment | 83.5 | 16.5 |
| RN4220 pML100::rnpA-A.S. | RnpA depletion | 13.9 | 86.1 |
Consistent with previous measures, the results revealed that most (89%) mRNA species are rapidly degraded (half-life, ≤5 min) in mock-treated exponential-phase S. aureus cells (28, 34, 35, 43). Conversely, pretreatment with a subinhibitory concentration of RNPA2000 was found to reduce the cellular mRNA degradation rate (5% of all transcripts displayed a half-life of ≤5 min), presumably by limiting the RNA-degrading activity of RnpA. While we cannot rule out the possibility that the agent affects other enzymes, including other cellular ribonucleases, the reduced RNA degradation properties of RNPA2000-treated cells closely resembled the RNA turnover properties of RnpA-depleted cells, in which only 13.9% of all transcripts were rapidly degraded (half-life, ≤5 min). Taken together, these results indicate that RNPA2000 reduces cellular mRNA degradation in a manner that is consistent with reduced RnpA function within S. aureus cells, suggesting that the agent limits the mRNA-degrading activity of the enzyme. Moreover, the reduced mRNA turnover properties of RNPA2000-treated cells mimicked the effects of cells treated with the known RnpA inhibitor RNPA1000 (28), indicating that the differing antimicrobial activities between the two compounds are not attributable to differences in their cellular RNA degradation inhibitory activities.
RNPA2000 inhibits RNase P-mediated tRNA processing within S. aureus.
Having established that RNPA1000 and RNPA2000 are likely to inhibit RnpA-mediated S. aureus cellular RNA degradation to a similar extent, two approaches were used to determine whether the molecules affect the cellular RnpA-mediated RNase P function(s) of the organism. First, we considered that two antimicrobials targeting independent steps of the same metabolic pathway can have combined antibacterial effects (44). For instance, sulfonamides and trimethoprim interfere with enzymes within the folic acid biosynthesis pathway (dihydropteroate synthetase and dihydrofolate, respectively) and act synergistically in combination (45). In that regard, RNase P (RnpA plus rnpB) and tRNA synthetases are thought to work in a sequential manner to generate mature charged tRNA molecules needed for translation. RNase P is required to process polycistronic and 5′-leader sequences of precursor tRNA species to generate mature tRNA species that are subsequently charged by tRNA synthetases. Thus, we performed an initial assessment of whether the RnpA inhibitors RNPA1000 and RNPA2000 are likely to inhibit RnpA-dependent RNase P precursor tRNA processing by determining whether they improve the potency of the isoleucyl-tRNA synthetase inhibitor mupirocin (46). Fractional inhibitory concentration (FIC) measures revealed a synergistic effect with RNPA2000 (FIC, 0.44) when combined with mupirocin, whereas no significant interaction was observed between RNPA2000 and the other classes of antibiotics tested, including vancomycin, daptomycin, erythromycin, or rifampin (47). Likewise, fractional inhibitory concentration testing did not reveal any significant activity between mupirocin and RNPA1000 (FIC, 0.73). Taken together, these results provided an initial indication that RNPA2000 may act within the same tRNA maturation pathway as mupirocin by inhibiting RnpA-dependent cellular RNase P activity, whereas RNPA1000 may not.
As a more definitive means of determining whether RNPA1000 and RNPA2000 inhibit S. aureus RNase P tRNA processing, we considered that the inhibition of cellular RNase P function would result in the accumulation of the enzyme's substrates, polycistronic tRNA species, and ptRNA within the cells, as previously described in studies of conditional lethal rnpA mutants in other bacterial species (22, 23, 48). Accordingly, Northern blotting was used to measure the cellular titers of precursor and polycistronic tRNATyr species of S. aureus cells treated with various concentrations (0× to 2× the MIC for 30 min) of RNPA1000, RNPA2000, or other classes of antibiotics. As shown in Fig. 5A, the treatment of S. aureus UAMS-1 with increasing concentrations of RNPA2000 elicited a dose-dependent increase in higher-molecular-weight species expected of polycistronic tRNATyr and ptRNATyr in comparison to the untreated cells. Indeed, 5′-rapid amplification of cDNA ends (RACE) using an amplification primer complementary to the 3′ terminus of tRNATyr confirmed that RNPA2000-treated cells accumulate at least two polycistronic tRNATyr species (tRNAPhe,Thr,Tyr and tRNAAsn,Glu,Val,Tyr) that were not detected in the mock-treated cells, confirming that the agent inhibits cellular tRNA maturation (see Fig. S6 in the supplemental material). Moreover, the observed inhibition of ptRNATyr and/or polycistronic tRNATyr processing appeared to be RNPA2000 specific, as Northern blotting and reverse transcription-quantitative PCR (qRT-PCR) using primers directed to the polycistronic tRNAPhe,Thr,Tyr species demonstrated that treatment with either RNPA1000, a third RnpA inhibitor identified in our initial screening campaign, RNPA3000, and the antibiotics erythromycin, mupirocin, vancomycin, ciprofloxacin, daptomycin, or rifampin failed to cause an accumulation of polycistronic tRNATyr and ptRNATyr within S. aureus UAMS-1 (Fig. 5B and C). While the other agents tested failed to mimic the tRNA maturation inhibitory effects of RNPA2000, S. aureus expressing antisense rnpA transcripts also displayed the accumulation of polycistronic tRNAPhe,Thr,Tyr molecules during growth under RnpA-depleted conditions (Fig. 5C, inset). Taken together, these results suggest that the RnpA inhibitor RNPA2000, but not RNPA1000 or antibiotics targeting other cellular processes, inhibits the cellular RNase P-mediated precursor tRNA processing.
FIG 5.

RNPA2000 treatment leads to ptRNATyr and polycistronic tRNATyr accumulation within S. aureus cells. (A) Northern blotting results for S. aureus tRNATyr following treatment with increasing concentrations of RNPA2000 (0×, 0.5×, 1×, and 2× the MIC). (B) Northern blotting results probing for tRNATyr species within S. aureus treated with the putative RnpA inhibitors RNPA1000, RNPA2000, and RNPA3000, as well as antibiotics affecting other cellular targets. (C) qRT-PCR-based quantification of polycistronic tRNAPhe, Thr, Tyr levels within S. aureus cells treated with 2× the MIC of RNPA1000, RNPA2000, RNPA3000, or the indicated RnpA-independent antibiotic. Levels of tRNAPhe, Thr, Tyr within RnpA depletion cells (pML100:: A.S.-rnpA; rnpA mRNA-directed antisense molecule) cultured for the indicated time under low-induction conditions (5 ng ml−1 ATc, inset) relative to vector-containing cells; RNPA2000-treated cells are also shown (160-fold increase relative to mock-treated S. aureus cells).
RNPA2000 displays antimicrobial activity toward drug efflux-deficient Gram-negative pathogens.
As previously stated, neither RNPA1000 nor RNPA2000 displays antimicrobial activity toward Gram-negative bacteria. We initially predicted that the compounds would have no effect on these organisms for two reasons. First, RnpA is not believed to play a major role in RNA turnover in Gram-negative bacterial species. Rather, global RNA degradation is thought to be mediated by the RNase E subunit of the Gram-negative degradosome, which has no significant sequence similarity to S. aureus RnpA; in fact, RNPA1000 and RNPA2000 do not inhibit E. coli RNase E in vitro (data not shown). Thus, the S. aureus RNA degradation inhibitory properties of RNPA1000 and RNPA2000 would not be likely to affect RNA degradation in Gram-negative bacteria and, consequently, would not affect viability. Second, while RnpA is well established to modulate precursor tRNA processing in Gram-negative bacteria, S. aureus RnpA shares low amino acid sequence identity (∼33%) to Gram-negative RnpA proteins. Therefore, we anticipated that RnpA inhibitors, such as RNPA2000, which inhibit the ptRNA processing activity of S. aureus RnpA, would be unlikely to affect the tRNA maturation function of the protein in Gram-negative species.
Nonetheless, during the course of our studies, Turrini et al. demonstrated that S. aureus RnpA can functionally complement Gram-negative E. coli organisms lacking their own RnpA, presumably by restoring precursor tRNA processing activity to otherwise phenotypically RNase P-negative cells (49). This finding also raised the possibility that small-molecule inhibitors of S. aureus RnpA-mediated ptRNA processing, such as RNPA2000, also inhibit E. coli RnpA tRNA maturation function and prompted us to reconsider why RNPA2000 does not exhibit activity against Gram-negative pathogens. In that regard, we recognized that the compound may not reach its cellular target in Gram-negative organisms; either RNPA2000 does not permeate the cell membrane and/or the compound is actively effluxed from these organisms.
As an initial test of these possibilities, antimicrobial testing was repeated using an E. coli imp tolC strain that is predicted to exhibit increased cell wall permeability and reduced efflux properties. As expected, RNPA1000, which inhibits S. aureus RnpA-mediated RNA degradation but does not affect cellular RnpA-mediated ptRNA processing activity, did not display significant antimicrobial properties (MIC, >256 μg ml−1) toward E. coli imp tolC cells. Conversely, RNPA2000 displayed antimicrobial activity (MIC, 2 μg ml−1) toward the double mutant strain. Similarly, RNPA2000 antimicrobial testing in the presence of the drug efflux pump inhibitor phenylalanine-arginine β-naphthylamide (PAβN) improved the activity of the compound toward E. coli strain TOP10 cells (MIC, 2 μg ml−1), Pseudomonas aeruginosa (MIC, 4 μg ml−1), Klebsiella pneumoniae (MIC, 64 μg ml−1), and Acinetobacter baumannii (MIC, 128 μg ml−1). Taken together, these results provide an initial indication that small-molecule inhibitors of S. aureus RnpA-mediated tRNA processing also inhibit Gram-negative RnpA tRNA processing activity, resulting in growth inhibition. Moreover, the efflux of RNPA2000, and not necessarily the evolutionary divergence of RnpA, is responsible for Gram-negative RNPA2000 resistance.
Northern blotting was used to determine whether the antimicrobial activity of RNPA2000 toward efflux-deficient E. coli cells correlates with the inhibition of cellular tRNA processing. RNPA2000-treated E. coli imp tolC cells (2× the MIC; 4 μg ml−1) were found to accumulate ptRNATyr and polycistronic tRNATyr species (Fig. 6A) but did not display aberrant cellular mRNA turnover (data not shown). Likewise, RNPA2000 treatment also led to the accumulation of polycistronic tRNAValV,ValW (Fig. 6B), which Mohanty and Kushner (48) previously established to being accumulating within E. coli RNase P-conditional lethal mutants under nonpermissive growth conditions. As expected, RNPA2000 treatment did not affect the mRNA degradation properties of cellular mRNA species within E. coli imp tolC cells, as measured by qRT-PCR of multiple transcripts (data not shown). Taken together, these results indicate that small-molecule inhibitors of S. aureus RnpA-associated precursor tRNA processing also affect the function of the protein in E. coli and, presumably, other Gram-negative species.
FIG 6.
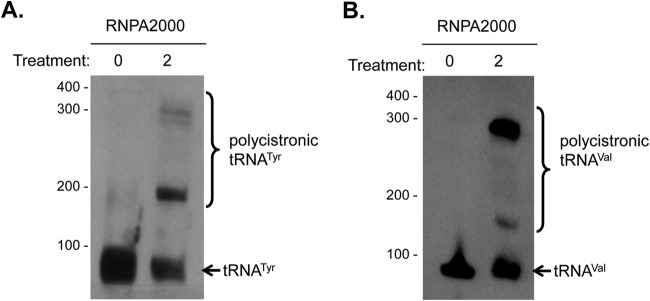
RNPA2000 inhibits tRNATyr and tRNAVal processing in efflux-deficient E. coli cells. Northern blotting probing for the E. coli tRNATyr (A) or tRNAVal (B) species following mock (0) or treatment with 2× the MIC RNPA2000.
DISCUSSION
As bacterial pathogens, such as S. aureus, continue to develop resistance to currently available antibiotics, new agents must be developed for the therapeutic intervention against infections by these organisms. In that regard, medicinal chemistry-based improvement of current classes of antimicrobials has provided next-generation antibiotics that have afforded a temporary reprieve. However, there is a ceiling to the amount of structural flexibility within an antimicrobial pharmacophore while retaining/improving antimicrobial efficacy. Moreover, enzymatic resistance determinants, such as β-lactamases, are already circulating and will likely rapidly evolve to inactivate the next generation of existing classes of antimicrobials. For these reasons, much effort has been devoted to the identification and validation of new antimicrobial targets, based on the premise that corresponding small-molecule inhibitors of these targets will provide structurally novel first-in-class antibiotics.
Toward that goal, we propose that S. aureus RnpA is a promising new antimicrobial target. Indeed, previous work revealed that RNPA1000, a small-molecule inhibitor of the RNA-degrading activity of the enzyme, is a structurally novel antimicrobial compound that displays the requisite cellular and physiochemical features worthwhile of consideration for further development (28, 29). In this study, we built upon earlier studies by expanding our assessment of the cellular mechanism of action of RNPA1000 along with another RnpA-mediated RNA degradation inhibitor, RNPA2000. In doing so, we show that S. aureus RnpA is likely to be a unique antimicrobial target, in the sense that it is a component of two holoenzyme complexes, each of which mediates a required cellular process. As a member of the RNA degradosome, RnpA catalyzes RNA digestion, presumably by providing substrate molecules necessary for the synthesis of RNA (19, 28). As a component of RNase P, RnpA catalyzes tRNA maturation and, consequently, is required for protein synthesis (22, 23). Thus, inhibitors of RnpA may serve as dual-threat antimicrobials, acting as multitarget ligands that eliminate the ability of the organism to undergo RNA decay and tRNA maturation.
As recently discussed by Silver (50), the majority (if not all) of successful monotherapeutic antimicrobials are multitarget ligands that either affect targets that are encoded by multiple genes (such as rRNAs) or simultaneously inhibit multiple essential cellular targets (such as type II topoisomerases) (50). A potential therapeutic benefit of the latter is that affecting multiple targets might impose an antimicrobial advantage compared to agents that affect a single process, due to the combined impact of simultaneously crippling two or more biological processes. A second feature that is believed to account for the therapeutic success of multitarget ligands is that endogenous high-level target-based resistance is slow to develop, requiring the stepwise accumulation of chromosomal mutations within each of the targets of the ligand, as opposed to a single-step mutation of a single target (reviewed in references 50 and 51). Indeed, high-level S. aureus fluoroquinolone resistance occurs by the stepwise acquisition of chromosomal mutations in each of the targets of the agent, the DNA gyrase GyrA subunit and the GrlA subunit of topoisomerase IV (52).
Our data support the hypothesis that S. aureus RnpA represents a unique and promising antimicrobial target, in part because small-molecule inhibitors of both the RNA-degrading and tRNA processing activities of the enzyme may phenotypically act as multitarget ligands. The phenotypic consequences of affecting both of the cellular roles of the enzyme may impose greater antimicrobial effects than do agents that inhibit a single RnpA function. Further, we anticipate that RnpA mutations that interfere with compound binding may be tolerated by one holoenzyme (i.e., RNA degradosome) but inactivate the second holoenzyme (i.e., RNase P); consequently, resistance to dual-function RnpA inhibitors may be slow to develop. The early comparison of the antimicrobial properties and mechanisms of action of the RnpA inhibitors RNPA1000 and RNPA2000 presented here support those predictions. More specifically, we found that RNPA1000, which predominantly inhibits the cellular RNA-degrading properties of the enzyme, selects for unstable mutants at a spontaneous resistance frequency of 3.7 × 10−13 and displays bacteriostatic activity against S. aureus (28). Conversely, RNPA2000, which inhibits both RnpA-mediated cellular RNA turnover and tRNA processing activities, displays no detectable spontaneous resistance frequency in S. aureus (<1 × 10−13) and is bactericidal. Admittedly, at present, we cannot rule out the possibility that the bactericidal nature of RNPA2000 is due directly to the ability of the agent to inhibit RnpA-mediated precursor tRNA processing as opposed to its combined effects on both cellular functions of the enzyme. However, the inhibition of precursor tRNA processing seems an unlikely mechanism given that RNPA2000 inhibits E. coli tRNA processing within efflux-deficient cells but is bacteriostatic toward that organism (data not shown). Thus, in the case of the S. aureus RnpA inhibitors investigated here, there does indeed seem to be an antimicrobial benefit associated with agents that inhibit both cellular processes of the enzyme in terms of reduced resistance frequency and bactericidal activity. From these perspectives, our data support the idea that developing agents that target multiple cellular processes may be a preferred design approach for future antimicrobial development campaigns.
Many investigators have discussed the importance of incorporating cell-based mechanism-of-action (MOA) studies into antimicrobial development paradigms as an early step to verify that the compounds of interest behave as expected in the context of the complexity of the cellular environment. Our data highlight the importance of such MOA studies. Indeed, while RNPA1000 was found to inhibit both RnpA-associated mRNA-degrading and RNase P precursor tRNA processing activities in vitro, the compound affected mRNA degradation only within bacterial cells. There are a multitude of potential reasons for this disconnect. For instance, our data establish that like other characterized RNase P complexes, S. aureus RnpA is required for RNase P activity at Mg concentrations expected within bacterial cells. Under these conditions, the protein is thought to facilitate transition state interactions between rnpB and substrate precursor tRNA molecules (53). It is possible (even likely) that within the cell, the binding affinities differ from those in our in vitro system, such that they favor the formation of active substrate-holoenzyme complexes over RNPA1000 binding. Alternatively, RNase P may interact with previously unappreciated factors within the cell in a manner that limits the binding and/or inhibitory properties of RNAP1000. Regardless, the compound may serve as an excellent chemical genetics probe for future studies designed to distinguish the cellular consequences of RnpA-mediated RNA degradation inhibition and provides a cautionary tale regarding the importance of cellular MOA studies.
While investigating the promise of RnpA as an antimicrobial target, we also considered that inhibitors of the tRNA maturation function of the protein may enhance the activities of currently available antibiotics that interfere with (an)other step(s) in the tRNA processing pathway. In direct support of that prediction, RNPA2000 was found to act synergistically with mupirocin, an antibiotic that inhibits isoleucyl-tRNA synthetase activity and has been used topically for decolonization and infection control purposes. Nonetheless, the effectiveness of mupirocin-based S. aureus decolonization has been called into question due, in part, to the emergence of low-level (and high-level) mupirocin-resistant isolates among patient populations (reviewed in reference 54). The combination of RnpA inhibitors together with mupirocin may be an avenue for future combination antimicrobial therapy that would ostensibly overcome mupirocin resistance issues.
The findings that the S. aureus RnpA inhibitor RNPA2000 inhibits tRNA processing and demonstrates antimicrobial activity toward efflux-deficient E. coli suggest that the enzyme can be exploited as a target for broad-spectrum antibiotics. Conceptually, RNPA2000 could be modified to overcome the efflux potential of Gram-negative species or be coadministered with efflux pump inhibitors (EPIs); however, EPI development and medicinal chemistry-based campaigns designed to overcome efflux properties of other antimicrobials have historically produced disappointing results. From these perspectives, it seems unlikely that the RNPA2000 scaffold will be easily exploited for the therapeutic intervention of Gram-negative pathogens. Rather, arguably, the more important observation from these studies is the finding that S. aureus RnpA inhibitors block the activities of evolutionarily diverse bacterial RnpA proteins, a feature that may be implemented in future screening campaigns that allow for the generation of broad-spectrum RnpA inhibitors.
Taken together, the results of our studies indicate that S. aureus RnpA can be considered a unique multifunctional target and that inhibitors of both the RNA-degrading and tRNA processing activities of the enzyme can be identified. Such inhibitors, as exemplified by RNPA2000, may exhibit improved therapeutic promise compared to agents targeting a single enzyme/function, by virtue of their propensity to simultaneously inhibit two cellular processes and a decreased likelihood of rapidly developing high-level endogenous target-based resistance in a single step. Moreover, RnpA inhibitors also offer the opportunity to engineer combination therapeutics that display synergistic antimicrobial properties when administered in conjunction with agents targeting other enzymes within the tRNA processing pathway, such as mupirocin. While the antimicrobial properties of RNPA2000 are relatively mild, the scaffold displays physicochemical properties (LogP = 2.14; aqueous solubility, >200 μM; cytochrome 450 IC50, >10 μM; <10% human ether-a-go-go-related gene [hERG] inhibition at 10 μM) that supports advancing the compound for scaffold refinement and optimization of an entirely novel class of new antimicrobial agents using medicinal chemistry-based approaches.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by the National Institutes of Allergy and Infectious Diseases awards AI103507 and AI073780, as well as funds from the University of Rochester Pilot Drug Discovery program and Annalise's Friends Foundation (to P.M.D.). T.M.E. was partially supported by a University of Nebraska Medical Center student assistantship.
We thank K. Korzekwa for providing physicochemical technical assistance.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.04352-14.
REFERENCES
- 1.Bayer AS, Schneider T, Sahl HG. 2013. Mechanisms of daptomycin resistance in Staphylococcus aureus: role of the cell membrane and cell wall. Ann N Y Acad Sci 1277:139–158. doi: 10.1111/j.1749-6632.2012.06819.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chang S, Sievert DM, Hageman JC, Boulton ML, Tenover FC, Downes FP, Shah S, Rudrik JT, Pupp GR, Brown WJ, Cardo D, Fridkin SK, Vancomycin-Resistant Staphylococcus aureus Investigative Team . 2003. Infection with vancomycin-resistant Staphylococcus aureus containing the vanA resistance gene. N Engl J Med 348:1342–1347. doi: 10.1056/NEJMoa025025. [DOI] [PubMed] [Google Scholar]
- 3.Dabul AN, Camargo IL. 2014. Molecular characterization of methicillin-resistant Staphylococcus aureus resistant to tigecycline and daptomycin isolated in a hospital in Brazil. Epidemiol Infect 142:479–483. doi: 10.1017/S0950268813001325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dantes R, Mu Y, Belflower R, Aragon D, Dumyati G, Harrison LH, Lessa FC, Lynfield R, Nadle J, Petit S, Ray SM, Schaffner W, Townes J, Fridkin S, Emerging Infections Program–Active Bacterial Core Surveillance MRSA Surveillance Investigators . 2013. National burden of invasive methicillin-resistant Staphylococcus aureus infections, United States, 2011. JAMA Int Med 173:1970–1978. doi: 10.1001/jamainternmed.2013.10423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gu B, Kelesidis T, Tsiodras S, Hindler J, Humphries RM. 2013. The emerging problem of linezolid-resistant Staphylococcus. J Antimicrob Chemother 68:4–11. doi: 10.1093/jac/dks354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carpousis AJ, Van Houwe G, Ehretsmann C, Krisch HM. 1994. Copurification of E. coli RNAase E and PNPase: evidence for a specific association between two enzymes important in RNA processing and degradation. Cell 76:889–900. doi: 10.1016/0092-8674(94)90363-8. [DOI] [PubMed] [Google Scholar]
- 7.Py B, Causton H, Mudd EA, Higgins CF. 1994. A protein complex mediating mRNA degradation in Escherichia coli. Mol Microbiol 14:717–729. doi: 10.1111/j.1365-2958.1994.tb01309.x. [DOI] [PubMed] [Google Scholar]
- 8.Py B, Higgins CF, Krisch HM, Carpousis AJ. 1996. A DEAD-box RNA helicase in the Escherichia coli RNA degradosome. Nature 381:169–172. doi: 10.1038/381169a0. [DOI] [PubMed] [Google Scholar]
- 9.Carpousis AJ. 2007. The RNA degradosome of Escherichia coli: an mRNA-degrading machine assembled on RNase E. Annu Rev Microbiol 61:71–87. doi: 10.1146/annurev.micro.61.080706.093440. [DOI] [PubMed] [Google Scholar]
- 10.Feng Y, Vickers TA, Cohen SN. 2002. The catalytic domain of RNase E shows inherent 3′ to 5′ directionality in cleavage site selection. Proc Natl Acad Sci U S A 99:14746–14751. doi: 10.1073/pnas.202590899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mackie GA. 1998. Ribonuclease E is a 5′-end-dependent endonuclease. Nature 395:720–723. doi: 10.1038/27246. [DOI] [PubMed] [Google Scholar]
- 12.Li Z, Deutscher MP. 2002. RNase E plays an essential role in the maturation of Escherichia coli tRNA precursors. RNA 8:97–109. doi: 10.1017/S1355838202014929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Z, Pandit S, Deutscher MP. 1999. RNase G (CafA protein) and RNase E are both required for the 5′ maturation of 16S ribosomal RNA. EMBO J 18:2878–2885. doi: 10.1093/emboj/18.10.2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin-Chao S, Wei CL, Lin YT. 1999. RNase E is required for the maturation of ssrA RNA and normal ssrA RNA peptide-tagging activity. Proc Natl Acad Sci U S A 96:12406–12411. doi: 10.1073/pnas.96.22.12406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mackie GA. 2013. RNase E: at the interface of bacterial RNA processing and decay. Nat Rev Microbiol 11:45–57. doi: 10.1038/nrmicro2930. [DOI] [PubMed] [Google Scholar]
- 16.McCullen CA, Benhammou JN, Majdalani N, Gottesman S. 2010. Mechanism of positive regulation by DsrA and RprA small noncoding RNAs: pairing increases translation and protects rpoS mRNA from degradation. J Bacteriol 192:5559–5571. doi: 10.1128/JB.00464-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Söderbom F, Svard SG, Kirsebom LA. 2005. RNase E cleavage in the 5′ leader of a tRNA precursor. J Mol Biol 352:22–27. doi: 10.1016/j.jmb.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 18.Commichau FM, Rothe FM, Herzberg C, Wagner E, Hellwig D, Lehnik-Habrink M, Hammer E, Volker U, Stulke J. 2009. Novel activities of glycolytic enzymes in Bacillus subtilis: interactions with essential proteins involved in mRNA processing. Mol Cell Proteomics 8:1350–1360. doi: 10.1074/mcp.M800546-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roux CM, DeMuth JP, Dunman PM. 2011. Characterization of components of the Staphylococcus aureus mRNA degradosome holoenzyme-like complex. J Bacteriol 193:5520–5526. doi: 10.1128/JB.05485-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chaudhuri RR, Allen AG, Owen PJ, Shalom G, Stone K, Harrison M, Burgis TA, Lockyer M, Garcia-Lara J, Foster SJ, Pleasance SJ, Peters SE, Maskell DJ, Charles IG. 2009. Comprehensive identification of essential Staphylococcus aureus genes using transposon-mediated differential hybridisation (TMDH). BMC Genomics 10:291. doi: 10.1186/1471-2164-10-291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eidem TM, Roux CM, Dunman PM. 2012. RNA decay: a novel therapeutic target in bacteria. Wiley Interdiscip Rev RNA 3:443–454. doi: 10.1002/wrna.1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spitzfaden C, Nicholson N, Jones JJ, Guth S, Lehr R, Prescott CD, Hegg LA, Eggleston DS. 2000. The structure of ribonuclease P protein from Staphylococcus aureus reveals a unique binding site for single-stranded RNA. J Mol Biol 295:105–115. doi: 10.1006/jmbi.1999.3341. [DOI] [PubMed] [Google Scholar]
- 23.Kazantsev AV, Pace NR. 2006. Bacterial RNase P: a new view of an ancient enzyme. Nat Rev Microbiol 4:729–740. doi: 10.1038/nrmicro1491. [DOI] [PubMed] [Google Scholar]
- 24.Buck AH, Dalby AB, Poole AW, Kazantsev AV, Pace NR. 2005. Protein activation of a ribozyme: the role of bacterial RNase P protein. EMBO J 24:3360–3368. doi: 10.1038/sj.emboj.7600805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guerrier-Takada C, Altman S. 1984. Catalytic activity of an RNA molecule prepared by transcription in vitro. Science 223:285–286. doi: 10.1126/science.6199841. [DOI] [PubMed] [Google Scholar]
- 26.Guerrier-Takada C, Gardiner K, Marsh T, Pace N, Altman S. 1983. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell 35:849–857. doi: 10.1016/0092-8674(83)90117-4. [DOI] [PubMed] [Google Scholar]
- 27.Niranjanakumari S, Kurz JC, Fierke CA. 1998. Expression, purification and characterization of the recombinant ribonuclease P protein component from Bacillus subtilis. Nucleic Acids Res 26:3090–3096. doi: 10.1093/nar/26.13.3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Olson PD, Kuechenmeister LJ, Anderson KL, Daily S, Beenken KE, Roux CM, Reniere ML, Lewis TL, Weiss WJ, Pulse M, Nguyen P, Simecka JW, Morrison JM, Sayood K, Asojo OA, Smeltzer MS, Skaar EP, Dunman PM. 2011. Small molecule inhibitors of Staphylococcus aureus RnpA alter cellular mRNA turnover, exhibit antimicrobial activity, and attenuate pathogenesis. PLoS Pathog 7:e1001287. doi: 10.1371/journal.ppat.1001287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eidem TM, Coughlan A, Towler MR, Dunman PM, Wren AW. 2013. Drug-eluting cements for hard tissue repair: a comparative study using vancomycin and RNPA1000 to inhibit growth of Staphylococcus aureus. J Biomater Appl 28:1235–1246. doi: 10.1177/0885328213503388. [DOI] [PubMed] [Google Scholar]
- 30.Clinical and Laboratory Standards Institute. 2013. Performance standards for antimicrobial susceptibility testing; 23rd informational supplement. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 31.Pearson RD, Steigbigel RT, Davis HT, Chapman SW. 1980. Method of reliable determination of minimal lethal antibiotic concentrations. Antimicrob Agents Chemother 18:699–708. doi: 10.1128/AAC.18.5.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taylor PC, Schoenknecht FD, Sherris JC, Linner EC. 1983. Determination of minimum bactericidal concentrations of oxacillin for Staphylococcus aureus: influence and significance of technical factors. Antimicrob Agents Chemother 23:142–150. doi: 10.1128/AAC.23.1.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Odds FC. 2003. Synergy, antagonism, and what the chequerboard puts between them. J Antimicrob Chemother 52:1. doi: 10.1093/jac/dkg301. [DOI] [PubMed] [Google Scholar]
- 34.Anderson KL, Roberts C, Disz T, Vonstein V, Hwang K, Overbeek R, Olson PD, Projan SJ, Dunman PM. 2006. Characterization of the Staphylococcus aureus heat shock, cold shock, stringent, and SOS responses and their effects on log-phase mRNA turnover. J Bacteriol 188:6739–6756. doi: 10.1128/JB.00609-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anderson KL, Roux CM, Olson MW, Luong TT, Lee CY, Olson R, Dunman PM. 2010. Characterizing the effects of inorganic acid and alkaline shock on the Staphylococcus aureus transcriptome and messenger RNA turnover. FEMS Immunol Med Microbiol 60:208–250. doi: 10.1111/j.1574-695X.2010.00736.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roberts C, Anderson KL, Murphy E, Projan SJ, Mounts W, Hurlburt B, Smeltzer M, Overbeek R, Disz T, Dunman PM. 2006. Characterizing the effect of the Staphylococcus aureus virulence factor regulator, SarA, on log-phase mRNA half-lives. J Bacteriol 188:2593–2603. doi: 10.1128/JB.188.7.2593-2603.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beenken KE, Dunman PM, McAleese F, Macapagal D, Murphy E, Projan SJ, Blevins JS, Smeltzer MS. 2004. Global gene expression in Staphylococcus aureus biofilms. J Bacteriol 186:4665–4684. doi: 10.1128/JB.186.14.4665-4684.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lei MG, Cue D, Roux CM, Dunman PM, Lee CY. 2011. Rsp inhibits attachment and biofilm formation by repressing fnbA in Staphylococcus aureus MW2. J Bacteriol 193:5231–5241. doi: 10.1128/JB.05454-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wegscheid B, Hartmann RK. 2007. In vivo and in vitro investigation of bacterial type B RNase P interaction with tRNA 3′-CCA. Nucleic Acids Res 35:2060–2073. doi: 10.1093/nar/gkm005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ji Y, Woodnutt G, Rosenberg M, Burnham MK. 2002. Identification of essential genes in Staphylococcus aureus using inducible antisense RNA. Methods Enzymol 358:123–128. doi: 10.1016/S0076-6879(02)58084-8. [DOI] [PubMed] [Google Scholar]
- 41.Donald RG, Skwish S, Forsyth RA, Anderson JW, Zhong T, Burns C, Lee S, Meng X, LoCastro L, Jarantow LW, Martin J, Lee SH, Taylor I, Robbins D, Malone C, Wang L, Zamudio CS, Youngman PJ, Phillips JW. 2009. A Staphylococcus aureus fitness test platform for mechanism-based profiling of antibacterial compounds. Chem Biol 16:826–836. doi: 10.1016/j.chembiol.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 42.Lin PH, Singh D, Bernstein JA, Lin-Chao S. 2008. Genomic analysis of mRNA decay in E. coli with DNA microarrays. Methods Enzymol 447:47–64. doi: 10.1016/S0076-6879(08)02203-9. [DOI] [PubMed] [Google Scholar]
- 43.Morrison JM, Anderson KL, Beenken KE, Smeltzer MS, Dunman PM. 2012. The staphylococcal accessory regulator, SarA, is an RNA-binding protein that modulates the mRNA turnover properties of late-exponential and stationary phase Staphylococcus aureus cells. Front Cell Infect Microbiol 2:26. doi: 10.3389/fcimb.2012.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Potter VR, Simonson H. 1951. Sequential blocking of metabolic pathways in vivo. Proc Soc Exp Biol Med 76:41–46. doi: 10.3181/00379727-76-18383. [DOI] [PubMed] [Google Scholar]
- 45.Bushby SR. 1975. Synergy of trimethoprim-sulfamethoxazole. Can Med Assoc J 112:63–66. [PMC free article] [PubMed] [Google Scholar]
- 46.Hughes J, Mellows G. 1980. Interaction of pseudomonic acid A with Escherichia coli B isoleucyl-tRNA synthetase. Biochem J 191:209–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.American Society for Microbiology. 2005. Instructions to authors. Antimicrob Agents Chemother 49:1–20. doi: 10.1128/aac.49.1.1-20.2005. [DOI] [Google Scholar]
- 48.Mohanty BK, Kushner SR. 2007. Ribonuclease P processes polycistronic tRNA transcripts in Escherichia coli independent of ribonuclease E. Nucleic Acids Res 35:7614–7625. doi: 10.1093/nar/gkm917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Turrini PC, Loveland JL, Dorit RL. 2012. By any other name: heterologous replacement of the Escherichia coli RNase P protein subunit has in vivo fitness consequences. PLoS One 7:e32456. doi: 10.1371/journal.pone.0032456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Silver LL. 2011. Challenges of antibacterial discovery. Clin Microbiol Rev 24:71–109. doi: 10.1128/CMR.00030-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.East SP, Silver LL. 2013. Multitarget ligands in antibacterial research: progress and opportunities. Expert Opin Drug Discov 8:143–156. doi: 10.1517/17460441.2013.743991. [DOI] [PubMed] [Google Scholar]
- 52.Ferrero L, Cameron B, Crouzet J. 1995. Analysis of gyrA and grlA mutations in stepwise-selected ciprofloxacin-resistant mutants of Staphylococcus aureus. Antimicrob Agents Chemother 39:1554–1558. doi: 10.1128/AAC.39.7.1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kurz JC, Fierke CA. 2002. The affinity of magnesium binding sites in the Bacillus subtilis RNase P·pre-tRNA complex is enhanced by the protein subunit. Biochemistry 41:9545–9558. doi: 10.1021/bi025553w. [DOI] [PubMed] [Google Scholar]
- 54.Abad CL, Pulia MS, Safdar N. 2013. Does the nose know? An update on MRSA decolonization strategies. Curr Infect Dis Rep 15:455–464. doi: 10.1007/s11908-013-0364-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gillaspy AF, Hickmon SG, Skinner RA, Thomas JR, Nelson CL, Smeltzer MS. 1995. Role of the accessory gene regulator (agr) in pathogenesis of staphylococcal osteomyelitis. Infect Immun 63:3373–3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Duthie ES, Lorenz LL. 1952. Staphylococcal coagulase; mode of action and antigenicity. J Gen Microbiol 6:95–107. doi: 10.1099/00221287-6-1-2-95. [DOI] [PubMed] [Google Scholar]
- 57.Begier EM, Frenette K, Barrett NL, Mshar P, Petit S, Boxrud DJ, Watkins-Colwell K, Wheeler S, Cebelinski EA, Glennen A, Nguyen D, Hadler JL, Connecticut Bioterrorism Field Epidemiology Response Team . 2004. A high-morbidity outbreak of methicillin-resistant Staphylococcus aureus among players on a college football team, facilitated by cosmetic body shaving and turf burns. Clin Infect Dis 39:1446–1453. doi: 10.1086/425313. [DOI] [PubMed] [Google Scholar]
- 58.Hiramatsu K, Hanaki H, Ino T, Yabuta K, Oguri T, Tenover FC. 1997. Methicillin-resistant Staphylococcus aureus clinical strain with reduced vancomycin susceptibility. J Antimicrob Chemother 40:135–136. doi: 10.1093/jac/40.1.135. [DOI] [PubMed] [Google Scholar]
- 59.Jacobs AC, Didone L, Jobson J, Sofia MK, Krysan D, Dunman PM. 2013. Adenylate kinase release as a high-throughput-screening-compatible reporter of bacterial lysis for identification of antibacterial agents. Antimicrob Agents Chemother 57:26–36. doi: 10.1128/AAC.01640-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jacobs AC, Hood I, Boyd KL, Olson PD, Morrison JM, Carson S, Sayood K, Iwen PC, Skaar EP, Dunman PM. 2010. Inactivation of phospholipase D diminishes Acinetobacter baumannii pathogenesis. Infect Immun 78:1952–1962. doi: 10.1128/IAI.00889-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Holloway BW, Morgan AF. 1986. Genome organization in Pseudomonas. Annu Rev Microbiol 40:79–105. doi: 10.1146/annurev.mi.40.100186.000455. [DOI] [PubMed] [Google Scholar]
- 62.Russo TA, Shon AS, Beanan JM, Olson R, MacDonald U, Pomakov AO, Visitacion MP. 2011. Hypervirulent K. pneumoniae secretes more and more active iron-acquisition molecules than “classical” K. pneumoniae thereby enhancing its virulence. PLoS One 6:e26734. doi: 10.1371/journal.pone.0026734. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.