

Abstract
The derivation of germ cells from human embryonic stem cells (hESCs) or human induced pluripotent stem (hIPS) cells represents a desirable experimental model and potential strategy for treating infertility. In the current study we developed a triple biomarker assay for identifying and isolating human primordial germ cells (PGCs) by first evaluating human PGC formation during the first trimester in vivo. Next, we applied this technology to characterizing in vitro derived PGCs (iPGCs) from pluripotent cells. Our results show that co-differentiation of hESCs on human fetal gonadal stromal cells significantly improves the efficiency of generating iPGCs. Furthermore, the efficiency was comparable between various pluripotent cell lines regardless of origin from the inner cell mass of human blastocysts (hESCs), or reprogramming of human skin fibroblasts (hIPS). In order to better characterize the iPGCs we performed Real time PCR, microarray and bisulfite sequencing. Our results show that iPGCs at day 7 of differentiation are transcriptionally distinct from the somatic cells, expressing genes associated with pluripotency and germ cell development while repressing genes associated with somatic differentiation (specifically multiple HOX genes). Using bisulfite sequencing, we show that iPGCs initiate imprint erasure from differentially methylated imprinted regions by day 7 of differentiation. However, iPGCs derived from hIPS cells do not initiate imprint erasure as efficiently. In conclusion, our results indicate that triple positive iPGCs derived from pluripotent cells differentiated on hFGS cells correspond to committed first trimester germ cells (before 9 weeks) that have initiated the process of imprint erasure.
Keywords: ESC germ cell, fetus, gonad, pluripotent
Introduction
Germ cells are responsible for the fidelity of DNA inheritance from one generation to the next. Errors in germ cell differentiation, especially during fetal life can result in adverse future outcomes that include infertility, germ cell tumors, or birth defects in progeny. However, due to a lack of models for human fetal germ cell development, most of the underlying cell and molecular defects from which these problems originate are not well understood. The use of human embryonic stem cells (hESCs) for differentiation into the human germ line has been proposed as an important model for studying human fetal germ cell development 1. Furthermore, there is potential for using gametes differentiated from either hESCs or human induced pluripotent stem (hIPS) cells as a therapy for couples with infertility 2.
In mammals, specification of the germ line has been evaluated most extensively in the mouse. Using the mouse as a model, it is clear that the murine germ line is specified directly from the epiblast during implantation 3, 4. Specification requires non-cell autonomous signaling by bone morphogenetic proteins (BMPs) from the extraembryonic ectoderm and visceral endoderm to the proximal epiblast 5-10. These newly derived germ cells, called primordial germ cells (PGCs), cluster outside the embryo before migrating through the hindgut endoderm and into the genital ridges. In mice, colonization of the genital ridges begins at embryonic (E) day E10.0, whereas in humans, colonization begins between 4-6 weeks of gestation [for review see 4, 11]. In mice, germ cells establish the chromatin framework during migration in order to generate the foundation for epigenetic reprogramming such as complete erasure of CpG methylation at imprinted genes during gonadal colonization. Therefore isolating PGCs at these stages early stages will be important for future work in understanding human germ line reprogramming. 12-18.
Generation of germ cells from murine ESCs has been well described 19-26. In contrast, differentiation of germ cells from hESCs has been achieved, but is less extensively studied 27-32. Furthermore, the capacity for hIPS cells to generate human germ cells in vitro is currently unknown. The most robust marker for identifying the presence of germ cells in a population is expression of the evolutionarily conserved RNA helicase VASA33. Although a highly specific marker for germ cells, in the absence of additional markers, VASA alone is not informative for defining specific stages of germ line development. Recently CXCR4 and SSEA1 were reported as novel cell surface markers for identifying human in vitro derived PGCs (iPGCs) during hESC differentiation 31, 32. However, in these original studies, male hESC lines were not evaluated. Furthermore, a direct comparison between multiple independently derived hESC lines to evaluate the general utility of surface biomarkers for identifying iPGCs has not been performed in detail.
In the current study we evaluated cell surface markers that correlated with VASA expression during germ cell formation in the first trimester of human development in vivo. Next we used these surface markers to isolate putative iPGCs from hESC and hIPS cell lines after differentiation on primary human fetal gonadal stromal (hFGS) cells in vitro. Our results show that co-culture of hESCs under differentiating conditions on hFGS cells significantly improves the efficiency of differentiating iPGCs, and that these cells correspond to first trimester PGCs that have initiated the process of imprint erasure.
Materials and Methods
Human Fetal Samples
Human embryos were obtained following elected termination and pathological evaluation at the UCLA Tissue Procurement Laboratory. All consented material was anonymous and carried no personal identifiers. Developmental age was determined by recall of the last menstrual period and subtraction of two weeks to account for time following fertilization to implantation. All human embryos used for this project were male. Use of human embryos was undertaken only after approval from the UCLA Institutional Review Board Approval for the protection of human subjects.
Human embryonic stem cells and Human Induced Pluripotent Stem Cells
Information regarding the hESCs, HSF-1 (NIH code UC01, 46XY), HSF-6 (UC06, 46XX) and H9 (NIH code WA09, 46XX) can be obtained at http://stemcells.nih.gov/stemcells. Undifferentiated hESC colonies were maintained as previously described 27. For all experiments hESCs were used between passages 40-50. The human induced pluripotent stem (hIPS) cell line 2 (hIPS2, 46XY) and hIPS1 (46XY) was cultured as previously described 34, except for the inclusion of primocin (InvivoGen, San Diego, CA). Human IPS2 cells were evaluated between passages 37-46 where as hIPS1 was evaluated between passages 52-53. Pluripotency was evaluated by teratoma analysis using transplantation into the testicles of severe combined immunodeficient (SCID) mice. All cell lines generated robust teratomas in the testis with contribution to all three embryonic lineages (ectoderm, mesoderm and endoderm). All hESC and hIPS experiments were conducted with prior approval from the UCLA Embryonic Stem Cell Research Oversight (ESCRO) committee. Teratoma analysis in SCID mice was conducted following approval by the UCLA Animal Research Committee (ARC).
Human fetal gonad stromal cells (hFGS cells)
Gonadal ridges from a 10-week-old human fetus was mechanically dissociated and incubated in 0.25% trypsin and 0.5mM EDTA (Gibco BRL) at 37°C for 10min. After complete dissociation, cells were plated on tissue culture plates coated with gelatin and grown in DMEM (Gibco BRL) supplemented with 15% fetal bovine serum (FBS, Hyclone), 0.1mM nonessential amino acids (Gibco BRL), 0.1mM β-mercaptoethanol (Gibco BRL), 2mM L-glutamine (Gibco BRL), and 1mM sodium pyruvate (Gibco BRL). Culture was grown at 5% CO2 and passaged every 4-5days by trypsinization with 0.25% trypsin and 0.5mM EDTA (Gibco BRL) at 37°C for 5min.
Human Placenta and Liver stromal cells
First trimester fetal liver (9 gestational week) and placenta (3.5 gestational week) were used to generate non-gonadal stromal cell lines. The names of the two non-gonadal stromal cell lines are PL137 (placenta) and FL128 (Liver). Stromal lines were cultured in 20% FBS (Hyclone), alpha-MEM (Gibco BRL), 1% penicillin/stremptomycin (Gibco BRL)
For inactivation of stromal cells, 10μg/ml of mitomycin-C (Calbiochem) was added to plate of confluent feeder cells, human placental or human fetal liver stromal cells and incubated at 37°C for 3 hours. After mitomycin-C incubation, cells were washed three times with DPBS and recovered in fresh media for overnight at 37°C.
Immunofluorescence
Immunoflourescence was performed on 5mm paraffin or frozen sections of male fetal gonads from 5-9 weeks of gestation (n=4). Paraffin sections were deparaffinized prior to immersion in Tris-EDTA buffer (10mM Tris base/1mM EDTA/0.05% Tween20, pH9.0) at 95°C for 40 min for antigen retrieval. Blocking was performed in 5% donkey serum in PBS for 1 hour prior to incubation with primary antibodies (Supplemental Table 1). All primary antibodies were detected with fluorescent dye-conjugated secondary antibodies (Supplemental Table 1). TO-PRO 3 iodide (Invitrogen) was used as a marker for cell nucleus. MACS-sorted cKIT positive cells were centrifuged at 350rpm for 5 min (Thermo electron corporation) and then stained with cKIT and VASA antibody after blocking with 5% donkey serum in PBS for 1 hour.
RNA extraction and PCR
Total RNA was extracted using the RNeasy kit (Qiagen) according to manufacturer’s instructions. Total RNA was quantified, and 1μg was used for cDNA synthesis using random primers (Invitrogen) under standard conditions. RT-PCR amplifications were conducted for 40cycles of 95°C, 30sec; 60°C, 30sec; and 72°C, 30sec. Primer sequences are as follows: GAPDH F 5’-acc aca gtc cat gcc atc ac-3’, GAPDH R 5’-tcc acc acc ctg ttg ctg ta-3’, SRY F 5’-aca gta aag gca acg tcc ag-3’, SRY R 5’-atc tgc ggg aag caa act gc-3’, AMH F 5’-acc tgg agg aag tga cct gg-3’, AMH R 5’-tac tca gcc ggg agt cct ct-3’, CYP19 F 5’-cta aat tgc ccc ctc tga ggt-3’, and CYP19 R 5’-cca cac caa gag aaa aag gcc-3’.
Quantitative Real time PCR was performed in duplicate for each sample from three separate experiments using TaqMan® Universial PCR master mix (Applied Biosystems). TaqMan PCR amplifications were initiated at 95°C for 10min followed by cycles of 95°C, 15sec and 72°C, 90sec. All primers/probe mixtures were purchased from Applied Biosystems; Human GAPDH (HS99999905_m1), Human VASA (HS00251859_m1), PRDM1 (HS01068508_m1), DPPA3 (HS01931905_g1), DAZL (HS00154706_m1), HOXA2 (HS00534579_m1), HOXC5 (HS00232747_m1).
Germ cell differentiation from hESCs
Differentiation was performed on inactivated stromal cells plated at 2×105 cells per well in a 6 well plate on 0.1% gelatin (Sigma). Differentiation media included DMEM/F12 (Gibco BRL) supplemented with 20% FBS (Gibco BRL), 0.1mM nonessential amino acids (Gibco BRL), 0.1mM β-mercaptoethanol (Gibco BRL), 1mM L-glutamine (Gibco BRL). Media was changed every 2 days during differentiation.
Flow Cytometry
Cells were dissociated with 0.25% trypsin and 0.5mM EDTA (Gibco BRL) at 37°C for 5min and collected by centrifugation at 1000 rpm for 5 min. Cells were incubated in 500μl of 1% BSA in PBS containing primary antibodies on ice for 20 minutes. Cells were washed and incubated in FITC (SSEA1)- or Cy5 (cKIT and PLAP)-conjugated secondary antibodies on ice for another 20 min. For intracellular staining of VASA protein, cells were permeablized in 250μl Fixation/Permeablization solution (BD Bioscience) on ice for 20min and then washed in 1x BD Perm/Wash™ buffer (BD Bioscience). Cells were labeled with Goat anti-human VASA antibody (R&D systems) in 1x BD Perm/Wash™ buffer for 30min. PE-conjugated donkey anti-goat IgG (Jackson ImmunoResearch Laboratory) was used as a secondary. Analysis was performed using an LSR II (Becton Dickinson) and FlowJo software (Tree Star Inc.).
Bisulfite Sequencing
Genomic DNA was extracted by ZR Genomic DNA II Kit™ (Zymo Research) and then bisulfite converted using the EZ DNA Methylation Kit™ (Zymo Research) according to manufacturer’s instructions. Amplification of differentially methylated regions were performed in the presence of 5mM MgCl2, 0.2mM dATP, dCTP, dGTP and dTTP, 10pmole each primer, and 1unit Platinum Taq polymerase (Invtrogen). PCR amplifications for H19 and PEG1 were initiated at 94°C for 3min followed by 45cycles of 94°C, 30sec; 58°C, 30sec; and 72°C, 30sec. Primer sequences are as follows 35, hH19F 5’-tgt ata gta tat ggg tat ttt tgg agg ttt-3’, hH19R 5’- tcc tat aaa tat cct att ccc aaa taa cc-3’, hPEG1F 5’-tyg ttg ttg gtt agt ttt gta ygg tt -3’, hPEG1R 5’-aaa aat aac acc ccc tcc tca aat-3’. The primary PCR amplifications for SNRPN were initiated at 94°C for 3min followed by 35cycles of 94°C, 60sec; 51°C, 60sec; and 72°C, 60sec and then 2.5uL of the primary PCR were diluted into a fresh 50μl reaction and reamplified for a further 25 cycles. Primer sequences are as follows 35 hSNRPNF1 5’-ctc caa aac aaa aaa ctt taa aac cca aat tc-3’, hSNRPNR1 5’-ggt ttt ttt tta ttg taa tag tgt tgt ggg g-3’, hSNRPNF2 5’-caa tac tcc aaa tcc taa aaa ctt aaa ata tc-3’ and hSNRPNF2 5’-ggt ttt agg ggt tta gta gtt ttt ttt ttt ttg g-3’. Amplified PCR products were cloned into pGEM-T Easy vector (Promega) and 12 clones were sequences for each sample. The sequenced clones were aligned by DNAStar software.
Microarray
HSF-1 and HSF-6 were differentiated on hFGS cells and sorted by FACS for cKIT/SSEA1 double positive and double negative cells at day 7. The positive and negative samples were sorted into samples of equivalent cell numbers (70,000 cells per replicate) directly into the first buffer of the RNA extraction kit. Total RNA was extracted immediately after sorting using the RNeasy micro kit (Qiagen). Once all samples were collected from HSF-1 and HSF-6, one-round amplification was performed in parallel on all samples using the MessageAmpII (Ambion) according to manufacturer’s instructions. Microarrays were performed using the Affymetrix human chip, U133plus2.0 array and processed using the Affymetrix Mas5.0 software. Selection criteria for differential expression between the positive and negative population at day 7 of differentiation was calculated as greater than or equal to 2 fold, with an absolute difference greater than or equal to 100 with a statistical cut-off of p <0.05. Real time PCR was used to validate candidate differentially expressed genes as described above. Microarray data is deposited at ArrayExpress http://www.ebi.ac.uk/microarray-as/ae/
Statistics
T-tests were performed to evaluate significance between two groups. Significance was accepted at p<0.05.
Results
Defining surface biomarkers that are co-expressed with VASA in the human germ line
VASA is the most reliable marker for distinguishing human PGCs from hESCs because it is differentially expressed between undifferentiated and differentiated hESCs 27, 28. In the first experiment (Figure 1), our goal was to co-localize VASA protein with previously characterized PGC surface markers to identify a combination of markers that could be used to quantify and isolate PGCs by flow cytometry and by FACS. Many human first trimester cell surface markers have previously been described including cKIT (CD117), Placental Alkaline Phosphatase (PLAP), Stage specific embryonic antigen (SSEA) 136-38. Unlike mouse PGC formation which is relatively homogeneous, human PGC development is remarkably heterogeneous, and PGCs can be subdivided into various subpopulations based on marker expression 36, 39. Given this heterogeneity our aim was to evaluate which surface markers overlapped with VASA and whether particular surface markers could delineate subpopulations of VASA positive PGCs during the first trimester.
Figure 1. Surface markers that define male human primordial germ cell (PGC) migration and colonization in the first trimester of human fetal life.
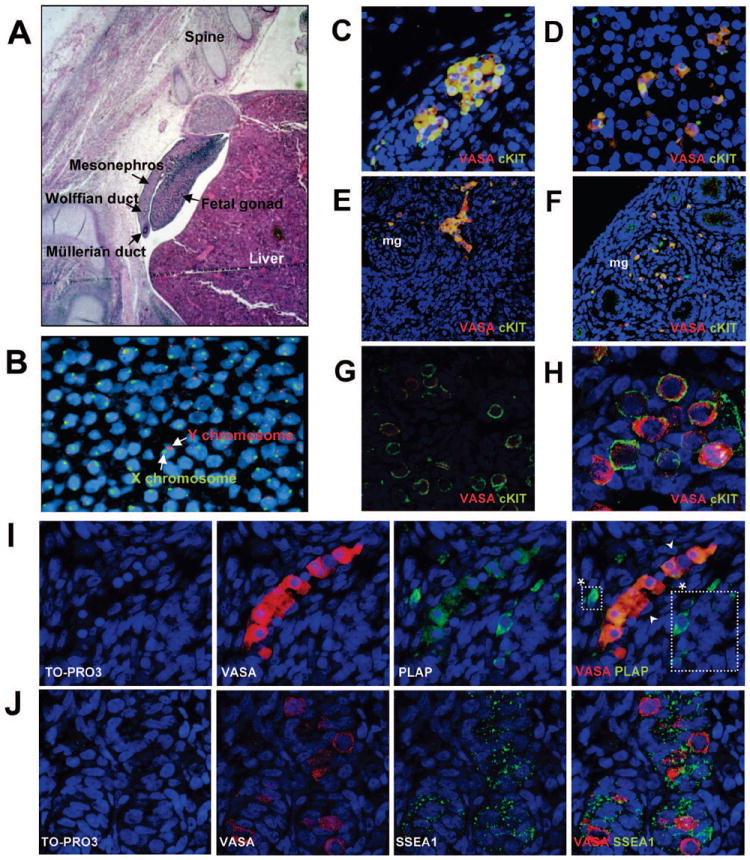
(A) Hematoxylin and Eosin staining of a 5 week human embryo. (B) Fluorescent in situ hybridization (FISH) with X chromosome-specific probe (green) and Y chromosome-specific probe (red) to determine embryo sex. (C-H) Immunofluorescent staining of overlay images showing the germ cell-specific marker VASA (red) and cKIT (green). Co-localization is seen as yellow. (C) VASA/cKIT co-expression (yellow) in PGC clusters exiting the hind gut (800x) (D) VASA/cKIT co-expression in PGCs migrating towards gonad through the mesonephros which contains mesonephric glomeruli (mg) (800x) (E) VASA/cKIT double positive PGCs entering into the gonad as a cluster from the mesonephros (400x) (F) VASA/cKIT double positive PGCs migrating through the mesonephric glomeruli (mg) during migration to the gonad (400x) (G,H) VASA/cKIT double positive PGCs in the fetal gonad (800x and 1600x) (I) Migrating VASA (red) positive PGCs with PLAP (green) (800x). (*) indicate PLAP positive VASA negative cells which were only observed during migration. (J) VASA (red) with SSEA1 (green) in colonized PGCs within the fetal gonad (1200x). TO-PRO 3 iodide was used as a marker for cell nucleus in all sections from B-J.
During the first 5 weeks of human development PGCs migrate into the fetal gonad. At this stage the gonad is intimately associated with a primitive kidney called the mesonephros (Figure 1A). FISH for the Y chromosome was used to determine that the sex of this embryo was male (Figure 1B). In the first trimester, we found that all migratory (Figure 1C-F) and early colonizing PGCs (Figure 1G-H) from 5-9 weeks of human gestation are double positive for VASA and cKIT. Next, we co-localized VASA with PLAP (Figure 1I). PLAP is not the same molecule as tissue non-specific alkaline phosphatase (TNAP), which is a characteristic cytoplasmic marker of undifferentiated ESCs that cannot be detected by surface staining using flow (Supplemental Figure 1A,B). PLAP is located on human chromosome 2q in a genomic cluster with tissue specific alkaline phosphatases including germ cell alkaline phosphatase and intestinal alkaline phosophatase 40. TNAP and PLAP share 50% homology and PLAP can be distinguished by antibody staining 41, 42. As a positive control for PLAP, we stained testicular cancer sections containing carcinoma in situ and seminoma which were positive (Supplemental Figure 1C) 41. In the human fetal samples, we found every migratory VASA positive cell was PLAP positive. However a few rare PLAP positive cells were VASA negative (Figure 1I, white boxes). At 9 weeks of gestation PLAP was not expressed in the fetal gonad (data not shown). This indicates that PLAP identifies a subset of less mature cKIT/VASA positive cells. The third cell surface marker, SSEA1 was similar to cKIT and was co-expressed with VASA during the first trimester of human life from 5-9 weeks (Figure 1J).
Given the unique speckled pattern of SSEA1 expression, we next co-localized SSEA1 with anti-mullerian hormone (AMH), which is a marker of sertoli cells (Figure 2A). We found that although the staining pattern of AMH and SSEA1 were very similar, the expression is mutually exclusive because we could identify red and green staining (not yellow) in the overlay of the confocal images. In order to confirm that SSEA1 was expressed on cKIT positive cells, we performed flow cytometry of human gonads at 7 weeks of gestation (Figure 2B) and determined that SSEA1 positive cells co-expressed cKIT.
Figure 2. SSEA1 co-localizes with cKIT positive cells in the first trimester fetal gonad.

(A) Human fetal testis at 9 weeks of gestation AMH (red) SSEA1 (green), TO-PRO 3 iodide stains the cell nucleus (1200x). Arrow indicates exclusive red and arrowhead indicates exclusive green cells. (B) Flow analysis of a 7-gestational week-old male gonad. Shown are negative controls (red lines) cKIT and SSEA1, (blue lines).
In vitro derivation of human PGCs (iPGCs) using cell surface biomarkers
In order to evaluate whether a human cell-based supportive niche would improve the derivation of iPGCs differentiated from hESCs we first generated a supporting cell layer from human fetal gonads (Figure 3). We have called this gonadal cell layer human fetal gonadal stromal cells (hFGS) (Figure 3A). Once the hFGS cell line was established (passage 2-5) we performed alkaline phosphatase staining (AP) to identify germ cells or embryonic germ cells. We found no AP staining in the hFGS cell line compared to hESCs. In order to characterize the hFGS cells we performed RT-PCR for makers of sertoli cells (SRY, AMH) and leydig cells (CYP19) as well as germ cells (VASA). The hFGS cells expressed high levels of CYP19 mRNA, indicating a major contribution of cells with a leydig cell identity. However, evidence of sertoli cells could be identified by low expression of AMH (fetal) and SRY (fetal and adult sertoli cells). VASA mRNA was not detected in the primary hFGS cell cultures. Next we performed flow cytometry on hFGS cells using TRA-1-85 (pan-human marker) and the germ cell surface biomarkers SSEA1, cKIT and PLAP (Figure 3C). These surface markers were not expressed on the hFGS cell line.
Figure 3. Isolation and characterization of stromal cells from first trimester human fetal gonads and comparison of different feeder layers for iPGC differentiation.

(A) hFGS cells grow as a monolayer and are negative for Alkaline Phosphatase (AP), whereas hESCs are positive for AP. (B) RT-PCR comparing hFGS cells and adult testis. (C) Flow cytometry of hFGS cells for TRA-1-85 (pan-human surface marker), SSEA1, cKIT and PLAP. (D) Percentages of cKIT/SSEA1/VASA triple positive cells in undifferentiated (Undiff.) HSF-1 and HSF-6 hESCs, and following differentiation for three days on matrigel, hFGS cells or matrigel with hFGS cell-conditioned media. (E) Comparison of iPGC differentiation in triplicate on hFGS cells, human placental stromal cells and human liver stromal cells after 7-day differentiation. Significance was achieved when comparing a to b and c to d (P<0.05).
To determine whether differentiation of hESC in a co-culture system with hFGS cells improved the efficiency for generating cKIT/SSEA1/VASA triple positive cells, we compared three alternate differentiation strategies for three days using HSF-1 and HSF-6 hESC lines (Figure 3D). These are, 1) adherent differentiation on matrigel, 2) co-culture with hFGS cells and 3) adherent differentiation on matrigel plus hFGS cell conditioned media. In undifferentiated hESCs, a small triple positive population corresponding to less than 1% of cells can be identified in both lines of hESCs. Following differentiation on matrigel, the proportion of SSEA1/KIT/VASA triple positive cells increases 1-2 fold on day 3 compared to day 0, but was still less than 1%. In contrast differentiation by co-culture on hFGS cells resulted in the generation of 2-4% triple positive cells by day 3, representing a significant improvement over matrigel for the generation of iPGCs (14-19 fold).
To evaluate whether this affect was due to factors released into the media, we differentiated hESCs for 3 days on matrigel in hFGS cell conditioned media (Figure 3D). We found that conditioned media returned the proportion of iPGCs to levels seen with matrigel, suggesting that cell-to-cell contact is important for this effect on the iPGCs. Finally, to determine whether non-gonadal sources of human fetal stromal cells could promote the differentiation of iPGCs from hESCs, we differentiated HSF-1 in triplicate for 7 days on hFGS cells, human placental stromal cells, or human liver stromal cells (Figure 3E). We found that all human stromal cells regardless of origin were capable of supporting iPGC differentiation from hESCs. However, differentiation on hFGS cells resulted in a significantly higher yield of iPGCs compared to the other stromal lines tested. Therefore, due to this significant increase in efficiency over the other strategies tested, all subsequent experiments were performed on hFGS cells.
To address the efficacy of using this triple biomarker strategy, we evaluated iPGC formation from three independently derived lines of hESC lines (HSF-1, HSF-6 and H9) on hFGS cells for 7 days and evaluated SSEA1/cKIT/VASA and PLAP/SSEA1/VASA triple positive expression by flow cytometry (shown are the flow plots of HSF-1). In undifferentiated hESCs, less than 1% of the population are positive for cKIT, SSEA1 PLAP or VASA (Figure 4A and B). However, after 7 days of differentiation, cKIT, SSEA1, PLAP and VASA positive cells can be identified. In the initial analysis, we first evaluated cKIT/SSEA1 double positive or PLAP/SSEA1 double positive and found that an equivalent proportion of cells were double positive for both surface biomarkers. To address whether the double positive cells had a germ cell identity, we performed a secondary analysis on the double positive population for VASA expression. Our results reveal that VASA protein is enriched in the double positive cells (blue peak) whereas the negative cells (red peak) were negative for VASA (Figure 4A). This type of flow analyses was repeated in triplicate for HSF-1, HSF-6 and H9. From this result it can be quantified that on day 7 of differentiation there is a significant increase in the proportion of triple positive cells from HSF-1, HSF-6 and H9 compared to the undifferentiated state. Furthermore, the proportion of triple positive iPGCs derived from each hESC line is very similar corresponding to 6-10% of the population. In order to determine whether iPGCs can be acquired at later stages, we performed flow cytometry at day 7, 14 and 21 days of differentiation in triplicate (Supplemental figure 2). This results shows that by day 14 of differentiation, 20% of cells are triple positive for the combinations of surface markers and the proportion of triple positive cells remains the same at day 21 of differentiation.
Figure 4. Derivation of iPGCs from HSF-1, HSF-6 and H9 lines of hESCs using hFGS cells consistently results in formation of triple positive cells at day 7 of differentiation.

(A) Flow cytometry of the HSF-1 line of hESCs. Red lines indicate negative controls (secondary antibody only) and blue lines indicate positive staining for each molecule as indicated above each plot. Following 7 days of differentiation, cKIT, SSEA1, PLAP and VASA positive cells could be identified above the negative control. Analysis of two channels reveals presence of double positive cKIT/SSEA1 and PLAP/SSEA1 cells. VASA expression was re-analyzed in the double positive (blue) and negative (red) populations after 7 days of differentiation. This demonstrates that VASA positive cells are only found within the cKIT/SSEA1 or PLAP/SSEA1 double positive population and not the netative. (B) Quantification of iPGC formation from HSF-1, HSF-6 and H9 lines of hESC lines at day 7 of differentiation. All experiments were performed in triplicate. Significance was achieved when comparing a to b and c to d (P<0.05). (C) Relative expression of VASA to GAPDH mRNA using Real-time PCR in double-positive (cKIT/SSEA1) cells sorted by FACS at day 7 of differentiation from HSF1 and HSF6 lines of hESCs (left panel). Middle panel shows a gel electrophoresis of the PCR products of VASA RT-PCR in the cKIT/SSEA1 double positive and double negative FACS sorted cells at day 7 of differentiation from HSF1 (middle). Immunofluorescent staining of HSF1-derived PGCs with cKIT (green) and VASA (red) after MACS-sorting and cytospin. VASA protein is expressed in the cytoplasm of cKIT-positive PGCs (1200x) (right panel).
To further characterize VASA expression in the iPGCs at day 7, we performed Real time PCR and immunofluorescence on sorted cells for VASA mRNA and protein (Figure 4C). Using FACS sorting for the double positive (SSEA1/cKIT) and negative population, we determined that VASA mRNA was expressed at equivalent levels in HSF-1 and HSF-6 positive cells relative to GAPDH. Furthermore, we can show that VASA mRNA is expressed in the double positive cells but was absent in the negative population. Finally, our results show that VASA is localized in the cytoplasm in the cKIT positive iPGCs (Figure 4C).
Given that that the double positive population of iPGCs from both HSF-1 and HSF-6 lines of hESCs was enriched in VASA we next performed microarray analysis comparing the HSF-1 and HSF-6 cKIT/SSEA1 double positive cells on day 7 of differentiation to the double negative cells (Figure 5A). We also performed Real time PCR to validate data observed in the microarray. We found that like VASA, the germ cell genes PRMD1, DPPA3 and DAZL are enriched in the iPGCs relative to the negative cells. Furthermore, we found that the iPGCs significantly repressed multiple HOX genes. Specifically, we found repression of the majority of genes transcribed from the HOXA cluster as well as repression of HOXB5, HOXC5, HOXC6, HOXD3, and HOXD4. Two of these genes, HOXA2 and HOXC5 were subsequently validated by Real time PCR as shown (Figure 5A).
Figure 5. Gene expression and CpG methylation status at imprinted genes in isolated iPGCs at day 7 of differentiation from HSF-1 and HSF-6.

(A) Heat map showing differentially expressed genes in HSF-1 and HSF-6 cKIT/SSEA1 double positive FACS sorted cells compared to the double negative cells. Real time PCR of germ cell-related genes (PRDM1, DPPA3, DAZL) and somatic genes (HOXA2, HOXC5) in double-positive (cKIT/SSEA1) and double-negative FACS sorted cells from HSF-1 at day 7 of differentiation. (B) CpGs of imprinted genes (H19, PEG1 and SNRPN DMRs) were analyzed following bisulfite sequencing from undifferentiated HSF-1 and HSF-6 hESCs and iPGCs derived at day 7 of differentiation. In vitro derived PGCs were isolated by FACS sorting for cKIT/SSEA1 double positive cells. Methylated CpGs are represented as filled circles, unmethylated CpGs are represented as open circles. Percentage methylation was calculated comparing the number of CpG methylated sites in undifferentiated hESCs before differentiation with the number of CpG methylated sites at the same locus in the iPGC population. Decreased methylation in iPGCs relative to undifferentiated hESCs is shown in green and no change shown in black. Abnormal methylation is shown in red.
Next we performed CpG methylation of imprinted genes in the HSF-1 and HSF-6 lines of hESCs (Figure 5B). First, we analyzed the undifferentiated hESC population. For HSF-6 we determined that H19, PEG1 and SNRPN imprinting control centers were differentially methylated whereas in HSF-1 only 2/3 loci exhibited differential methylation. The one abnormally methylated locus in HSF-1 was PEG1, which showed almost complete methylation with 95.7% of CpGs methylated in its imprinting control center. With regard to iPGCs derived from HSF-1 and HSF-6, we found that imprinting control centers that were differentially methylated in undifferentiated hESCs (H19 and SNRPN in HSF-1 and H19, PEG1 and SNRPN in HSF-6), initiated the process of imprint erasure by day 7 in the iPGCs (green). In contrast, the hypermethylated PEG1 locus remained fully methylated in iPGCs (97.7%, red) (Figure 5B).
Given that hESCs generated consistent numbers of iPGCs with differentiation, and these iPGCs demonstrated the capacity to initiate imprint erasure, we were next interested in evaluating hIPS cells because their origin is not from a human blastocyst, and therefore it is not clear whether these cells will have the capacity for germ cell formation with differentiation. For this analysis we used two of the recently reported human IPS cell lines (hIPS1 and hIPS2) 34. Similar to previous experiments, we analyzed iPGC formation by flow cytometry in triplicate (Figure 6A). We found that the proportion of triple positive iPGC cells (cKIT/SSEA1/VASA or PLAP/SSEA1/VASA) derived from hIPS1 and hIPS2 corresponded to 8-10% of the population on day 7. This was comparable to the proportion of iPGCs differentiated from independently derived lines of hESCs. In order to determine whether these iPGCs were capable of initiating imprint erasure similar to hESCs, we first evaluated CpG methylation in the starting fibroblast population and the reprogrammed hIPS2 line (Figure 6B). We found that during the process of reprogramming 2/3 loci remained differentially methylated at the imprinting control centers in hIPS2 whereas H19 became hypermethylated (from 55.4% to 92.6% methylated CpGs as shown in red). Next we generated iPGCs for 7 days, and evaluated CpG methylation in the positive and negative population. CpG methylation in the negative population remained consistent with less than 10% of CpGs changing methylation status relative to undifferentiated hIPS cells. Similar to hESCs, we found that the fully methylated locus (this time H19) was incapable of initiating imprint erasure in hIPS-iPGCs. In contrast only 1 of the 2 differentially methylated loci (PEG1) showed a decrease in CpG methylation in iPGCs (-17.5%) whereas the other locus (SNRPN) remained unchanged. This result indicates that hIPS2 line of iPGCs may be compromised in its ability to undergo erasure of CpG methylation at imprinted genes. Furthermore, the fact that the hypermethylated H19 locus did not undergo imprint erasure in hIPS-iPGCs reinforces the idea that the epigenetic state of the undifferentiated pluripotent cells regardless of origin from the inner cell mass or through reprogramming of fibroblasts may be critical for defining the ability of iPGCs to appropriately differentiate and undergo imprint erasure.
Figure 6. Derivation of iPGCs from hIPS cells and status of CpG methylation at imprinted genes in the hIPS-derived iPGCs.

(A) Quantification of iPGC derived from two hIPS lines (hIPS1, hIPS2) following flow analysis. Significance was achieved when comparing a to b and c to d (P<0.05). (C) CpGs of imprinted genes (H19, PEG1 and SNRPN DMRs) were analyzed by bisulfite sequencing from fibroblasts before reprogramming, undifferentiated hIPS2 and iPGCs sorted at day 7 of differentiation using cKIT/SSEA1 surface markers. The cKIT/SSEA1 double negative population was used as a control. Methylated CpGs are represented as filled circles, unmethylated CpGs are represented as open circles. Percentage methylation was calculated by comparing the number of CpG methyl groups in the starting population verses the test group. For example fibroblast verses hIPS2; hIPS2 verses cKIT/SSEA1 FACS sorted double positive iPGC; and hIPS2 verses the double negative population. Decreased methylation is shown in green no change shown in black and abnormal methylation is shown in red.
Discussion
In the current study we demonstrate that human SSEA1/cKIT/PLAP positive PGCs can be isolated from hESCs and hIPS cells. Furthermore, we demonstrate that the efficiency of deriving triple positive cells on day 7 of differentiation is consistent between lines regardless of origin from the inner cell mass of blastocysts or the reprogramming of human fibroblasts using OCT4, SOX2, KLF4 and c-MYC. As part of this work, we used results from the human embryo in vivo to instruct the acquisition and staging of germ cell development in vitro from pluripotent cells. Access to human fetal samples was invaluable for staging iPGC formation from pluripotent cells as the same cell surface markers used for judging PGC formation in vivo were subsequently used for the in vitro experiments. Combining information on surface markers, together with gene transcription as well as methylation status at imprinted genes, we can classify the iPGCs acquired at day 7 of differentiation as cKIT/SSEA1/PLAP positive PGCs that have initiated the process of imprint erasure. Furthermore, transcriptional analysis would suggest that iPGCs with this surface biomarker identity are not meiotic given the absence of transcription of meiotic genes. In sum, we propose that day 7 of differentiation on hFGS cells corresponds to a developmental window after specification and prior to 9 weeks of human gestation.
Staging of iPGC formation on hFGS cells was based initially on the combination of cell surface markers expressed by PGCs in vivo. Specifically, positive expression of SSEA1, PLAP and cKIT. SSEA1 has been a controversial marker of human PGC formation, as some studies have reported it as positive, while other studies have reported it as a negative marker37, 38, 43. Our results agree with Kerr et al., 37, 38 that SSEA1 faithfully marks VASA positive human fetal germ cells from 5 weeks of gestation. Furthermore, we also show using double staining for cKIT/VASA and SSEA1/VASA that the PGC population from 5-9 weeks of gestation is relatively homogeneous. In particular every cKIT positive cell was VASA positive by immunofluorescence and every SSEA1 positive cell was VASA positive by immunofluorescence. Therefore, the presence of double positive cKIT/SSEA1 cells that express VASA are highly suggestive of first trimester human PGCs. It remains to be determined how these surface markers can be used to study second trimester PGC development which is marked by significant heterogeneity in PGC protein expression36, 39.
PLAP has previously been reported as positive to human PGCs in the first trimester, declining in the second trimester and is absent from germ cells in the third trimester 36. In the current study we confirmed that PLAP was expressed in first trimester PGCs. However, our results demonstrate that PLAP expression begins to decline in PGCs at 7 weeks of gestation, and is absent from VASA expressing PGCs by 9 weeks of gestation. This disparity from previously published results may reflect differences in staining procedures give that we did not use amplification methods to increase the signal intensity36. In the current study, we found no difference in the percentage of SSEA1/cKIT double positive cells or PLAP/SSEA1 double positive cells emerging with differentiation from hESCs or hIPS cells. This result indicates, that at day 7 of differentiation the SSEA1/cKIT positive iPGCs are relatively immature, and are most likely also PLAP positive. Evaluation of iPGCs at later stages of differentiation (14 and 21 days) determined that the proportion of PLAP/SSEA1 cells remained equivalent to cKIT/SSEA1 positive cells. This suggests that increasing the time of differentiation on hFGS cells (up to 21 days) did not result in a switch to a PLAP negative (post 9 weeks of gestation) iPGC development.
In further support of the immature stage of PGC formation, we determined that the iPGCs do not transcribe genes associated with meiosis, or spermatogenesis. Instead, using microarray and Real time PCR we found that the iPGCs on day 7 were enriched in DPPA3, PLAP, PRDM1 (BLIMP1), DAZL and VASA mRNA together with significant repression of multiple HOX genes. This result is in strong agreement with the developmental dynamics that occur in the initial phases of murine PGC formation soon after specification in which Hox gene expression is significantly reduced 44. This result adds further evidence to the immature state of these iPGCs at day 7 of differentiation. Isolation of iPGCs after three weeks in adherent culture without any supporting cell layer has been performed by one group using the female H9 lines of hESCs 31. Unlike the current study in which the iPGCs at 21 days remain relatively immature and express PLAP, the iPGCs derived by Tilgner et al.31 expressed markers of meiosis, but were incapable of assembling correct synaptonemal complexes. This difference between the previous study and the results described here may be due to male-female differences, as we would not expect PGCs to enter meiosis and particularly immature PGCs that are PLAP positive. Furthermore, it could be hypothesized that fetal gonadal stromal cells derived from first trimester human gonads may prevent precocious meiosis of iPGCs.
In the current report, we determined that hFGS cells improved the efficiency of differentiating iPGCs compared to adherent culture, conditioned media and also non-gonadal first trimester human stromal cells. Although hFGS cells generated the highest yields of iPGCs, the non-gonadal stromal cells, from placenta and fetal liver also supported iPGC differentiation. Given that PGCs do not normally migrate through the fetal liver or placenta in vivo, the success of these gonadal and non-gonadal cell lines for supporting iPGC formation indicates that it is not the specificity of the hFGS cells per se that supports specification of iPGCs from pluripotent cells. Instead, the most likely hypothesis is that each of these cell lines produces common iPGC survival and/or self-renewal factor/s. These factors could either that acts directly on the differentiating iPGCs, or indirectly by coordinately differentiating the somatic cells. It is interesting to note that all supportive cell lines were derived from first trimester human fetal material. In future work, the effect of second or third trimester gonadal and non-gonadal cell lines can be evaluated for a role in executing later stage germ cell specific events such as completing imprint erasure, inducing mitotic arrest or initiation of correct meiosis. Two recent reports evaluated niche environments for the differentiation of human PGCs from hESCs 32, 45. The first demonstrated that hESCs differentiated on mouse embryonic fibroblasts promoted an increase in expression of germ cell-specific genes in a mixed population of differentiating hESCs 45. Our results with human fetal cells across multiple hESC lines support this finding. A second report described the differentiation of small sized female H9 hESC colonies on laminin for 10 days. This resulted in the induction of both sertoli cells and iPGCs32.
In the current study we evaluated CpG methylation at three imprinted genes during iPGC formation from three pluripotent cell lines, HSF-6, HSF-1 and hIPS2. In somatic cells derived from pluripotent cells, which do not undergo imprint erasure, we found that CpG methylation fluctuated less than 10% (1.7% to 8.8%) from the starting undifferentiated hESC population. Therefore, any change in more than 10% of CpG dinucleotides at a given locus was considered to be occurring above background. Our results show that differentially methylated loci initiate imprint erasure in iPGCs where as hypermethylated loci did not. Hypermethylation of PEG1 in HSF-1 was most likely culture induced in our laboratory as previous reports have found this locus to be accurately methylated in many hESC lines but sensitive to perturbation in culture46. With regard to H19 in hIPS2, our results indicate that hypermethylation of its imprinting control center occurred during the process of reprogramming. H19 has previously been reported as a one of the more sensitive loci to perturbation in pluripotent human cells 47, therefore it is not surprising that loss of imprinting occurred at this particular locus. SNRPN in contrast was differentially methylated in hESCs as well as hIPS2. This is consistent with the finding that SNRPN is one of the most stable differentially methylated imprinted loci in pluripotent cells 47. We found the iPGCs derived from HSF-1 and HSF-6 were both capable of initiating imprint erasure at SNRPN, however iPGCs derived form hIPS2 were not. Given that only 1/3 imprinted loci initiated imprint erasure in hIPS2-derived iPGCs, this may indicate a problem with epigenetic reprogramming of iPGCs derived following induced reprogramming. However, more hIPS cell lines need to be evaluated to determine whether the reduced efficiency of CpG erasure is consistent between hIPS cell lines compared to hESCs.
Conclusion/Summary
The derivation of iPGCs from hESCs constitutes both a desirable model for reproductive geneticists, and a potential method for treating couples with infertility due to germ cell defects. Our results show that derivation of iPGCs from pluripotent cells following 7 days of differentiation on hFGS results in the generation of immature PGCs corresponding to a developmental stage in vivo between specification and less than 9 weeks of gestation. Our results suggest that the ability to initiate imprint erasure was dependent upon the epigenetic status of the undifferentiated pluripotent cell population from which the iPGC population was generated. Therefore, in future studies more detailed attention must paid to the genome-wide epigenetic landscape of the pluripotent cell population prior to iPGC differentiation as this will assist in the analysis and future use of iPGCs for research or cell-based therapy.
Supplementary Material
Acknowledgments
The authors would like to thank Raymond Kwan for assistance with hESC culture, Dr Nagesh Rao PhD at the UCLA Clinical Cytogenetics lab for Fluorescence in situ hybridization, Dr. Angela Chen from UCLA OB/GYN for human tissues, Xinmin Li at the UCLA clinical microarray core facility, Jing Wen Tan and Dr Rachel Kim of the UCLA Broad Stem Cell Center hESC core facility. This work is supported by funds from the UCLA Department of Molecular Cell and Developmental Biology, STOP Cancer Foundation and NIH P01 GM081621-01A1 (ATC, ZG, MAT, HKAM and KP).
Footnotes
Tae Sub Park: Concept and Design, Collection and assembly of data, Data analysis and interpretation
Zoran Galic: Collection and/or assembly of data
Anne Conway: Collection and/or assembly of data
Anne Lindgren: Collection and/or assembly of data
Benjamin Van Handel: Collection and/or assembly of data
Mattias Magnusson: Collection and/or assembly of data
Laura Richter: Collection and/or assembly of data
Michael Teitell: Data analysis and interpretation, Manuscript writing
Hanna Mikkola: Collection and/or assembly of data
William Lowry: Collection and/or assembly of data, Manuscript writing
Kathrin Plath: Collection and/or assembly of data
Amander Clark: Concept and Design, Collection and/or assembly of data, Data analysis and interpretation and Manuscript writing
References
- 1.Daley GQ. Gametes from embryonic stem cells: a cup half empty or half full? Science. 2007 Apr 20;316(5823):409–410. doi: 10.1126/science.1138772. [DOI] [PubMed] [Google Scholar]
- 2.Holden C. Sperm from Skin Becoming a Reality. ScienceNOW daily News. 2008;415:2. [Google Scholar]
- 3.Surani MA. Germ cells: the eternal link between generations. C R Biol. 2007 Jun-Jul;330(6-7):474–478. doi: 10.1016/j.crvi.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 4.Hayashi K, De Sousa Lopes S, Surani M. Germ cell specification in mice. Science. 2007;316:394–396. doi: 10.1126/science.1137545. [DOI] [PubMed] [Google Scholar]
- 5.Ying Y, L XM, Marble A, Lawson KA, Zhao GQ. Requirement of Bmp8b for the generation of primordial germ cells in the mouse. Mol Endocrinol. 2000;14:1053–1063. doi: 10.1210/mend.14.7.0479. [DOI] [PubMed] [Google Scholar]
- 6.Ying Y, Zhao Q-G. Cooperation of Endoderm-Derived BMP2 and Extraembryonic Ectoderm-Derived BMP4 in Primordial germ cell generation in the mouse. Dev Biol. 2001;232:484–492. doi: 10.1006/dbio.2001.0173. [DOI] [PubMed] [Google Scholar]
- 7.Fujiwara T, Dunn N, Hogan B. Bone morphogenetic protein 4 in the extraembryonic mesoderm is required for allantois development and the localization and survival of primordial germ cells. Proc Natl Acad Sci U S A. 2001;98:13739–14744. doi: 10.1073/pnas.241508898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lawson K, D NR, Roelen BA, Zeinstra LM, Davis AM, Wright CV, Korving JP, Hogan BL. Bmp4 is required for the generation of primordial germ cells in the mouse embryo. Genes Dev. 1999;13:373–376. doi: 10.1101/gad.13.4.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Sousa Lopes S, Roelen M, Monteiro R, et al. BMP signaling mediated by ALK2 in the visceral endoderm is necessary for the generation of primordial germ cells in the mouse embryo. Genes Dev. 2004;18:1838–1849. doi: 10.1101/gad.294004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Sousa Lopes SM, Hayashi K, Surani MA. Proximal visceral endoderm and extraembryonic ectoderm regulate the formation of primordial germ cell precursors. BMC Dev Biol. 2007;7:140. doi: 10.1186/1471-213X-7-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clark A. Establishment and differentiation of human embryonic stem cell derived germ cells. Soc Reprod Fertil Suppl. 2007;63:77–86. [PubMed] [Google Scholar]
- 12.Yamazaki Y, Low E, Marikawa Y, et al. Adult mice cloned from migrating primordial germ cells. Proc Natl Acad Sci USA. 2005;102:11361–11366. doi: 10.1073/pnas.0504943102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yamazaki Y, Mann M, Lee S, et al. Reprogramming of primordial germ cells begins before migration into the genital ridges, making these cells inadequate donors for reproductive cloning. Proc Natl Acad Sci U S A. 2003;100:12207–12212. doi: 10.1073/pnas.2035119100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Szabo P, Hubner K, Scholer H, Mann J. Allele-specific expression of imprinted genes in mouse migratory primordial germ cells. Mech Dev. 2002;115:157–160. doi: 10.1016/s0925-4773(02)00087-4. [DOI] [PubMed] [Google Scholar]
- 15.Hajkova P, Ancelin K, Waldmann T, et al. Chromatin dynamics during epigenetic reprogramming in the mouse germ line. Nature. 2008 Mar 19; doi: 10.1038/nature06714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hajkova P, Erhardt S, Lane N, et al. Epigenetic reprogramming in mouse primordial germ cells. Mech Dev. 2002;117:15–23. doi: 10.1016/s0925-4773(02)00181-8. [DOI] [PubMed] [Google Scholar]
- 17.Monk M, McLaren A. X-chromosome activity in fetal germ cells of the mouse. J Embryol Exp Morphol. 1981;63:75–84. [PubMed] [Google Scholar]
- 18.Monk M, Boubelik M, Lehnert S. Temporal and regional changes in DNA methylation in the embryonic and extraembryonic and germ cell lineages during mouse embryo development. Development. 1987;99:371–382. doi: 10.1242/dev.99.3.371. [DOI] [PubMed] [Google Scholar]
- 19.Hubner K, Fuhrmann G, Christenson L, et al. Derivation of oocytes from mouse embryonic stem cells. Science. 2003 doi: 10.1126/science.1083452. [DOI] [PubMed] [Google Scholar]
- 20.Toyooka Y, Tsunekawa N, Akasu R, Noce T. Embryonic stem cells can form germ cells in vitro. PNAS. 2003;100(20):11457–11462. doi: 10.1073/pnas.1932826100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Geijsen N, Horoschak M, Kim K, Grilbnau J, Eggan K, Daley G. Derivation of embryonic germ cells and male gametes from embryonic stem cells. Nature. 2004;427:148–154. doi: 10.1038/nature02247. [DOI] [PubMed] [Google Scholar]
- 22.Lacham-Kaplan O, Chy H, Trounson A. Testicular cell conditioned medium supports differentiation of embryonic stem (ES) cells into ovarian structures containing oocytes. Stem Cells. 2005 Aug 18; doi: 10.1634/stemcells.2005-0204. [DOI] [PubMed] [Google Scholar]
- 23.Nayernia K, Nolte J, Michelmann H, et al. In vitro differentiated embryonic stem cells give rise to male gametes that can generate offspring mice. Dev Cell. 2006;11:125–132. doi: 10.1016/j.devcel.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 24.Payer B, Chuva de Sousa Lopes S, Barton S, Lee C, Saitou M, Surani M. Generation of stella-GFP transgenic mice: a novel tool to study germ cell development. Genesis. 2006;44:75–83. doi: 10.1002/gene.20187. [DOI] [PubMed] [Google Scholar]
- 25.Salvador L, Silva C, Kostetskii I, Radice G, Strauss Jr. The promoter of the oocyte-specific gene, Gdf9, is active in population of cultured mouse embryonic stem cells with an oocyte-like phenotype. Methods. 2008;45:172–181. doi: 10.1016/j.ymeth.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ohinata Y, Sano M, Shigeta M, Yamanaka K, Saitou M. A comprehensive, non-invasive visualization of primordial germ cell development in mice by the Blimp1-mVenus and stella-ECFP double transgenic reporter. Reproduction. 2008 doi: 10.1530/REP-08-0053. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 27.Clark AT, Bodnar MS, Fox M, et al. Spontaneous differentiation of germ cells from human embryonic stem cells in vitro. Hum Mol Genet. 2004;13:727–739. doi: 10.1093/hmg/ddh088. [DOI] [PubMed] [Google Scholar]
- 28.Kee K, Gonsalves J, Clark A, RP RA. Bone morphogenetic Proteins induce germ cell differentiation from human embryonic stem cells. Stem Cells and Differentiation. 2006;15:831–837. doi: 10.1089/scd.2006.15.831. [DOI] [PubMed] [Google Scholar]
- 29.Chen H, Kuo H, Chien C, et al. Derivation characterization and differentiation of human embryonic stem cells: comparing serum-containing versus serum free media and evidence of germ cell differentiation. Hum Reprod. 2007;2:567–577. doi: 10.1093/humrep/del412. [DOI] [PubMed] [Google Scholar]
- 30.Mikkola M, Olsson C, Palgi J, et al. Distinct differentiation characteristics of individual human embryonic stem cell lines. BMC Dev Biol. 2006;6:40. doi: 10.1186/1471-213X-6-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tilgner K, Atkinson S, Golebiewska A, Stojkovic M, Lako M, Armstrong L. Isolation of primordial germ cells from differentiating human embryonic stem cells. Stem Cells. 2008 doi: 10.1634/stemcells.2008-0289. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 32.Bucay N, Yebra M, Cirulli V, et al. A novel Approach for the derivation of putative promordial germ cells and sertoli cells from human embryonic stem cells. Stem Cells. 2008 doi: 10.1634/stemcells.2007-1018. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 33.Castrillon DH, Quade BJ, Wang TY, Quigley C, Crum CP. The human VASA gene is specifically expressed in the germ cell lineage. PNAS. 2000;97:9585–9590. doi: 10.1073/pnas.160274797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lowry WE, Richter L, Yachechko R, et al. Generation of human induced pluripotent stem cells from dermal fibroblasts. Proc Natl Acad Sci U S A. 2008 Feb 26;105(8):2883–2888. doi: 10.1073/pnas.0711983105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Judson H, Hayward B, Sheridan E, Bonthron D. A global disorder of imprinting in the human female germ line. Nature. 2002;41:539–542. doi: 10.1038/416539a. [DOI] [PubMed] [Google Scholar]
- 36.Gaskell TL, Esnal A, Robinson LL, Anderson RA, Saunders PT. Immunohistochemical Profiling of Germ Cells Within the Human Fetal Testis: Identification of Three Subpopulations. Biol Reprod. 2004 Aug 18; doi: 10.1095/biolreprod.104.028381. [DOI] [PubMed] [Google Scholar]
- 37.Kerr CL, Hill CM, Blumenthal PD, Gearhart JD. Expression of Pluripotent Stem Cell Markers in the Human Fetal Testis. Stem Cells. 2007 Nov 15; doi: 10.1634/stemcells.2007-0605. [DOI] [PubMed] [Google Scholar]
- 38.Kerr CL, Hill CM, Blumenthal PD, Gearhart JD. Expression of pluripotent stem cell markers in the human fetal ovary. Hum Reprod. 2008 Jan 17; doi: 10.1093/humrep/dem411. [DOI] [PubMed] [Google Scholar]
- 39.Gjerstorff M, Kock K, Nielsen O, Ditzel H. MAGE-A1, GAGE and NY-ESO-1 cancer/testis antigen expression during human gonadal development. Human Reprod. 2007;22:953–960. doi: 10.1093/humrep/del494. [DOI] [PubMed] [Google Scholar]
- 40.Millan J. Mammalian Alkaline Phosphatases: From Biology to pplications in Medicine and Biotechnology. Wiley-VCH; 2006. [Google Scholar]
- 41.Damjanov P, Lange P, Harris H. Immunohistochemical localization of Placental Like Alkaline Phosphatase in testis and germ cell tumors using monoclonal antibodies. Am J Pathol. 1983;111:156–165. [PMC free article] [PubMed] [Google Scholar]
- 42.Le Du M, Milan J. Structural evidence of functional divergence in human alkaline phosphatases. Journal of biological Chemistry. 2002;277:49808–49814. doi: 10.1074/jbc.M207394200. [DOI] [PubMed] [Google Scholar]
- 43.Liu S, Liu H, Tang S, et al. Characterization of stage-specific embryonic antigen-1 expression during early stages of human embryogenesis. Oncol Report. 2004;12:1251–1256. [PubMed] [Google Scholar]
- 44.Seki Y, Yamaji M, Yabuta Y, et al. Cellular dynamics associated with the genome-wide epigenetic reprogramming in migrating primordial germ cells in mice. Development. 2007;134 doi: 10.1242/dev.005611. Epub 2007 Jun 2013. [DOI] [PubMed] [Google Scholar]
- 45.West F, Machacek D, Boyd N, Pandiyan K, Robbins K, Stice S. Enrichment and differentiation of human germ-like cells mediated by feeder cells and basic fibroblast growth factor signaling. Stem Cells. 2008;26:2768–2776. doi: 10.1634/stemcells.2008-0124. [DOI] [PubMed] [Google Scholar]
- 46.Kim K, Thurston A, Mummery C, et al. Gene-specific vulnerability to imprinting variability in human embryonic stem cell lines. Genome Res. 2007;17 doi: 10.1101/gr.6609207. Epub 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rugg-Gunn P, Ferguson-Smith A, Pedersen R. Status of genomic imprinting in human embryonic stem cells as revealed by a large cohort of independently derived and maintained lines. Hum Mol Genet. 2007;(Spec No.):R243–R251. doi: 10.1093/hmg/ddm245. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.