

Abstract
In heterothallic ascomycetes, mating is controlled by two nonallelic idiomorphs that determine the ‘sex’ of the corresponding strains. We recently discovered mating-type loci and a sexual life cycle in the penicillin-producing fungus, Penicillium chrysogenum. All industrial penicillin production strains worldwide are derived from a MAT1-1 isolate. No MAT1-2 strain has been investigated in detail until now. Here, we provide the first functional analysis of a MAT1-2 locus from a wild-type strain. Similar to MAT1-1, the MAT1-2 locus has functions beyond sexual development. Unlike MAT1-1, the MAT1-2 locus affects germination and surface properties of conidiospores and controls light-dependent asexual sporulation. Mating of the MAT1-2 wild type with a MAT1-1 high penicillin producer generated sexual spores. We determined the genomic sequences of parental and progeny strains using next-generation sequencing and found evidence for genome-wide recombination. SNP calling showed that derived industrial strains had an uneven distribution of point mutations compared with the wild type. We found evidence for meiotic recombination in all chromosomes. Our results point to a strategy combining the use of mating-type genes, genetics, and next-generation sequencing to optimize conventional strain improvement methods.
Introduction
In fungi, mating and sexual propagation are controlled by chromosomal regions, known as mating-type loci, which determine the two opposite sexes. Mating-type loci have been studied most thoroughly in Saccharomyces cerevisiae. In this ascomycetous yeast, ‘α’ and ‘a’ strains carry transcriptionally active mating-type loci called ‘MATα’ and ‘MATa’. The MATα mating-type locus comprises two genes, α1 and α2, which encode MATα1 and MATα2 proteins (Haber, 2012). MATα1 is a transcription factor (TF) with a characteristic α-box DNA-binding domain to promote expression of α-specific genes, including genes that express the α-factor mating pheromone and the Ste2 pheromone receptor for the opposite mating pheromone (the a-factor) (Herskowitz, 1989; Galgoczy et al., 2004; Ni et al., 2011). MATα2 encodes a homeodomain protein, which cooperates with Mcm1 to repress a-specific genes that produce the a-factor and the Ste3 pheromone receptor (Bardwell, 2005). Similarly, the MATa locus encodes two proteins. The first, MATa1, is a homeodomain TF with a highly conserved DNA binding domain; the second, MATa2, is a protein of unknown function (Herskowitz et al., 1992).
In diploid cells, MATa1 and MATα2 form a heterodimer that switches off haploid-specific but supports diploid-specific gene expression (Booth et al., 2010). Unlike filamentous ascomycetes, S. cerevisiae has the ability to switch mating type. Two intact but silent copies of the mating-type alleles, HMLα (Hidden MAT Left) and HMRa (Hidden MAT Right), are located at opposite ends of the same chromosome that harbors MAT. These alleles serve as donors that allow a MATa cell to switch to MATα, or vice versa (Butler et al., 2004).
Unlike ascomycetous yeasts, the structure of the mating-type loci is different in filamentous ascomycetes, and no mating-type switching has been observed to date (Debuchy et al., 2010). Instead, heterothallic strains carry one of two nonallelic idiomorphs called MAT1-1 or MAT1-2. These mating-type loci determine the ‘sex’ of the corresponding strains (Debuchy and Turgeon, 2006; Pöggeler, 2007; Ni et al., 2011; Haber, 2012). The two mating-type idiomorphs typically carry one to several mating-type genes (Glass et al., 1990; Staben and Yanofsky, 1990). All idiomorphs encode conserved TFs, which act as major regulators of sexual communication and mating (Nelson, 1996; Debuchy and Turgeon, 2006). The conserved TF encoded in MAT1-1 is characterized by an α-domain, which shows high similarity to the α-domain TF in the S. cerevisiae mating-type locus. The conserved TF encoded by MAT1-2 contains a high mobility group (HMG) domain that has no similarity to the mating-type TFs in yeast, but it is found in TFs of diverse lower or higher eukaryotes. Functional analyses of mating-type loci have shown that the encoded TFs are involved in both mating and sexual development (Kronstad and Staben, 1997; Ni et al., 2011).
We previously discovered mating-type loci in diverse strains of the penicillin producer, Penicillium chrysogenum. Although Alexander Fleming initially discovered a P. chrysogenum strain that carries a MAT1-2 locus, all currently used production strains, derived from a contaminated cantaloupe, exhibit the MAT1-1 locus (Hoff et al., 2008). As just described, these MAT loci encode either an α domain or HMG TF. We recently described new functions for the MAT1-1-encoded α-domain TF. Functional analyses provided further evidence that, in addition to sexual mating, MAT1-1 controls developmental processes of biotechnological relevance, such as penicillin biosynthesis, conidiation and hyphal morphology (Böhm et al., 2013).
Here, we functionally characterized a MAT1-2 locus from a wild-type strain that like other wild-type isolates produces a rather low penicillin titer compared with recently characterized industrial P. chrysogenum strains (Böhm et al., 2013). Our data revealed that the mating-type locus not only affects sexual development but also asexual development and morphology. We also found that the characterized MAT1-2 wild-type strain was able to mate with an industrial penicillin producer strain carrying a deviant karyotype. Genome sequencing of parental strains and ascospore progeny, when compared with recently published genome sequences (van den Berg et al., 2008; Specht et al., 2014), indicated that even highly developed industrial strains, which have undergone several rounds of mutagenesis, have retained the capacity for sexual recombination. Based on genome sequencing data and SNP calling, we derived a genome-wide recombination map for ascospore progeny. Our data highlighted a potential for using sexual recombination to generate industrial strains with novel genetic properties.
Results
Construction of MAT1-2-1 deletion, complementation and overexpression strains
Previously, we identified MAT1-1 and MAT1-2 loci in different strains of P. chrysogenum (Hoff et al., 2008). All production strains derived from the Wisconsin lineage (NRRL1951) carry the MAT1-1 locus, but derivatives of the original Fleming strain and diverse wild-type isolates contain the MAT1-2 locus. One of the latter isolates is Pc3 (IB 08/921), which was shown to undergo sexual reproduction with an improved MAT1-1 strain that was a direct descendent of a wild-type isolate (Böhm et al., 2013). For further characterization, we amplified the MAT1-2 locus from Pc3, and the derived sequence showed high similarity to MAT loci in the Fleming isolates, NRRL1249B21 and NRRL824 (Hoff et al., 2008). The identified open reading frame encoded a protein with the conserved HMG domain, which comprised 13 putative DNA-binding sites (Fig. S1). The HMG coding sequence contains a conserved intron after the first nucleotide of the invariant serine codon, which is present in all known ascomycetous MAT1-2-1 genes (Debuchy and Turgeon, 2006).
Sequences adjacent to both MAT idiomorphs are highly conserved in most filamentous ascomycetes, with more than 95% nucleotide identity. They are flanked by SLA2 and APN2, as previously described for other filamentous ascomycetes (Dyer, 2007; Hoff et al., 2008; Pöggeler et al., 2011; Kück and Böhm, 2013). The deletion construct, pKOMAT (Table S1), was transformed into Pc3 to substitute the native MAT1-2-1 with the phleomycin resistance cassette. Four independent transformants were generated, and the precise deletion of the MAT1-2-1 gene was verified by Southern analysis (Fig. S2A). Using the deletion strain ΔMAT1-2-1 T10 as host, we constructed complementation strains (ΔMAT1-2-1::MAT1-2-1) by ectopically integrating the wild-type MAT1-2-1 gene copy, combined with a nourseothricin (nat) resistance marker, into the genome. On plasmid pKompMAT2, the MAT1-2 locus is under the control of its native promoter. The successful rescue of the deletion was analyzed by Southern hybridization using radiolabeled MAT1-2-1 specific probes (Fig. S2B). We investigated nine transformants and identified two strains with a single copy of the MAT1-2-1 gene (T24, T26), which were chosen for further functional analyses (Fig. S2B).
In reverse, we also studied MAT1-2 function by constructing MAT1-2-1 overexpression strains. Besides wild-type Pc3, we chose P2niaD18, a high penicillin-producing MAT1-1 strain, as hosts for transformation experiments. Plasmid pPgpd-MAT-2-ptrA (Fig. S3A) with the MAT1-2 locus under the control of the strong constitutive gpd promoter from Aspergillus nidulans was ectopically integrated into the genomes of both strains, and copy numbers of pPgpd-MAT-2-ptrA were determined in the two hosts by Southern hybridization (Fig. S3B and C). For further studies, we selected six strains carrying different copies of the transgene. As derivatives of P2niaD18, we selected P2MAT2-OE T1 (multiple copies of MAT1-2), and P2MAT2-OE T2 and T5 (both having two copies), while Pc3MAT2-OE T20 (multiple copies), Pc3MAT2-OE T26 (three copies) and Pc3MAT2-OE T23 (single copy) were derivatives of Pc3. Subsequent quantitative real-time polymerase chain reaction (qRT-PCR) analyses demonstrated that the extra copies of MAT1-2-1 resulted in enhanced transcriptional expression of MAT1-2-1 in all Pc3MAT2-OE strains. Compared with the reference wild-type Pc3, the overexpression strains showed elevated transcriptional expression of MAT1-2-1, with the highest value of 169-fold transcriptional expression for a single multicopy strain (Pc3MAT2-OE T26; Fig. S3D).
MAT1-2 controls light-dependent sporulation
We have already demonstrated that the MAT1-1 locus of P. chrysogenum regulates transcription of a wide range of genes; thus, it controls processes such as penicillin biosynthesis, hyphal morphology and asexual sporulation (Böhm et al., 2013). To determine the involvement of the MAT1-2 locus in asexual spore formation, we counted conidiospores in a defined area during long-term light illumination. These numbers were compared among Pc3, P2niaD18, and all recombinant MAT1-2 and recombinant MAT1-1 strains, revealing that Pc3 exhibited light-dependent sporulation after 168 h of growth (Fig. 1). The number of conidiospores generated in light (4.2 × 108 spores cm−2) was approximately twice the number generated in darkness (1.8 × 108 spores cm−2). In contrast, the MAT1-2-1 deletion strains showed increased sporulation in darkness, reaching levels (3.4 × 108 spores cm−2) nearly equivalent to those observed in light (3.6 × 108 spores cm−2). This suggests that repression of conidiation in darkness is abolished in ΔMAT1-2-1 strains. Moreover, the MAT1-2-1 complementation strains exhibited a restored phenotype; the light–dark effect on asexual sporulation was comparable with that observed in wild-type Pc3. Similarly, MAT1-2-1 overexpression resulted in light-dependent sporulation in both host strains (Fig. 1).
Figure 1.
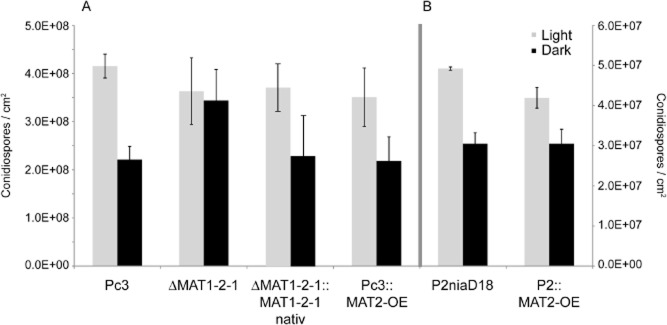
Quantification of light-dependent conidiospore formation of MAT1-2 (A) and MAT1-1 (B) strains. All strains were grown under constant light or in darkness for 168 h. From all recombinant transformants, three independent strains were chosen for analysis. Error bars represent mean ± SD (n = 3) from three independent experiments. Note the calibration of both axis is differently for both experiments.
Based on these data, we conclude that MAT1-2-1 acts as a repressor of light-dependent sporulation. The next question was whether MAT1-2-1 expression is light dependent in wild-type or overexpression strains. As determined by qRT-PCR, none of the strains investigated showed up- or down-regulation of MAT1-2-1 after long-term illumination (Fig. S4A). Likewise, during 168 h of illumination, genes for TF PcBrlA and diverse light receptors were not differentially expressed in wild-type or MAT1-2-1 deletion strains (Fig. S4B).
MAT1-2 controls germination and surface properties of conidiospores
We then tested whether spore germination and surface properties were affected by MAT1-2-1.
Plotting germ tube emergence over time revealed that the recombinant MAT1-2-1 strains derived from Pc3 germinated faster than wild type (Pc3). After 6 h of incubation, about 10% of all conidiospores derived from Pc3 had germinated, whereas 17% of the conidiospores from the MAT1-2-1 deletion had germinated. Thus, deletion of MAT1-2-1 led to an enhanced germination rate that correlated with a higher number of germ tubes. In the complementation strains, the germination rate was comparable with the wild-type level.
All strains showed a germination rate of 100% after 9 h of incubation (Fig. 2), which is also visible in the images of Fig. 3. A different picture was observed with P2niaD18 and its derivatives. The reference strain showed a germination rate of only 50% after 15 h of incubation, while the MAT1-2-1 overexpression strains had a rate of only 10% after 15 h (Fig. 2). This difference is also seen in Fig. 3, where half of the conidiospores are not swollen and thus fail to germinate. Our results suggest that in the MAT1-1 host, MAT1-2-1 acts as a repressor of germination and regulates the number of germ tubes. In contrast, in the wild-type strain, MAT1-2-1, had an activating effect on germination. Thus, depending on the mating type of the host strain, MAT1-2-1 plays a role in spore germination. Another observation was that the increased germination rate correlated with the appearance of two germ tubes at single conidiospores. Mostly single germ tubes were observed in wild-type Pc3, whereas in Pc2MAT2-OE, the majority of spores showed two bipolar germination tubes (Fig. S5).
Figure 2.
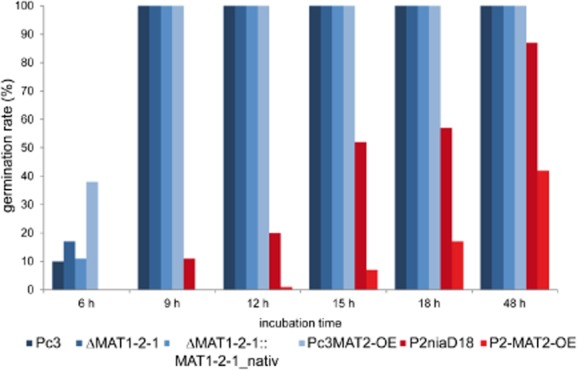
Time plot of conidial germ-tube emergence. Recombinant MAT1-2 and reference strains were grown on solid media as indicated. From all recombinant transformants, three independent strains were chosen for analysis, except for the complementation strains, where two independent transformants were used. In all cases, 400 conidia from each strain were investigated for determination of germination rate as given in percentages. Bars in blue and red indicate the two different host strains, namely Pc3 and P2niaD18, and represent mean values (n = 3).
Figure 3.
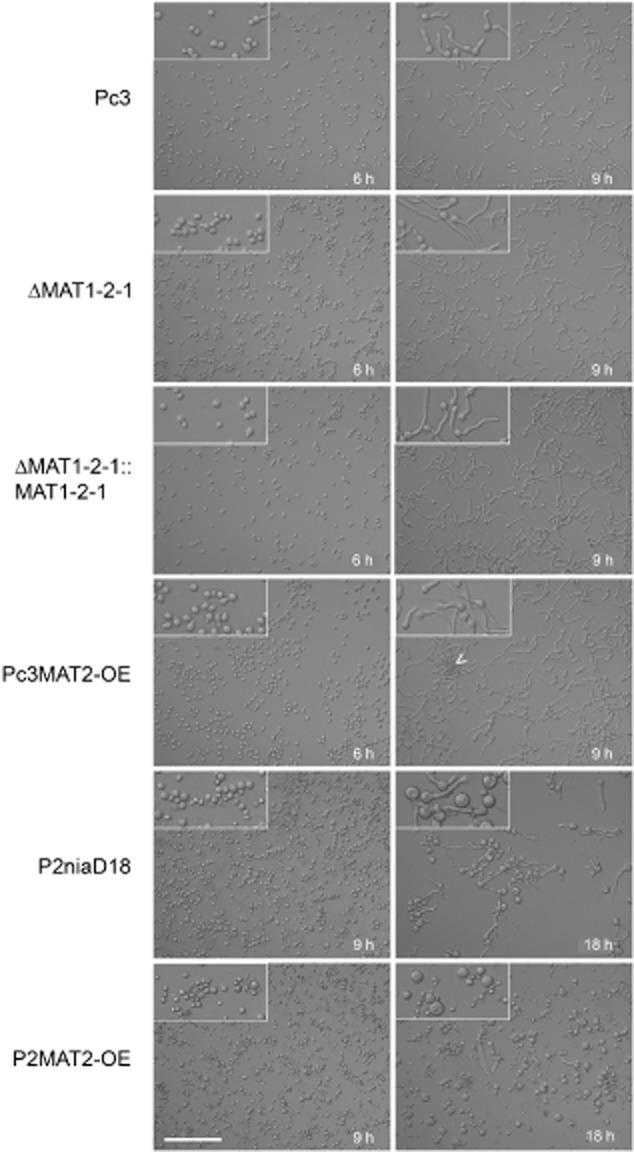
Micrographs of germinating conidia and formation of spore clusters in strains as indicated. Insets show enlargements (twofold) of images, and the arrowhead points to agglutinated conidiospores. Scale bar corresponds to 100 μm in all images.
Another phenomenon observed during microscopic investigation of MAT1-2-1 overexpression strains was enhanced conidiospore agglutination, where conidiospores attached to each other just before germination, and the spores germinated simultaneously (arrowhead in Fig. 3). This phenomenon was observed in Pc3MAT2-OE and in recombinant strains derived from P2niaD18. Despite vortexing the spore suspensions before inoculation, the agglutination effect was still observed (data not shown). The strains formed clusters of up to 20 spores attached to each other. Agglutination was observed shortly before germination and led to the formation of pellet-like structures, a morphological feature probably responsible for the observed pellet phenotypes in diverse MAT1-2-1 overexpression strains.
To further analyze the impact of MAT1-2-1 on pellet formation, we conducted time course experiments in liquid shaking cultures. Under these growth conditions, MAT1-2-1 is transcriptionally expressed as confirmed by qRT-PCR (Fig. S4B). Reference and recombinant MAT1-2 and MAT1-1 strains were incubated for 48, 72, 96 and 168 h in liquid CCM, and 240 pellets of each strain at each time point were analyzed. Pellet phenotypes were distinct and consistent for the strains investigated. After 48 h of incubation, 85% of the pellets from strain Pc3 were 2000–4000 μm in diameter (Fig. 4A, Fig. S4B). Over the time course of 168 h, the pellet sizes increased, and 65% of the final pellets were 4000–6500 μm in diameter. MAT1-2-1 deletion strains did not show changes in pellet formation; the pellets displayed a similar size distribution pattern at each time point (Fig. S7). In contrast, the overexpression strains in Pc3 generated larger pellets than the reference strain, even after only 72 h of incubation (Fig. 4A). At this time point, only 5% of Pc3 pellets were larger than 5000 μm, but in overexpression strains, 40% of the pellets were larger than 5000 μm, with a maximum diameter of about 7700 μm. At the same time, small pellets of about 2000 μm were also apparent, leading to a heterogeneous distribution of pellet sizes, which was more apparent in P2MAT2-OE strains. P2niaD18 exhibited pellets no larger than 1500 μm, even after 96 h of incubation (Fig. 4B, Fig. S4B). Although the overexpression strains generated pellets of about 2000 μm in diameter after only 48 h of incubation, 40% of the pellets were smaller than 500 μm. After 96 h, the distribution pattern shifted towards larger pellets, where 40% of the pellets were 2000–4000 μm in size, and only 10% of pellets were about 500 μm in diameter.
Figure 4.
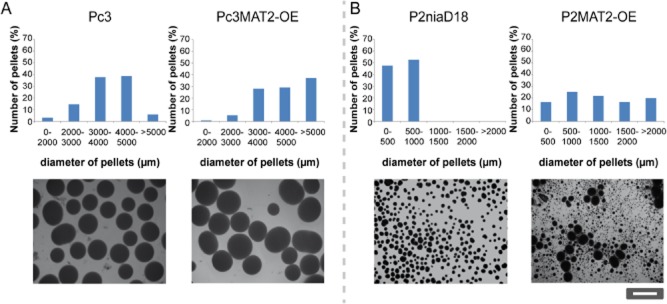
Pellet formation in liquid shaking cultures formed by reference and recombinant MAT1-2 and recombinant MAT1-1 strains. Error bars represent mean ± SD of 240 random pellets from three independent strains.A. Quantification of pellet diameter in MAT1-2-1 overexpression strains in the Pc3 background grown for 72 h in liquid CCM culture.B. Quantification of pellet diameter in MAT1-2-1 overexpression strains in the P2niaD18 background after 72 h of growth in liquid CCM. All pellet phenotypes are illustrated in representative micrographs.
These results demonstrate that MAT1-2-1 has an impact on conidiospore morphology and affects pellet morphology, independently of the genetic background of the host. We propose that the observed pellet morphologies arose as a consequence of conidiospore agglutination. Single germinating spores will form rather small pellets. However, the pellet diameter varies with the number of attached spores; thus, pellets of every size are formed.
Mating between wild-type and industrial producer strains
Here and in a previous investigation, we showed that MAT loci in P. chrysogenum controlled developmental processes not related to sexual reproduction. Furthermore, a previous study showed that the MAT1-1 locus is not required for fruiting body but for ascospore formation and therefore necessary for the completion of the sexual life cycle (Böhm et al., 2013). The next question was whether the MAT1-2 locus is necessary for crossings between MAT1-2 and MAT1-1 strains.
The wild-type P. chrysogenum isolate from Peoria (Illinois) is a MAT1-1 strain, designated NRRL1951, and the progenitor of Q176 and all derived industrial strains, including the sequenced Wisconsin 54-1255 (Fig. 5) (van den Berg et al., 2008). Classical mutation of Wisconsin 54-1255 and selection programs resulted in a series of strains with at least an 85-fold increase in penicillin titer (Nielsen, 1997). Nippon Kayaku Co. (Japan) provided the producer P2, characterized by an amplified gene cluster carrying the three penicillin biosynthesis genes (Lein, 1986; Nielsen, 1997). Finally, UV mutagenesis resulted in a nitrate reductase-deficient strain, called P2niaD18 (Hoff et al., 2008). The recently published genomic sequence of P2niaD18 (Specht et al., 2014) shows at least two chromosomal rearrangements compared with the previously sequenced Wisconsin 54-1255 strain (van den Berg et al., 2008).
Figure 5.
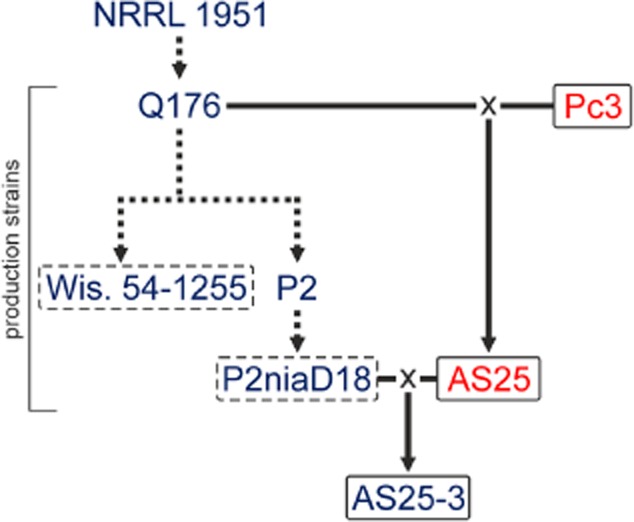
Genealogy of penicillin production strains of P. chrysogenum. Dashed lines indicate conventional mutagenesis during strain improvement programs. Matings (×) of low-penicillin producer Q176 with wild-type Pc3 were performed, and ascospore isolate AS25 was obtained (Böhm et al., 2013). AS25 was further crossed with the industrial high-penicillin production strain P2niaD18. The MAT locus of each strain is indicated by blue for MAT1-1 or red for MAT1-2.
To investigate the function of MAT1-2-1 in sexual development, we crossed Q176 with ΔMAT1-2-1 and Pc3 in two independent sets of experiments with at least 10 mating plates each. After 16 weeks of incubation, we observed cleistothecia only when the intact MAT1-2 locus was present in the parental strain. In ΔMAT1-2-1 strains, we never observed fruiting body formation; this indicated that the encoded gene product is a prerequisite for completion of the sexual life cycle, as was shown for other ascomycetes (Ferreira et al., 1998; Desjardins et al., 2004; Pöggeler et al., 2006; Paoletti et al., 2007).
Next, we crossed diverse MAT1-2 strains with the production strain P2niaD18 (MAT1-1), which has undergone several chromosomal rearrangements compared with its progenitor strain Q176 (Böhm et al., 2013). The MAT1-2 strains included Pc3, the ascospore isolate AS25 (Böhm et al., 2013) and the Fleming strain (Hoff et al., 2008). After 5 weeks of incubation, 10 crosses were investigated for cleistothecia formation. Crosses with the Fleming strain formed no fruiting bodies after this time of incubation, while crosses with Pc3 produced infrequent cleistothecia. The most efficient crossing was obtained with AS25 (Fig. 5). Therefore, further analyses were conducted with isolates from this crossing. Mature cleistothecia were collected for further ascospore isolation using a considerably modified procedure compared with previous reports (Böhm et al., 2013) in order to remove contaminating conidiospores. Cleistothecia were maintained at 60°C for 30 min to prevent germination of conidiospores; ascospores are known to be more resistant against heat in various Penicillium and Aspergillus species (Pitt and Hocking, 2009). To isolate asci and ascospores, cleistothecia were crushed by vortexing with glass beads for 5 min.
Isolated ascospores were screened for recombination at the molecular level by probing for 11 marker genes in a RFLP analysis and identifying the mating-type locus with Southern hybridization, as described previously (Böhm et al., 2013). Out of 100 isolates screened, we detected nine with a recombinant genotype. As discussed below, this rather low frequency of recombinant isolates can be explained by heterogeneity of the karyotypes of both parental strains and the incidence of recombination hot spots. To further investigate meiotic recombination at the genomic scale, we chose AS25-3, which carried marker genes obtained from both parental strains.
Genome sequencing, mapping and SNP detection
We determined the genomic sequence of the wild-type strain Pc3 and two recombinant ascospore isolates, AS25 and AS25-3 (Fig. 5) with next generation sequencing, and compared them to the recently published sequence of P2niaD18 (Specht et al., 2014). The Pc3 genome was sequenced in paired-end, 100 nt reads on an Illumina HiSeq 2000 system. In total, 13,751,610 reads were obtained, which represented a nominal coverage of ∼ 31-fold across the 32.4 Mb P2niaD18 genome. The processed reads were assembled de novo into 1151 scaffolds, with a minimum contig length of 1 kb and an N50 of 142,118 bp. All scaffolds represented 32,239,728 nt, which resulted in a ∼ 99% nominal coverage of the currently assembled P2niaD18 genome sequence. The main features of the Pc3 genome assembly are shown in Table 1. The scaffolds were aligned to the improved genomic sequence of P2niaD18 to identify differences in the chromosomal architecture between the strains (Specht et al., 2014).
Table 1.
Main features of the P. chrysogenum Pc3 genome assembly
| Size of final assembly | 32.2 Mb |
| Number of scaffolds | 1151 |
| Nmax in bases | 663,571 |
| Scaffold N50 in bases | 142,118 |
| Number of gaps | 1202 |
| Total gap length percentage | 0.1 |
| GC percentage | 49.0 |
Compared with P2niaD18, we detected at least one chromosomal rearrangement in both contig 24 and contig 35 of Pc3. PCR analysis showed that sequences from contig 24 and contig 35 are translocated to opposite ends of chromosome I of P2niaD18 (Fig. S8). Remarkably, this translocation was confirmed for all tested strains of the Wisconsin lineage, including Q176 and Wisconsin 54-1255. However, NRRL 1951, the wild-type ancestor of all penicillin-producing strains had the same chromosomal architecture at this site as Pc3. To investigate genomic recombination among the ascospore isolates, the genomic sequences of AS25 and AS25-3 (for origins, see Fig. 5) were determined by next-generation sequencing using 50 nt reads produced with the Illumina HiSeq 2000 system. After filtering, we obtained 15,860,919 reads for AS25 and 20,726,343 reads for AS25-3, resulting in nominal coverages of 22.2 × for AS25 and 27.8 × for AS25-3 (Table 2). In a first comparison, SNP calling was performed with Pc3, the progenitor strain of AS25 and P2niaD18. We mapped 82.1% of the obtained reads for Pc3 to the reference genome sequence of P2niaD18, which represented a mean mapping quality score of 38.3- and 31.4-fold coverage. We identified 27,271 high-quality SNPs as defined in the Experimental Procedures section. This high number of SNP demonstrates the ability to distinguish strains and therefore differently inherited sequences on the genome of ascospore isolates.
Table 2.
Comparative analyses of parental and progeny strains of P. chrysogenum.
| Compared strains reads of mapped to | Resulting coverage | Analysis performed | High-quality SNPsa | |
|---|---|---|---|---|
| Pc3 | P2niaD18 | 31.4 | SNP calling and construction of Pc3 consensus sequence | 27,271 |
| AS25 | P2niaD18 | 22.2 | SNP calling to predict parental SNPs of AS25–3 | 21,133 |
| AS25 | Pc3 scaffolds | 22.8 | SNP calling | 397 |
| AS25-3 | P2niaD18 | 27.8 | SNP calling and construction of AS25–3 consensus sequence | 22,430 |
| AS25-3 | AS25 consensus | 28.0 | SNP calling | 11,816 |
SNPs with a quality score greater than 50 and an allele frequency of 1.0.
The de novo assembled genomic sequence of Pc3 was then used as a reference for SNP calling with the reads obtained from AS25. We mapped 92.0% of the processed AS25 reads to the 1118 de novo assembled scaffolds of Pc3, and only 397 high-quality SNPs were predicted. Similarly, the processed short reads from AS25 were mapped to the reference sequence of P2niaD18. The distribution of 21,133 high-quality SNPs between AS25 and P2niaD18 indicates a high genomic divergence of both strains. Sliding window analysis, counting all SNPs between AS25 and P2niaD18, shows an uneven distribution of all SNPs across the four chromosomes (Fig. S9). In addition, we constructed a consensus sequence of AS25 based on the P2niaD18 chromosome model. The AS25 consensus sequences contained 7497 gaps with a total gap size of 968,572 nt. The consensus sequence was further used as a reference to compare the genome sequences of ascospore isolate AS25-3 with its parental strain AS25. We succeeded in mapping 90.8% of all AS25-3 reads on the consensus sequence of AS25 and achieved 28.0-fold coverage. Thus, the overall SNP calling analysis showed a rather high similarity between Pc3 and AS25 and, at the same time, high sequence divergence with P2niaD18. These results are consistent with the observation that AS25 share most of the phenotypic properties of Pc3 (Böhm et al., 2013). This includes the rather low penicillin titer that depends on a large number of factors (Brakhage et al., 2004) that were modified in current production strains.
SNP calling was also applied to detect recombinant genomic regions in ascospore isolates. To achieve this, AS25-3 reads were mapped to the parental P2niaD18 sequence. In total, 86.8% of the processed short reads from AS25-3 were mapped to P2niaD18, resulting in 27.8-fold coverage. From this, we obtained a consensus sequence for AS25-3 that included 6135 gaps, with a total size of 887,230 nt. In addition, 1,252,628 nt were filtered out, due to insufficient coverage. The remaining consensus sequence represented 93.4% of the nuclear genome and showed sufficiently high quality for SNP calling analysis (Nielsen et al., 2011). In a comparison with the P2niaD18 sequence, we identified 22,430 high-quality SNPs in the AS25-3 sequence. To facilitate comparison of the different SNP analyses performed in this study, an overview of the results is shown in Table 2. To verify the SNP calling results, DNA was randomly selected from genomic regions of all analyzed strains and amplified for Sanger resequencing. By comparing amplicons with consensus sequences, we tested and verified 77 SNPs, indicating that the results of the SNP calling were highly reliable.
Identification of differently inherited genomic regions in ascospore isolates
To identify the different inherited regions in the AS25-3 genome, SNPs from both parental strains, P2niaD18 and AS25 (Fig. S10), were compared with the AS25-3 consensus sequence. We found that 72.7% of the high-quality SNPs could be unambiguously allocated to AS25, and 18.9% seemed to originate from P2niaD18. Based on these results, we performed sliding window analysis to uncover the distribution of recombinant regions on the AS25-3 genome (Fig. 6A). Different colors indicate recombinations due to crossover, crossover-associated gene conversion or noncrossover gene conversion, as discussed below. For optimal resolution, we used 10 kb steps for a genome-wide scale and 500 bp steps for a detailed view in the sliding window analysis, and determined the frequency and thus distribution of SNPs. The majority of SNPs in AS25-3 were unique to AS25, and most of the windows that represented SNPs inherited from P2niaD18 were clustered in distinct regions. Areas with a low number of SNPs could not be assigned to either Pc3 or AS25 and are shown in white. Using a 10 kb sliding-window analysis, we calculated 301 recombination events within the 32.4 Mb genome of AS25-3.
Figure 6.

Genome-wide distribution of recombination events in AS25-3.A. High-quality SNPs of the parental strains P2niaD18 and AS25 were compared with the progeny AS25-3. For a genome-wide scale, sliding window analysis with 10 kb windows were performed to display the origin of the sequence. Regions clearly assigned to P2niaD18 are blue; regions inherited from AS25 are red.B. To validate selected crossing over sites in detail, 500 bp sliding window analysis and Sanger resequencing was performed. As an example, a recombination site within a 7 kb region, containing 27 SNPs, from chromosome II was verified (position is indicated by triangle). The predicted region of recombination, framed in yellow, is located between the two adjacent SNPs that are differently inherited.
To verify recombinant sequences further, we amplified DNA from three recombinant regions for Sanger sequencing (Fig. 6, Fig. S11). From this analysis, we found that all selected regions contained DNA derived from both parental strains. In Fig. 6B, a representative recombination site is shown with 27 SNPs on a 7 kb region between nucleotides 10,154,500–10,161,500 on chromosome II. Our data showed that the ascospore isolate AS25-3 was highly recombinant and that meiotic recombination had occurred during mating between the industrial MAT1-1 penicillin-producer P2niaD18 and MAT1-2 ascospore isolate AS25. Furthermore, our data provide evidence that backcrossing of an ascospore isolate with P2niaD18 increased the number of sequences derived from the industrial MAT1-1-1 strain, and as discussed below, this has also been observed with other fungi carrying dissimilar karyotypes.
Discussion
MAT locus-encoded TFs are involved in regulating mating and sexual development in filamentous fungi. These TFs are critical for MAT identity; they also govern the expression of pheromone and pheromone receptor genes and control meiosis-inducing genes (van Heeckeren et al., 1998; Debuchy and Turgeon, 2006). We recently demonstrated that a MAT1-1-1 encoded TF had adopted additional functions in fungal development (Böhm et al., 2013). Here, we show that a MAT1-2-1 encoded TF harboring an HMG domain regulating light-dependent conidiosporogenesis and the frequency of conidiospore germination. In contrast to the MAT1-2-1 wild type, MAT1-2-1 deletion strains had equivalent sporulation levels in light and darkness and a higher spore germination rate. Therefore, we propose that MAT1-2-1, albeit not differentially expressed upon long-term light illumination, acts as a repressor of light-dependent sporulation and conidiospore germination.
The HMG mating-type TF promotes sexual and asexual developmental processes
Filamentous fungi grow by forming a dense network of hyphae, which develops by hyphal tip extension and branching. In response to different environmental stimuli, such as light, nutrients or signaling peptides, fungi develop characteristic structures for either asexual or sexual reproduction. In P. chrysogenum, the balance between asexual and sexual development is controlled by light. Asexual development and conidiospore production typically takes places under light conditions and is repressed in darkness. Wild-type strains produce twice as many asexual spores in light compared with growth in complete darkness. However, sexual reproduction with the formation of fruiting bodies is induced in darkness and abolished by light (Böhm et al., 2013). The latter process resembles the light-controlled development observed in other filamentous fungi, e.g. A. nidulans or A. fumigatus (O'Gorman et al., 2009; Sarikaya Bayram et al., 2010). In A. nidulans red light perceived by the phytochrome, FphA regulates the transition from asexual to sexual development. Red light activates asexual development with the production of conidiospores and stops sexual development (Idnurm and Heitman, 2005). In darkness, the sexual pathway is enhanced. Light sensing in fungi requires an appropriate system of receptors for detecting the different incoming light qualities (Blumenstein et al., 2005; Purschwitz et al., 2008; Bayram et al., 2010).
A similar role for regulating the balance of asexual and sexual development is also described for VeA (Calvo, 2008). This protein seems to act as a link between light sensing and development in A. nidulans (Bayram et al., 2008). In P. chrysogenum, VelA and VelB, two subunits of a homologue of the velvet complex control light-dependent asexual sporulation (Hoff et al., 2010a; Kopke et al., 2013). The fact that deletion of MAT1-2-1 shows the same phenotype as deletion of velvet subunits suggests that MAT1-2-1 and velvet subunits (TFs) act together in an identical signaling pathway for light-dependent sporulation. For Fusarium verticillioides, MAT1-2-1 deletion strains showed a reduction in asexual sporulation under diurnal light conditions or in complete darkness (Bodor et al., 2012). Thus, depending on the fungal species, MAT1-2-1 appears to control various fungal developmental programs, and in P. chrysogenum probably ensures that sexual development takes place during darkness by repressing conidiosporogenesis.
Depending on whether the host strain carries the MAT1-1 or MAT1-2 locus, MAT1-2-1 controls spore and germ tube formation differently. In the presence of MAT1-1-1, the introduction of MAT1-2-1 leads to a radical reduction in spore germination. Based on these results, one could speculate that the two MAT TFs form a heterodimer, which changes their DNA-binding activity and/or specificity. It is described for many TFs and sex-determining proteins of various species from yeasts to humans that homo or heterodimer formation changes their function in regulating developmental processes. In diverse yeasts, the heterodimeric a1-α2 complex represses haploid-specific gene expression and supports diploid-specific gene expression, which promotes meiosis (Booth et al., 2010). Similarly, in Ustilago maydis, bE and bW heterodimeric complexes promote a switch from the haploid yeast phase to the pathogenic dikaryotic phase with filament growth (Kämper et al., 1995). The human SOX (sex-determining region [SRY]-type HMG box) family of TFs is known to determine cell fate during organ development. Homodimers of SOX proteins inhibit DNA binding, while heterodimers support DNA-binding functions (Kasimiotis et al., 2000).
Independent of the host strain, we observed the formation of pellets with heterogeneous sizes. We hypothesized that this effect was due to enhanced conidiospore agglutination, which indicates that the MAT1-2 locus regulates the surface properties of asexual spores. Pellet formation depends on several morphological processes. Pellets typically result from (i) aggregation of spores before germination, (ii) clustering of germ tubes, or (iii) aggregation of young mycelia. The process depends on the physicochemical and physiological characteristics of spores and hyphae (Prosser, 1995). Pellet size is affected by environmental factors, such as the concentration of the spore inoculum, the pH and composition of the growth medium; however, size is also affected by the genetic properties of particular fungal strains (Fomina and Gadd, 2002). Based on this and our previous results, we concluded that pellet size resulted from the length of the germ tubes (Böhm et al., 2013) and the surface properties of conidiospores. We hypothesize that the agglutination of spores results in larger-sized pellets, which reached a diameter of nearly 7000 μm. It was previously shown that the aggregation of spores is dependent on their surface properties, such as their electrical charge and hydrophobicity, and that aggregation leads to pellet formation (Clement et al., 1994; Jones, 1994). Additionally, the stability of the formed agglomerates depends on electrostatic forces, hydrophobic interactions and chemical interactions, such as polysaccharide bridging (Dynesen and Nielsen, 2003). The aggregated conidiospores attach to other germinating or nongerminated conidiospores, and the resulting aggregates accumulate to form so-called superaggregates (Dynesen and Nielsen, 2003). This phenomenon probably occurred with the MAT1-2-1 overexpression strains described here. We propose that the MAT TF alters the surface properties of conidiospores, possibly by changing their hydrophobicity. The significant effects on pellet formation suggest that manipulating MAT genes might provide a novel strategy for strain improvement. Pellet formation is of great interest in industrial fermentation processes as pellet aggregation reduces the viscosity of the culture broth; lower viscosity facilitates shearing and aeration of the culture and thus promotes productivity due to enhanced supplies of oxygen and nutrients.
Our functional characterization of the MAT1-2 locus from P. chrysogenum demonstrated that MAT1-2-1, similar to MAT1-1-1, functions in a broad spectrum of asexual developmental processes. However, the function of MAT1-2 was clearly distinct from the opposite mating-type locus. For example, we observed no effect on penicillin production in any of the recombinant strains (Böhm, unpublished). In other ascomycetes, MAT1-2-1 is mainly described as a regulator of ascospore formation and fruiting body development, and regulates the expression of genes involved in sexual development or in the pheromone system. In basidiomycetes, the mating-type locus was already implicated in morphological changes. In Cryptococcus neoformans, the α allele of the MAT locus enhanced hyphal growth during fruiting compared with the a allele. Thus, isolates possessing the a allele produced shorter hyphae than isolates with the α allele (Lin et al., 2006). In the plant pathogenic basidiomycete U. maydis, the mating-type locus-encoded homeodomain TFs bE and bW are responsible for fungal proliferation in the plant host (Wahl et al., 2010).
Despite chromosomal divergences of parental strains, ascospore isolates carry highly recombinant chromosomes
Our mating experiments clearly demonstrate that the MAT1-2 locus is a prerequisite for completing a sexual life cycle. The MAT1-2-1 deletion strain was unable to form cleistothecia in crossings with a MAT1-1 strain. Our data are consistent with a recent report on the homothallic fungus, Sordaria macrospora (Pöggeler et al., 2006), where deletion of the gene for MAT1-2-1 (Smta-1) encoding a homologous HMG TF resulted in mutant strains that lacked fruiting body formation. Similarly, other studies showed that sterile strains were obtained from A. nidulans and A. fumigatus when the homologous genes were deleted (Paoletti et al., 2007; Szewczyk and Krappmann, 2010).
In contrast to our previous crossing experiments (Böhm et al., 2013), we used strains for mating that are distinguished by deviant karyotypes. Recently, we showed that parental strain P2niaD18 has undergone several rounds of mutagenesis, leading to two major chromosomal rearrangements differing from the Q176 progenitor strain of the Wisconsin lineage (Specht et al., 2014). A further chromosomal rearrangement was observed when the derived Wisconsin strains were compared with the primary wild-type isolate (NRRL 1951). At this chromosomal site, NRRL 1951 resembles the chromosomal organization of the Pc3 MAT1-2 wild type (Fig. S7).
Genome sequencing in concert with SNP calling allowed us to determine recombination events on the AS25-3 genome that might be due to crossovers, crossover-associated gene conversions or noncrossover gene conversions. The rather complex pattern of recombinant sequences and the high amount of recombination sites can be explained in several ways. Recent high-resolution mapping of meiotic recombination events in yeast showed that differences occurring in the distribution of crossovers and noncrossovers are other than expected by chance (Mancera et al., 2008).
Studies in yeast calculated 9.5 double recombination events per megabase within the progeny of two karyotypic similar yeast strains (Mancera et al., 2008). Taking a 10 kb sliding-window analysis, we found 301 recombinant regions, corresponding to 9.3 recombination events per megabase. Our finding of differently inherited SNPs is very similar to recombination mapping data in the malaria parasite Plasmodium falciparum (Samarakoon et al., 2011). Using high-throughput sequencing followed by a sliding window analysis, these authors showed a highly dissimilar distribution of differently inherited SNPs on chromosomes of sibling parasite clones. It was further shown that complex crossing over breakpoints are accompanied by a conversion tract, which is defined by rapid allele (SNP) changes.
Furthermore, early work with yeast showed that multiple heterozygosities significantly alter the outcome of recombination events, reducing the frequency of crossovers and simultaneously increasing the frequency of gene conversions. This lead to a high number of exceptional tetrads (Borts and Haber, 1987). The effect of heterozygosity on recombination was further studied by crossing two yeast strains selected due to their substantial genetic diversity, leading to elevated mitotic mutation rates in segregants of intraspecies yeast hybrids (Mancera et al., 2011). The rather high levels of heterozygosity within the two P. chrysogenum strains used for mating were demonstrated by genome sequencing (Specht et al., 2014) and identifcation of chromosomal rearrangements (Fig. S8). This let us to conclude that the high levels of heterozygosity between mating partners from P. chrysogenum are responsible for the mosaic and nonrandom distribution of parental genome sequences.
Compared with AS25, the genome of AS25-3 contained four-fold the number of SNPs assigned to the MAT-1-1-1 parental strain. Thus, we demonstrate here that backcrossing products of a progeny strain with an industrial penicillin producer can cause an increase in the number of sequences derived from the industrial parent. We speculate that the increased recombination rate was caused by a change in chromosome architecture during the first mating. Most likely greater chromosomal similarity promotes a higher chromosomal recombination rate during meiosis. The elevated exchange of genomic sequences implies that further backcrossing could be used to combine strain-specific traits. It was previously suggested that the occurrence and extent of chromosome length polymorphisms within a species inversely correlate with the frequency of meiosis (Kistler and Miao, 1992). This hypothesis was based on the observation that extreme karyotypic variation was only found in species that reproduce asexually. Experimental proof was provided from crossing experiments with the model ascomycete, S. macrospora. In this ascomycete, strains of various geographic origins possessing a high degree of chromosome variability were crossed, which, depending on the chromosomal configuration, reduced fertility in sexual crossings (Pöggeler et al., 2000).
Our data on chromosomal recombination are relevant to the constant commercial demand for constructing novel production strains. For instance, genes that control hyphal morphology, mycotoxin production and antifungal resistance are of particular economic importance (Anderson, 2005; Keller et al., 2005). Furthermore, our work provides options for combining genome biology with conventional genetic and culturing techniques and, thus, has implications for optimizing penicillin production in industrial strains.
In conclusion, this study has extended the concept of using mating-type loci for classical strain improvement (Pöggeler, 2001). Moreover, the new technique of crossing industrial producers combined with rapidly acquired genomic information will provide a basis for future targeted searches to identify genes relevant for penicillin biosynthesis. Thus, our results point to future strategies that integrate available molecular tools for P. chrysogenum and applying sexual lifecycle selection. This approach, previously called ‘concerted breeding’ (Esser, 1977), will increase our knowledge about complex metabolic pathways and provide solutions for improving industrial strains.
Experimental procedures
Strains and culture conditions
The Escherichia coli strain, K12 XL1-Blue, was used for general plasmid construction and maintenance (Bullock et al., 1987; Sambrook and Russell, 2001). Standard protocols were used for cloning and propagation of recombinant plasmids (Sambrook and Russell, 2001). All wild-type and recombinant P. chrysogenum strains investigated in this study are summarized in Table S2. Strains were grown at 27°C with shaking (120 r.p.m.) in liquid complete culture medium (CCM). For solid media, we used CCM or M322, as previously described (Minuth et al., 1982; Hoff et al., 2010b). All liquid and solid media were inoculated with 107 spores from freshly prepared spore suspensions derived from cultures grown on M322 medium for 3–4 days.
Transformation
DNA-mediated transformation of P. chrysogenum strains was performed as recently described (Hoff et al., 2010b).
Construction of MAT1-2-1 deletion strains
For characterization and in vitro recombination, the complete MAT locus of Pc3 was amplified and sequenced with primer pairs 3 × 4, 5 × 9 and 6 × 7 (Table S3). For MAT1-2-1 deletion mutants (ΔMAT1-2-1), we used a previously described deletion vector (Böhm et al., 2013; Table S1), which was amenable, due to the highly conserved flanking regions of MAT1-1-1 and MAT1-2-1. This vector was designed to substitute the native MAT1-2-1 gene in Pc3 with the phleomycin resistance cassette (Fig. S4B). The plasmid was linearized with the restriction enzymes, MluI and NotI, and the resulting 4.0 kb fragment was used to transform Pc3. The resulting transformants were screened and analyzed, as described previously (Hoff et al., 2010b). Purified DNA from single ΔMAT1-2-1 spore isolates was used to verify the complete lack of the MAT1-2-1 gene by Southern hybridization analysis (Fig. S4B).
Plasmid and strain construction of complementation strains
For construction of the MAT1-2-1 complementation plasmid, 1 156 bp of the 5′ region together with the entire MAT1-2 locus was amplified by PCR using primer pair 5FMAT2inf_f and 5FMAT2inf_r. Fusion via In-Fusion cloning (Clontech® Laboratories Inc., USA) with the PstI-BamHI fragment of plasmid pKOku70 resulted in the construction of vector pKOku70MAT2. Subsequently, 1168 bp of the 3′ region amplified with primer pair 3FMAT2inf_f and 3FMAT2inf_r was fused via In-Fusion cloning with the SalI fragment of plasmid pKOku70MAT2. The resulting plasmid pKompMAT2 carried the MAT1-2-1 gene, under the control of the native promoter, and the NAT gene as a selectable marker. For complementation analysis, the ΔMAT1-2-1 T10 strain was transformed with plasmid, pKompMAT2. The successful rescue of the MAT1-2-1 gene was verified by Southern hybridization, and the corresponding strains were designated ΔMAT1-2-1::MAT1-2-1 (Fig. S4B).
Construction of MAT1-2-1 overexpression strains
For generating MAT1-2-1 overexpression strains, the pPgpd-MAT-2-ptrA plasmid was transferred into both P2niaD18 and Pc3. The resulting transformants were selected with pyrithiamine-supplemented agar plates because the construct carried the ptrA resistance gene. Positive transformants were named P2MAT2-OE or Pc3MAT2-OE respectively. Copy numbers of integrated plasmids were determined by Southern hybridization with a 32P-radiolabeled MAT1-2-1 probe (Fig. S3A and B).
Spore quantification assay
To quantify conidiospores, for each strain, four Petri dishes with solid CCM were inoculated with 200 μl of a suspension containing 107 spores. Two dishes were incubated in constant light, and two in complete darkness for 168 h. From each plate, a defined area of about 1 cm2 was cut out, and the spores were counted with a hemocytometer.
Light microscopy
Microscopic observations of spore germination, hyphal morphology and pellet quantification assays were performed as previously described (Hoff et al., 2010a; Böhm et al., 2013). To quantify spore germination, in all cases, 400 conidia from each strain were analyzed with light microscopy after growth for 6, 9, 12, 15, 18 and 48 h on CCM-slides.
Nucleic acid isolation, cDNA synthesis and qRT-PCR
Nucleic acid preparations, hybridizations, cDNA synthesis and qRT-PCR analyses were carried out as described recently (Hoff et al., 2010a). qRT-PCR was conducted with the following modifications: the PromegaGoTaq® qPCR Master Mix was used as recommended by the manufacturer, and the incubation cycles were performed on a StepOnePlus™ Real-Time PCR System (Applied Biosystems, Life Technologies, Darmstadt, Germany) with the primers listed in Table S3.
Crossing experiments
Crossing strains with different mating types were conducted as previously described, with some modifications (Böhm et al., 2013). After isolation, the fruiting bodies were added to 200 μl sterile water supplemented with glass beads. The samples were then heated at 60°C for 30 min and vortexed for 5 min; then, aliquots (50 μl) were plated on oatmeal agar with biotin, incubated at 27°C and examined daily for germinating ascospores. Isolated ascospores were characterized and analyzed by RFLP as previously described (Böhm et al., 2013). Progeny of the cross between AS25 and P2niaD18 were named AS25- with continuous numbers.
DNA sequencing and analysis
Genomic DNA was obtained from the P. chrysogenum strains Pc3, AS25 and AS25-3, as previously described, and sequenced with an Illumina HiSeq 2000 at GATC Biotech AG (Konstanz, Germany). The wild-type strain, Pc3, was sequenced with a paired-end sequencing strategy, in 100 nt reads, and an expected insert size of ∼ 300 bp. The progeny strains, AS25 and AS25-3, were sequenced in 50 nt single-end reads. Raw sequencing data from next-generation sequencing have been deposited in the NCBI sequence read archive (http://www.ncbi.nlm.nih.gov/sra) under the accession numbers SRR1462342 for Pc3, SRR1462340 for AS25 and SRR1462341 for AS25-3. The genomic sequences of P. chrysogenum P2niaD18 is available at DDBJ/EMBL/GenBank under the accession no. JMSF00000000 (Specht et al., 2014) and the de novo assembled scaffolds of strain Pc3 (IB 08/921) has been deposited under the accession no. JPDR00000000. The MultAlign program (http://multalin.toulouse.inra.fr/multalin/) was used for sequence alignment, and GeneDoc (http://iubio.bio.indiana.edu/soft/molbio/ibmpc/genedoc-readme.html) was used for display.
Genome assembly and mapping
The raw sequence reads were processed with quality score-based filtering and trimming with Trimmomatic v0.30 (Bolger et al., 2014). The processed reads of Pc3 were used for de novo assembly with the Velvet assembler v1.2.10 (Zerbino and Birney, 2008). Detailed descriptions of the parameters used with Trimmomatic and Velvet programs are provided (File S1). To detect chromosomal architecture variations between the Pc3 and P2niaD18 strains, the resulting Pc3 scaffolds were aligned to the genomic sequence of P2niaD18 with BLAST and CONTIGuator v2.7.3 (Altschul et al., 1997; Galardini et al., 2011). The filtered and trimmed reads of all three sequenced strains were mapped against a reference genome with bowtie2 v2.1.0 (Langmead and Salzberg, 2012). The resulting SAM files were processed with SAMtools v0.1.19 (File S1; Li et al., 2009).
SNP discovery and sequence comparisons
SNPs were extracted from high-throughput DNA-sequencing data with mpileup, available in the SAMtools package (Li et al., 2009). For this purpose, the processed reads were mapped to a parental reference genomic sequence; the parameters for mpileup and further information are given in File S1. To characterize AS25, SNP discovery was performed with the de novo assembled scaffolds of Pc3 as reference. For isolate AS25-3, the P2niaD18 genome sequence was used as reference (Specht et al., 2014). In addition, the processed reads of AS25 were used for SNP calling in the P2niaD18 genome.
The resulting SNPs were used to identify the origin of genomic regions in the progeny strains. For this purpose, consensus sequences of Pc3, AS25-3 and AS25 were assembled with the mpileup consensus function, and a custom-made Perl script was used to compare SNPs between parental sequences and a descendant consensus sequence (Fig. S10). All SNPs with a minimum quality score of 50 and an allele frequency of 1.0 were considered high-quality SNPs, and they were divided into three categories. The first two categories represented SNPs that could be assigned to one of the two parental strains; the third category represented SNPs that could not be assigned to either of the parents, or were ambiguous, due to low coverage. To guarantee reliable results, we further considered SNPs certain when they were located in the consensus sequence, but as unclear in regions filtered out by varFilter due to insufficient read depth. To identify differently inherited regions in the progeny genome, we performed a comparison between the progeny consensus sequence and each parental SNP. Furthermore, we implemented a sliding window analysis with a custom-made Perl script. To estimate background noise and to clearly identify differentially inherited genomic regions, we integrated all uniquely allocated parental SNPs and the vague or inconsistent results into our analysis. For this purpose, we computed a quality score for each sliding window, based on the number of total SNPs and the number of SNPs that could be allocated to one of the parental strains. All SNPs assigned to a parental strain were assigned the value of −1 or 1, and for each window separately, the sum of these values was divided by the total number of analyzed SNPs. The resulting score for each window was taken as an indication of the genomic origin, and only windows with scores ranging between −1.0 and −0.5 or 0.5 and 1.0, respectively, were considered as reliable.
SNP validation
To validate the accuracy of SNP predictions, PCR analyses were performed, and the products were sequenced with an ABI 3130xl (Department of Chemistry and Biochemistry, Ruhr-University, Bochum, Germany). In addition, we designed primers (Table S3) to verify meiotic crossing-over events. In total, 77 SNPs were validated, and three putative recombinant loci were amplified from parental and progeny strains and Sanger sequenced. To evaluate the resulting sequences, alignments for multiple sequences were performed with Clustal-Omega (Sievers et al., 2011) and analyzed manually.
Acknowledgments
We acknowledge the excellent technical support by Katerina Koutsantas, Ingeborg Godehardt, Susanne Schlewinski and Stefanie Mertens. We are thankful for PD Dr. Minou Nowrousian and Dominik Terfehr for helpful comment on the manuscript; Rebecca Brekau for her help with some of the experiments; Drs. Hubert Kürnsteiner, Thomas Specht and Ivo Zadra for their continuous interest; and Prof. Dr. David Catcheside (Flinders University, Australia) and Prof. Dr. Michael Freitag (Oregon State University, USA) for advice to discuss meiotic recombinations. J.B. and T.A.D. are members of the RUB Research School. This work was supported by the Christian Doppler-Society (Vienna, Austria) and Sandoz GmbH (Kundl, Austria). The authors declare no competing financial interests.
Supporting Information
Additional supporting information may be found in the online version of this article at the publisher's web-site.
References
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W. Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson JB. Evolution of antifungal-drug resistance: mechanisms and pathogen fitness. Nat Rev Microbiol. 2005;3:547–556. doi: 10.1038/nrmicro1179. [DOI] [PubMed] [Google Scholar]
- Bardwell L. A walk-through of the yeast mating pheromone response pathway. Peptides. 2005;26:339–350. doi: 10.1016/j.peptides.2004.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayram Ö, Krappmann S, Ni M, Bok JW, Helmstaedt K, Valerius O, et al. VelB/VeA/LaeA complex coordinates light signal with fungal development and secondary metabolism. Science. 2008;320:1504–1506. doi: 10.1126/science.1155888. [DOI] [PubMed] [Google Scholar]
- Bayram Ö, Braus GH, Fischer R. Rodriguez-Romero J. Spotlight on Aspergillus nidulans photosensory systems. Fungal Genet Biol. 2010;47:900–908. doi: 10.1016/j.fgb.2010.05.008. [DOI] [PubMed] [Google Scholar]
- van den Berg MA, Albang R, Albermann K, Badger JH, Daran JM, Driessen AJ, et al. Genome sequencing and analysis of the filamentous fungus Penicillium chrysogenum. Nat Biotechnol. 2008;26:1161–1168. doi: 10.1038/nbt.1498. [DOI] [PubMed] [Google Scholar]
- Blumenstein A, Vienken K, Tasler R, Purschwitz J, Veith D, Frankenberg-Dinkel N. Fischer R. The Aspergillus nidulans phytochrome FphA represses sexual development in red light. Curr Biol. 2005;15:1833–1838. doi: 10.1016/j.cub.2005.08.061. [DOI] [PubMed] [Google Scholar]
- Bodor Á, Nagygyörgy E. Hornok L. Inactivation of the mat1-2-1 mating type gene results in reduced asexual sporulation and con10 expression in Fusarium verticillioides. Acta Phytopathol Entomol Hungar. 2012;47:7–15. [Google Scholar]
- Bolger AM, Lohse M. Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth LN, Tuch BB. Johnson AD. Intercalation of a new tier of transcription regulation into an ancient circuit. Nature. 2010;468:959–963. doi: 10.1038/nature09560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borts RH. Haber JE. Meiotic recombination in yeast: alteration by multiple heterozygosities. Science. 1987;237:1459–1465. doi: 10.1126/science.2820060. [DOI] [PubMed] [Google Scholar]
- Böhm J, Hoff B, O'Gorman CM, Wolfers S, Klix V, Binger D, et al. Sexual reproduction and mating-type-mediated strain development in the penicillin-producing fungus Penicillium chrysogenum. Proc Natl Acad Sci USA. 2013;110:1476–1481. doi: 10.1073/pnas.1217943110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brakhage AA, Spröte P, Al-Abdallah Q, Gehrke A, Plattner H. Tuncher A. Regulation of penicillin biosynthesis in filamentous fungi. Adv Biochem Eng Biotechnol. 2004;88:45–90. doi: 10.1007/b99257. [DOI] [PubMed] [Google Scholar]
- Bullock WO, Fernandez JM. Short JM. XL1-Blue: a high-efficiency plasmid transforming recA Escherichia coli strain with ß-galactosidase selection. Biotechniques. 1987;5:376–379. [Google Scholar]
- Butler G, Kenny C, Fagan A, Kurischko C, Gaillardin C. Wolfe KH. Evolution of the MAT locus and its Ho endonuclease in yeast species. Proc Natl Acad Sci USA. 2004;101:1632–1637. doi: 10.1073/pnas.0304170101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo AM. The VeA regulatory system and its role in morphological and chemical development in fungi. Fungal Genet Biol. 2008;45:1053–1061. doi: 10.1016/j.fgb.2008.03.014. [DOI] [PubMed] [Google Scholar]
- Clement JA, Porter R, Butt TM. Beckett A. The role of hydrophobicity in attachment of urediniospores and sporelings of Uromyces viciae-fabae. Mycol Res. 1994;98:1217–1228. [Google Scholar]
- Debuchy R. Turgeon B. Mating-type structure, evolution, and function in Euascomycetes. In: Esser K, editor; Growth, Differentiation and Sexuality. Heidelberg: Springer; 2006. pp. 293–323. (ed.)., and. [Google Scholar]
- Debuchy R, Berteaux-Lecellier V. Silar P. Mating systems and sexual morphogenesis in ascomycetes. In: Ebbole DJ, editor; Borkovich KA, editor. Cellular and Molecular Biology of Filamentous Fungi. Washington, DC: ASM Press; 2010. pp. 501–535. and (eds)., and. [Google Scholar]
- Desjardins AE, Brown DW, Yun SH, Proctor RH, Lee T, Plattner RD, et al. Deletion and complementation of the mating type (MAT) locus of the wheat head blight pathogen Gibberella zeae. Appl Environ Microbiol. 2004;70:2437–2444. doi: 10.1128/AEM.70.4.2437-2444.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer PS. Sexual reproduction and significance of MAT in the Aspergilli. In: Casselton LA, editor; Heitman J, Kronstad JW, Taylor JW, editors. Sex in Fungi: Molecular Determination and Evolutionary Implications. Washington, DC: ASM Press; 2007. pp. 123–142. and (eds). [Google Scholar]
- Dynesen J. Nielsen J. Surface hydrophobicity of Aspergillus nidulans conidiospores and its role in pellet formation. Biotechnol Prog. 2003;19:1049–1052. doi: 10.1021/bp0340032. [DOI] [PubMed] [Google Scholar]
- Esser K. Concerted breeding in fungi and its biotechnological application. Endeavour. 1977;1:143–148. [Google Scholar]
- Ferreira AV, An Z, Metzenberg RL. Glass NL. Characterization of mat A-2mat A-3 and ΔmatA mating-type mutants of Neurospora crassa. Genetics. 1998;148:1069–1079. doi: 10.1093/genetics/148.3.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fomina M. Gadd GM. Influence of clay minerals on the morphology of fungal pellets. Mycol Res. 2002;106:107–117. [Google Scholar]
- Galardini M, Biondi EG, Bazzicalupo M. Mengoni A. CONTIGuator: a bacterial genomes finishing tool for structural insights on draft genomes. Source Code Biol Med. 2011;6:11. doi: 10.1186/1751-0473-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galgoczy DJ, Cassidy-Stone A, Llinas M, O'Rourke SM, Herskowitz I, DeRisi JL. Johnson AD. Genomic dissection of the cell-type-specification circuit in Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 2004;101:18069–18074. doi: 10.1073/pnas.0407611102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass NL, Grotelueschen J. Metzenberg RL. Neurospora crassa A mating-type region. Proc Natl Acad Sci USA. 1990;87:4912–4916. doi: 10.1073/pnas.87.13.4912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber JE. Mating-type genes and MAT switching in Saccharomyces cerevisiae. Genetics. 2012;191:33–64. doi: 10.1534/genetics.111.134577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Heeckeren WJ, Dorris DR. Struhl K. The mating-type proteins of fission yeast induce meiosis by directly activating mei3 transcription. Mol Cell Biol. 1998;18:7317–7326. doi: 10.1128/mcb.18.12.7317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herskowitz I. A regulatory hierarchy for cell specialization in yeast. Nature. 1989;342:749–757. doi: 10.1038/342749a0. [DOI] [PubMed] [Google Scholar]
- Herskowitz I, Rine J. Strathern J. Mating-type determination and mating-type interconversion in Saccharomyces cerevisiae. Cold Spring Harb Monogr Arch. 1992;21:583–656. [Google Scholar]
- Hoff B, Pöggeler S. Kück U. Eighty years after its discovery, Fleming's Penicillium strain discloses the secret of its sex. Eukaryot Cell. 2008;7:465–470. doi: 10.1128/EC.00430-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoff B, Kamerewerd J, Sigl C, Mitterbauer R, Zadra I, Kürnsteiner H. Kück U. Two components of a velvet-like complex control hyphal morphogenesis, conidiophore development, and penicillin biosynthesis in Penicillium chrysogenum. Eukaryot Cell. 2010a;9:1236–1250. doi: 10.1128/EC.00077-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoff B, Kamerewerd J, Sigl C, Zadra I. Kück U. Homologous recombination in the antibiotic producer Penicillium chrysogenum: strain ΔPcku70 shows up-regulation of genes from the HOG pathway. Appl Microbiol Biotechnol. 2010b;85:1081–1094. doi: 10.1007/s00253-009-2168-4. [DOI] [PubMed] [Google Scholar]
- Idnurm A. Heitman J. Light controls growth and development via a conserved pathway in the fungal kingdom. PLoS Biol. 2005;3:e95. doi: 10.1371/journal.pbio.0030095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones E. Fungal adhesion. Mycol Res. 1994;98:961–981. [Google Scholar]
- Kasimiotis H, Myers MA, Argentaro A, Mertin S, Fida S, Ferraro T, et al. Sex-determining region Y-related protein SOX13 is a diabetes autoantigen expressed in pancreatic islets. Diabetes. 2000;49:555–561. doi: 10.2337/diabetes.49.4.555. [DOI] [PubMed] [Google Scholar]
- Kämper J, Reichmann M, Romeis T, Bölker M. Kahmann R. Multiallelic recognition: nonself-dependent dimerization of the bE and bW homeodomain proteins in Ustilago maydis. Cell. 1995;81:73–83. doi: 10.1016/0092-8674(95)90372-0. [DOI] [PubMed] [Google Scholar]
- Keller NP, Turner G. Bennett JW. Fungal secondary metabolism – from biochemistry to genomics. Nat Rev Microbiol. 2005;3:937–947. doi: 10.1038/nrmicro1286. [DOI] [PubMed] [Google Scholar]
- Kistler HC. Miao VP. New modes of genetic change in filamentous fungi. Annu Rev Phytopathol. 1992;30:131–153. doi: 10.1146/annurev.py.30.090192.001023. [DOI] [PubMed] [Google Scholar]
- Kopke K, Hoff B, Bloemendal S, Katschorowski A, Kamerewerd J. Kück U. Members of the Penicillium chrysogenum velvet complex play functionally opposing roles in the regulation of penicillin biosynthesis and conidiation. Eukaryot Cell. 2013;12:299–310. doi: 10.1128/EC.00272-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronstad JW. Staben C. Mating type in filamentous fungi. Annu Rev Genet. 1997;31:245–276. doi: 10.1146/annurev.genet.31.1.245. [DOI] [PubMed] [Google Scholar]
- Kück U. Böhm J. Mating type genes and cryptic sexuality as tools for genetically manipulating industrial molds. Appl Microbiol Biotechnol. 2013;97:9609–9620. doi: 10.1007/s00253-013-5268-0. [DOI] [PubMed] [Google Scholar]
- Langmead B. Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein J. The Panlabs penicillin strain improvement program. In: Hostalek Z, editor; Vanek Z, editor. Overproduction of Microbial Metabolites; Strain Improvement and Process Control Strategies. Boston: Butterworth Publishers; 1986. pp. 105–139. and (eds). [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Huang JC, Mitchell TG. Heitman J. Virulence attributes and hyphal growth of C. neoformans are quantitative traits and the MATα allele enhances filamentation. PLoS Genet. 2006;2:e187. doi: 10.1371/journal.pgen.0020187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancera E, Bourgon R, Brozzi A, Huber W. Steinmetz LM. High-resolution mapping of meiotic crossovers and non-crossovers in yeast. Nature. 2008;454:479–485. doi: 10.1038/nature07135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancera E, Bourgon R, Huber W. Steinmetz LM. Genome-wide survey of post-meiotic segregation during yeast recombination. Genome Biol. 2011;12:R36. doi: 10.1186/gb-2011-12-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minuth W, Tudzynski P. Esser K. Extrachromosomal genetics of Cephalosporium acremonium. Curr Genet. 1982;25:34–40. doi: 10.1007/BF00435215. [DOI] [PubMed] [Google Scholar]
- Nelson MA. Mating systems in ascomycetes: a romp in the sac. Trends Genet. 1996;12:69–74. doi: 10.1016/0168-9525(96)81403-x. [DOI] [PubMed] [Google Scholar]
- Ni M, Feretzaki M, Sun S, Wang X. Heitman J. Sex in fungi. Annu Rev Genet. 2011;45:405–430. doi: 10.1146/annurev-genet-110410-132536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen J. Physiological Engineering Aspects of Penicillium Chrysogenum. Singapore: World Scientific Publishing; 1997. [Google Scholar]
- Nielsen R, Paul JS, Albrechtsen A. Song YS. Genotype and SNP calling from next-generation sequencing data. Nat Rev Genet. 2011;12:443–451. doi: 10.1038/nrg2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Gorman CM, Fuller HT. Dyer PS. Discovery of a sexual cycle in the opportunistic fungal pathogen Aspergillus fumigatus. Nature. 2009;457:471–474. doi: 10.1038/nature07528. [DOI] [PubMed] [Google Scholar]
- Paoletti M, Seymour FA, Alcocer MJ, Kaur N, Calvo AM, Archer DB. Dyer PS. Mating type and the genetic basis of self-fertility in the model fungus Aspergillus nidulans. Curr Biol. 2007;17:1384–1389. doi: 10.1016/j.cub.2007.07.012. [DOI] [PubMed] [Google Scholar]
- Pitt JI. Hocking AD. Fungi and Food Spoilage. Dordrecht, Heidelberg, London, New York: Springer; 2009. [Google Scholar]
- Pöggeler S. Mating-type genes for classical strain improvements of ascomycetes. Appl Microbiol Biotechnol. 2001;56:589–601. doi: 10.1007/s002530100721. [DOI] [PubMed] [Google Scholar]
- Pöggeler S. MAT and its role in the homothallic ascomycete Sordaria macrospora. In: Casselton LA, editor; Heitman J, Kronstad JW, Taylor JW, editors. Sex in Fungi: Molecular Determination and Evolutionary Implications. Washington, DC: ASM Press; 2007. pp. 171–188. and (eds). [Google Scholar]
- Pöggeler S, Masloff S, Jacobsen S. Kück U. Karyotype polymorphism correlates with intraspecific infertility in the homothallic ascomycete Sordaria macrospora. J Evol Biol. 2000;13:281–289. [Google Scholar]
- Pöggeler S, Nowrousian M, Ringelberg C, Loros JJ, Dunlap JC. Kück U. Microarray and real-time PCR analyses reveal mating type-dependent gene expression in a homothallic fungus. Mol Genet Genomics. 2006;275:492–503. doi: 10.1007/s00438-006-0107-y. [DOI] [PubMed] [Google Scholar]
- Pöggeler S, O'Gorman CM, Hoff B. Kück U. Molecular organization of the mating-type loci in the homothallic Ascomycete Eupenicillium crustaceum. Fungal Biol. 2011;115:615–624. doi: 10.1016/j.funbio.2011.03.003. [DOI] [PubMed] [Google Scholar]
- Prosser J. Kinetics of filamentous growth and branching. In: Gadd GM, editor; Gow NAR, editor. The Growing Fungus. London: Springer; 1995. pp. 301–318. and (eds.). [Google Scholar]
- Purschwitz J, Müller S, Kastner C, Schöser M, Haas H, Espeso EA, et al. Functional and physical interaction of blue- and red-light sensors in Aspergillus nidulans. Curr Biol. 2008;18:255–259. doi: 10.1016/j.cub.2008.01.061. [DOI] [PubMed] [Google Scholar]
- Samarakoon U, Regier A, Tan A, Desany BA, Collins B, Tan JC, et al. High-throughput 454 resequencing for allele discovery and recombination mapping in Plasmodium falciparum. BMC Genomics. 2011;12:116. doi: 10.1186/1471-2164-12-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J. Russell DW. Molecular Cloning. A Laboratory Manual. New York: Cold Spring Harbor Laboratory Press, Cold Spring Harbor; 2001. [Google Scholar]
- Sarikaya Bayram Ö, Bayram Ö, Valerius O, Park HS, Irniger S, Gerke J, et al. LaeA control of velvet family regulatory proteins for light-dependent development and fungal cell-type specificity. PLoS Genet. 2010;6:e1001226. doi: 10.1371/journal.pgen.1001226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Specht T, Dahlmann TA, Zadra I, Kürnsteiner H. Kück U. Complete sequencing and chromosome-scale genome assembly of the industrial progenitor strain P2niaD18 from the penicillin producer Penicillium chrysogenum. Genome Announc. 2014;2:e00577-14. doi: 10.1128/genomeA.00577-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staben C. Yanofsky C. Neurospora crassa a mating-type region. Proc Natl Acad Sci USA. 1990;87:4917–4921. doi: 10.1073/pnas.87.13.4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szewczyk E. Krappmann S. Conserved regulators of mating are essential for Aspergillus fumigatus cleistothecium formation. Eukaryot Cell. 2010;9:774–783. doi: 10.1128/EC.00375-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl R, Zahiri A. Kämper J. The Ustilago maydis b mating type locus controls hyphal proliferation and expression of secreted virulence factors in planta. Mol Microbiol. 2010;75:208–220. doi: 10.1111/j.1365-2958.2009.06984.x. [DOI] [PubMed] [Google Scholar]
- Zerbino DR. Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.