

ABSTRACT
Excessive weight and obesity are associated with the development of diabetes mellitus type 2 (DMII) in humans. They also pose high risks of Staphylococcus aureus colonization and overt infections. S. aureus causes a wide range of severe illnesses in both healthy and immunocompromised individuals. Among S. aureus virulence factors, superantigens are essential for pathogenicity. In this study, we show that rabbits that are chronically exposed to S. aureus superantigen toxic shock syndrome toxin-1 (TSST-1) experience impaired glucose tolerance, systemic inflammation, and elevated endotoxin levels in the bloodstream, all of which are common findings in DMII. Additionally, such DMII-associated findings are also seen through effects of TSST-1 on isolated adipocytes. Collectively, our findings suggest that chronic exposure to S. aureus superantigens facilitates the development of DMII, which may lead to therapeutic targeting of S. aureus and its superantigens.
IMPORTANCE
Obesity has a strong correlation with type 2 diabetes, in which fatty tissue, containing adipocytes, contributes to the development of the illness through altered metabolism and chronic inflammation. The human microbiome changes in persons with obesity and type 2 diabetes, including increases in Staphylococcus aureus colonization and overt infections. While the microbiome is essential for human wellness, there is little understanding of the role of microbes in obesity or the development of diabetes. Here, we demonstrate that the S. aureus superantigen toxic shock syndrome toxin-1 (TSST-1), an essential exotoxin in pathogenesis, induces inflammation, lipolysis, and insulin resistance in adipocytes both in vitro and in vivo. Chronic stimulation of rabbits with TSST-1 results in impaired systemic glucose tolerance, the hallmark finding in type 2 diabetes in humans, suggesting a role of S. aureus and its superantigens in the progression to type 2 diabetes.
INTRODUCTION
The occurrence of diabetes mellitus type 2 (DMII) is an uncontrolled pandemic. In 2013, nearly 350 million people were diagnosed with DMII worldwide, and the number is expected to be more than 500 million by 2035 (1). Approximately 5 to 10% of the total health care budget has been used to manage DMII in many countries (1, 2). Once the disease progresses, DMII may result in severe complications, including congenital heart failure, renal failure, blindness, arterial diseases, and diabetic foot ulcers (3). DMII is often considered a syndrome of disordered metabolism, with abnormally high blood glucose levels.
Common symptoms of DMII include hyperglycemia, excessive urine production, compensatory thirst, increased fluid intake, blurred vision, unexplained weight loss, lethargy, and changes in energy metabolism. The disease is primarily characterized by impaired glucose tolerance due to deficiencies in insulin action and/or insulin secretion (4). However, chronic inflammation and high bloodstream levels of endotoxin have also consistently been observed in individuals with DMII (5).
While there is a strong connection between DMII and inheritable genetics, obesity contributes to approximately 55% of DMII cases (4). In obesity, the disparity between fatty acid uptake and oxidation results in excessive accumulation of triacylglycerol and fatty acid metabolites in the skeletal muscle (6), which can lead to decreases in insulin signaling and glucose disposal rates. In addition, with the expansion of adipose tissue as obesity progress, there may be an upregulation of proinflammatory cytokine production by adipocytes after exposure to endotoxin and environmental cues (5, 7). Such prolonged stimulation leads to chronic subclinical inflammation, as well as insulin resistance, which may ultimately contribute to the development of DMII.
Interestingly, the microbiome is also altered in obesity. In the gut, there are decreases in Bacteriodetes and increases in Firmicutes; the latter phylum is dominated by Gram-positive organisms like Staphylococcus species (8, 9). It is hypothesized that such changes in the microbiome correlate with increased energy extracted from the diet, which in turn promotes the development of obesity (8, 10, 11). Furthermore, the rate of Staphylococcus aureus nasal colonization is also increased in men and women with a high body mass index (12). In addition, S. aureus skin infection is also more prevalent in overweight and obese individuals than in lean subjects (12, 13). Although often considered an opportunistic pathogen, S. aureus causes numerous life-threatening infections in humans, resulting in menstrual toxic shock syndrome (TSS), pneumonia, sepsis, osteomyelitis, and endocarditis (14). Considering the strong correlation between obesity and DMII and the suggested roles of microbes in the pathophysiology of obesity, it is possible that the presence of S. aureus in obese individuals has an impact on the development of DMII.
Within the S. aureus virulence factor repertoire, superantigens (SAgs) are associated with major infections caused by the organism (14). All human S. aureus pathogenic isolates produce one or more SAgs, and 22 S. aureus SAgs have been identified (14). The hallmark SAg activity is the ability to induce systemic inflammation by stably cross-bridging the variable region of the β chain of T cell receptors (Vβ-TCRs) and α and/or β chains of major histocompatibility complex class II (MHC-II) molecules of antigen-presenting cells (14). Recent studies in rabbits show that interference with SAgs successfully prevents serious infections associated with S. aureus (15–17).
In a previous study, we showed that the S. aureus SAg toxic shock syndrome toxin-1 (TSST-1), alone and in combination with endotoxin, induces the production of the proinflammatory cytokines interleukin-6 (IL-6) and IL-8 in immortalized human adipocytes. Since inflammation has a major role in DMII, for example, by inducing insulin resistance in liver, skeletal muscle, and adipose tissues (7), we hypothesized that SAgs could contribute to development of DMII, either alone or by increasing circulating endotoxin. Therefore, in this study, we examined whether chronic exposure to TSST-1 has an impact on the development of DMII.
RESULTS
TSST-1 induced impaired glucose tolerance in vivo.
To examine the relationship between SAgs and DMII, we challenged New Zealand White rabbits (5/group) chronically with a sublethal dose of TSST-1 or phosphate-buffered saline (PBS; control) for 6 weeks. Weekly, rabbits were challenged with glucose subcutaneously, and the ability to metabolize glucose was monitored by use of a conventional glucose meter. TSST-1-treated rabbits exhibited gradually reduced abilities to metabolize glucose in comparison with its metabolization in control animals (Fig. 1A). Even though the glucose metabolism deficiency was not observed in the first 2 weeks of TSST-1 treatment, once it occurred, the deficiency persisted and worsened until the end of the experiment, suggesting that the influence of TSST-1 on impaired glucose tolerance resulted from chronic exposure.
FIG 1 .

Chronic exposure to TSST-1 induces impaired glucose metabolism. (A) Blood glucose levels in two groups of rabbits (5/group) over time, as assayed weekly. Statistically significant differences were determined by two-way analysis of variance (ANOVA). (B) Relative pancreatic insulin transcript levels in the same two groups of animals, analyzed immediately after the glucose challenge test at the end of the 6-week experiment. Statistically significant differences were determined by Student’s t test. Error bars show standard deviations.
Immediately after the glucose tolerance test at the end of the sixth week, the rabbits were sacrificed and their pancreases were isolated. For each pancreas, three random areas were selected for total RNA extraction. Quantitative reverse transcription PCRs (RT-qPCRs) were then performed to analyze the levels of insulin transcript in the two groups of animals. TSST-1-treated rabbits had higher insulin transcript levels than the control animals (Fig. 1B), indicating normal β-cell function in SAg-treated rabbits. Considering that these rabbits also experienced impaired glucose tolerance, these data collectively suggest that chronic exposure to TSST-1 induced insulin resistance in the animals, resulting in deficient glucose metabolism.
TSST-1 induced systemic inflammation and elevated bloodstream endotoxin in rabbits.
Despite the difference in abilities to clear blood glucose, there was no significant difference in body weight between TSST-1- and PBS-treated animals (Fig. S1A in the supplemental material), and there was no apparent sign of distress in the TSST-1-treated rabbits over the course of the experiment. However, gross anatomy analysis showed that the spleens of TSST-1-treated animals were larger and collectively heavier than those of the controls (Fig. 2A and B). Furthermore, examination of plasma revealed that the TSST-1-treated rabbits also had significantly higher levels of tumor necrosis factor alpha (TNF-α) in the circulation (Fig. 2C and D) than did controls, indicating that chronic systemic inflammation in vivo was present in SAg-treated animals.
FIG 2 .

TSST-1 induces systemic inflammation and elevated circulating endotoxin levels in vivo. (A) Representative images of the spleens harvested from TSST-1 and PBS animals after 1 week and 6 weeks of treatment. (B) Average weights of spleens from TSST-1- and PBS-treated animals after 6 weeks of treatment. (C) TNF-α levels in plasma collected from the two groups of animals each week. (D) Average TNF-α levels in plasma collected from two groups of animals throughout the experiment. (E) Image shows representative results of the Limulus amebocyte lysate assay performed on collected plasma. (F) Average endotoxin (lipopolysaccharide [LPS]) levels in the circulation of two groups of animals after 6 weeks of treatment. Statistically significant differences were determined by Student’s t test (*, P < 0.05; **, P < 0.01; ***, P < 0.001). Error bars show standard deviations.
The endotoxin levels in the circulation were also elevated in TSST-1-treated animals in comparison with the levels in the controls (Fig. 2E and F). These observations are consistent with DMII in humans, in which both TNF-α and endotoxin are elevated in the bloodstream. Since endotoxin is proinflammatory both in vitro and in vivo and synergizes with TSST-1, the presence of endotoxin in the blood would significantly exacerbate systemic inflammation in the TSST-1-treated animals.
TSST-1 induced liver damage both in vitro and in vivo.
Under normal conditions, the liver is the main site for endogenous endotoxin clearance (18). Hence, detectable endotoxin in the circulation could suggest liver damage. Histopathology of the liver tissue from TSST-1-treated rabbits showed microvesicle formation, suggesting hepatocyte necrosis (Fig. 3A). Moreover, in vitro treatment with TSST-1 directly induced cytotoxicity in HepG2 cells (a human hepatocyte cell line) (Fig. 3B and C); this effect was significantly increased by endotoxin (Fig. 3D). Therefore, TSST-1 may promote the elevation of endogenous endotoxin levels in vivo through inducing liver damage.
FIG 3 .

TSST-1 induces liver damage. (A) Hematoxylin-and-eosin (H&E)-stained livers from TSST-1- and PBS-treated rabbits after 6 weeks of treatment. Circles indicate areas of microvesicle formation. (B) Frequencies of HepG2 cells labeled with annexin V antibody after treatment with TSST-1 (100 µg/ml) or PBS at various time points. (C) Percentages of HepG2 cells surviving after 24 and 48 h of treatment with TSST-1 (100, 50, and 10 µg/ml) or PBS. (D) Percentages of HepG2 cells surviving after 48 h of treatment with TSST-1 (50 µg/ml) alone, LPS (endotoxin; 10 and 100 ng/ml) alone, or TSST-1 and LPS together. Statistically significant differences were analyzed by Student’s t test (*, P < 0.05; **, P < 0.01; ***, P < 0.001). Error bars show standard deviations.
TSST-1 induced proinflammatory cytokine and chemokine production in adipocytes in vitro and in vivo.
Prolonged exposure of rabbits to TSST-1 induced chronic inflammation, elevated the endotoxin levels in the circulation, and impaired glucose tolerance, all of which are conditions seen in DMII patients. Next, to understand the possible mechanisms of action, we explored the direct effect of TSST-1 on adipocytes, noting the strong correlation between obesity and DMII. In vitro, TSST-1 significantly induced proinflammatory cytokine (IL-6) production in both human and rabbit adipocytes in dose- and time-dependent manners (Fig. 4A; Fig. S1B in the supplemental material), and the effect was significantly enhanced by TNF-α and endotoxin (Fig. 4B and C). Previously, we showed that there was no difference in the ability of TSST-1 to induce proinflammatory cytokine production in adipocytes isolated from either DMII or nondiabetic individuals, suggesting that the observed TSST-1 effect is independent of metabolic alterations in adipocytes in the progression to DMII.
FIG 4 .

TSST-1 induces inflammation in adipocytes. (A) IL-6 levels in human adipocyte supernatants after 24 and 48 h of treatment with TSST-1 (100, 50, 10, and 1 µg/ml) or PBS. (B) IL-6 levels in human adipocyte supernatants after 24 h of treatment with TSST-1 (50 µg/ml) alone, TNF-α (1 ng/ml) alone, or TSST-1 and TNF-α together. (C) IL-6 levels in human adipocyte supernatants after 6 h of treatment with TSST-1 (50 µg/ml) alone, LPS (endotoxin; 5 ng/ml) alone, or TSST-1 and LPS together. (D and E) IL-6, IL-8, TNF-α, MCP-1, and adiponectin transcript levels in adipocytes isolated from two groups of animals ex vivo after 1 week (D) or 6 weeks (E) of treatment. Statistically significant differences were determined by Student’s t test (*, P < 0.05; **, P < 0.01; ***, P < 0.001). Error bars show standard deviations.
To examine the influence of TSST-1 on adipocytes in vivo, adipocytes were tested ex vivo after treatment of rabbits with TSST-1, and the levels of IL-6, IL-8, TNF-α, monocyte chemoattractant protein-1 (MCP-1), and adiponectin transcripts were analyzed by RT-qPCR. After the first week, IL-6, IL-8, TNF-α, and MCP-1 transcript levels were significantly upregulated in adipocytes from TSST-1-treated animals in comparison with their levels in the controls (Fig. 4D). Although there were reductions in IL-6, IL-8, and MCP-1 transcript levels at the end of the sixth week, the TNF-α transcript level remained elevated, which is consistent with the observed chronic systemic inflammation in these SAg-treated animals (Fig. 4E). There was no significant difference in adiponectin transcript levels in adipocytes isolated from TSST-1- or PBS-treated animals. Collectively, TSST-1 induced proinflammatory signal (IL-6, IL-8, and TNF-α) production in adipocytes, which could contribute directly to the development and maintenance of the chronic systemic inflammation observed in rabbits. At the early time points (1 week), TSST-1 treatment also induced MCP-1 expression in adipocytes. MCP-1 and IL-8 are both chemoattractants that could further recruit macrophages and neutrophils to inflamed adipose tissue, where TSST-1 could then stimulate these immune cells to produce more proinflammatory cytokines. Ultimately, this process could greatly enhance the systemic inflammation.
TSST-1 induced insulin resistance in adipocytes ex vivo.
Since the TSST-1-treated rabbits experienced impaired glucose tolerance without observed deficiency in the ability to produce insulin, it is possible that chronic stimulation with TSST-1 induces insulin resistance. Hence, we examined the ability of adipocytes to take up glucose after chronic exposure to TSST-1. After 6 weeks of treatment, adipocytes from TSST-1- and PBS-treated rabbits were isolated ex vivo, induced with identical amounts of insulin, and tested for the ability to take up 2-deoxy-d-glucose (2DG). Adipocytes from TSST-1-treated rabbits absorbed significantly less 2DG than those from the control rabbits (Fig. 5B). Since the two groups of adipocytes were induced with the same amount of insulin, the results indicate that chronic exposure to TSST-1 leads to insulin resistance in adipocytes in vivo.
FIG 5 .
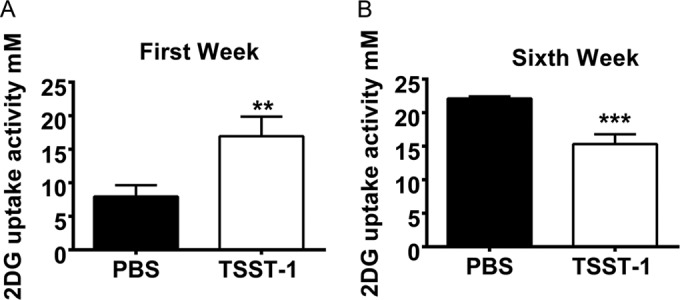
TSST-1 induces insulin resistance in adipocytes ex vivo. 2DG uptake ability of adipocytes isolated from two groups of animals after 1 week (A) and 6 weeks (B) of treatment ex vivo. Statistically significant differences were determined by Student’s t test (**, P < 0.01; ***, P < 0.001). Error bars show standard deviations.
Interestingly, the same effect was not observed in adipocytes isolated from TSST-1-treated rabbits at earlier time points. In fact, the glucose uptake ability was upregulated in rabbits after 1 week of treatment with TSST-1 (Fig. 5A); such an outcome might result from the ability of adipocytes to compensate for the acute effects of systemic inflammation, which was confirmed by in vitro data. Acute treatment with TNF-α also increased glucose uptake in adipocytes (Fig. S1C in the supplemental material). Together, these data further support the idea that insulin resistance in adipocytes could be achieved through chronic exposure to TSST-1 rather than acute stimulation.
TSST-1 induced lipolysis.
Considering that skeletal muscles are responsible for 70% of glucose uptake in the body (19), insulin resistance in adipose tissue alone cannot account for the peripheral insulin resistance observed in the TSST-1-treated rabbits. However, chronic inflammation can disrupt insulin signaling in muscles, resulting in reduced insulin sensitivity (7). Not only inflammation but excessive fatty acid levels in the circulation could result in insulin resistance in skeletal muscle via inducing the accumulation of intramuscular triglycerides and lipids (20). Since adipocytes are the main sites for lipolysis, the breakdown of triglycerides into glycerol and fatty acids (21), it is possible that TSST-1 contributes to the development of peripheral insulin resistance through directly inducing lipolysis in adipocytes. Treatment with TSST-1 significantly induced lipolysis in both human and rabbit adipocytes in a dose-dependent manner (Fig. 6A; Fig. S1D in the supplemental material), which was consistent with the upregulation of transcript levels of adipose triglyceride lipase (ATGL), hormone-sensitive lipase (HSL), and monoacylglycerol lipase (MGL), the three main enzymes involved in adipocyte lipolysis (Fig. 6B; Fig. S1E). Furthermore, the upregulation of HSL expression levels was also observed in ex vivo adipocytes from TSST-1-treated rabbits, suggesting in vivo lipolysis (Fig. 6C). Of note, there are no predicted genes for ATGL and MGL in the rabbit genome, so these were not tested.
FIG 6 .
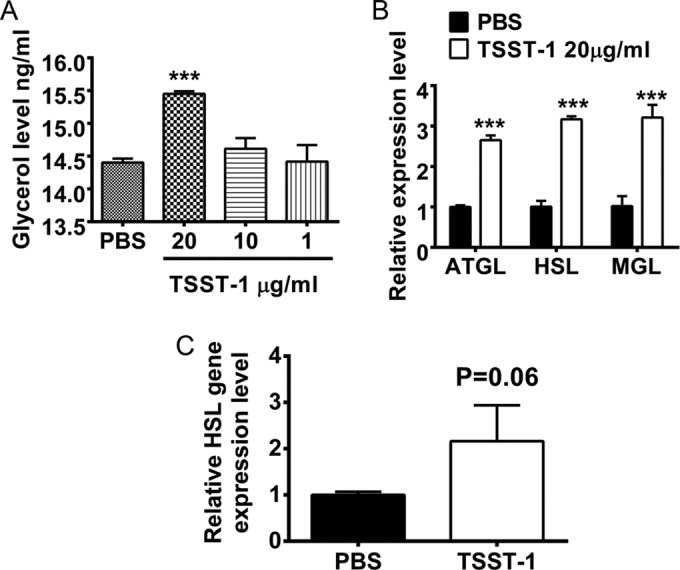
TSST-1 induces lipolysis in adipocytes. (A) Glycerol levels in NDAD supernatants after 6 h of treatment with TSST-1 (20, 10, and 1 µg/ml) or PBS. (B) Relative expression levels of ATGL, HSL, and MGL genes in human adipocytes after 6 h of treatment with TSST-1 (20 µg/ml) or PBS. (C) HSL gene expression levels in ex vivo adipocytes isolated after 6 weeks of treatment with TSST-1 or PBS. Statistically significant differences were determined by Student’s t test (*, P < 0.05; **, P < 0.01; ***, P < 0.001). Error bars show standard deviations.
TSST-1 production in vivo.
Previously, we and others have shown that women with menstrual TSS or healthy women with TSST-1-producing S. aureus vaginally and present on tampons (up to 1011 CFUs/tampon) may have up to 100 µg of TSST-1 per tampon (22, 23). Additionally, SAgs have been demonstrated in the lungs of rabbits with necrotizing pneumonia (17), and vaccination against SAgs protects rabbits from lethal pneumonia (15), further suggesting that SAgs are produced in vivo. All of these data suggest that SAgs are produced in vivo by colonizing or infecting S. aureus.
We evaluated S. aureus directly from the skin of 4 patients with diabetes, two with DMI and two with DMII. The skin of palms and forearms was swabbed in 2-cm by 2-cm areas, and S. aureus and SAgs were quantified directly. Based on all four patients being positive for S. aureus both on palms and forearms, we extrapolated the amounts of S. aureus and SAgs that may be present assuming uniform colonization of the skin; we assumed the average skin area of humans to be 1.75 m2. These 4 patients had an estimated 1011 to 1013 S. aureus cells on their skin surface. Two of the S. aureus strains produced TSST-1, and two produced staphylococcal enterotoxin C (SEC). The lower limit of TSST-1 or SEC detection in our Western blot assay is 800 pg/cm2. Assuming these 4 patients had the same amount of SAg over all of their skin as the tested palms and forearms, they would have been continually exposed to at least 1 µg of SAg. Our experience from unpublished rabbit studies is that the hide permeability of SAgs is approximately 1/10 to 1/100 the amount applied over a 24-h period. Thus, our calculations indicate that these 4 persons were possibly continuously exposed to 10 to 100 ng subcutaneously. Previously, we have shown that rabbits are susceptible to SAgs to approximately the same extent as humans (24). In our current studies, rabbits were exposed to 250 ng of TSST-1 per hour from miniosmotic pumps continually over the 6-week period, similar to the exposure suggested for humans.
DISCUSSION
DMII is a chronic metabolic disorder whose prevalence has been increasing rapidly worldwide (Fig. 7) (1, 4). Along with genetic determinants, obesity is significantly correlated with the development of DMII (25). Once DMII is established and when it is not under control, the disease can progress to serious complications, including diabetic foot ulcers (3). Obese persons and persons with diabetic foot ulcers have high incidences of S. aureus colonization and infections (12), and in the case of diabetic foot ulcer infections, S. aureus isolates have been shown to carry higher numbers of SAg genes than normal colonization strains (26). In addition, rabbits which have S. aureus infective endocarditis experience impaired glucose tolerance, the main symptom of DMII (data not shown). Considering the essential roles of SAgs in S. aureus pathogenesis, we examined the potential effects of SAgs on the development of DMII.
FIG 7 .

Model of the possible role of SAgs in the development of DMII. As obesity progresses (42), the rates of S. aureus colonization and infection increase [1]. Since all pathogenic human S. aureus isolates produce SAgs, obese individuals have a high risk of frequent exposure to SAgs [2]. In vivo, SAgs induce proinflammatory cytokine (IL-6, IL-8, and TNF-α) production in adipocytes [3], which may contribute to the development of systemic inflammation [4]. By producing chemoattractants, such as MCP-1 and IL-8, adipocytes can further recruit immune cells to adipose tissue [5]. Once recruited, these immune cells can also be stimulated by SAgs to produce additional proinflammatory signals [6]. SAgs induce liver damage (hepatocyte apoptosis) [8], which leads to a reduction in the endotoxin clearance function of the liver, resulting in elevated circulating endotoxin (LPS) levels [9]. Together with the proinflammatory signals from the recruited immune cells, endotoxin in the circulation (perhaps spilling over from the intestinal microbiota) can further enhance and maintain ongoing systemic inflammation [7 and 10]; chronic systemic inflammation has previously been shown to have an important role in the development of peripheral insulin resistance [11]. TSST-1 induces lipolysis in adipocytes [12]. Free fatty acids can be taken up by skeletal muscle and subsequently converted into intramuscular lipids, which are strongly associated with the development of insulin resistance [13]. Chronic exposure to SAgs in vivo can lead to insulin resistance in adipocytes [14]. Altogether, chronic insulin resistance in multiple tissues results in impaired glucose tolerance, the hallmark of DMII [15]. Numbers in brackets indicate the representative order of the process.
Since mouse models have been reported to be inadequate to study SAg effects (24) and, additionally, human inflammatory diseases (27), we chose to study rabbits, which were challenged with sublethal doses (yet doses expected in humans) of TSST-1 and tested for impaired glucose tolerance. Collectively, our data indicate that TSST-1 treatment gradually reduces the ability of rabbits to clear blood glucose over time. Deficiencies in glucose clearance might result from peripheral insulin resistance that diminishes insulin receptor activation in multiple tissues, resulting in reduction in insulin signaling cascades and insulin-dependent actions, such as transporting glucose into the cells via glucose transporters (25). Another possibility for impaired glucose removal from blood is that there may not be enough insulin produced by β-islet cells in the pancreas (25). Analysis comparing the ability to produce insulin between TSST-1- and PBS-treated groups of animals showed that there is more insulin transcript in the pancreas of TSST-1-treated rabbits than in the pancreas of PBS-treated rabbits. This outcome suggests that TSST-1-treated animals are not deficient in the ability to produce insulin.
It is possible that chronic exposure to SAgs could induce protective antibodies during natural S. aureus infection. However, many humans appear unable to produce protective antibodies to any given SAg, as demonstrated most recently in a study in which 20% of adult women, regardless of country, were found to lack antibodies to TSST-1 and, thus, remained susceptible to menstrual TSS (28). Different S. aureus strains carry different subsets of SAgs, and while their roles in pathogenesis can be redundant, neutralizing antibodies to SAgs often do not provide cross protection. Finally, it is well established that chronic exposure to SAgs has greater proinflammatory effects in rabbits than a single bolus exposure with the same dose. Thus, obese humans, with high colonization/infection by S. aureus, are likely to be continually exposed and susceptible to one or more SAgs, with an inability to make neutralizing antibodies.
Inflammation has a significant role in the development of insulin resistance (7). Previous studies have shown that endotoxin is able to induce proinflammatory cytokine production in adipocytes, and chronic exposure may lead to insulin resistance and promote the development of DMII (7). Our findings indicate that TSST-1 can also have the same effect on adipocytes and that the effect can be enhanced with endotoxin and other proinflammatory signals, like TNF-α. It is well known that SAgs amplify the effects of endotoxin by up to 106-fold (29).
TSST-1 stimulation of rabbits elevates the levels of endotoxin in the circulation. Under conditions of normal health, low levels of endotoxin are thought to leak through the intestinal wall into the circulation and are cleared by the liver’s Kupffer cells (18). As a result, there is no persistent inflammatory effect. However, with TSST-1 exposure, histopathological analysis of rabbit livers indicates cellular damage. In vitro treatment with TSST-1 also directly induces hepatocyte apoptosis. Previous studies reported that acute treatment of rabbits with SAgs inhibits Kupffer cell RNA synthesis and reduces the ability of the liver to clear endotoxin from the circulation (30); this effect may explain in part the ability of SAgs to enhance the effects of endotoxin in animals. These data suggest that TSST-1 induces damage to the liver over time, as seen in TSS patients, which leads to decreases in the endotoxin clearance function of the liver, resulting in increased levels of endotoxin in the circulation. The correlations between liver disease and DMII in humans, such as high risks of DMII development among alcoholic individuals, patients with nonalcoholic liver disease, and patients with liver steatosis, have been well established (31). In addition, endotoxin has consistently been shown to have a major role in the development of peripheral insulin resistance via its proinflammatory nature (5). Therefore, by increasing the circulating endotoxin level via liver damage in vivo, TSST-1 could potentially have a significant impact on the development of insulin resistance in DMII.
We examined adipocytes ex vivo after treatment of rabbits with TSST-1 to further assess the effects of SAgs in vivo. Our data show that proinflammatory cytokine expression is upregulated in adipocytes upon in vivo TSST-1 exposure. This may contribute to chronic inflammation, which was seen in the rabbits. In addition, proinflammatory cytokines can recruit macrophages to adipose tissue, in which these cells can also be stimulated by SAgs to produce more proinflammatory signals.
Normal adipocytes may also have anti-inflammatory functions. Adiponectin is an anti-inflammatory adipokine, which is produced by adipocytes to dampen either local or systemic inflammation (7). However, there was no difference in the adiponectin transcript levels in adipocytes isolated from the TSST-1-treated and PBS-treated rabbits, regardless of the duration of the treatment. This outcome suggests that TSST-1 stimulation may have a negative effect on adiponectin activation cascades.
Peripheral insulin resistance generally results from insulin insensitivity in the skeletal muscles and adipose tissues (20). Our findings reveal that chronic instead of acute exposure to TSST-1 can result in insulin resistance in adipocytes. In addition, TSST-1 treatment induces adipocyte lipolysis, wherein lipolysis products, such as glycerol and fatty acids, can have major roles in the development of insulin resistance in skeletal muscles through lipid accumulation (20). It is noteworthy that S. aureus enterotoxin A (SEA) has also been shown to induce lipolysis and to reduce glucose uptake in primary adipocytes in vitro (32), which further supports the role of SAgs in the development of insulin resistance. Thus, TSST-1 and perhaps SAgs in general have multiple ways to contribute to the development of peripheral insulin resistance in vivo, i.e., through (i) causing systemic inflammation and affecting insulin signaling, (ii) direct effects on adipocyte insulin resistance, and (iii) indirect effects on skeletal muscle insulin resistance through lipolysis.
Collectively, our findings show that prolonged exposure to SAgs, such as TSST-1, possibly happening through frequent S. aureus colonization and infection in obese individuals, may result in impaired glucose metabolism, the hallmark of DMII. This outcome can be traced back to the ability of SAgs to cause insulin resistance through inducing chronic systemic inflammation and lipolysis. Throughout history, microbial pathogens have surprised us with their contributions to chronic diseases, such as Reiter’s syndrome, gastrointestinal ulcers/cancer, and cervical cancer (33). Now, based on our findings, the possibility exists that S. aureus could influence the development of DMII. Therapies targeting S. aureus colonization and SAgs may become important for prevention and treatment.
MATERIALS AND METHODS
Cells.
Immortalized human preadipocytes from healthy human donors were characterized previously (34). Rabbit primary preadipocytes and adipocytes were isolated from adipose tissues surrounding the kidneys, following a published protocol (35). Rabbit pancreases were isolated immediately after the last glucose challenge test at the end of the 6th week of treatment. Three random areas in each pancreas were selected for total RNA extraction and analysis of insulin transcript levels.
In vitro preadipocytes were grown to confluence in preadipocyte medium (Lonza, Allendale, NJ) and then changed to differentiation medium (Lonza, Allendale, NJ) prepared according to the manufacturer’s instructions. The cells were maintained in this medium for 10 days to allow differentiation to adipocytes. The differentiated human cells are referred to herein as nondiabetic differentiated adipocytes (NDADs).
Animal models.
All animal experiments were performed under an approved University of Iowa IACUC protocol. Alzet miniosmotic pumps (purchased to contain 200-µl total volumes and release SAgs at a constant rate of 0.15 µl/h for 6 weeks) (Alza Corp., Palo Alto, CA) containing either TSST-1 (1.6 mg/ml, 200 µl) or PBS (200 µl) were implanted subcutaneously through small, midline incisions in the lumbar regions of New Zealand White rabbits (36). The wounds were closed with sutures, and the animals were monitored for 6 weeks. Although we did not assess the release of TSST-1 by the miniosmotic pumps over the 6-week test period, we have previously shown that miniosmotic pumps from Alza Corp release SAgs in the expected amounts
Each week, after overnight fasting, rabbits were administered d-glucose (1 g/kg of body weight) subcutaneously at the same total volume. A blood glucose meter and TRUEtest strips (Nipro Diagnostics, Fort Lauderdale, FL) were used to monitor blood glucose levels every 15 min.
Toxin purification.
The SAg purification method is detailed elsewhere (37). In brief, S. aureus RN450Δhlb::pCE107 (for TSST-1) was cultured in beef heart (BH) medium (with high aeration) for 48 h. The cultures were then treated with absolute ethanol (80% final volume) for toxin precipitation. SAgs were resolubilized in distilled water and purified to homogeneity by thin-layer isoelectric focusing.
Endotoxin was purified from Salmonella enterica serovar Typhimurium by using the hot phenol extraction method (38). The endotoxin preparation was demonstrated to be biologically active by using the Limulus amebocyte assay and pyrogen testing (the minimum pyrogenic dose was 0.01 µg/ml).
Western immunoblotting.
We tested three humans for the presence of the SAgs TSST-1 and SEC by a Western immunoblot technique. Swabs wetted with PBS were rolled in two directions across 2-cm by 2-cm squares of palm and forearm skin; the swabs hold 0.1 ml of PBS. The swabs were then placed into 0.2 ml of PBS and vortexed for 1 min. The swabs were removed, and the mixture was clarified by centrifugation (14,000 × g for 5 min). The clarified supernatants (10-µl volumes) were spotted onto polyvinylidene difluoride (PVDF) membranes (Bio-Rad Laboratories, Hercules, CA). Additionally, 10-µl volumes of serially diluted, purified TSST-1 or SEC were added to the membranes to establish a standard curve. The PVDF membranes were developed with antibodies to the corresponding SAg, antibodies against rabbit antibodies, and substrate (39).
ELISAs for cytokines.
NDADs and primary rabbit adipocytes were treated with TSST-1 (1, 10, 20, and 50 µg/ml), endotoxin (5 ng/ml), or TNF-α (1 ng/ml) for 6, 24, or 48 h at 37°C in 5% CO2. The culture supernatants were collected for IL-6 quantification by enzyme-linked immunosorbent assay (ELISA) (human IL-6 DuoSet kit; R&D Systems, Minneapolis, MN).
For analysis of cytokine levels in the circulation, blood was collected from rabbit anterior ear veins into heparin weekly and spun down at 450 × g for 10 min to collect plasma. Circulating TNF-α in plasma was measured by ELISA (rabbit TNF-α DuoSet kit; R&D Systems, Minneapolis, MN).
Quantification of transcript levels by RT-qPCR.
Total RNA was extracted from cells using Trizol reagent (Invitrogen, Grand Island, NY) and chloroform extraction, followed by purification with RNeasy minicolumns (Qiagen, Redwood City, CA). RNA was reverse transcribed using a high-capacity cDNA reverse transcription kit (Applied Biosystems, Grand Island, NY). qPCR was performed with SYBR Select master mix (Life Technology, Grand Island, NY). The primers used are detailed in Table 1. The target gene transcript levels were normalized to the transcript levels of the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and peroxisome proliferator-activated receptor-gamma (PPARγ) genes for adipocytes or the histone deacetylase-9 (HDAC9) gene for pancreatic β-cells.
TABLE 1 .
qPCR primer sequences
| RNA source | Gene producta | Direction | Nucleotide sequence |
|---|---|---|---|
| Rabbit | IL-6 | Forward | GTCAACTGCATGAACAGAAAGG |
| Reverse | AGCAGGCAGGTCTCATTATTC | ||
| IL-8 | Forward | CCTGCTGTCTCTGACTCTTTG | |
| Reverse | AGGTGTGGAGTGTGTCTTTATG | ||
| TNF-α | Forward | CCCAAACAACCTCCATCTAGTC | |
| Reverse | CACTTGCGGGTTTGCTACTA | ||
| MCP-1 | Forward | AGTCACCTGCTGCTATACATTC | |
| Reverse | ACAGCTTCTTTGGGACACTT | ||
| Adiponectin | Forward | TCCTACCACATCACCGTCTAT | |
| Reverse | CACGTTCTTGTCCTGGTACTG | ||
| PPARγ | Forward | CCGAGAAGGAGAAGCTGTTG | |
| Reverse | CGGGAAGGACTTGATGTATGAG | ||
| Insulin | Forward | AGCTGGCCCTGCAGAAG | |
| Reverse | CCCTAGTTGCAGTAGTTCTCC | ||
| HDAC9 | Forward | GGCTGTGAAGATAAAGGAGGAA | |
| Reverse | AGGAAGGGCTGTTGCATAAA | ||
| GAPDH | Forward | GACCACTTTGTGAAGCTCATTTC | |
| Reverse | GTGGTTTGAGGGCTCTTACTC | ||
| HSL | Forward | CTAGCTCAACAGGAAGCTGAA | |
| Reverse | GCTCTCTCTTGGGATGTAGATTG | ||
| Human | ATGL | Forward | TGTCTGCAGCGGTTTCAT |
| Reverse | CTCATAGAGTGGCAGGTTGTC | ||
| HSL | Forward | GATGGAAGTGCTATCGTCTCTG | |
| Reverse | AGTCAGTGGCATCTCAAAGG | ||
| MGL | Forward | AGGACACCAGACTGCAATAAC | |
| Reverse | ACAGCAACCACCTCATCAC | ||
| GAPDH | Forward | GGTGTGAACCATGAGAAGTATGA | |
| Reverse | GAGTCCTTCCACGATACCAAAG |
MCP-1, monocyte chemoattractant protein-1; PPARγ, peroxisome proliferator-activated receptor gamma; HDAC9, histone deacetylase-9; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; HSL, hormone-sensitive lipase; ATGL, adipose triglyceride lipase; MGL, monoglyceride lipase.
Detection of lipolysis.
In vitro NDADs and primary rabbit adipocytes were treated with TSST-1 (1, 10, and 20 µg/ml) in adipocyte-growing medium (Lonza) without the presence of serum for 6 h. The culture supernatants were collected for quantification of glycerol levels with the triglyceride (GPO [glycerol phosphate oxidase]) reagent set (Pointe Scientific, Canton, MI).
2DG uptake assay.
Details of the 2DG uptake assay have been published previously (40); some modifications were made for ex vivo rabbit adipocytes. In short, isolated rabbit primary adipocytes were treated with 1 µM recombinant human insulin (Sigma Aldrich, St. Louis, MO) for 30 min at 37°C, washed twice, and challenged with 2 mM 2-deoxy glucose (2DG) (Sigma Aldrich, St. Louis, MO) for 20 min at 37°C. The cells were then washed extensively, resuspended in 0.1 M NaOH, and snap-frozen at −80°C overnight. The next day, samples were thawed, sonicated, neutralized with 0.1 M HCl and triethanolamine TEA buffer (200 mM, pH 8.1), and spun down at 12,000 × g for 5 min. The collected supernatants were then used to measure the intracellular 2DG levels.
Limulus amebocyte lysate assay.
After 6 weeks of treatment, plasma endotoxin was isolated with the chloroform extraction method and quantified with E-Toxate (Sigma Aldrich, St. Louis, MO) at an absorbance wavelength of 600 nm. Purified Salmonella endotoxin was used to construct a standard curve.
Cytotoxicity assay.
After treatment with toxins, HepG2 cells were washed and resuspended in PBS. CellTiter 96 aqueous one solution (Promega, Madison, WI) was used to monitor metabolically active cells (indicated by a color change, a property that is lost with dead cells). The colorimetric reaction was measured at a 492-nm wavelength.
In other experiments, treated cells were washed and stained with an annexin V cell death/apoptosis kit (Life Technology, Grand Island, NY) according to the manufacturer’s instructions. The levels of labeled annexin V were then quantified and analyzed with the BD FACSCalibur (BD Biosciences, San Jose, CA) and FlowJo software (FlowJo, Ashland, OR). (41).
SUPPLEMENTAL MATERIAL
(A) Weights of TSST-1- and PBS-treated rabbits throughout the 6-week experiment. (B) IL-6 transcript levels in rabbit adipocytes after 6 and 24 h of treatment with TSST-1 (50 µg/ml) or PBS. (C) 2DG uptake ability of human adipocytes after 24 h of treatment with TNF-α (10 and 1 ng/ml) or PBS. (D) Glycerol levels in rabbit adipocyte supernatants after 6 h of treatment with TSST-1 (20, 10, and 1 µg/ml) or PBS in vitro. (E) HSL (hormone-sensitive lipase) gene expression level in rabbit adipocytes after 6 h of treatment with TSST-1 (20 µg/ml) or PBS in vitro. Download
ACKNOWLEDGMENTS
This work was supported by a Mark Stinski developmental grant (A.J.K.), Department of Microbiology startup funds (P.M.S. and W.S.-P.), and a Carver Trust Collaborative pilot grant (A.J.K. and P.M.S.).
Footnotes
Citation Vu BG, Stach CS, Kulhankova K, Salgado-Pabón W, Klingelhutz AJ, Schlievert PM. 2015. Chronic superantigen exposure induces systemic inflammation, elevated bloodstream endotoxin, and abnormal glucose tolerance in rabbits: possible role in diabetes. mBio 6(2):e02554-14. doi:10.1128/mBio.02554-14.
REFERENCES
- 1.International Diabetes Federation 2013. Global burden of diabetes. Diabetic atlas, 6th ed Brussels, Belgium: Accessed 17 October 2014 http://www.idf.org/diabetesatlas. [Google Scholar]
- 2.Zimmet P, Alberti KG, Shaw J. 2001. Global and societal implications of the diabetes epidemic. Nature 414:782–787. doi: 10.1038/414782a. [DOI] [PubMed] [Google Scholar]
- 3.Nathan DM. 1993. Long-term complications of diabetes mellitus. N Engl J Med 328:1676–1685. doi: 10.1056/NEJM199306103282306. [DOI] [PubMed] [Google Scholar]
- 4.Olokoba AB, Obateru OA, Olokoba LB. 2012. Type 2 diabetes mellitus: a review of current trends. Oman Med J 27:269–273. doi: 10.5001/omj.2012.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Creely SJ, McTernan PG, Kusminski CM, Fisher FM, Da Silva NF, Khanolkar M, Evans M, Harte AL, Kumar S. 2007. Lipopolysaccharide activates an innate immune system response in human adipose tissue in obesity and type 2 diabetes. Am J Physiol Endocrinol Metab 292:E740–E747. doi: 10.1152/ajpendo.00302.2006. [DOI] [PubMed] [Google Scholar]
- 6.Venables MC, Jeukendrup AE. 2009. Physical inactivity and obesity: links with insulin resistance and type 2 diabetes mellitus. Diabetes Metab Res Rev 25(Suppl 1):S18–S23. doi: 10.1002/dmrr.983. [DOI] [PubMed] [Google Scholar]
- 7.Piya MK, McTernan PG, Kumar S. 2013. Adipokine inflammation and insulin resistance: the role of glucose, lipids and endotoxin. J Endocrinol 216:T1–T15. doi: 10.1530/JOE-12-0498. [DOI] [PubMed] [Google Scholar]
- 8.Ley RE, Turnbaugh PJ, Klein S, Gordon JI. 2006. Microbial ecology: human gut microbes associated with obesity. Nature 444:1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- 9.Walters WA, Xu Z, Knight R. 2014. Meta-analyses of human gut microbes associated with obesity and IBD. FEBS Lett 588:4223–4233. doi: 10.1016/j.febslet.2014.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gill SR, Pop M, Deboy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, Gordon JI, Relman DA, Fraser-Liggett CM, Nelson KE. 2006. Metagenomic analysis of the human distal gut microbiome. Science 312:1355–1359. doi: 10.1126/science.1124234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bäckhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, Semenkovich CF, Gordon JI. 2004. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A 101:15718–15723. doi: 10.1073/pnas.0407076101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Olsen K, Danielsen K, Wilsgaard T, Sangvik M, Sollid JU, Thune I, Eggen AE, Simonsen GS, Furberg AS. 2013. Obesity and Staphylococcus aureus nasal colonization among women and men in a general population. PLoS One 8:e63716. doi: 10.1371/journal.pone.0063716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Early GJ, Seifried SE. 2012. Risk factors for community-associated Staphylococcus aureus skin infection in children of Maui. Hawaii J Med Public Health 71:218–223. [PMC free article] [PubMed] [Google Scholar]
- 14.Spaulding AR, Salgado-Pabón W, Kohler PL, Horswill AR, Leung DY, Schlievert PM. 2013. Staphylococcal and streptococcal superantigen exotoxins. Clin Microbiol Rev 26:422–447. doi: 10.1128/CMR.00104-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spaulding AR, Salgado-Pabón W, Merriman JA, Stach CS, Ji Y, Gillman AN, Peterson ML, Schlievert PM. 2014. Vaccination against Staphylococcus aureus pneumonia. J Infect Dis 209:1955–1962. doi: 10.1093/infdis/jit823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spaulding AR, Lin YC, Merriman JA, Brosnahan AJ, Peterson ML, Schlievert PM. 2012. Immunity to Staphylococcus aureus secreted proteins protects rabbits from serious illnesses. Vaccine 30:5099–5109. doi: 10.1016/j.vaccine.2012.05.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Strandberg KL, Rotschafer JH, Vetter SM, Buonpane RA, Kranz DM, Schlievert PM. 2010. Staphylococcal superantigens cause lethal pulmonary disease in rabbits. J Infect Dis 202:1690–1697. doi: 10.1086/657156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crispe IN. 2003. Hepatic T cells and liver tolerance. Nat Rev Immunol 3:51–62. doi: 10.1038/nri981. [DOI] [PubMed] [Google Scholar]
- 19.Björnholm M, Zierath JR. 2005. Insulin signal transduction in human skeletal muscle: identifying the defects in type II diabetes. Biochem Soc Trans 33:354–357. doi: 10.1042/BST0330354. [DOI] [PubMed] [Google Scholar]
- 20.Samuel VT, Shulman GI. 2012. Mechanisms for insulin resistance: common threads and missing links. Cell 148:852–871. doi: 10.1016/j.cell.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zechner R, Zimmermann R, Eichmann TO, Kohlwein SD, Haemmerle G, Lass A, Madeo F. 2012. Fat signals—lipases and lipolysis in lipid metabolism and signaling. Cell Metab 15:279–291. doi: 10.1016/j.cmet.2011.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schlievert PM, Nemeth KA, Davis CC, Peterson ML, Jones BE. 2010. Staphylococcus aureus exotoxins are present in vivo in tampons. Clin Vaccine Immunol 17:722–727. doi: 10.1128/CVI.00483-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rosten PM, Bartlett KH, Chow AW. 1987. Detection and quantitation of toxic shock syndrome toxin 1 in vitro and in vivo by noncompetitive enzyme-linked immunosorbent assay. J Clin Microbiol 25:327–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Salgado-Pabón W, Schlievert PM. 2014. Models matter: the search for an effective Staphylococcus aureus vaccine. Nat Rev Microbiol 12:585–591. doi: 10.1038/nrmicro3308. [DOI] [PubMed] [Google Scholar]
- 25.Lin Y, Sun Z. 2010. Current views on type 2 diabetes. J Endocrinol 204:1–11. doi: 10.1677/JOE-09-0260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vu BG, Stach CS, Salgado-Pabón W, Diekema DJ, Gardner SE, Schlievert PM. 2014. Superantigens of Staphylococcus aureus from patients with diabetic foot ulcers. J Infect Dis 210:1920–1927. doi: 10.1093/infdis/jiu350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald-Smith GP, Gao H, Hennessy L, Finnerty CC, López CM, Honari S, Moore EE, Minei JP, Cuschieri J, Bankey PE, Johnson JL, Sperry J, Nathens AB, Billiar TR, West MA, Jeschke MG, Klein MB, Gamelli RL, Gibran NS, Brownstein BH, Miller-Graziano C, Calvano SE, Mason PH, Cobb JP, Rahme LG, Lowry SF, Maier RV, Moldawer LL, Herndon DN, Davis RW, Xiao W, Tompkins RG, Inflammation and Host Response to Injury, Large Scale Collaborative Research Program . 2013. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A 110:3507–3512. doi: 10.1073/pnas.1222878110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parsonnet J, Hansmann MA, Delaney ML, Modern PA, Dubois AM, Wieland-Alter W, Wissemann KW, Wild JE, Jones MB, Seymour JL, Onderdonk AB. 2005. Prevalence of toxic shock syndrome toxin 1-producing Staphylococcus aureus and the presence of antibodies to this superantigen in menstruating women. J Clin Microbiol 43:4628–4634. doi: 10.1128/JCM.43.9.4628-4634.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schlievert PM. 1982. Enhancement of host susceptibility to lethal endotoxin shock by staphylococcal pyrogenic exotoxin type C. Infect Immun 36:123–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fujiwaka H, Igarashi H, Usami H, Tanaka S, Tamura H. 1986. Clearance of endotoxin from blood of rabbits injected with staphylococcal toxic shock syndrome toxin-1. Infect Immun 52:134–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Levinthal GN, Tavil AS. 1999. Liver disease and diabetes mellitus. Clin Diabetes http://journal.diabetes.org/clinicaldiabetes/v17n21999/Pg73.htm.
- 32.Banke E, Rödström K, Ekelund M, Dalla-Riva J, Lagerstedt JO, Nilsson S, Degerman E, Lindkvist-Petersson K, Nilson B. 2014. Superantigen activates the gp130 receptor on adipocytes resulting in altered adipocyte metabolism. Metab Clin Exp 63:831–840. doi: 10.1016/j.metabol.2014.03.004. [DOI] [PubMed] [Google Scholar]
- 33.Gargano LM, Hughes JM. 2014. Microbial origins of chronic diseases. Annu Rev Public Health 35:65–82. doi: 10.1146/annurev-publhealth-032013-182426. [DOI] [PubMed] [Google Scholar]
- 34.Vu BG, Gourronc FA, Bernlohr DA, Schlievert PM, Klingelhutz AJ. 2013. Staphylococcal superantigens stimulate immortalized human adipocytes to produce chemokines. PLoS One 8:e77988. doi: 10.1371/journal.pone.0077988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McTernan PG, Anderson LA, Anwar AJ, Eggo MC, Crocker J, Barnett AH, Stewart PM, Kumar S. 2002. Glucocorticoid regulation of p450 aromatase activity in human adipose tissue: gender and site differences. J Clin Endocrinol Metab 87:1327–1336. doi: 10.1210/jcem.87.3.8288. [DOI] [PubMed] [Google Scholar]
- 36.Parsonnet J, Gillis ZA, Richter AG, Pier GB. 1987. A rabbit model of toxic shock syndrome that uses a constant, subcutaneous infusion of toxic shock syndrome toxin 1. Infect Immun 55:1070–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blomster-Hautamaa DA, Schlievert PM. 1988. Preparation of toxic shock syndrome toxin-1. Methods Enzymol 165:37–43. [DOI] [PubMed] [Google Scholar]
- 38.Westphal O, Luderitz O, Keiderling W. 1952. Effects of bacterial toxins; biochemical analysis of inflammation. Zentralbl Bakteriol Parasitenkd Infektionskr Hyg 158:152–160. [PubMed] [Google Scholar]
- 39.Blake MS, Johnston KH, Russell-Jones GJ, Gotschlich EC. 1984. A rapid, sensitive method for detection of alkaline phosphatase-conjugated anti-antibody on Western blots. Anal Biochem 136:175–179. doi: 10.1016/0003-2697(84)90320-8. [DOI] [PubMed] [Google Scholar]
- 40.Yamamoto N, Ueda M, Sato T, Kawasaki K, Sawada K, Kawabata K, Ashida H. 2011. Measurement of glucose uptake in cultured cells. Curr Protoc Pharmacol 12:14.1–14.22. doi: 10.1002/0471141755.ph1214s55. [DOI] [PubMed] [Google Scholar]
- 41.Roggiani M, Stoehr JA, Olmsted SB, Matsuka YV, Pillai S, Ohlendorf DH, Schlievert PM. 2000. Toxoids of streptococcal pyrogenic exotoxin A are protective in rabbit models of streptococcal toxic shock syndrome. Infect Immun 68:5011–5017. doi: 10.1128/IAI.68.9.5011-5017.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.World Health Organization. 2008. Overweight/obesity, 2008. Prevalence of obesity, ages 20+, age standardized: both sexes. http://gamapserver.who.int/gho/interactive_charts/ncd/risk_factors/overweight_obesity/atlas.html?indicator=i1&date=bothsexes.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Weights of TSST-1- and PBS-treated rabbits throughout the 6-week experiment. (B) IL-6 transcript levels in rabbit adipocytes after 6 and 24 h of treatment with TSST-1 (50 µg/ml) or PBS. (C) 2DG uptake ability of human adipocytes after 24 h of treatment with TNF-α (10 and 1 ng/ml) or PBS. (D) Glycerol levels in rabbit adipocyte supernatants after 6 h of treatment with TSST-1 (20, 10, and 1 µg/ml) or PBS in vitro. (E) HSL (hormone-sensitive lipase) gene expression level in rabbit adipocytes after 6 h of treatment with TSST-1 (20 µg/ml) or PBS in vitro. Download