

Abstract
Activation of trimeric G proteins has been traditionally viewed as the exclusive job of G protein-coupled receptors (GPCRs). This view has been challenged by the discovery of non-receptor activators of trimeric G proteins. Among them, GIV (a.k.a. Girdin) is the first for which a guanine nucleotide exchange factor (GEF) activity has been unequivocally associated with a well defined motif. Here we discuss how GIV assembles alternative signaling pathways by sensing cues from various classes of surface receptors and relaying them via G protein activation. We also describe the dysregulation of this mechanism in disease and how its targeting holds promise for novel therapeutics.
Keywords: Guanine Nucleotide Exchange Factor (GEF), Heterotrimeric G Protein, Receptor Tyrosine Kinase, Src Homology 2 Domain (SH2 Domain), Tumor Metastasis, Liver Fibrosis, Nephrotic Syndrome
Introduction
Heterotrimeric (henceforth trimeric) G proteins work as molecular switches that control the flow of information from extracellular cues perceived by G protein-coupled receptors (GPCRs)4 at the cell surface to a wide array of intracellular effector proteins that control cell behavior (1, 2). Resting (GDP-bound) Gα subunits in complex with Gβγ are activated by ligand-occupied GPCRs, which are guanine nucleotide exchange factors (GEFs) and promote the exchange of GDP for GTP on the α subunit (1). Signaling is turned off by the intrinsic GTPase activity of Gα, leading to reassociation of Gα with Gβγ. This well studied sequence of reactions is commonly referred to as the “G protein cycle” and represents the core components and events of this signal transduction mechanism. Extensive work during the last decades has revealed that this signaling mechanism is dysregulated in major human diseases such as cancer, fibrosis, neurodegeneration, diabetes, and cardiovascular disease. In fact, GPCRs represent the target for 30–50% of marketed drugs (3).
A less well studied aspect of G protein signaling pertains to the role of the so-called “accessory proteins” (4). These refer to a still emerging heterogeneous set of proteins capable of modulating the activity of G proteins in various ways. Detailed descriptions of these accessory proteins or some of their subfamilies have been the subject of extensive reviews elsewhere (4–8). Here we will focus on reviewing recent discoveries on a particular G protein activator called GIV (a.k.a. Girdin). We will discuss how these recent discoveries provide a new perspective on how we understand trimeric G protein signaling and its cross-talk with other signaling pathways, and how this impacts a variety of cellular processes. We will also discuss the impact of GIV-mediated signaling in the progression of human diseases and the future perspectives that this opens for therapeutics.
Accessory Proteins in G Protein Signaling
Historically, the first accessory proteins in G protein signaling (and the best characterized to date) were the RGS proteins (9–11). Soon after, a group of GoLoco/GPR proteins was also identified (12, 13). Although both RGS and GoLoco/GPR proteins work as inhibitors of Gα subunits, the molecular mechanisms that they use are different; RGS proteins are GTPase-activating proteins (GAPs) that accelerate the intrinsic GTPase activity of Gα (9, 10), whereas GoLoco/GPR proteins are guanine nucleotide dissociation inhibitors (GDIs) that block nucleotide exchange (12, 14, 15). Of note, these groups of regulators are structurally well defined by shared signature motifs or domains. The “GoLoco/GPR motif” (∼20–30 aa) (16, 17) and the “RGS box” (∼120 aa) (10, 18–20) are sufficient to exert guanine nucleotide dissociation inhibitor or GTPase-activating protein activity, respectively, on Gα subunits.
Although the identity of tractable domains has propelled the biological characterization of RGS and GoLoco/GPR proteins and incentivized efforts to pharmacologically target them (21, 22), the characterization of a third group of accessory proteins called non-receptor GEFs has progressed at a slower pace. Non-receptor proteins such as AGS1 (23), Ric-8A (24), Ric-8B (25), Arr4/Get3 (26), or CSPα (27), among some others (4), have been described to mimic the action of GPCRs by virtue of their GEF activity toward different Gα subunits. However, these examples represent a heterogeneous group of proteins, and no signature domain or motif was described as responsible for their GEF activity. This precluded the design of tools, such as GEF-deficient mutants, to unequivocally link the biological functions of these proteins (26, 28, 29) to their GEF activity instead of to other functional domains that they may have. In this regard, the discovery of the first defined GEF motif in GIV (30) has provided a unique opportunity to further our understanding of non-receptor GEFs.
GIV, a Non-receptor GEF for Trimeric G Proteins That Works via a Defined Motif
GIV is a large (1870-aa) multidomain protein (Fig. 1A) capable of binding to multiple cellular components (e.g. actin filaments, phosphoinositides, trimeric G proteins, etc.). The identification of GIV, as well as its initial characterization as a signaling molecule, was originally reported by several independent groups (31–35) before the discovery of its GEF motif. Anai et al. (33) provided the first evidence directly linking GIV expression to the enhancement of the PI3K-Akt pathway, which was confirmed by us (36) and others (37). These and other studies (31, 38–40) indicated that GIV played a critical role in the response of different cell types to receptor tyrosine kinase (RTK) and GPCR stimulation. In the current review, we will focus on the functions of GIV specifically associated with its ability to bind and modulate G proteins. We direct the readers to other recent reviews covering other functions of GIV (41).
FIGURE 1.

GIV is a multi-modular protein that activates Gαi via its C-terminal GEF motif by assembling a unique GIV-Gαi protein-protein interface. A, schematic representation of the domain organization of GIV. MT, microtubule; GBD, GTPase-binding; PI4P, phosphatidylinositol 4-phosphate; NT, N terminus; CT, C terminus. B, left, homology model of the GEF sequence of GIV (orange) bound to Gαi3 (blue, green, and red) generated as described in Ref. 30. Green denotes the switch II (SwII) region, and red denotes the α3 helix. Right, same view as is the left panel with a space-filling surface representation of Gαi3 colored by hydrophobicity (red to blue scale indicates increasing hydrophobicity). Three hydrophobic residues in GIV (Leu-1682, Phe-1685, and Leu-1686) are predicted to dock onto a hydrophobic cleft on Gαi3.
GIV was originally identified as a Gαi3-binding protein in a yeast two-hybrid screen (34). GIV can bind robustly to the Gi family members Gαi1, Gα2, and Gα3 and to a lesser extent to Gαs. No significant binding is observed to the representative members of other G protein subfamilies, such as Gα12 and Gαq (34). A critical realization (30) came from the identification of sequence similarity between a stretch of ∼25 evolutionarily conserved amino acids in the C-terminal domain of GIV and KB-752. KB-752 is a synthetic peptide with GEF activity toward Gαi proteins but presumed to have no similarity to any known G protein regulator at the time (42). A series of studies provided the biochemical basis to establish GIV as a bona fide GEF for Gαi subunits and also described details on the structural basis for its binding to G proteins (30, 32, 43–45). Enzymatic assays with purified components demonstrated that GIV does not affect the rate of catalysis of GTP hydrolysis by Gαi3 but instead accelerates the rate of nucleotide exchange, leading to Gα subunit activation consistent with a GEF activity (30, 44). Another feature shared with other known GEFs is the inability of GIV to bind Gα subunits in the active conformation (GTP-bound) (30, 32). This ensures the directionality of the reaction toward signaling activation: i.e. GIV engages Gα-GDP as a substrate and facilitates the nucleotide exchange reaction, and once GTP is loaded onto the G protein, the complex dissociates to allow binding of the active G protein to its effectors, leaving GIV free for a new round of activation.
These studies also provided important structural insights into the assembly of the GIV-Gα complex by using a combination of homology modeling (based on the x-ray structure of the KB-752 peptide bound to Gαi1 (42)) and site-directed mutagenesis (30, 45). The conclusion of these studies indicates that conserved hydrophobic residues that align on one side of a short aliphatic helix in GIV dock onto a hydrophobic cleft between the switch II and the α3 helix of Gαi (Fig. 1B). This mode of binding explains the inability of GIV to bind active Gαi because the conformation of the switch II helix in Gαi-GTP occludes the predicted binding site (46). Another important implication of this mode of binding is that the docking site of GIV on Gαi overlaps with the Gβγ binding region (30). Although it is not known whether GIV can directly activate a Gαi-βγ trimer in vitro, it was shown that GIV can displace Gβγ from a preformed Gαi-βγ trimer in vitro and enhance Gβγ-dependent signaling (e.g. PI3K-Akt) in cells via its GEF motif (30). A question that remains open is how much of the action of GIV is mediated by Gβγ subunits released from Gα purely by physical displacement or by activation of Gαi.
GIV Links Multiple Classes of Surface Receptors to G Protein Activation
Mapping for the first time the specific motif and residues in a non-receptor GEF required to bind and activate G proteins has provided a unique advantage over other known non-receptor GEFs. Designing surgical mutations that prevent the coupling of GIV to Gα has served not only to validate that its GEF activity in vitro is mediated by a defined motif but also to characterize the biological functions specifically associated with this activity in cells (see below). Initial experiments dissected a signaling mechanism in which Gβγ subunits released upon GIV-mediated G protein activation resulted in activation of PI3K-Akt (30), a pathway previously reported to be modulated by GIV via an unknown mechanism (33). Later work has dramatically expanded the repertoire of intracellular signals controlled by the GEF activity of GIV (Fig. 2), including PKA/CREB, ERK1/2, Src, STAT3, mTOR (mammalian target of rapamycin), and SMAD2/3, among others (36, 47–49).
FIGURE 2.

The GEF motif of GIV modulates key signaling networks downstream of diverse classes of receptors. Top, a schematic summarizing the diverse classes of receptors that converge upon GIV and have been shown to require the GEF function of GIV to transduce downstream signaling. Solid lines connecting the receptor (i.e. RTK) to GIV represent direct coupling by physical interaction, whereas the dotted lines represent coupling by an unknown mechanism. InsR, insulin receptor; PDGFR, PDGF receptor; VEGFR, VEGF receptor; fMLPR, formylmethionylleucylphenylalanine receptor; LPAR, lysophosphatidic acid receptor; TGFβR, transforming growth factor β receptor; mTOR, mammalian target of rapamycin; pCREB, phosphorylated CREB; pTyr, phosphotyrosine. Bottom, summary of different signaling pathways that are either enhanced (green upward arrow) or suppressed (red downward arrow) by the GEF activity of GIV. Numbers indicate the reference number in the text for the publication where the original finding was reported.
What became apparent early on while investigating the function of GEF-deficient GIV mutants was that GIV-dependent G protein activation was not important exclusively for signaling pathways triggered by stimulation of GPCRs. Instead, the GEF activity of GIV is required to signal downstream of multiple RTKs (30, 36, 43, 44, 48). More recently, it was also shown for Toll-like receptors (TLRs) (47) and transforming growth factor β receptors (TGFβRs) (47). These findings have important implications because they place activation of trimeric G proteins as a critical signal transmission step in the context of signaling pathways not traditionally believed to utilize them. Interestingly, it has been recently reported that Ric-8A, another non-receptor GEF for trimeric G proteins, is required for efficient RTK signaling (50, 51). However, it has recently been shown that Ric-8 proteins are G protein chaperones (52–54), and it is controversial whether the effects observed for Ric-8 in cells are mediated by its GEF or its chaperone activity (55, 56). Thus, the picture that starts to emerge is one in which GIV, and maybe other non-receptor GEFs, works as a common platform on which inputs from different surface receptors converge to be subsequently transmitted via G protein activation (Fig. 2). This mechanism is likely to underlie the signaling rewiring mediated by the GEF activity of GIV in different pathological conditions (see below).
However, how does GIV become engaged with these different surface receptors? This question still remains incompletely answered. Of all surface receptor classes GIV is linked to, the molecular mechanisms of coupling are best understood for RTKs. GIV was first shown (36) to directly bind the tyrosine-phosphorylated intracellular tail of EGFR, the prototypical RTK. Subsequent work demonstrated that this mode of binding is conserved for other RTKs (including insulin receptor β and VEGFR2) (57), suggesting its generality. However, the structural basis for this has been elucidated only recently (57). A stretch of ∼110 aa in the C-terminal domain of GIV appears to display structural plasticity: i.e. it is capable of transitioning from a disordered state to an SH2-like folded domain capable of binding phosphotyrosine ligands. When a critical residue in this domain is mutated, binding to phosphotyrosines is lost and GIV no longer transduces signals downstream of RTKs, even in the presence of an intact GEF motif (57). Taken together, these findings delineate a signal transduction mechanism in which trimeric G proteins become activated by RTKs via GIV: i.e. autophosphorylated RTK tails recruit the SH2-like domain of GIV, which in turn activates G proteins via its GEF motif (Fig. 3).
FIGURE 3.
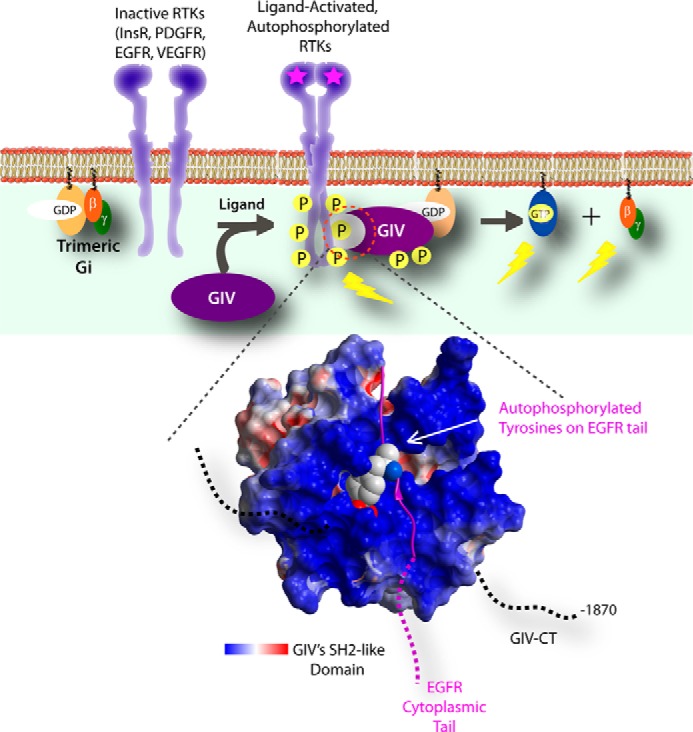
GIV directly binds multiple ligand-activated RTKs via an SH2-like domain in its C terminus. Top, a schematic summarizing the sequence of events triggered by growth factors (such as EGF) is shown. Upon ligand stimulation, RTK dimerization and autophosphorylation of the cytoplasmic tail are triggered. Specific phosphotyrosines within the RTK tail (e.g. Tyr-1148 and Tyr-1173 on EGFR) serve as sites for the recruitment of GIV. Such recruitment requires recognition of phosphotyrosine ligands by an ∼110-aa stretch within the C terminus of GIV that folds into an SH2-like domain that stably docks onto autophosphorylated RTK tail. Close proximity to EGFR facilitates efficient phosphorylation of GIV on critical tyrosines that bind and activate Class 1 PI3-kinases. InsR, insulin receptor; PDGFR, PDGF receptor; VEGFR, VEGF receptor. Bottom, molecular modeling of the interface between the SH2 domain of GIV (red, white, and blue) and EGFR-derived phosphotyrosine peptide (purple) corresponding to Tyr(P)-1148 and its flanking residues, a high-affinity binding site for GIV on the EGF receptor. The acidic, neutral, and basic potentials are displayed in red, white, and blue, respectively. The electrostatic surface potential of the phosphotyrosine recognition and binding pocket of the SH2 domain of GIV is mostly basic. GIV CT, GIV C terminus.
The GEF Motif of GIV Is Crucial for the Regulation of Diverse Biological Processes
Considering the variety of surface receptors GIV is coupled to and the many targets that exist for G proteins, the wide array of cellular processes GIV is involved in is not surprising. To date the G protein modulatory function of GIV has been reported to regulate cell motility and tissue invasion (30, 36, 44, 49, 58), mitosis (36, 59), autophagy (43), cell survival (47, 48), and intracellular protein trafficking (36, 59).
Cell Motility
When the GEF motif of GIV was initially identified, the best characterized cellular function of GIV was cell migration. GIV had been shown to be required for cell motility due to regulation of the actin cytoskeleton remodeling (60). Cells without GIV showed defects in directional migration and failed to form actin stress fibers. The observation that depletion of Gαi3 or expression of inactive Gαi3 mutants phenocopied GIV depletion (32) prompted the investigation of the G protein regulatory function of GIV in this process. Cells engineered to express exclusively GEF-deficient GIV failed to migrate, to form actin stress fibers, and to activate pro-migratory PI3K signals via Gβγ (30). This was originally observed in multiple cancer cell lines but has been subsequently demonstrated to be true for other cell types including hepatic stellate cells (47) and kidney podocytes (48). It is tempting to speculate that the G protein regulatory function of GIV is also required in other cell types such as endothelial cells, leukocytes, non-transformed fibroblasts, and smooth muscle cells, which require GIV for cell motility (32, 40, 60).
Autophagy
The investigation of the role of GIV in autophagy was prompted by two seemingly unconnected mechanisms of regulation of this process. On the one hand, autophagy is well known to be inhibited upon RTK stimulation (e.g. insulin), and on the other hand, it has been suggested that G protein activation may also inhibit autophagy (61, 62). GIV was found to be required for the inhibition of autophagy upon insulin stimulation, and this required an intact GEF motif (43), connecting the two previously unrelated mechanisms. This is an example of how the ability of GIV to assemble alternative G protein signaling pathways (e.g. triggered by an RTK) helps explain previously unappreciated mechanisms of control of cell behavior.
Cell Survival
In certain cell types such as hepatic stellate cells (47) and kidney podocytes (48), activation of G proteins by GIV is an intermediate and required step in pro-survival pathways. In these cell types, disruption of the GEF motif of GIV triggers apoptosis, presumably also via impairment of PI3K-Akt signaling. These findings not only expand the repertoire of the cellular functions of GIV but also suggest that the signaling networks downstream of GIV may be cell-specific.
Intracellular Trafficking and Mitosis
Two independent groups initially described GIV as a protein that can localize to endomembranes (34, 35). The biological significance of this localization was substantiated by the discovery of the role of GIV in the intracellular trafficking of EGFR (36, 59). Cells expressing GIV mutants that cannot bind G proteins accumulate EGFR in early endosomes after ligand stimulation. As a consequence, EGFR signaling is reprogrammed such that pro-mitotic signals (e.g. ERK) emanating from endosomes are enhanced, whereas pro-migratory signals at the plasma membrane (e.g. PI3K) are inhibited, resulting in a faster rate of proliferation. Interestingly, this trafficking mechanism controlled by GIV seems to be mediated by its interaction with Gαs and not with Gαi (59). Although GIV binds in vitro more robustly to Gαi than to Gαs, the functionality of the GIV-Gαs complex in cells suggests that this weaker interaction is sufficient to drive biological processes and/or that modifications occurring in cells enhance the coupling of GIV to Gαs. It is still not known whether GIV acts as a GEF on Gαs or has any other effect on its intrinsic activity.
Implications of GIV and Its GEF Activity in Disease
GIV expression is dysregulated in different diseases such as cancer, fibrosis, and nephrotic syndrome. A common theme observed in all these diseases is that GIV expression is up-regulated and its coupling to G proteins triggers phenotypic changes in key cell types that contribute to disease progression (Fig. 4).
FIGURE 4.

The GEF up-regulation of GIV is directly linked to multiple human diseases. A model depicting the common theme for the role of GIV in disease (from top to bottom) is shown. Up-regulation of GIV expression promotes its coupling to G proteins and enhancement of downstream signaling events. This altered pattern of signaling triggers phenotypic changes in key cell types, thereby contributing to disease progression. Examples of this mechanism of the action of GIV have been described in hepatic stellate cells during liver fibrosis (47), tumor cells during metastatic progression (30, 32, 36, 58), and kidney podocytes upon nephrotic injury (48).
Cancer Metastasis
The importance of GIV in cancer metastasis has been established by us and others based on experiments carried out in cultured cells and murine models of tumor invasion, as well as human cancers (32, 36, 39, 45, 49, 58, 63–67). GIV is expressed at very low levels in non-transformed epithelial tissues, but it is up-regulated in highly invasive cancers of many types (colon, breast, pancreas, etc.) (32, 36, 39, 45, 49, 58, 63–67). Consistently, depletion of GIV impairs the prometastatic behavior of invasive tumor cells in culture and cancer metastasis in murine models (39). The differential expression of GIV in tumors is also clinically significant because its expression serves as a bona fide biomarker for metastasis: i.e. we and others have reported that GIV expression in tumors in situ correlates with cancer metastasis and predicts patient death in different cancers including breast, colorectal, and esophagus, among others (58, 65–67). Although GIV is a multidomain protein, its GEF motif is the critical element controlling the behavior of highly invasive tumor cells. The mechanistic model suggested by these findings is that up-regulation of GIV expression in advanced metastatic cancers favors its coupling to G proteins, which in turn promotes signaling hyperactivation that enhances invasiveness.
Hepatic Fibrosis
Somewhat equivalent observations have been recently reported in liver fibrosis (47). Liver fibrosis is a multi-receptor-driven disease in which a healthy liver undergoes a “scarification” process due to abnormal deposition of extracellular matrix components that cause an increase in the stiffness of the tissue and eventual loss of organ function. Although GIV expression is very low in the healthy liver, it is dramatically up-regulated as it progresses toward the fibrotic state. GIV expression in hepatocytes, the most abundant cell type in the liver, remains undetectable, whereas it is increased manyfold in hepatic stellate cells (47), the main cell type responsible for driving liver fibrosis via collagen deposition among other mechanisms. In hepatic stellate cells, the GEF function of GIV serves as a central hub within the fibrogenic signaling network initiated by diverse classes of receptors. GIV enhances the profibrotic pathways (PI3K-Akt-FoxO1 and TGFβ-SMAD) and inhibits the anti-fibrotic pathway (cAMP-PKA-pCREB, where pCREB indicates phosphorylated CREB) to skew the signaling network in favor of fibrosis, all via activation of Gαi (47). An aspect of this mechanism that remains uninvestigated is the possible role of Gαs regulation by GIV in this context. As mentioned above, GIV can bind to Gαs (34, 59), although the specific consequences of this interaction on the intrinsic activity of the G protein are known. Considering the key role of cAMP as an anti-fibrotic signal (68–70), modulation of Gαs by GIV may have significant implications in the progression of the disease.
Nephrotic Syndrome
A variation on the theme is found in nephrotic syndrome, which is caused by the loss of the kidney's filtration function. In the normal kidney, the initial filtration step in the glomerulus is carried out in part by a specialized cell type, the podocyte, which forms cell interdigitations in which special cell-cell junctions contribute to the filtration barrier. In the case of nephrotic syndrome, GIV is also up-regulated, but here it serves as an adaptive response to nephrotic injury that protects podocytes against apoptosis (48). This contrasts with the observations in metastasis and fibrosis, in which GIV up-regulation actually promotes the disease progression. GIV utilizes its GEF function to activate the pro-survival PI3K-Akt pathway in response to VEGF and compensate for the down-regulation of this pathway caused by the loss of nephrin during early stages of nephrosis.
Future Perspectives
The recent advances described above indicate that GIV assembles alternative signaling pathways by perceiving cues from different classes of receptors and relaying them via G protein activation. This mechanism is critical in the progression of different diseases, many of which represent a huge burden for public health. It makes a compelling argument for the further development of the GIV-Gαi interface as a novel and attractive target for therapeutic intervention in many of these diseases. Because GIV coupling to G proteins can promote diseases such as metastasis and fibrosis, disruption of the GIV-G protein interaction should prove helpful in ameliorating the clinical outcome of these diseases. The rational design of pharmacological agents would be greatly aided by the tractability of the GIV-G protein interface and availability of structural detail.
Although we have focused here on describing cellular processes and diseases for which the GEF activity of GIV has been demonstrated to play a role, this activity may also contribute to other processes. For example, GIV has been shown to modulate the response of endothelial cells (38) and vascular smooth muscle cells (40) to growth factors, contributing to the regulation of angiogenesis and vascular remodeling upon injury (38, 40, 71). GIV has also been shown to be required for proper neuronal migration and postnatal brain development (72, 73) by a mechanism involving GIV-dependent activation of Akt (37). Although it is tempting to speculate that the GEF activity of GIV may contribute to these processes, further investigation in these areas is needed to clarify its involvement.
Despite the progress made, a number of questions still remain open. For example, does GIV mediate G protein activation downstream of additional classes of surface receptors, or are additional biological processes controlled by GIV? Neither of these possibilities seems impossible or far-fetched considering the wide spectrum of receptors already described to utilize GIV as a convergence platform (Fig. 2) for transactivation of G protein signaling. A related complexity resides in the fact that we do not fully understand how GIV can couple to such a diverse group of surface receptors. Although the molecular coupling to RTKs has been studied in more detail, it is challenging, but not impossible, to envision a common theme in the mechanism by which GIV couples to other receptor subclasses. One possibility is that different molecular mechanisms have evolved to allow the integration of GIV into different signaling pathways. For example, with regard to GPCRs, one could envision several models by which GIV could contribute to enhanced G protein activation using mechanisms that are similar to that shown in the case of class II AGS proteins (7). This class of accessory proteins in G protein signaling is proposed to associate with Gαi subunits after GPCR activation but before reassociation with Gβγ (7), which leads to prolonged signaling via “free” Gβγ. Such a mode of action would be compatible with GIV. Another proposed model (7) is that AGS-G protein complexes are direct substrates for GPCRs. One could speculate that analogous GIV-G protein complexes could also be substrates for GPCRs and thereby contribute to overall G protein activation by GPCRs.
Another important question that has been explored only tangentially is whether other proteins contain a GEF sequence similar to that found in GIV. There is evidence supporting this notion because Calnuc and NUCB2, two proteins sharing significant sequence similarity, have been described to possess a “Gα binding and activating” (GBA) motif similar to the GEF sequence found in GIV (74). Although this GBA motif in Calnuc and NUCB2 is required for binding to G proteins and can promote modest activation of Gαi subunits in vitro, it is still unknown what biological functions it may have. Future efforts trying to systematically identify “GBA proteins” and characterize their biological functions would lead us close to answering exciting questions related to the generality of this mechanism of signal transduction, its implications in controlling cell behavior, and the suitability of these GBA proteins as potential therapeutic targets.
This work was supported, in whole or in part, by National Institutes of Health Grants R01GM108733 (to M. G.-M.), R01CA160911 and R01099226 (to P. G.), and R01CA100768 and R37DK17724 (to M. G. F.) This work was also supported by the American Cancer Society Grant RSG-13-362-01-TBE (to M. G.-M.) and the Burroughs Wellcome Foundation (to P. G.). This is the third article in the Thematic Minireview series “Cell Biology of G Protein Signaling.”
- GPCR
- G protein-coupled receptor
- GEF
- guanine nucleotide exchange factor
- RGS
- regulator of G protein signaling
- AGS
- activator of G protein signaling
- GBA
- Gα binding and activating motif
- GPR
- G protein regulator
- RTK
- receptor tyrosine kinase
- SH2
- Src homology 2
- CREB
- cAMP-response element-binding protein
- aa
- amino acid(s)
- EGFR
- epidermal growth factor receptor.
REFERENCES
- 1. Gilman A. G. (1987) G proteins: transducers of receptor-generated signals. Annu. Rev. Biochem. 56, 615–649 [DOI] [PubMed] [Google Scholar]
- 2. Morris A. J., Malbon C. C. (1999) Physiological regulation of G protein-linked signaling. Physiol. Rev. 79, 1373–1430 [DOI] [PubMed] [Google Scholar]
- 3. Hopkins A. L., Groom C. R. (2002) The druggable genome. Nat. Rev. Drug Discov. 1, 727–730 [DOI] [PubMed] [Google Scholar]
- 4. Sato M., Blumer J. B., Simon V., Lanier S. M. (2006) Accessory proteins for G proteins: partners in signaling. Annu. Rev. Pharmacol. Toxicol. 46, 151–187 [DOI] [PubMed] [Google Scholar]
- 5. Siderovski D. P., Willard F. S. (2005) The GAPs, GEFs, and GDIs of heterotrimeric G-protein α subunits. Int. J. Biol. Sci. 1, 51–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ross E. M., Wilkie T. M. (2000) GTPase-activating proteins for heterotrimeric G proteins: regulators of G protein signaling (RGS) and RGS-like proteins. Annu. Rev. Biochem. 69, 795–827 [DOI] [PubMed] [Google Scholar]
- 7. Blumer J. B., Oner S. S., Lanier S. M. (2012) Group II activators of G-protein signalling and proteins containing a G-protein regulatory motif. Acta Physiol. (Oxf.) 204, 202–218 [DOI] [PubMed] [Google Scholar]
- 8. De Vries L., Zheng B., Fischer T., Elenko E., Farquhar M. G. (2000) The regulator of G protein signaling family. Annu. Rev. Pharmacol. Toxicol. 40, 235–271 [DOI] [PubMed] [Google Scholar]
- 9. Watson N., Linder M. E., Druey K. M., Kehrl J. H., Blumer K. J. (1996) RGS family members: GTPase-activating proteins for heterotrimeric G-protein α-subunits. Nature 383, 172–175 [DOI] [PubMed] [Google Scholar]
- 10. Berman D. M., Wilkie T. M., Gilman A. G. (1996) GAIP and RGS4 are GTPase-activating proteins for the Gi subfamily of G protein α subunits. Cell 86, 445–452 [DOI] [PubMed] [Google Scholar]
- 11. De Vries L., Elenko E., Hubler L., Jones T. L., Farquhar M. G. (1996) GAIP is membrane-anchored by palmitoylation and interacts with the activated (GTP-bound) form of Gαi subunits. Proc. Natl. Acad. Sci. U.S.A. 93, 15203–15208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. De Vries L., Fischer T., Tronchère H., Brothers G. M., Strockbine B., Siderovski D. P., Farquhar M. G. (2000) Activator of G protein signaling 3 is a guanine dissociation inhibitor for Gαi subunits. Proc. Natl. Acad. Sci. U.S.A. 97, 14364–14369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Peterson Y. K., Bernard M. L., Ma H., Hazard S., 3rd, Graber S. G., Lanier S. M. (2000) Stabilization of the GDP-bound conformation of Giα by a peptide derived from the G-protein regulatory motif of AGS3. J. Biol. Chem. 275, 33193–33196 [DOI] [PubMed] [Google Scholar]
- 14. Bernard M. L., Peterson Y. K., Chung P., Jourdan J., Lanier S. M. (2001) Selective interaction of AGS3 with G-proteins and the influence of AGS3 on the activation state of G-proteins. J. Biol. Chem. 276, 1585–1593 [DOI] [PubMed] [Google Scholar]
- 15. Natochin M., Lester B., Peterson Y. K., Bernard M. L., Lanier S. M., Artemyev N. O. (2000) AGS3 inhibits GDP dissociation from Gα subunits of the Gi family and rhodopsin-dependent activation of transducin. J. Biol. Chem. 275, 40981–40985 [DOI] [PubMed] [Google Scholar]
- 16. Kimple R. J., Kimple M. E., Betts L., Sondek J., Siderovski D. P. (2002) Structural determinants for GoLoco-induced inhibition of nucleotide release by Gα subunits. Nature 416, 878–881 [DOI] [PubMed] [Google Scholar]
- 17. Peterson Y. K., Hazard S., 3rd, Graber S. G., Lanier S. M. (2002) Identification of structural features in the G-protein regulatory motif required for regulation of heterotrimeric G-proteins. J. Biol. Chem. 277, 6767–6770 [DOI] [PubMed] [Google Scholar]
- 18. De Vries L., Mousli M., Wurmser A., Farquhar M. G. (1995) GAIP, a protein that specifically interacts with the trimeric G protein Gαi3, is a member of a protein family with a highly conserved core domain. Proc. Natl. Acad. Sci. U.S.A. 92, 11916–11920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Druey K. M., Blumer K. J., Kang V. H., Kehrl J. H. (1996) Inhibition of G-protein-mediated MAP kinase activation by a new mammalian gene family. Nature 379, 742–746 [DOI] [PubMed] [Google Scholar]
- 20. Srinivasa S. P., Watson N., Overton M. C., Blumer K. J. (1998) Mechanism of RGS4, a GTPase-activating protein for G protein α subunits. J. Biol. Chem. 273, 1529–1533 [DOI] [PubMed] [Google Scholar]
- 21. Blazer L. L., Roman D. L., Chung A., Larsen M. J., Greedy B. M., Husbands S. M., Neubig R. R. (2010) Reversible, allosteric small-molecule inhibitors of regulator of G protein signaling proteins. Mol. Pharmacol. 78, 524–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kimple A. J., Yasgar A., Hughes M., Jadhav A., Willard F. S., Muller R. E., Austin C. P., Inglese J., Ibeanu G. C., Siderovski D. P., Simeonov A. (2008) A high throughput fluorescence polarization assay for inhibitors of the GoLoco motif/G-α interaction. Comb Chem. High Throughput Screen. 11, 396–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cismowski M. J., Ma C., Ribas C., Xie X., Spruyt M., Lizano J. S., Lanier S. M., Duzic E. (2000) Activation of heterotrimeric G-protein signaling by a ras-related protein: implications for signal integration. J. Biol. Chem. 275, 23421–23424 [DOI] [PubMed] [Google Scholar]
- 24. Tall G. G., Krumins A. M., Gilman A. G. (2003) Mammalian Ric-8A (synembryn) is a heterotrimeric Gα protein guanine nucleotide exchange factor. J. Biol. Chem. 278, 8356–8362 [DOI] [PubMed] [Google Scholar]
- 25. Chan P., Gabay M., Wright F. A., Tall G. G. (2011) Ric-8B is a GTP-dependent G protein αs guanine nucleotide exchange factor. J. Biol. Chem. 286, 19932–19942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee M. J., Dohlman H. G. (2008) Coactivation of G protein signaling by cell-surface receptors and an intracellular exchange factor. Curr. Biol. 18, 211–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Natochin M., Campbell T. N., Barren B., Miller L. C., Hameed S., Artemyev N. O., Braun J. E. (2005) Characterization of the Gαs regulator cysteine string protein. J. Biol. Chem. 280, 30236–30241 [DOI] [PubMed] [Google Scholar]
- 28. Vaidyanathan G., Cismowski M. J., Wang G., Vincent T. S., Brown K. D., Lanier S. M. (2004) The Ras-related protein AGS1/RASD1 suppresses cell growth. Oncogene 23, 5858–5863 [DOI] [PubMed] [Google Scholar]
- 29. Tõnissoo T., Lulla S., Meier R., Saare M., Ruisu K., Pooga M., Karis A. (2010) Nucleotide exchange factor RIC-8 is indispensable in mammalian early development. Dev. Dyn. 239, 3404–3415 [DOI] [PubMed] [Google Scholar]
- 30. Garcia-Marcos M., Ghosh P., Farquhar M. G. (2009) GIV is a nonreceptor GEF for Gαi with a unique motif that regulates Akt signaling. Proc. Natl. Acad. Sci. U.S.A. 106, 3178–3183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Enomoto A., Murakami H., Asai N., Morone N., Watanabe T., Kawai K., Murakumo Y., Usukura J., Kaibuchi K., Takahashi M. (2005) Akt/PKB regulates actin organization and cell motility via Girdin/APE. Dev. Cell 9, 389–402 [DOI] [PubMed] [Google Scholar]
- 32. Ghosh P., Garcia-Marcos M., Bornheimer S. J., Farquhar M. G. (2008) Activation of Gαi3 triggers cell migration via regulation of GIV. J. Cell Biol. 182, 381–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Anai M., Shojima N., Katagiri H., Ogihara T., Sakoda H., Onishi Y., Ono H., Fujishiro M., Fukushima Y., Horike N., Viana A., Kikuchi M., Noguchi N., Takahashi S., Takata K., Oka Y., Uchijima Y., Kurihara H., Asano T. (2005) A novel protein kinase B (PKB)/AKT-binding protein enhances PKB kinase activity and regulates DNA synthesis. J. Biol. Chem. 280, 18525–18535 [DOI] [PubMed] [Google Scholar]
- 34. Le-Niculescu H., Niesman I., Fischer T., DeVries L., Farquhar M. G. (2005) Identification and characterization of GIV, a novel Gαi/s-interacting protein found on COPI, endoplasmic reticulum-Golgi transport vesicles. J. Biol. Chem. 280, 22012–22020 [DOI] [PubMed] [Google Scholar]
- 35. Simpson F., Martin S., Evans T. M., Kerr M., James D. E., Parton R. G., Teasdale R. D., Wicking C. (2005) A novel Hook-related protein family and the characterization of Hook-related protein 1. Traffic 6, 442–458 [DOI] [PubMed] [Google Scholar]
- 36. Ghosh P., Beas A. O., Bornheimer S. J., Garcia-Marcos M., Forry E. P., Johannson C., Ear J., Jung B. H., Cabrera B., Carethers J. M., Farquhar M. G. (2010) A Gαi-GIV molecular complex binds epidermal growth factor receptor and determines whether cells migrate or proliferate. Mol. Biol. Cell 21, 2338–2354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kim J. Y., Duan X., Liu C. Y., Jang M. H., Guo J. U., Pow-anpongkul N., Kang E., Song H., Ming G. L. (2009) DISC1 regulates new neuron development in the adult brain via modulation of AKT-mTOR signaling through KIAA1212. Neuron 63, 761–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kitamura T., Asai N., Enomoto A., Maeda K., Kato T., Ishida M., Jiang P., Watanabe T., Usukura J., Kondo T., Costantini F., Murohara T., Takahashi M. (2008) Regulation of VEGF-mediated angiogenesis by the Akt/PKB substrate Girdin. Nat. Cell Biol. 10, 329–337 [DOI] [PubMed] [Google Scholar]
- 39. Jiang P., Enomoto A., Jijiwa M., Kato T., Hasegawa T., Ishida M., Sato T., Asai N., Murakumo Y., Takahashi M. (2008) An actin-binding protein Girdin regulates the motility of breast cancer cells. Cancer Res. 68, 1310–1318 [DOI] [PubMed] [Google Scholar]
- 40. Miyake H., Maeda K., Asai N., Shibata R., Ichimiya H., Isotani-Sakakibara M., Yamamura Y., Kato K., Enomoto A., Takahashi M., Murohara T. (2011) The actin-binding protein Girdin and its Akt-mediated phosphorylation regulate neointima formation after vascular injury. Circ. Res. 108, 1170–1179 [DOI] [PubMed] [Google Scholar]
- 41. Weng L., Enomoto A., Ishida-Takagishi M., Asai N., Takahashi M. (2010) Girding for migratory cues: roles of the Akt substrate Girdin in cancer progression and angiogenesis. Cancer Sci. 101, 836–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Johnston C. A., Willard F. S., Jezyk M. R., Fredericks Z., Bodor E. T., Jones M. B., Blaesius R., Watts V. J., Harden T. K., Sondek J., Ramer J. K., Siderovski D. P. (2005) Structure of Gαi1 bound to a GDP-selective peptide provides insight into guanine nucleotide exchange. Structure 13, 1069–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Garcia-Marcos M., Ear J., Farquhar M. G., Ghosh P. (2011) A GDI (AGS3) and a GEF (GIV) regulate autophagy by balancing G protein activity and growth factor signals. Mol. Biol. Cell 22, 673–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Garcia-Marcos M., Ghosh P., Ear J., Farquhar M. G. (2010) A structural determinant that renders Gαi sensitive to activation by GIV/Girdin is required to promote cell migration. J. Biol. Chem. 285, 12765–12777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Garcia-Marcos M., Kietrsunthorn P. S., Pavlova Y., Adia M. A., Ghosh P., Farquhar M. G. (2012) Functional characterization of the guanine nucleotide exchange factor (GEF) motif of GIV protein reveals a threshold effect in signaling. Proc. Natl. Acad. Sci. U.S.A. 109, 1961–1966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Coleman D. E., Berghuis A. M., Lee E., Linder M. E., Gilman A. G., Sprang S. R. (1994) Structures of active conformations of Giα1 and the mechanism of GTP hydrolysis. Science 265, 1405–1412 [DOI] [PubMed] [Google Scholar]
- 47. Lopez-Sanchez I., Dunkel Y., Roh Y. S., Mittal Y., De Minicis S., Muranyi A., Singh S., Shanmugam K., Aroonsakool N., Murray F., Ho S. B., Seki E., Brenner D. A., Ghosh P. (2014) GIV/Girdin is a central hub for profibrogenic signalling networks during liver fibrosis. Nat. Commun. 5, 4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang H., Misaki T., Taupin V., Eguchi A., Ghosh P., Farquhar M. G. (2014) GIV/Girdin links vascular endothelial growth factor signaling to Akt survival signaling in podocytes independent of nephrin. J. Am. Soc. Nephrol. 10.1681/ASN.2013090985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dunkel Y., Ong A., Notani D., Mittal Y., Lam M., Mi X., Ghosh P. (2012) STAT3 protein up-regulates Gα-interacting vesicle-associated protein (GIV)/Girdin expression, and GIV enhances STAT3 activation in a positive feedback loop during wound healing and tumor invasion/metastasis. J. Biol. Chem. 287, 41667–41683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang L., Guo D., Xing B., Zhang J. J., Shu H. B., Guo L., Huang X. Y. (2011) Resistance to inhibitors of cholinesterase-8A (Ric-8A) is critical for growth factor receptor-induced actin cytoskeletal reorganization. J. Biol. Chem. 286, 31055–31061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xing B., Wang L., Guo D., Huang J., Espenel C., Kreitzer G., Zhang J. J., Guo L., Huang X. Y. (2013) Atypical protein kinase Cλ is critical for growth factor receptor-induced dorsal ruffle turnover and cell migration. J. Biol. Chem. 288, 32827–32836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Thomas C. J., Briknarová K., Hilmer J. K., Movahed N., Bothner B., Sumida J. P., Tall G. G., Sprang S. R. (2011) The nucleotide exchange factor Ric-8A is a chaperone for the conformationally dynamic nucleotide-free state of Gαi1. PLoS One 6, e23197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gabay M., Pinter M. E., Wright F. A., Chan P., Murphy A. J., Valenzuela D. M., Yancopoulos G. D., Tall G. G. (2011) Ric-8 proteins are molecular chaperones that direct nascent G protein α subunit membrane association. Sci. Signal. 4, ra79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chan P., Thomas C. J., Sprang S. R., Tall G. G. (2013) Molecular chaperoning function of Ric-8 is to fold nascent heterotrimeric G protein α subunits. Proc. Natl. Acad. Sci. U.S.A. 110, 3794–3799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Papasergi M. M., Patel B. R., Tall G. G. (2015) The G protein α chaperone Ric-8 as a potential therapeutic target. Mol. Pharmacol. 87, 52–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tall G. G., Patel B. R., Chan P. (2013) Ric-8 folding of G proteins better explains Ric-8 apparent amplification of G protein-coupled receptor signaling. Proc. Natl. Acad. Sci. U.S.A. 110, E3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lin C., Ear J., Midde K., Lopez-Sanchez I., Aznar N., Garcia-Marcos M., Kufareva I., Abagyan R., Ghosh P. (2014) Structural basis for activation of trimeric Gi proteins by multiple growth factor receptors via GIV/Girdin. Mol. Biol. Cell 25, 3654–3671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Garcia-Marcos M., Jung B. H., Ear J., Cabrera B., Carethers J. M., Ghosh P. (2011) Expression of GIV/Girdin, a metastasis-related protein, predicts patient survival in colon cancer. FASEB J. 25, 590–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Beas A. O., Taupin V., Teodorof C., Nguyen L. T., Garcia-Marcos M., Farquhar M. G. (2012) Gαs promotes EEA1 endosome maturation and shuts down proliferative signaling through interaction with GIV (Girdin). Mol. Biol. Cell 23, 4623–4634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Enomoto A., Ping J., Takahashi M. (2006) Girdin, a novel actin-binding protein, and its family of proteins possess versatile functions in the Akt and Wnt signaling pathways. Ann. N.Y. Acad. Sci. 1086, 169–184 [DOI] [PubMed] [Google Scholar]
- 61. Ogier-Denis E., Couvineau A., Maoret J. J., Houri J. J., Bauvy C., De Stefanis D., Isidoro C., Laburthe M., Codogno P. (1995) A heterotrimeric Gi3-protein controls autophagic sequestration in the human colon cancer cell line HT-29. J. Biol. Chem. 270, 13–16 [DOI] [PubMed] [Google Scholar]
- 62. Ogier-Denis E., Houri J. J., Bauvy C., Codogno P. (1996) Guanine nucleotide exchange on heterotrimeric Gi3 protein controls autophagic sequestration in HT-29 cells. J. Biol. Chem. 271, 28593–28600 [DOI] [PubMed] [Google Scholar]
- 63. Ghosh P., Garcia-Marcos M., Farquhar M. G. (2011) GIV/Girdin is a rheostat that fine-tunes growth factor signals during tumor progression. Cell Adh. Migr. 5, 237–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jun B. Y., Kim S. W., Jung C. K., Cho Y. K., Lee I. S., Choi M. G., Choi K. Y., Oh S. T. (2013) Expression of Girdin in human colorectal cancer and its association with tumor progression. Dis. Colon Rectum 56, 51–57 [DOI] [PubMed] [Google Scholar]
- 65. Ling Y., Jiang P., Cui S. P., Ren Y. L., Zhu S. N., Yang J. P., Du J., Zhang Y., Liu J. Y., Zhang B. (2011) Clinical implications for Girdin protein expression in breast cancer. Cancer Invest. 29, 405–410 [DOI] [PubMed] [Google Scholar]
- 66. Liu C., Xue H., Lu Y., Chi B. (2012) Stem cell gene Girdin: a potential early liver metastasis predictor of colorectal cancer. Mol. Biol. Rep. 39, 8717–8722 [DOI] [PubMed] [Google Scholar]
- 67. Liu C., Zhang Y., Xu H., Zhang R., Li H., Lu P., Jin F. (2012) Girdin protein: a new potential distant metastasis predictor of breast cancer. Med. Oncol. 29, 1554–1560 [DOI] [PubMed] [Google Scholar]
- 68. Kisseleva T., Brenner D. A. (2008) Mechanisms of fibrogenesis. Exp. Biol. Med. (Maywood) 233, 109–122 [DOI] [PubMed] [Google Scholar]
- 69. Swaney J. S., Roth D. M., Olson E. R., Naugle J. E., Meszaros J. G., Insel P. A. (2005) Inhibition of cardiac myofibroblast formation and collagen synthesis by activation and overexpression of adenylyl cyclase. Proc. Natl. Acad. Sci. U.S.A. 102, 437–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Solís-Herruzo J. A., Hernández I., De la Torre P., García I., Sánchez J. A., Fernández I., Castellano G., Muñoz-Yagüe T. (1998) G proteins are involved in the suppression of collagen α1(I) gene expression in cultured rat hepatic stellate cells. Cell. Signal. 10, 173–183 [DOI] [PubMed] [Google Scholar]
- 71. Ito T., Komeima K., Yasuma T., Enomoto A., Asai N., Asai M., Iwase S., Takahashi M., Terasaki H. (2013) Girdin and its phosphorylation dynamically regulate neonatal vascular development and pathological neovascularization in the retina. Am. J. Pathol. 182, 586–596 [DOI] [PubMed] [Google Scholar]
- 72. Enomoto A., Asai N., Namba T., Wang Y., Kato T., Tanaka M., Tatsumi H., Taya S., Tsuboi D., Kuroda K., Kaneko N., Sawamoto K., Miyamoto R., Jijiwa M., Murakumo Y., Sokabe M., Seki T., Kaibuchi K., Takahashi M. (2009) Roles of disrupted-in-schizophrenia 1-interacting protein Girdin in postnatal development of the dentate gyrus. Neuron 63, 774–787 [DOI] [PubMed] [Google Scholar]
- 73. Wang Y., Kaneko N., Asai N., Enomoto A., Isotani-Sakakibara M., Kato T., Asai M., Murakumo Y., Ota H., Hikita T., Namba T., Kuroda K., Kaibuchi K., Ming G. L., Song H., Sawamoto K., Takahashi M. (2011) Girdin is an intrinsic regulator of neuroblast chain migration in the rostral migratory stream of the postnatal brain. J. Neurosci. 31, 8109–8122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Garcia-Marcos M., Kietrsunthorn P. S., Wang H., Ghosh P., Farquhar M. G. (2011) G protein binding sites on Calnuc (nucleobindin 1) and NUCB2 (nucleobindin 2) define a new class of Gαi-regulatory motifs. J. Biol. Chem. 286, 28138–28149 [DOI] [PMC free article] [PubMed] [Google Scholar]