

Background: Presenilin TMD8 contains a conserved AXXXAXXXG motif, involved in transmembrane protein interactions.
Results: Mutation of PS AXXXAXXXG motifs strongly impacts γ-secretase activity.
Conclusion: The geometry and activity of γ-secretase depend on the integrity of the conserved AXXXAXXXG motifs in TMD8.
Significance: The PS AXXXAXXXG motif is a key determinant for understanding the structure, assembly, and function of the γ-secretase complex.
Keywords: Alzheimer Disease, Amyloid Precursor Protein (APP), gamma-Secretase, Notch Receptor, Presenilin, Transmembrane Domain
Abstract
Understanding the molecular mechanisms controlling the physiological and pathological activity of γ-secretase represents a challenging task in Alzheimer disease research. The assembly and proteolytic activity of this enzyme require the correct interaction of the 19 transmembrane domains (TMDs) present in its four subunits, including presenilin (PS1 or PS2), the γ-secretase catalytic core. GXXXG and GXXXG-like motifs are critical for TMDs interactions as well as for protein folding and assembly. The GXXXG motifs on γ-secretase subunits (e.g. APH-1) or on γ-secretase substrates (e.g. APP) are known to be involved in γ-secretase assembly and in Aβ peptide production, respectively. We identified on PS1 and PS2 TMD8 two highly conserved AXXXAXXXG motifs. The presence of a mutation causing an inherited form of Alzheimer disease (familial Alzheimer disease) in the PS1 motif suggested their involvement in the physiopathological configuration of the γ-secretase complex. In this study, we targeted the role of these motifs on TMD8 of PSs, focusing on their role in PS assembly and catalytic activity. Each motif was mutated, and the impact on complex assembly, activity, and substrate docking was monitored. Different amino acid substitutions on the same motif resulted in opposite effects on γ-secretase activity, without affecting the assembly or significantly impairing the maturation of the complex. Our data suggest that AXXXAXXXG motifs in PS TMD8 are key determinants for the conformation of the mature γ-secretase complex, participating in the switch between the physiological and pathological functional conformations of the γ-secretase.
Introduction
Aβ3 is produced by the sequential cleavage of the amyloid precursor protein (APP) by β- and γ-secretase. The amyloidogenic C-terminal fragment generated by β-secretase (β-CTF or C99) is processed by γ-secretase at the ϵ-site, leading to the release of the APP intracellular domain (AICD) (1–3). The remaining Aβ fragment is further trimmed by γ-secretase until the Aβ peptides of various lengths (Aβ38 to Aβ43) are released (4–7). The progressive accumulation and deposition of Aβ40 and Aβ42, the latter isoform being more aggregation-prone, triggers a cascade of events central to AD (8, 9). This general processing mechanism also applies to other γ-secretase substrates, such as Notch, where it leads to the release of the transcriptionally active Notch intracellular domain (NICD) (10, 11).
γ-Secretase is an aspartyl intramembrane cleaving protease (12). Presenilins (PSs) form the catalytic core of γ-secretase. In mammals, there are two homologous presenilins (PS1 and PS2). The assembly of the γ-secretase complex requires the association of either PS1 or PS2 with three other membrane proteins: nicastrin (NCT), anterior pharynx defective 1 (APH-1a or APH-1b), and presenilin enhancer 2 (PEN-2) (13, 14). Presenilins have nine transmembrane domains (TMDs) (15), and the two catalytic aspartic acid residues are located on its TMD6 and -7, both required for substrate hydrolysis. γ-Secretase assembly is accompanied by an activating step in which presenilin is endoproteolysed and converted into a heterodimer formed by its N- and C-terminal fragments (PS-NTF/PS-CTF) (16–18). In addition to this activating step, γ-secretase complex has two catalytic activities: (i) endoproteolytic, releasing intracellular domains from type I integral membrane protein substrates, and (ii) carboxypeptidase, trimming the remaining TM domains from the substrate until the final product is released (19).
Interactions between the TMDs within presenilins and its binding partners are essential for γ-secretase assembly and maturation as well as substrate docking and cleavage (14). The majority of AD inherited cases linked to presenilin mutations (185 for PS1 and 13 for PS2) are clustered in the PS1 (∼62%) and PS2 (∼70%) TMDs. Sequence integrity of each TMD is critical for its integration and folding in the membrane as well as for the structure of the active site pocket (20, 21). Recently, the moderate resolution structure of the γ-secretase complex has been determined by electron cryomicroscopy (22, 23). The structure reveals a horseshoe-shaped complex of the TM helices and confirms the 1:1:1:1 stoichiometry of the four subunits but is not at high enough resolution to make specific TM helix assignments.
The current structural model and biochemical data on the γ-secretase complex provide a foundation for probing the assembly and function of the TM helices within the complex. Specific helix-helix interactions that guide membrane protein folding and assembly are often mediated by the GXXXG motif. Studies on the dimerizing TM domain of the glycophorin A (GpA) showed that by creating a flat surface, or interface, GXXXG motifs allow the close approach of the polar backbone of opposing helices (24–26). Stabilizing helix interactions take the form of interhelical CαH hydrogen bonds mediated by the glycines of the motif (27). Following the characterization of this motif by many groups, it became clear that the motif extends to residues with small/polar side chains in the GXXXG-like motif, with the configuration of [small]XXX[small], where small residues, including alanine, serine, and threonine, are separated by three amino acids (28–32). Serine, alanine, and glycine have the highest packing values in membrane proteins (28), and are the most prevalent residues in tightly packed helix interfaces (29). Additionally in a GXXXG-like motif, serines or threonines mediate the formation of C–OH hydrogen bonds that strengthen the packing properties of the motif (28, 31, 32). The γ-secretase complex provides a fascinating system in which to understand how helix association influences enzyme activity, assembly, and substrate interactions because both the helices of the proteins in the complex and (often) the substrates contain GXXXG or GXXXG-like motifs (33). APP harbors an extended GXXXG interface (621GXXXGXXXGXXXG633, APP695 numbering) that controls APP homodimerization and is a major determinant in the structural features of the JM/TM regions playing a key role in amyloidogenic processing and Aβ release (34–37). GXXXG motifs have been described in APH-1 TMD4 (122GXXXGXXXGXXXG130). Their interactive properties were found to be critical for the stability of the protein and for γ-secretase assembly, by controlling the interaction of APH-1 with other subunits (38–40).
The orientation of TM helices can be probed through the occurrence of GXXXG and GXXXG-like motifs (41), which in turn can provide insights into membrane protein structure and assembly. By sequence analysis, we found several motifs occurring in PS1 and PS2 compatible with the GXXXG-like motif sequences. There are AXXXS (PS1) and SXXXA (PS1 and PS2) motifs in TMD2 and TMD5, respectively. TMD7 has two motifs (GXXXG and SXXXGXXXA), which lie on each side of the catalytic Asp. Both of the motifs in TMD7 contain sites of FAD mutations, which might be expected because TMD7 forms a portion of the catalytic core of the presenilins, and a change in helix packing may modulate enzyme activity. TMD8 and TMD9 are both unusual in having two sequential motifs: AXXXAXXXG (PS1, 409AXXXAXXXG417; PS2, 390AXXXAXXXG408) in TMD8 and AXXXSXXXG (PS1, 434AXXXSXXXG442; PS2, 415AXXXSXXXG423) in TMD9. In both sequences, the small residues are separated by three other amino acids, in analogy with the interactive motif of the GpA, strongly supporting their involvement in tight packing interactions.
TMD8 is a highly conserved hydrophobic (PS1/PS2 90.5% identity) 20-amino acid TMD (42, 43). This domain is involved in the formation and stability of the complex (44, 45) and in the membrane integration of TMD7 into the membrane bilayer by “forced transmembrane orientation” (43). In PS1, both motifs include two critical amino acids: Gly-417, having a pivotal role in γ-secretase activity (46), and Ala-409, whose substitution with Thr (PS1 A409T) is described as a FAD mutation (47). The alanine to threonine substitution has the potential to strengthen helix interactions through interhelical hydrogen bonding that is permitted by close helix packing (28, 32).
On the basis of the helix interactions observed in other membrane proteins containing GXXXG and GXXXG-like motifs, one would anticipate that conserved AXXXAXXXG motifs in TMD8 play a role in γ-secretase function. Specifically, the expectation is that glycine or alanine to leucine mutations would disrupt the helix packing interactions, whereas the A409T mutation would strengthen helix interactions. Mutations of AXXXAXXXG interactive motifs on helices that are not part of the catalytic active site might be expected to impair enzyme activity as a result of disrupting helix-helix association or assembly of the complex. To test this proposal, we specifically mutated the motif on PS1 and PS2 and studied the effects on complex assembly and activity with respect to amyloidogenic processing and liberation of the APP and Notch intracellular domains. These studies form the foundation for determining the role of the AXXXAXXXG motifs on the other TM helices of the presenilins and understanding the interplay between helix interactions, enzyme activity, and mechanism(s) underlying the FAD mutations.
EXPERIMENTAL PROCEDURES
Chemicals and Reagents
All culture media and reagents, Nu-Page® Novex® 4–12% BisTris, and NativePAGETM Novex® 4–16% BisTris gels were from Life Technologies. Analytical grade solvents and salts were purchased from Sigma-Aldrich. Transfection reagents Trans-IT2020 and Trans-IT293 were from Mirus Bio Corp. (Madison, WI). CHAPS was obtained from Merck. N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester (DAPT) was from Calbiochem. Protease inhibitor mixture was purchased from Roche Applied Science. Reagents for the luciferase assay (Dual-Glo Luciferase Assay kit) were purchased from Promega (Madison, WI). Nitrocellulose and PVDF membranes were obtained from GE Healthcare and EMD Millipore (Billerica, MA), respectively. ECL reagents were obtained from Perkin Elmer Life Sciences. The following antibodies were used: Mab1563 anti-human PS1-NTF (EMD Millipore); PA1-758 anti NCT (Affinity BioReagents, Neshanic Station, NJ); anti-APP C-ter (generous gift of N. Sergeant, INSERM U422, Lille, France); PA1–2010 anti-APH-1 (Thermo Scientific); and D39D1 anti-PS1-CTF, D30G3 anti-PS2-CTF, D2G6 anti-PEN-2, anti-Myc, and anti-HA tags (Cell Signaling, Danvers, MA). Secondary antibodies were obtained from Amersham Biosciences. The applied dilutions for each antibody were according to the manufacturer's instructions.
Cells Lines and Cell Culture
Chinese hamster ovary (CHO) cell lines were grown in Ham's F-12 medium. Mouse embryonic fibroblast (MEF) wild type (MEF+/+) and knock-out cell lines for both PS1 and PS2 (MEFPSdKO) as well as MEFPSdKO rescued with PS1/PS2 wild type or mutant forms (MEFPSdKOr) were cultured in DMEM/F-12. Human embryonic kidney (HEK293T and HEK293) cell lines were grown in DMEM high glucose medium. All media were supplemented with 10% fetal bovine serum (FBS) (Thermo Scientific) and penicillin-streptomycin solution (10 units, 10 μg). Puromycin (2.5 μg/ml) was used for MEFdKOr.
Plasmids and Site-directed Mutagenesis
Original plasmids expressing human PS1 were described previously (48). The coding sequence of human PS2 was amplified by RT-PCR and cloned in the same backbone. QuikChange site-specific mutagenesis (Stratagene, La Jolla, CA) was applied on PS1 and PS2 plasmids as described previously (49). APH-1, PEN-2, and NCT cDNAs were retro-transcribed from HEK293 cells and amplified by PCR using specific primers. PCR products were cloned into pCDNA-3.1 V5 vector (Invitrogen). All constructs were confirmed by full sequencing (Macrogen Inc. Europe, Amsterdam, Netherlands). For reporter gene analysis, the following plasmids have been used: Hairy/Enhancer of Split1 (HES1) luciferase reporter gene (pHes1(2.5k)-luc) (50), Renilla luciferase vector (pRL-TK, Promega, Madison, WI), pCS2 Notch1 ΔEMV-6MT (NotchΔE) (51) (plasmid 41737, Addgene, Cambridge, MA), APP695Gal4 (pMst-APPGal4), Gal4RE-luc (pG5E1B-luc), and Fe65 (pCMV5-Fe65) (52, 53).
Western Blotting
Total cell lysates were prepared in lysis buffer (125 mm Tris, pH 6.8, 2% SDS, 10% glycerol). Proteins were separated using Nu-Page® Novex® 4–12% gradient gels and transferred on a nitrocellulose or PVDF membrane, blocked with nonfat dry milk (Merck) (5% in PBS, 0.01% Tween 20), and incubated overnight at 4 °C with the primary antibodies. Membranes were then washed and incubated with secondary antibodies coupled to horseradish peroxidase prior to ECL detection. Quantification of band signal was performed using the Quantity One® software coupled to the Gel Doc 2000 device (Bio-Rad).
Cell Transfection
Cells were transfected 24 h after seeding using Trans-IT2020 for CHO and MEF cells and Trans-IT293 for HEK293T cells, according to the manufacturer's instructions. Cell lysates and culture media were harvested for analysis 48 h after transfection.
Electrochemiluminescence Immunoassay (ECLIA)
Aβ40 and Aβ42 peptides were quantified in the cell medium (34) by the Aβ multiplex ECLIA (Meso Scale Discovery, Gaithersburg, MD). One day after plating (MEFs) or transfection (CHO cells), cells were conditioned in serum-free medium for 16 or 8 h, respectively. Cell medium was then collected, and Aβ was quantified according to the manufacturer's instructions. Two Aβ multiplex assays, 4G8 and 6E10, have been used to measure rodent and human Aβ, respectively (54).
Co-immunoprecipitation
Co-immunoprecipitation was carried out based on Shiraishi et al. (44) and Xia et al. (55). 48 h after transfection, CHO cells were washed with cold PBS and homogenized in lysis buffer A (50 mm Tris, pH 7.6, 150 mm NaCl, 2 mm EDTA, 1% CHAPS, and protease inhibitor mixture). Cell debris was discarded by low speed centrifugation (800 × g for 10 min) at 4 °C. 100 μg of solubilized membranes were precleared with 20% Protein G-Sepharose (Amersham Biosciences) for 3 h at 4 °C. Cleared lysates were then incubated overnight at 4 °C under gentle rocking with anti-human PS1 antibody (Mab1563) in the presence of fresh 20% Protein G-Sepharose. The day after, beads were washed once with buffer B (50 mm Tris, pH 7.6, 500 mm NaCl, and protease inhibitor mixture), twice with buffer A (50 mm Tris, pH 7.6, 150 mm NaCl, 2 mm EDTA, protease inhibitor mixture), and once with 50 mm Tris-HCl, pH 7.6. Proteins were finally detached using NuPAGE® LDS sample buffer (Invitrogen) supplemented with 50 μm DTT and analyzed by Western blotting.
Lentiviral Constructions and MEF Rescued Cell Lines
PS1 and PS2 mutants generated in plasmid vectors (see above) were subcloned in the pLenti CMV/TO Puro lentiviral vector (56) (plasmid 17482, Addgene). Lentiviruses were produced in HEK293T cells (49, 57) and used to infect MEFPSdKO cells. Selection started 72 h after infection by adding 5 μg/ml puromycin (Sigma-Aldrich). Recombinant cell lines were further subcultivated in culture medium containing 2.5 μg/ml puromycin, and the PS expression profile was monitored by Western blotting (see above).
In Vitro γ-Secretase Activity Assay
A cell-free in vitro γ-secretase activity assay was applied for measuring the activity of γ-secretase with a fluoroprobe mimicking the APP γ-cleavage site (54, 58), performed as previously described (54). All of the steps were carried out at 4 °C unless otherwise specified. Cells were seeded in 60-cm2 dishes, washed in cold PBS, and harvested 1 day after. Cell lysis was performed in buffer A (5 mm Tris-HCl, pH 7.4, 5 mm EDTA, 5 mm EGTA, protease inhibitor mixture), cell debris was discarded by centrifugation (800 × g for 10 min), and remaining supernatants were further centrifuged (2,500 × g for 1 h). Membrane pellets were resuspended in buffer B (50 mm Tris-HCl, pH 6.8, 2 mm EDTA, 150 mm KCl, protease inhibitor mixture), and membrane proteins were diluted to 1 μg/μl using the reaction buffer C (50 mm Tris-HCl, pH 6.8, 2 mm EDTA, 150 mm KCl, 0.25% CHAPS, protease inhibitor mixture) and solubilized for 1 h by gentle rotation. Samples containing 30 μg of solubilized membrane proteins and 6 μm APP-mimicking fluoroprobe (Nma-GGVVIATVK(Dnp)RRR-NH2; Merck) were incubated in 100 μl (of buffer B) overnight at 37 °C. Fluorescent signal (excitation/emission, 335/440 nm) was measured on a Victor X3 Multilabel Plate Reader (PerkinElmer Life Sciences).
AICD Monitoring in Cell-free Assays
A cell-free assay to monitor AICD production has been applied as described previously (54). All steps were performed at 4 °C unless otherwise specified. Confluent cells were washed with PBS and harvested with MOPS buffer (1 mm MOPS, 1 mm KCl, pH 7.0). After incubation for 10 min on ice, membranes were homogenized, and cell debris were discarded by centrifugation (1,000 × g for 15 min). The remaining supernatants were further centrifuged (16,000 × g for 40 min), and the membranes in the pellet were solubilized in a 150 mm sodium citrate, pH 6.4, buffer. After 2 h of incubation at 37 °C, membranes were put on ice and sonicated in Laemmli buffer for Western blotting analysis.
Dual-Luciferase Reporter Gene Assay
NICD and AICD release were measured by reporter gene assays as described previously (52, 59). For NICD production, MEFs were transfected 24 h after seeding with the HES1 luciferase reporter gene, Renilla luciferase vector, and NotchΔE or with the corresponding empty (mock) vector (54). Transfected cells were collected and analyzed by Western blotting, to monitor NotchΔE expression and processing; NICD release was measured by a luciferase assay (see below). For AICD release, MEFs were transfected with APP695Gal4, Fe65, Gal4RE-luc, and Renilla luciferase vectors (52, 53). Luciferase activity was measured 48 h after transfection by the Dual-Glo® luciferase assay (Promega, Madison, WI) on a Sirius luminometer (Berthold). Luciferase activity corrected for transfection efficiencies was calculated as the firefly/Renilla luciferase ratio (59).
Blue Native PAGE
Cells were harvested on ice 24 h after seeding. Treatment with γ-secretase inhibitors (1 μm DAPT or L-685,458) was applied for 16 h. Cells were briefly centrifuged at 4 °C. The cell pellet was homogenized on ice using Native Sample Buffer 4× (Invitrogen) supplemented with 1% n-dodecyl β-d-maltoside (Sigma-Aldrich) and the protease inhibitor mixture. Samples were then centrifuged (100,000 × g) for 1 h at 4 °C. 20 μg of solubilized membrane proteins were separated in native PAGE 4–16% gel, transferred onto PVDF membranes, and incubated with primary and secondary antibodies followed by ECL detection.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism (GraphPad Software, San Diego, CA) and post hoc tests as indicated.
RESULTS
Disruption of PS1 AXXXAXXXG Motif Impairs Presenilin Maturation and APP Cleavage
Amino acid sequence analysis of γ-secretase subunits revealed the presence of AXXXAXXXG in both PS1 and PS2 TMD8 (PS1, 409AXXXAXXXG417; PS2, 390AXXXAXXXG408) (Fig. 1A). By a site-directed mutagenesis approach, we substituted the small residues of the motif, Ala and/or Gly, by Leu, as done previously for APP GXXXG motifs (34). The mutants were designated PS1-AA (A409L/A413L), PS1-AG (A413L/G417L), PS2-AA (A390L/A394L), and PS2-AG (A394L/G398L) (Fig. 1A). Additional mutants were generated: the PS1-AT (PS1 A409T), a FAD mutant (47), the corresponding engineered PS2-AT (PS2 A390T), and the inactive PS1-DA (PS1 D385A) and PS2-DA (PS2 D366A), where one catalytic Asp was substituted by Ala (16, 60, 61). Considering that PS1-dependent γ-secretase is known as the major γ-secretase complex responsible for Aβ production (62), we first analyzed the effects of those mutations on PS1 in CHO cells, a widely used cellular model for studying APP processing and γ-secretase activity (34, 60, 63).
FIGURE 1.

Mutations in PS1 AXXXAXXXG motif influence APP metabolism in CHO cells. A, schematic representation of PS topology. The endoproteolytic site in the cytoplasmic loop (scissors), the catalytic aspartates in TMD6 and TMD7 (red dots), and the TMD8 AXXXAXXXG motifs (blue dot) are indicated. Amino acid sequence alignments of TMD7 and TMD8 are shown for PS1/PS2-WT and mutant isoforms. Catalytic Asp in TMD7 (PS1 Asp-385 and PS2 Asp-366) and the corresponding DA inactive mutant (red) are underlined. In TMD8, Gly or Ala residues from the AXXXAXXXG interface and corresponding mutants (blue) are underlined. B, CHO cells were transiently transfected with human PS1 isoforms (WT, AA, AG, AT, and DA). Expression and maturation of the exogenous PS was revealed by an anti-hPS1-NTF antibody. Full-length PS1 (PS1-FL) and the N-terminal fragment produced upon endoproteolysis (PS1-NTF) are indicated by arrows. C, Western blot of cell lysate from CHO cells transiently co-transfected with human PS1 isoforms (WT, AA, AG, AT, and DA) and HA-tagged APP β-CTF (C99HA). C99HA as well as the APP α-CTF (C83HA), produced from C99HA α-cleavage, were detected by using an anti-HA antibody and are indicated by arrows. The C83HA/C99HA ratio was calculated as an indicator of C99HA processing by γ-secretase. Results (means ± S.E. (error bars)) are represented as a percentage of the C83HA/C99HA ratio measured in PS1-WT-expressing cells (n = 5). D, results (means ± S.E.) from quantification of Aβ40 and Aβ42 in C are given as a percentage of PS1-WT (n = 3). E, quantification (means ± S.E.) of the Aβ42/Aβ40 ratio in PS1-WT and PS1-AT cells in C is expressed as a percentage of PS1-WT (n = 3). The statistical analyses in C and D were performed by one-way ANOVA followed by Dunnett's post hoc test. *, p < 0.05; **, p < 0.001; ***, p < 0.0001 in E by Mann-Whitney test.
Full-length PS1 was identified in all overexpressing cells by Western blotting with a specific anti-hPS1-NTF antibody (Mab1563) (Fig. 1B). However, the PS1-NTF, produced upon PS1 endoproteolysis, was detected only for PS1-WT and PS1-AT. The absence of the endoproteolytic fragment in PS1-AA-, PS1-AG-, and PS1-DA-expressing cells indicated that the AXXXAXXXG motif is required for PS1 maturation.
Because PS1-AA, PS1-AG, and PS1-DA showed similar profiles, we decided to further analyze the role of the PS1 AXXXAXXXG motif in APP processing. With this aim, we used CHO cells co-transfected with the PS1 mutants and an HA-tagged APP β-CTF (C99HA) and quantified the Aβ peptides produced and the intracellular accumulation of both APP-CTFs (C99HA and C83HA) (Fig. 1, C and D). Aβ quantification was performed by ECLIA using a specific antibody recognizing human Aβ (6E10) (54). Compared with PS1-WT, the levels of Aβ40 and Aβ42 released from PS1-AA, PS1-AG, and PS1-DA were reduced (Fig. 1D). However, in PS1-AT-expressing cells, Aβ40 and Aβ42 production was significantly increased, indicating that the FAD mutation on the same structural motif had opposite effects. Intracellular accumulation of APP-CTFs (C99HA and C83HA) is related to γ-secretase activity in the cell. Indeed, an increase in the C83HA/C99HA ratio can be considered as an accurate indicator of γ-secretase activity impairment in C99-expressing CHO cells (34, 54). C83HA/C99HA was increased for PS1-AA, PS1-AG, and PS1-DA, whereas it was decreased for PS1-AT (Fig. 1C). These results were in line with the Aβ peptide quantification. Interestingly, a trend in increased Aβ42/Aβ40 ratio was observed for PS1-AT mutant, a known feature of FAD PS mutants (Fig. 1E). Reduced Aβ peptide production can result from decreased γ-secretase activity, impaired association (or docking) of the substrate with the complex. The PS1 AXXXAXXXG motif could indeed mediate the interaction with the three in-register GXXXG motifs in APP, already known to be critical for Aβ production (34–37), thus explaining the results on APP processing. Alternatively, the PS motif might be involved in inducing a certain geometry of the complex via tight TMD packing that does not contribute to the overall substrate association. Our co-immunoprecipitation (co-IP) data revealed that the C99HA/PS1 interaction was not altered by mutation of the PS1 AXXXAXXXG motif (Fig. 2, A and B). No co-IP was observed in the absence of the anti-PS1 IP antibody (Mab1563), indicating that co-IP experiments were not biased by the sticky nature of γ-secretase substrates. This observation is also consistent with our results showing that mutation of the APP GXXXG motifs did not modify the APP/PS1 interaction (data not shown) (see Ref. 34).
FIGURE 2.

Mutations of PS AXXXAXXXG motif do not impair the interaction of PS with APP substrate and the other γ-secretase subunits. Association of PSs with APP substrate and subunits was analyzed by co-IP. PS isoforms (WT, AA, AG, AT, and DA) were transiently co-transfected in CHO cells with C99HA (A) or V5-tagged human γ-secretase subunits (NCT, PEN-2, and APH-1) (C). A, Western blots of cell extracts (inputs) were revealed by an anti-hPS1-NTF or anti-HA antibody. PS1 (PS1-FL and PS1-NTF) and C99HA and the derived C83HA were detected at the expected molecular size and indicated by arrows. B, the control co-IP was performed in PS1-WT- and C99HA-co-expressing cells, in the absence of the immunoprecipitating antibody (anti-hPS1-NTF). The PS1-WT and C99HA in the input and in the control co-IP were detected by an anti-hPS1-NTF or anti-HA antibody, respectively. Equal amounts of protein from A were subjected to co-IP using the anti-hPS1-NTF antibody and revealed by Western blotting. The presence of PS1 (PS1-FL and PS1-CTF) was detected by the anti-PS1-CTF antibody, and the co-immunoprecipitated C99HA and C83HA were detected by the anti-HA antibody and indicated by the arrows. C, Western blots of cell extracts (inputs) revealed by an anti-hPS1-NTF or anti-V5 antibody. PS1 (PS1-FL and PS1-NTF) and γ-secretase subunits were detected by the anti-hPS1-NTF or anti-V5 tag antibody. PS1 (PS1-FL and PS1-NTF), mature and immature NCT (mNCT and imNCT), APH-1, and PEN-2 were detected at the expected molecular size and are indicated by arrows. D, the control co-IP was performed in PS1-WT- and V5-tagged substrate-co-expressing cells, in the absence of the immunoprecipitating antibody (anti-hPS1-NTF). The PS1-WT and the V5-tagged subunit in the input and in the control co-IP were detected by an anti-hPS1-NTF or anti-V5 antibody, respectively. Equal amounts of protein from C were subjected to co-immunoprecipitation using an anti-hPS1-NTF antibody and revealed by Western blotting. The presence of PS1 (PS1-FL and PS1-CTF) was detected by an anti-PS1-CTF antibody, and the co-immunoprecipitated γ-secretase subunits were detected by an anti-V5 tag antibody. Bands corresponding to mature (mNCT) and immature (imNCT) nicastrin, APH-1, and PEN-2 are indicated by arrows.
Together, these results support the role of the PS AXXXAXXXG motif in PS1 maturation and formation of an active γ-secretase complex. This led us to further analyze the involvement of the PS AXXXAXXXG motif in the assembly of the γ-secretase complex.
PS1 AXXXAXXXG Motif Is Not Involved in the Assembly with the APH-1/NCT Heterodimer
To verify the role of the PS1 motif in mediating the interaction with other components of the complex, we reconstituted an exogenous (human) γ-secretase complex suitable for co-IP studies in CHO cells. Vectors expressing tagged (V5) NCT, APH-1, and PEN-2 were built and co-transfected with the different PS1 constructs. The PS1 expression and maturation profile was not influenced by the exogenous co-expression of the other γ-secretase cofactors (Fig. 2C). All of the exogenous subunits, including mature and immature NCT, APH-1, and PEN-2, were expressed and detected by an anti-V5 antibody at the expected sizes (Fig. 2C). The specificity of the V5 signal was confirmed by antibodies directed against NCT, APH-1, or PEN-2 (not shown). Co-IP experiments performed using the anti-hPS1-NTF (Mab1563) antibody showed that all PS1 forms, including the PS1-AA and PS1-AG mutants, co-immunoprecipitated with all of the other human γ-secretase subunits (Fig. 2D). No co-IP was observed in the absence of the anti-hPS1-NTF IP antibody, confirming the reliability of these results. These results suggested that the PS1 AXXXAXXXG motif was not involved in the packing of the complex, namely in the interaction of PS with the APH-1/NCT heterodimer (14) or with PEN-2.
Expression of PS1 or PS2 in MEFPSdKO Cell Lines Yields Similar Results
One limitation of the transient co-expression experiments in CHO cells comes from the competition of endogenous PS1 and PS2 with the exogenous human presenilins. We decided to further analyze PS1 and PS2 mutants in the MEF PS1 and PS2 null backgrounds, known as the MEFPSdKO cell line (50, 64). A big asset here is that these cell lines allow rebuilding of different γ-secretase complexes, depending exclusively on the PS forms re-expressed (PS rescued). All of the PS forms (PS1 and PS2) were stably expressed by lentiviral infection in rescued MEFPSdKO cell lines, referred to hereafter as MEFPSdKOr (Fig. 3). PS expression and endoproteolysis were monitored by Western blotting (Fig. 3, A and B). The N- and C-terminal fragments of PS1-WT and PS2-WT were detected, but both PS1-DA and PS2-DA failed to undergo endoproteolysis, as reported previously (18, 65, 66), causing the accumulation of full-length PS in the cell. Similar to previous results in CHO cells, in PS-AA and PS-AG mutants, no endoproteolytic products were detected despite corresponding full-length PS accumulated in the cells, confirming the role of the AXXXAXXXG motif in PS1 endoproteolysis and extending this observation to PS2. This strongly supported the conserved role of the motifs in PSs. In contrast, the endoproteolysis of PS1-AT and PS2-AT was preserved, as shown by the generation of the corresponding PS-NTF and -CTF (Fig. 3, A and B). As for the PS1-WT form, all of the pool of PS1-AT was converted by endoproteolysis in the rescued model (full-length protein was barely detectable), whereas the full-length PS2-WT and PS2-AT were still detectable.
FIGURE 3.

Characterization of MEFs rescue cell lines stably expressing PS mutants. PS1 and PS2 KO mouse embryonic fibroblasts (MEFPSdKO) stably re-expressing the different PS1 and PS2 isoforms were generated. PS expression and maturation profile and maturation of the γ-secretase complex indicated by maturation of NCT were studied by Western blotting. Western blots of cell lysates (MEFPSdKO (dKO), MEF+/+ (+/+), and MEFPSdKO stably rescued with the indicated PS1 or PS2 isoforms) were revealed with anti-PS1-CTF or anti-hPS1-NTF antibody (A), anti-PS2-CTF antibody (B), and anti-NCT antibody (A and B). Bands corresponding to full-length PS (PS-FL), endoproteolytic fragments (NTF or CTF), and mature and immature NCT (mNCT and imNCT) are indicated by arrows. C, the native conformation of γ-secretase was studied in MEFs in non-denaturating conditions (blue native PAGE) and compared with the native conformation of human γ-secretase isolated from HEK293 cells. The presence of PS1 (above) and PS2 (below) in the native γ-secretase complex was revealed in MEFPSdKO (dKO), MEF+/+ (+/+), and HEK293 by using specific anti-PS1-CTF or anti-PS2-CTF antibodies. D, the same experiments were carried out on MEF+/+ (+/+) and MEFPSdKO (dKO) cell lines to confirm the presence of the other γ-secretase subunits in the high molecular weight complexes by using anti-NCT, anti-APH-1, and anti-PEN-2 antibodies. Black arrows, bands corresponding to a high molecular weight γ-secretase complex recognized by anti-PS, anti-NCT, anti-APH-1, and anti-PEN-2 antibodies. E, blue native PAGE performed on MEFPSdKO cells stably expressing PS1 (left) and PS2 (right) isoforms revealed by anti-PS1-CTF or anti-PS2-CTF antibodies. Black arrows, bands corresponding to the expected molecular size of a full γ-secretase complex. F, the same experiments were carried out on MEF+/+ (+/+) in the absence or presence of DAPT or L-685,458 (L685). The arrows indicate bands corresponding to γ-secretase complex.
Profile of the γ-Secretase Complex in MEFPSdKOr
NCT maturation occurs as a final step in γ-secretase assembly (50, 67–69) and results from extensive glycosylation of its ectodomain. This process does not occur in the absence of PSs (50, 67–69). Restoration of PS expression in MEFPSdKOr cell lines (Fig. 3, A and B), rescued NCT maturation, albeit to a lesser extent when compared with MEF+/+, with quantitative differences observed between the rescued cell lines, particularly for PS2-AG. These results showed that exogenous PS forms in MEFdKOr cells could be integrated into a mature γ-secretase complex, but only PS-WT and PS1-AT appeared to form fully mature complex. This observation strongly supports the requirement of the structural properties of the PS AXXXAXXXG motif for PS endoproteolysis but not for PS integration into a mature γ-secretase complex and led us to further analyze γ-secretase activity in these cell lines.
The fact that different amino acid substitutions on the same structural determinant result in opposite effects on PS activity while generally preserving the maturation of the complex suggests that the PS AXXXAXXXG motif could be important for the conformation of the mature γ-secretase complex (70). Non-denaturating electrophoresis analyses were carried out (blue native PAGE), with HEK293 taken as a reference for the formation of the native γ-secretase complex (70). MEF+/+ and HEK293 cells, but not MEFPSdKO, showed two high molecular mass bands, between 242 and 480 kDa, both positive for PS1 and PS2, compatible with the previously described high molecular mass band (∼400 kDa) observed for fully assembled γ-secretase (45, 70) (Fig. 3C). These two bands were detected in MEF+/+ with antibodies against NCT, PEN-2, and APH-1 (Fig. 3D). In MEFPSdKO rescued cells expressing the active PS forms (WT and AT), the doublet observed in MEF+/+ and HEK293 cells was restored. In contrast, only the upper ∼480 kDa band was detected in the non-active PS mutants (DA, AA, and AG) (Fig. 3E). A similar profile correlation was observed when MEF+/+ cells were treated with two specific γ-secretase inhibitors: DAPT and L-685,458. The treatment with both inhibitors caused a reduction of the lower band (above 242 kDa), all of the signal shifting to the higher apparent molecular mass complex (above 242 kDa) (Fig. 3F). These inhibitors are either allosteric inhibitor (DAPT) or transition state analog (L-685,458), known to bind active γ-secretase and thereby probably changing its spatial conformation (71, 72). In addition, this clearly suggested a correlation between the native profile and the activity state of the complex. By controlling the native conformation of the complex, PS AXXXAXXXG motifs might control its activity.
Influence of PS AXXXAXXXG Motifs on Substrate Processing by γ-Secretase
Processing of endogenous APP was studied in the MEF cell lines. APP-CTFs were monitored by Western blotting, and Aβ peptides were quantified by ECLIA (54) using a specific anti-mouse Aβ antibody (4G8). Endogenous AICD production is very difficult to detect and was therefore monitored in a cell-free assay on isolated cell membranes (54). Endogenous APP expression was comparable in all tested cell lines. In MEFPSdKO cells, APP CTFs accumulated in the cells (Fig. 4A), and the release of Aβ peptides as well as of AICD was completely abolished (Figs. 4, B and C), as reported previously (64). By rescuing with wild-type PS1 and PS2, CTF and Aβ production was restored to levels comparable with those observed in MEF+/+ cells. In PS-AA, PS-AG, and PS-DA cells, intracellular levels of APP-CTFs were similar to those observed in the MEFPSdKO cell line, and Aβ peptides and AICD were undetectable or at very low levels (Fig. 4, B and C). In PS1 and PS2-AT cells, no APP-CTFs accumulated. In PS1-AT, both Aβ40 and Aβ42 production was significantly increased when compared with PS1-WT-expressing cells, whereas in PS2-AT, Aβ levels were comparable with those observed in PS2-WT (Fig. 4B). It is important to note here that in both PS1-AT and PS2-AT mutants, the Aβ42/Aβ40 ratio was increased when compared with the corresponding PS-WT forms (Fig. 4B). The cell-free assay revealed that AICD production was restored in PS1-WT- and PS2-WT-expressing cells, whereas no AICD was detected in the AA and AG mutants. Strikingly in PS1AT, which showed and increased Aβ, no AICD fragment was detected after incubation. On the contrary, under the same conditions, AICD was detected in PS2AT-expressing cells. These results support the previous observations in CHO cells, indicating that PS-WT and PS-AT contribute to the formation of an active γ-secretase complex, whereas PS-AA and PS-AG were part of an inactive γ-secretase complex similar to the PS-DA mutants. Here again, the effects of mutations of PS AXXXAXXXG motifs appeared dual, depending on the nature of the mutations introduced. Increased Aβ production in PS1-AT can result from increased efficiency of γ-secretase to process APP at its γ-site (7).
FIGURE 4.

Effects of PS AXXXAXXXG motifs mutations on endogenous APP processing Aβ production and AICD release. A, Western blot of cell lysates from MEFPSdKO PS1 (left) and PS2 (right) rescued cells revealed with an anti-APP C-ter antibody, compared with MEF+/+ (+/+) and MEFPSdKO (dKO). Bands corresponding to APP and APP α- or β-C-terminal fragments (APP-CTFs) are indicated by arrows. B, quantification of Aβ40 and Aβ42 concentrations in the extracellular media and the corresponding Aβ42/Aβ40 reported in PS-WT and PS-AT constructs. Results (means ± S.E. (error bars)) are given as a percentage of Aβ or Aβ42/Aβ40 measured in PS-WT cell medium (n = 3). nd, non-detectable; ***, p < 0.0001 as compared with +/+; ###, p < 0.0001 as compared with PS-WT. Statistical analyses on Aβ quantification were performed by one-way ANOVA followed by Dunnett's post hoc test, whereas analyses of Aβ42/Aβ40 ratio were performed by the Mann-Whitney test. C, AICD production measured by cell-free assays on solubilized membranes from MEFPSdKO (dKO), MEF+/+ (+/+), and MEFPSdKO stably rescued with the indicated PS1 or PS2 isoforms. AICD release was monitored before (0 h) and after 2 h of incubation by Western blotting using an anti-APP C-ter antibody. Lower (low exp.) and higher (high exp.) exposures of the same blots are shown.
In order to measure directly γ-secretase activity, we used an in vitro γ-secretase assay as described previously (54, 58). In this assay, solubilized membranes were incubated with a fluoroprobe mimicking the APP γ-cleavage sites (Fig. 5A). When MEFPSdKO and MEF+/+ cells were tested in the absence or presence of DAPT, γ-secretase activity was reduced by ∼40% compared with the corresponding non-treated MEF+/+ (Fig. 5B). The remaining background signal was in accordance with previous results (54, 58). The same assay was applied to solubilized membranes from MEFPSdKOr cells. When PS-AA and PS-AG mutants were compared with the corresponding PS wild type forms (PS1-WT or PS2-WT) (Fig. 5B), they showed an average reduction in γ-secretase activity of ∼58 and ∼49%, respectively, similar to the corresponding catalytically inactive forms (PS1-DA and PS2-DA). PS1-AT, however, showed a 2.5-fold increased activity compared with PS1-WT, whereas PS2-AT showed a similar activity compared with PS2-WT. These results indicate first that the AXXXAXXXG motif in PS can control γ-secretase activity in opposite ways; leucine substitutions abolished in vitro γ-secretase activity, whereas an FAD-causing mutation (PS1-AT) was associated with an increase in in vitro γ-secretase activity. Results obtained in the in vitro γ-secretase activity assay were in agreement with those obtained on Aβ production (Fig. 4B) but showed some differences from the measurements on AICD release (Fig. 4C). It is important to note here that the fluoroprobe used harbors the APP γ-sites but the ϵ-site.
FIGURE 5.
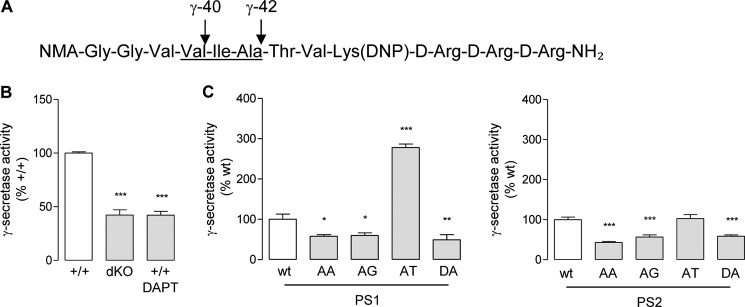
In vitro γ-secretase assays. A, schematic representation of the fluoroprobe used to assay in vitro γ-secretase activity. NMA and DNP, the fluorescent and the quenching group, respectively. The amino acids corresponding to positions 40–42 of the APP γ-cleavage site are underlined. Membranes from MEFs were isolated and incubated for 16 h with the fluoroprobe. B, the γ-secretase assay was performed on membranes of MEF+/+ (+/+) cells, MEFPSdKO (dKO) cells, and MEF+/+ cells treated with 10 μm DAPT (+/+ DAPT), a reference γ-secretase inhibitor. Results (means ± S.E. (error bars)) are given as a percentage of γ-secretase activity measured in MEF+/+ cells (n = 8). C, γ-secretase activity was measured on MEFPSdKO rescued by PS1 (left) and PS2 (right) isoforms. Results (means ± S.E.) are given as a percentage of γ-secretase activity measured in PS-WT-expressing cells (n = 6). All statistical analyses were performed by one-way ANOVA followed by Dunnett's post test. *, p < 0.05; **, p < 0.001; ***, p < 0.0001.
To further investigate the effect of PS AXXXAXXXG motifs on substrate cleavage at ϵ-site, we carried out a transactivation assay as described previously (52, 73). Contrary to cell-free assays, this assay monitors the release of functional AICD by its ability to activate transcription of a reporter gene (Fig. 6A). The signal measured in MEFPSdKO cells was identical in the presence or absence of Fe65, confirming that luciferase activity depends only on AICD release (Fig. 6B). In MEFPSdKOr cells, PS1- and PS2-WT AICD-dependent transcriptional activity was increased when compared with MEFPSdKO (Fig. 6C), with PS2-WT higher than PS1-WT. The luciferase signal from PS-AA, PS-AG, and PS-DA was <50% of the activity measured in PS-WT forms, in line with the AICD profile previously obtained in the cell-free assay (Fig. 4C). Interestingly, the luciferase signal from PS1-AT was also roughly 50% of the PS1-WT signal despite much higher levels of Aβ production, suggesting that the ϵ-cleavage activity was much lower than the subsequent cleavage efficiency or activity to produce the Aβ peptides. Surprisingly, for the PS2-AT mutant, the transactivation assay indicated a lower AICD release when compared to PS2-WT, whereas comparable amounts of AICD bands were detected in cell-free assays (Fig. 4C).
FIGURE 6.

Effects of PS AXXXAXXXG motifs on AICD release. A, schematic representation of the transactivation assay used to monitor AICD release. Cells were co-transfected with APPGal4, Fe65, Gal4RE-luc reporter gene, and pRL-TK Renilla vector. When APPGal4 is cleaved by γ-secretase at the ϵ-site, AICDGal4 is released and translocates (with Fe65) into the nucleus, where the AICDGal4-Fe65-Tip60 complex transactivates the Gal4RE reporter gene. Luciferase activity was calculated as the firefly luciferase/Renilla luciferase ratio. B, transactivation assays were carried out on MEF+/+ (+/+) and MEFPSdKO (dKO) cells in the presence or absence of Fe65. Results (means ± S.E. (error bars)) were normalized and shown as percentage of luciferase activity measured in +/+ cells (n = 6). ns, non-significant; ***, p < 0.0001 as compared with +/+; ###, p < 0.0001 as compared with Fe65-expressing +/+ cells. C, the same assay was performed in +/+, dKO, and MEFPSdKO rescued with PS1-WT or PS2-WT cells in the presence of Fe65. Results (means ± S.E.) are normalized and given as percentage luciferase activity measured in +/+ (n = 6). ns, non-significant; **, p < 0.01; ***, p < 0.0001 as compared with +/+; ###, p < 0.0001 as compared with PS1-WT. D, transactivation assay in MEFPSdKO rescued with PS1 (left) or PS2 (right) isoforms. Results (means ± S.E.) are normalized and shown as a percentage of corresponding WT controls (PS1-WT or PS2-WT); ***, p < 0.0001 as compared with respective WT control. n = 7 for PS1 and n = 8 for PS2. All statistical analyses were performed by one-way ANOVA, followed by Dunnett's post hoc test.
Another key γ-secretase substrate is the Notch receptor. Notch is cleaved at the S3 site within its TM domain by γ-secretase to release the transcriptionally active NICD (65). The S3 site corresponds to the APP ϵ-site, and NICD production is critical in developmental cell signaling (1, 62). The functional effects of the PS AXXXAXXXG motifs in Notch processing were addressed in a transactivation assay (54, 59) (Fig. 7A), in which the cleavage of a truncated Notch construct (NotchΔE) (51) by γ-secretase and subsequent NICD release transactivates a HES1 luciferase reporter gene (74). Expression of NotchΔE in this model is necessary because in its absence, transcriptional activity of the HES1 luciferase reporter gene was low, and only a slight decrease was measured in MEFPSdKO cells (Fig. 7B). NICD (measured by Western blotting or transactivation assays) is not produced from NotchΔE in MEFPSdKO as expected (Fig. 7B). In MEFPSdKOr cell lines, both PS1 and PS2 restored NICD production, as shown by transactivation assays and Western blotting. PS1-WT rescue was more efficient than PS2-WT (Fig. 7C). The PS-AA and -AG mutants did not generate the NICD fragment and showed a significantly reduced signal in transactivation assays when compared with the relative PS-WT forms (PS1-WT or PS2-WT) (Fig. 7D). The results were similar to those observed for PS-DA mutants. Interestingly, PS1-AT mutants were associated with higher NICD production when compared with PS1-WT, contrary to the results obtained with AICD release (Fig. 7D), suggesting that, in these rescued cells, there is specificity for substrate processing of the PS1-dependent γ-secretase at the ϵ-site (for APP) or S3 site (for Notch). In the PS2-AT mutant, the NICD production was only very slightly and non-significantly increased when compared with PS2-WT.
FIGURE 7.

Effects of PS AXXXAXXXG motifs in Notch γ-cleavage and NICD release. A, schematic representation of gene transactivation assay used to monitor NICD production. LP, Notch leader peptide. Other transcriptional co-activators associated with NICD (e.g. CSL and MAML1) are represented by a dotted circle. Cells were co-transfected with a HES1 luciferase reporter gene, with pRL-TK Renilla plasmid, and, where specified, with a Myc-tagged ΔE-Notch construct (NotchΔE). When overexpressed, NotchΔE is cleaved by γ-secretase at the S3 site, and the Myc-tagged NICD released translocates into the nucleus, where it transactivates the HES1 reporter gene. B, MEF+/+ (+/+) and MEFPSdKO (dKO) were co-transfected in the presence or absence of NotchΔE as specified. Western blots (top) of cell extracts were revealed with anti-Myc antibody. The bands corresponding to NotchΔE and its derived NICD are indicated by arrows. Luciferase activity (bottom) was measured in the same cell extracts. Results (means ± S.E.) are given as percentage of luciferase activity measured in +/+ cells. ns, non significant; ***, p < 0.0001, as compared with +/+; ###, p < 0.0001 as compared with NotchΔE-expressing +/+ cells (n = 4). C, the same assay was performed on +/+, dKO, and PS1-WT and PS2-WT cells. Results (means ± S.E. (error bars)) are given as a percentage of luciferase activity measured in +/+ cells. **, p < 0.01; ***, p < 0.0001, as compared with +/+; ###, p < 0.0001 as compared with PS1-WT (n = 4). D, analysis of NICD production and release of MEFPSdKO rescued for PS1 (left) and PS2 isoforms (right). Western blots of cell extracts revealed by anti-Myc antibody. The bands corresponding to NotchΔE and its derived NICD are indicated by arrows. Luciferase activity (bottom) was measured in the same cell extracts. Results (means ± S.E.) are given as a percentage of activity measured in the WT controls (PS1-WT or PS2-WT); *, p < 0.05; **, p < 0.01; ***, p < 0.0001 (PS1, n = 3; PS2, n = 5). All statistical analyses were performed by one-way ANOVA followed by Dunnett's post hoc test.
DISCUSSION
In this study, we targeted the role of the AXXXAXXXG motif that we identified in TMD8 of PS, focusing in particular on its involvement in γ-secretase proteolytic properties and assembly. Using site-specific mutagenesis, we generated different PS mutants: the PS-AA and PS-AG, where the leucine mutations (LXXXL) are expected to disrupt close helix packing (34); the FAD mutant PS1-AT and the corresponding PS2-AT, where the first alanine of the motif is substituted by a threonine (PS1A409T and PS2A390T); and PS-DA, serving as a catalytically inactive form of PS (12). Our major finding is that the activity of the γ-secretase complex depends on the integrity of the AXXXAXXXG motif in TMD8 of PS1 and PS2. LXXXL mutations results in loss of γ-secretase activity, despite unaltered association with the other proteins of the complex. Thus, small residues, which probably promote close helix packing of TMD8 along the motif, are required for normal function of the γ-secretase complex.
In contrast, mutation of the first alanine residue of the first GXXXG-like sequence to threonine had an enhancing although contrasting effect on γ-secretase activity, suggesting that this substitution strengthens the association of TMD8 within the complex, impacting catalytic properties that can be discriminated between PS1 and PS2.
We can make a distinction here between mature and functional (or active) γ-secretase complexes (14, 19, 75). Maturation is related to NCT glycosylation. Our results confirmed pervious work showing that NCT glycosylation does not occur in the absence of PSs (69, 76). In other terms, and although the stepwise process of γ-secretase assembly is not fully understood, NCT is extensively glycosylated once the four core subunits (PS, APH-1, PEN-2, and NCT) are assembled (14). γ-Secretase activation occurs when PS undergoes endoproteolysis, leading to the release from the active site of the inhibitory cytosolic loop, thereby activating the enzyme (66, 77). When expressed with the other γ-secretase components, all PS forms, including the PS-AA and PS-AG mutants, were able to generate a fully assembled γ-secretase. In particular, the interaction of APH-1, which contains three sequential GXXXG motifs in TMD4 (122GXXXGXXXGXXXG130) and is expected to interact with the C terminus of PS (38–40, 78), was not affected. Therefore, close helix association of the PS AXXXAXXXG motifs in TMD8 was not required for assembly of the complex. Some mutants, in particular PS2-AG, reduced the level of NCT glycosylation, indicating a possible alteration of γ-secretase maturation that might be related to an altered trafficking of the complex for these mutants. Biotinylation assays (not shown) revealed that the pool of PS reaching the cell surface was not modified, suggesting that trafficking impairment by the mutations (if any) might only be very subtle and with a low impact on complex assembly and maturation.
γ-Secretase cleaves its substrates by different cleaving events (3, 7, 11). An initial hypothesis is that AXXXAXXXG motifs are involved in docking of substrates to the γ-secretase, a critical step in their cleavage. Indeed, several, but not all (e.g. Notch receptor), γ-secretase substrates contain GXXXG or GXXXG-like motifs (33), suggesting a key role of these motifs in geometry, substrate specificity, and processing. In the case of APP, several studies have clearly demonstrated the involvement of GXXXG motifs (621GXXXGXXXGXXXG633, APP695 numbering) in Aβ production (34–37). Because APP was able to generate stable interactions with all PS forms, including PS-AA and PS-AG, it is clear that the PS AXXXAXXXG motifs are not required for APP docking, suggesting that these motifs are not involved in substrate recruitment. Although the interaction with the substrate was preserved, the disruption of the AXXXAXXXG by leucine substitutions abolished γ-secretase activity in terms of Aβ peptide production as well as AICD and NICD release (51–54, 73). This result supports the fundamental role of these motifs in TMD8 in the overall activity of the complex.
In contrast to the LXXXL mutants, AT mutation in both PS1 (A409T) and PS2 (A390T) differentially affected γ-secretase activity. We suggest that these catalytic effects result from the strengthening of the association of TMD8 within the complex, due to Ala to Thr substitution. PS1 FAD mutation (A409T) leads to a complex modification of amyloidogenic processing via mechanisms that are still largely unknown but consistent with their impact on AD onset. PS1 AD mutants are generally associated with an increase in the Aβ42/Aβ40 ratio, often due to reduced Aβ40 production, increased Aβ42 release, or an overall increased production of both amyloid peptides (21, 68, 79). Interestingly, for the PS1-AT mutant, the production of both Aβ peptides (Aβ40 and Aβ42) was restored, with a slight but significant increase in Aβ42/Aβ40 ratio. Both Aβ quantification and in vitro γ-secretase assays showed that PS1-AT processes APP γ-sites more efficiently than the non-mutated PS1. Much less is known about the role of PS2-dependent γ-secretase complexes in AD. Our engineered PS2-AT mutation, corresponding to the PS1 FAD mutant, restored Aβ production to a lesser extent than the PS1-AT mutant, consistent with the results obtained in in vitro γ-secretase assays. However, the PS2 AT mutant, like PS1 AT, increased the Aβ42/Aβ40 ratio. This is in line with the idea that PS2 is a minor contributor in Aβ production when compared with PS1 (62) but indicates that AT mutations in PS1 and PS2 result in conformational changes shifting γ-secretase toward pathological APP processing.
To better understand the influence of the AT mutation on processing, we analyzed the production of the AICD from APP in PS-WT and PS-AT mutants. The initial cleavage of APP by γ-secretase occurs at the ϵ-site, releasing AICD, and the remaining membrane fragment is C-terminally trimmed to the γ-site to generate Aβ (80). We applied specific assays to evaluate the impact of the mutation on AICD production: an in vitro cell-free assay monitoring the release of the APP C-terminal fragment (1, 54, 81) and a functional assay measuring AICD-dependent gene transactivation (52, 73). In PS1-AT, we measured opposite effects on Aβ production (increase) and AICD release (decrease), both in cell-free and transactivation assays. One possible explanation for the lower than expected production of AICD in the PS1-AT mutant is that the mutation shifts the first cleavage site for APP, possibly starting upstream to the ϵ-site. If this were the case, the residual C-terminal fragment would retain several hydrophobic residues and would not detach from the membrane; hence, an inhibition of AICD-induced transcription would occur. Further mass spectrometry studies will be required to identify the precise position where the AT-mutated PS1 initiates trimming of the β-CTF. Contrary to PS1-AT, the PS2-AT produced AICD in cell-free assays but surprisingly showed lower transactivation properties than non-mutated PS2. This observation could suggest that PS mutation may impact not only the AICD production per se but also the transcriptional property of this intracellular fragment of APP. Testing this hypothesis is far beyond the scope of the present work; it requires further studies to confirm that AICDs produced from PS1- and PS2-dependent γ-secretases display different transcriptional properties and that mutation of PS1 or PS2 could affect by itself AICD-dependent transcription.
To further decipher the influence of PS mutation on γ-secretase properties and intracellular domain function, we compared the production of the AICD with that of the NICD from Notch receptor proteolysis. All of the mutations abolishing γ-secretase activity (AA, AG, and DA) blocked AICD/NICD release and AICD/NICD-dependent transcription. Interestingly, the PS1-AT mutation induces opposite effects on APP and Notch processing. The increase in NICD production in the AT mutant compared with the wild-type PS is in agreement with the overall increase in catalytic activity. The PS2-AT the mutation did not affect Notch processing when compared with the corresponding PS2-WT form. We should first keep in mind here that Notch processing is mainly operated by PS1-dependent γ-secretase because PS1 KO is lethal at the embryonic stage and displays a Notch-deficient phenotype (62). The mechanisms underlying selectivity of PS1- and PS2-dependent γ-secretases toward their substrates are very poorly understood. It appears also from our study that similar mutations of PS1 and PS2 can have different effects on the processing of a given substrate.
We next investigated the role of the PS AXXXAXXXG sequence as a structural determinant in adoption by γ-secretase of a native conformational state using native gels. The profile of the PS-AA and PS-AG mutants was comparable with that of PS-DA, exhibiting one conformational state, which correlated with decreased or abolished activity. Importantly, the conformational state adopted by these mutants was identical to the one observed after treatment by γ-secretase inhibitors. These inhibitors bind to the active γ-secretase, and our results indicated that they can shift γ-secretase to an inactive or blocked conformation similar to that adopted by inactive mutants. In contrast, PS-AT and PS-WT exhibited two conformational states. These data indicate that the PS AXXXAXXXG motif represents a structural determinant for the final native (active) conformation of the γ-secretase complex. Taken together, the data suggest a strong correlation between the integrity of the PS AXXXAXXXG motif and adoption of the native (active) conformation of the complex. It is worth noting that this motif was required in PS1 for association with γ-secretase subunits that contain GXXXG motifs themselves, namely APH-1, suggesting either that GXXXG motifs regulate geometry of the complex postassociation or that they fulfill their role by forming interactions with other similar motifs in other PS TMDs. The conserved role of the PS motif of TMD8 in the native conformation of the complex is underlined by the identical conformational effects of LXXXL and AT mutants in both PS1 and PS2. Considering the intimate relation between TMD7 and TMD8 during PS integration into the membrane and the results presented here, it is tempting to speculate that the AXXXAXXXG sequence in TMD8 creates a network of interactions with TMD7, which contains two similar motifs. This interaction could ensure the correct integration and orientation of the catalytic Asp residue on TMD7, thereby influencing the catalytic activity of the complex. Further studies will be required to explore this hypothesis.
Finally, in this study, we have initiated a new approach for understanding the structure, assembly, and function of the γ-secretase complex, namely targeting a structural determinant belonging to the GXXXG-like motifs that have generally been found to mediate helix interactions in membrane proteins. The recent EM structure of the complex reveals a horseshoe-shaped geometry and confirms the 1:1:1:1 stoichiometry of the PS, NCT, APH-1, and PEN-2 subunits (82). The most intensively studied region of PS is the C-terminal four helices (TMD6–TMD9). The hydrophilic catalytic core is composed of TMD6 and -7, containing the catalytic Asp residues, along with TMD1 and -9. TMD9 forms part of the substrate-binding site. TMD8, the target of the current study, must pack on one or more of these helices due to the relatively short extramembraneous loops that connect TMD8 with TMD7 and TMD9. The current model of the complex indicates that helices 6–9 line an interior cavity that is capped by NCT. TMD6 and TMD9 are modeled to be at the front of the cavity, and TMD7 and TMD8 are modeled at the rear of the cavity (22, 23). One possible assignment of the TM helices allows PEN-2 to pack against TMD7 and TMD8, suggesting that TMD8 is in a central position within the complex rather than on the periphery. The influence of the AXXXAXXXG motifs on γ-secretase activity is consistent with this location. However, the ability of the complex to assemble in the presence of the PS-AA and PS-AG mutations suggests that the AXXXAXXXG motifs are probably oriented toward TMD7 or TMD9 rather than PEN-2. Additional mutational studies on TMD8 and other TM helices of the complex may allow discrimination of different possible arrangements of the TM helices consistent with the EM structure.
Acknowledgments
We are grateful to Bart de Strooper (KULeuven) for the kind gift of pHES2-luciferase plasmid and MEF cells and to Nicolas Sergeant (INSERM-Lille) for the generous gift of anti-APP C-ter antibody. Laetitia El Haylani is acknowledged for skillful technical support.
This work was supported, in whole or in part, by National Institutes of Health Grant AG27317 (to S. O. S.). This work was also supported by grants from the Fondation Recherche Alzheimer, the Foundation for Research on Alzheimer's Disease (to C. M. and P. K. C.), the Commission du Patrimoine of Université Catholique de Louvain (to C. M.), and the Interuniversity Attraction Pole Programme-Belgian Sate-Belgian Science Policy (P7/16-Belgian Medical Genetics Initiative) (to J. N. O., P. K. C., and S. N. C.). This work was also supported by the Salus Sanguinis Foundation, the Action de Recherche Concertée ARC10/15-027 of the University Catholique de Louvain, and the Special Fund for Research (FSR, UCL) (to S. N. C.).
- Aβ
- β-amyloid peptide
- AD
- Alzheimer disease
- AICD
- APP intracellular domain
- APP
- amyloid precursor protein
- PS
- presenilin
- CTF
- C-terminal fragment
- NTF
- N-terminal fragment
- DAPT
- N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester
- ECLIA
- electrochemiluminescence immunoassay
- HEK
- human embryonic kidney
- MEF
- mouse embryonic fibroblast
- MEFPSdKO
- MEF double knockout for PS1 and PS2
- MEFPSdKOr
- MEFPSdKO re-expressing either PS1 or PS2
- NICD
- Notch intracellular domain
- TMD
- transmembrane domain
- IP
- immunoprecipitation
- FL
- full-length
- ANOVA
- analysis of variance
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
REFERENCES
- 1. Sastre M., Steiner H., Fuchs K., Capell A., Multhaup G., Condron M. M., Teplow D. B., Haass C. (2001) Presenilin-dependent γ-secretase processing of β-amyloid precursor protein at a site corresponding to the S3 cleavage of Notch. EMBO Rep. 2, 835–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gu Y., Misonou H., Sato T., Dohmae N., Takio K., Ihara Y. (2001) Distinct intramembrane cleavage of the β-amyloid precursor protein family resembling γ-secretase-like cleavage of Notch. J. Biol. Chem. 276, 35235–35238 [DOI] [PubMed] [Google Scholar]
- 3. Pardossi-Piquard R., Checler F. (2012) The physiology of the β-amyloid precursor protein intracellular domain AICD. J. Neurochem. 120, 109–124 [DOI] [PubMed] [Google Scholar]
- 4. Funamoto S., Morishima-Kawashima M., Tanimura Y., Hirotani N., Saido T. C., Ihara Y. (2004) Truncated carboxyl-terminal fragments of β-amyloid precursor protein are processed to amyloid β-proteins 40 and 42. Biochemistry 43, 13532–13540 [DOI] [PubMed] [Google Scholar]
- 5. Qi-Takahara Y., Morishima-Kawashima M., Tanimura Y., Dolios G., Hirotani N., Horikoshi Y., Kametani F., Maeda M., Saido T. C., Wang R., Ihara Y. (2005) Longer forms of amyloid beta protein: implications for the mechanism of intramembrane cleavage by γ-secretase. J. Neurosci. 25, 436–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Takami M., Nagashima Y., Sano Y., Ishihara S., Morishima-Kawashima M., Funamoto S., Ihara Y. (2009) γ-Secretase: successive tripeptide and tetrapeptide release from the transmembrane domain of β-carboxyl terminal fragment. J. Neurosci. 29, 13042–13052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Matsumura N., Takami M., Okochi M., Wada-Kakuda S., Fujiwara H., Tagami S., Funamoto S., Ihara Y., Morishima-Kawashima M. (2014) γ-Secretase associated with lipid rafts: multiple interactive pathways in the stepwise processing of β-carboxyl-terminal fragment. J. Biol. Chem. 289, 5109–5121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hardy J., Selkoe D. J. (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356 [DOI] [PubMed] [Google Scholar]
- 9. Hardy J. A., Higgins G. A. (1992) Alzheimer's disease: the amyloid cascade hypothesis. Science 256, 184–185 [DOI] [PubMed] [Google Scholar]
- 10. Kopan R., Ilagan M. X. (2009) The canonical Notch signaling pathway: unfolding the activation mechanism. Cell 137, 216–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kopan R. (2012) Notch signaling. Cold Spring Harbor Perspect. Biol. 10.1101/cshperspect.a011213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wolfe M. S. (2009) Intramembrane-cleaving proteases. J. Biol. Chem. 284, 13969–13973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dries D. R., Yu G. (2008) Assembly, maturation, and trafficking of the γ-secretase complex in Alzheimer's disease. Curr. Alzheimer Res. 5, 132–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Spasic D., Annaert W. (2008) Building γ-secretase: the bits and pieces. J. Cell Sci. 121, 413–420 [DOI] [PubMed] [Google Scholar]
- 15. Laudon H., Hansson E. M., Melén K., Bergman A., Farmery M. R., Winblad B., Lendahl U., von Heijne G., Näslund J. (2005) A nine-transmembrane domain topology for presenilin 1. J. Biol. Chem. 280, 35352–35360 [DOI] [PubMed] [Google Scholar]
- 16. Thinakaran G., Borchelt D. R., Lee M. K., Slunt H. H., Spitzer L., Kim G., Ratovitsky T., Davenport F., Nordstedt C., Seeger M., Hardy J., Levey A. I., Gandy S. E., Jenkins N. A., Copeland N. G., Price D. L., Sisodia S. S. (1996) Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron 17, 181–190 [DOI] [PubMed] [Google Scholar]
- 17. Kimberly W. T., Xia W., Rahmati T., Wolfe M. S., Selkoe D. J. (2000) The transmembrane aspartates in presenilin 1 and 2 are obligatory for γ-secretase activity and amyloid β-protein generation. J. Biol. Chem. 275, 3173–3178 [DOI] [PubMed] [Google Scholar]
- 18. Wolfe M. S., Xia W., Ostaszewski B. L., Diehl T. S., Kimberly W. T., Selkoe D. J. (1999) Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and γ-secretase activity. Nature 398, 513–517 [DOI] [PubMed] [Google Scholar]
- 19. Wolfe M. S. (2013) Toward the structure of presenilin/γ-secretase and presenilin homologs. Biochim. Biophys. Acta 1828, 2886–2897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chau D. M., Crump C. J., Villa J. C., Scheinberg D. A., Li Y. M. (2012) Familial Alzheimer disease presenilin-1 mutations alter the active site conformation of γ-secretase. J. Biol. Chem. 287, 17288–17296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wanngren J., Lara P., Ojemalm K., Maioli S., Moradi N., Chen L., Tjernberg L. O., Lundkvist J., Nilsson I., Karlström H. (2014) Changed membrane integration and catalytic site conformation are two mechanisms behind the increased Aβ42/Aβ40 ratio by presenilin 1 familial Alzheimer-linked mutations. FEBS Open Bio 4, 393–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lu P., Bai X. C., Ma D., Xie T., Yan C., Sun L., Yang G., Zhao Y., Zhou R., Scheres S. H., Shi Y. (2014) Three-dimensional structure of human γ-secretase. Nature 512, 166–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wolfe M. S., Selkoe D. J. (2014) γ-Secretase: a horseshoe structure brings good luck. Cell 158, 247–249 [DOI] [PubMed] [Google Scholar]
- 24. Lemmon M. A., Flanagan J. M., Hunt J. F., Adair B. D., Bormann B. J., Dempsey C. E., Engelman D. M. (1992) Glycophorin A dimerization is driven by specific interactions between transmembrane α-helices. J. Biol. Chem. 267, 7683–7689 [PubMed] [Google Scholar]
- 25. Smith S. O., Song D., Shekar S., Groesbeek M., Ziliox M., Aimoto S. (2001) Structure of the transmembrane dimer interface of glycophorin A in membrane bilayers. Biochemistry 40, 6553–6558 [DOI] [PubMed] [Google Scholar]
- 26. Senes A., Engel D. E., DeGrado W. F. (2004) Folding of helical membrane proteins: the role of polar, GxxxG-like and proline motifs. Curr. Opin. Struct. Biol. 14, 465–479 [DOI] [PubMed] [Google Scholar]
- 27. Senes A., Ubarretxena-Belandia I., Engelman D. M. (2001) The Cα–H·O hydrogen bond: a determinant of stability and specificity in transmembrane helix interactions. Proc. Natl. Acad. Sci. U.S.A. 98, 9056–9061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Eilers M., Shekar S. C., Shieh T., Smith S. O., Fleming P. J. (2000) Internal packing of helical membrane proteins. Proc. Natl. Acad. Sci. U.S.A. 97, 5796–5801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Eilers M., Patel A. B., Liu W., Smith S. O. (2002) Comparison of helix interactions in membrane and soluble α-bundle proteins. Biophys. J. 82, 2720–2736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dawson J. P., Weinger J. S., Engelman D. M. (2002) Motifs of serine and threonine can drive association of transmembrane helices. J. Mol. Biol. 316, 799–805 [DOI] [PubMed] [Google Scholar]
- 31. Adamian L., Liang J. (2002) Interhelical hydrogen bonds and spatial motifs in membrane proteins: polar clamps and serine zippers. Proteins 47, 209–218 [DOI] [PubMed] [Google Scholar]
- 32. Smith S. O., Eilers M., Song D., Crocker E., Ying W., Groesbeek M., Metz G., Ziliox M., Aimoto S. (2002) Implications of threonine hydrogen bonding in the glycophorin A transmembrane helix dimer. Biophys. J. 82, 2476–2486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Beel A. J., Sanders C. R. (2008) Substrate specificity of γ-secretase and other intramembrane proteases. Cell. Mol. Life Sci. 65, 1311–1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kienlen-Campard P., Tasiaux B., Van Hees J., Li M., Huysseune S., Sato T., Fei J. Z., Aimoto S., Courtoy P. J., Smith S. O., Constantinescu S. N., Octave J. N. (2008) Amyloidogenic processing but not amyloid precursor protein (APP) intracellular C-terminal domain production requires a precisely oriented APP dimer assembled by transmembrane GXXXG motifs. J. Biol. Chem. 283, 7733–7744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sato T., Tang T. C., Reubins G., Fei J. Z., Fujimoto T., Kienlen-Campard P., Constantinescu S. N., Octave J. N., Aimoto S., Smith S. O. (2009) A helix-to-coil transition at the ϵ-cut site in the transmembrane dimer of the amyloid precursor protein is required for proteolysis. Proc. Natl. Acad. Sci. U.S.A. 106, 1421–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tang T. C., Hu Y., Kienlen-Campard P., El Haylani L., Decock M., Van Hees J., Fu Z., Octave J. N., Constantinescu S. N., Smith S. O. (2014) Conformational changes induced by the A21G Flemish mutation in the amyloid precursor protein lead to increased Aβ production. Structure 22, 387–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Munter L. M., Voigt P., Harmeier A., Kaden D., Gottschalk K. E., Weise C., Pipkorn R., Schaefer M., Langosch D., Multhaup G. (2007) GxxxG motifs within the amyloid precursor protein transmembrane sequence are critical for the etiology of Aβ42. EMBO J. 26, 1702–1712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Araki W., Saito S., Takahashi-Sasaki N., Shiraishi H., Komano H., Murayama K. S. (2006) Characterization of APH-1 mutants with a disrupted transmembrane GxxxG motif. J. Mol. Neurosci. 29, 35–43 [DOI] [PubMed] [Google Scholar]
- 39. Lee S. F., Shah S., Yu C., Wigley W. C., Li H., Lim M., Pedersen K., Han W., Thomas P., Lundkvist J., Hao Y. H., Yu G. (2004) A conserved GXXXG motif in APH-1 is critical for assembly and activity of the γ-secretase complex. J. Biol. Chem. 279, 4144–4152 [DOI] [PubMed] [Google Scholar]
- 40. Niimura M., Isoo N., Takasugi N., Tsuruoka M., Ui-Tei K., Saigo K., Morohashi Y., Tomita T., Iwatsubo T. (2005) Aph-1 contributes to the stabilization and trafficking of the γ-secretase complex through mechanisms involving intermolecular and intramolecular interactions. J. Biol. Chem. 280, 12967–12975 [DOI] [PubMed] [Google Scholar]
- 41. Liu W., Eilers M., Patel A. B., Smith S. O. (2004) Helix packing moments reveal diversity and conservation in membrane protein structure. J. Mol. Biol. 337, 713–729 [DOI] [PubMed] [Google Scholar]
- 42. Sobhanifar S., Schneider B., Löhr F., Gottstein D., Ikeya T., Mlynarczyk K., Pulawski W., Ghoshdastider U., Kolinski M., Filipek S., Güntert P., Bernhard F., Dötsch V. (2010) Structural investigation of the C-terminal catalytic fragment of presenilin 1. Proc. Natl. Acad. Sci. U.S.A. 107, 9644–9649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Oh Y. S., Turner R. J. (2005) Topology of the C-terminal fragment of human presenilin 1. Biochemistry 44, 11821–11828 [DOI] [PubMed] [Google Scholar]
- 44. Shiraishi H., Marutani T., Wang H. Q., Maeda Y., Kurono Y., Takashima A., Araki W., Nishimura M., Yanagisawa K., Komano H. (2006) Reconstitution of γ-secretase by truncated presenilin (PS) fragments revealed that PS C-terminal transmembrane domain is critical for formation of γ-secretase complex. Genes Cells 11, 83–93 [DOI] [PubMed] [Google Scholar]
- 45. Watanabe N., Takagi S., Tominaga A., Tomita T., Iwatsubo T. (2010) Functional analysis of the transmembrane domains of presenilin 1: participation of transmembrane domains 2 and 6 in the formation of initial substrate-binding site of γ-secretase. J. Biol. Chem. 285, 19738–19746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sato C., Takagi S., Tomita T., Iwatsubo T. (2008) The C-terminal PAL motif and transmembrane domain 9 of presenilin 1 are involved in the formation of the catalytic pore of the γ-secretase. J. Neurosci. 28, 6264–6271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Aldudo J., Bullido M. J., Valdivieso F. (1999) DGGE method for the mutational analysis of the coding and proximal promoter regions of the Alzheimer's disease presenilin-1 gene: two novel mutations. Hum. Mutat. 14, 433–439 [DOI] [PubMed] [Google Scholar]
- 48. Octave J. N., Essalmani R., Tasiaux B., Menager J., Czech C., Mercken L. (2000) The role of presenilin-1 in the γ-secretase cleavage of the amyloid precursor protein of Alzheimer's disease. J. Biol. Chem. 275, 1525–1528 [DOI] [PubMed] [Google Scholar]
- 49. Ben Khalifa N., Tyteca D., Marinangeli C., Depuydt M., Collet J. F., Courtoy P. J., Renauld J. C., Constantinescu S., Octave J. N., Kienlen-Campard P. (2012) Structural features of the KPI domain control APP dimerization, trafficking, and processing. FASEB J. 26, 855–867 [DOI] [PubMed] [Google Scholar]
- 50. Hébert S. S., Serneels L., Tolia A., Craessaerts K., Derks C., Filippov M. A., Müller U., De Strooper B. (2006) Regulated intramembrane proteolysis of amyloid precursor protein and regulation of expression of putative target genes. EMBO Rep. 7, 739–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kopan R., Schroeter E. H., Weintraub H., Nye J. S. (1996) Signal transduction by activated mNotch: importance of proteolytic processing and its regulation by the extracellular domain. Proc. Natl. Acad. Sci. U.S.A. 93, 1683–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cao X., Südhof T. C. (2001) A transcriptionally [correction of transcriptively] active complex of APP with Fe65 and histone acetyltransferase Tip60. Science 293, 115–120 [DOI] [PubMed] [Google Scholar]
- 53. Huysseune S., Kienlen-Campard P., Octave J. N. (2007) Fe65 does not stabilize AICD during activation of transcription in a luciferase assay. Biochem. Biophys. Res. Commun. 361, 317–322 [DOI] [PubMed] [Google Scholar]
- 54. Hage S., Marinangeli C., Stanga S., Octave J. N., Quetin-Leclercq J., Kienlen-Campard P. (2014) γ-Secretase inhibitor activity of a Pterocarpus erinaceus extract. Neurodegener. Dis. 14, 39–51 [DOI] [PubMed] [Google Scholar]
- 55. Xia W., Zhang J., Perez R., Koo E. H., Selkoe D. J. (1997) Interaction between amyloid precursor protein and presenilins in mammalian cells: implications for the pathogenesis of Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 94, 8208–8213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Campeau E., Ruhl V. E., Rodier F., Smith C. L., Rahmberg B. L., Fuss J. O., Campisi J., Yaswen P., Cooper P. K., Kaufman P. D. (2009) A versatile viral system for expression and depletion of proteins in mammalian cells. PloS One 4, e6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Salmon P., Trono D. (2007) Production and titration of lentiviral vectors. Curr. Protoc. Hum. Genet. 10.1002/0471142905.hg1210s54 [DOI] [PubMed] [Google Scholar]
- 58. Wang L. F., Zhang R., Xie X. (2009) Development of a high-throughput assay for screening of γ-secretase inhibitor with endogenous human, mouse or Drosophila γ-secretase. Molecules 14, 3589–3599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Berechid B. E., Kitzmann M., Foltz D. R., Roach A. H., Seiffert D., Thompson L. A., Olson R. E., Bernstein A., Donoviel D. B., Nye J. S. (2002) Identification and characterization of presenilin-independent Notch signaling. J. Biol. Chem. 277, 8154–8165 [DOI] [PubMed] [Google Scholar]
- 60. Esler W. P., Kimberly W. T., Ostaszewski B. L., Diehl T. S., Moore C. L., Tsai J. Y., Rahmati T., Xia W., Selkoe D. J., Wolfe M. S. (2000) Transition-state analogue inhibitors of γ-secretase bind directly to presenilin-1. Nat. Cell Biol. 2, 428–434 [DOI] [PubMed] [Google Scholar]
- 61. Wolfe M. S., De Los Angeles J., Miller D. D., Xia W., Selkoe D. J. (1999) Are presenilins intramembrane-cleaving proteases? Implications for the molecular mechanism of Alzheimer's disease. Biochemistry 38, 11223–11230 [DOI] [PubMed] [Google Scholar]
- 62. De Strooper B., Saftig P., Craessaerts K., Vanderstichele H., Guhde G., Annaert W., Von Figura K., Van Leuven F. (1998) Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 391, 387–390 [DOI] [PubMed] [Google Scholar]
- 63. Berezovska O., Lleo A., Herl L. D., Frosch M. P., Stern E. A., Bacskai B. J., Hyman B. T. (2005) Familial Alzheimer's disease presenilin 1 mutations cause alterations in the conformation of presenilin and interactions with amyloid precursor protein. J. Neurosci. 25, 3009–3017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Herreman A., Serneels L., Annaert W., Collen D., Schoonjans L., De Strooper B. (2000) Total inactivation of γ-secretase activity in presenilin-deficient embryonic stem cells. Nat. Cell Biol. 2, 461–462 [DOI] [PubMed] [Google Scholar]
- 65. De Strooper B., Annaert W., Cupers P., Saftig P., Craessaerts K., Mumm J. S., Schroeter E. H., Schrijvers V., Wolfe M. S., Ray W. J., Goate A., Kopan R. (1999) A presenilin-1-dependent γ-secretase-like protease mediates release of Notch intracellular domain. Nature 398, 518–522 [DOI] [PubMed] [Google Scholar]
- 66. Fukumori A., Fluhrer R., Steiner H., Haass C. (2010) Three-amino acid spacing of presenilin endoproteolysis suggests a general stepwise cleavage of γ-secretase-mediated intramembrane proteolysis. J. Neurosci. 30, 7853–7862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Edbauer D., Winkler E., Haass C., Steiner H. (2002) Presenilin and nicastrin regulate each other and determine amyloid β-peptide production via complex formation. Proc. Natl. Acad. Sci. U.S.A. 99, 8666–8671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bentahir M., Nyabi O., Verhamme J., Tolia A., Horré K., Wiltfang J., Esselmann H., De Strooper B. (2006) Presenilin clinical mutations can affect γ-secretase activity by different mechanisms. J. Neurochem. 96, 732–742 [DOI] [PubMed] [Google Scholar]
- 69. Nyabi O., Pype S., Mercken M., Herreman A., Saftig P., Craessaerts K., Serneels L., Annaert W., De Strooper B. (2002) No endogenous Aβ production in presenilin-deficient fibroblasts. Nat. Cell Biol. 4, E164; author reply E165–E166 [DOI] [PubMed] [Google Scholar]
- 70. Osenkowski P., Li H., Ye W., Li D., Aeschbach L., Fraering P. C., Wolfe M. S., Selkoe D. J., Li H. (2009) Cryoelectron microscopy structure of purified γ-secretase at 12 Å resolution. J. Mol. Biol. 385, 642–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Dovey H. F., John V., Anderson J. P., Chen L. Z., de Saint Andrieu P., Fang L. Y., Freedman S. B., Folmer B., Goldbach E., Holsztynska E. J., Hu K. L., Johnson-Wood K. L., Kennedy S. L., Kholodenko D., Knops J. E., Latimer L. H., Lee M., Liao Z., Lieberburg I. M., Motter R. N., Mutter L. C., Nietz J., Quinn K. P., Sacchi K. L., Seubert P. A., Shopp G. M., Thorsett E. D., Tung J. S., Wu J., Yang S., Yin C. T., Schenk D. B., May P. C., Altstiel L. D., Bender M. H., Boggs L. N., Britton T. C., Clemens J. C., Czilli D. L., Dieckman-McGinty D. K., Droste J. J., Fuson K. S., Gitter B. D., Hyslop P. A., Johnstone E. M., Li W. Y., Little S. P., Mabry T. E., Miller F. D., Audia J. E. (2001) Functional γ-secretase inhibitors reduce β-amyloid peptide levels in brain. J. Neurochem. 76, 173–181 [DOI] [PubMed] [Google Scholar]
- 72. Shearman M. S., Beher D., Clarke E. E., Lewis H. D., Harrison T., Hunt P., Nadin A., Smith A. L., Stevenson G., Castro J. L. (2000) L-685,458, an aspartyl protease transition state mimic, is a potent inhibitor of amyloid β-protein precursor γ-secretase activity. Biochemistry 39, 8698–8704 [DOI] [PubMed] [Google Scholar]
- 73. Cao X., Südhof T. C. (2004) Dissection of amyloid-β precursor protein-dependent transcriptional transactivation. J. Biol. Chem. 279, 24601–24611 [DOI] [PubMed] [Google Scholar]
- 74. De Strooper B., Simons M., Multhaup G., Van Leuven F., Beyreuther K., Dotti C. G. (1995) Production of intracellular amyloid-containing fragments in hippocampal neurons expressing human amyloid precursor protein and protection against amyloidogenesis by subtle amino acid substitutions in the rodent sequence. EMBO J. 14, 4932–4938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Leem J. Y., Saura C. A., Pietrzik C., Christianson J., Wanamaker C., King L. T., Veselits M. L., Tomita T., Gasparini L., Iwatsubo T., Xu H., Green W. N., Koo E. H., Thinakaran G. (2002) A role for presenilin 1 in regulating the delivery of amyloid precursor protein to the cell surface. Neurobiol. Dis. 11, 64–82 [DOI] [PubMed] [Google Scholar]
- 76. Herreman A., Van Gassen G., Bentahir M., Nyabi O., Craessaerts K., Mueller U., Annaert W., De Strooper B. (2003) γ-Secretase activity requires the presenilin-dependent trafficking of nicastrin through the Golgi apparatus but not its complex glycosylation. J. Cell Sci. 116, 1127–1136 [DOI] [PubMed] [Google Scholar]
- 77. Knappenberger K. S., Tian G., Ye X., Sobotka-Briner C., Ghanekar S. V., Greenberg B. D., Scott C. W. (2004) Mechanism of γ-secretase cleavage activation: is γ-secretase regulated through autoinhibition involving the presenilin-1 exon 9 loop? Biochemistry 43, 6208–6218 [DOI] [PubMed] [Google Scholar]
- 78. Jozwiak K., Krzysko K. A., Bojarski L., Gacia M., Filipek S. (2008) Molecular models of the interface between anterior pharynx-defective protein 1 (APH-1) and presenilin involving GxxxG motifs. ChemMedChem 3, 627–634 [DOI] [PubMed] [Google Scholar]
- 79. Weggen S., Beher D. (2012) Molecular consequences of amyloid precursor protein and presenilin mutations causing autosomal-dominant Alzheimer's disease. Alzheimers Res. Ther. 4, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Weidemann A., Eggert S., Reinhard F. B., Vogel M., Paliga K., Baier G., Masters C. L., Beyreuther K., Evin G. (2002) A novel ϵ-cleavage within the transmembrane domain of the Alzheimer amyloid precursor protein demonstrates homology with Notch processing. Biochemistry 41, 2825–2835 [DOI] [PubMed] [Google Scholar]
- 81. Pinnix I., Musunuru U., Tun H., Sridharan A., Golde T., Eckman C., Ziani-Cherif C., Onstead L., Sambamurti K. (2001) A novel γ-secretase assay based on detection of the putative C-terminal fragment-γ of amyloid β protein precursor. J. Biol. Chem. 276, 481–487 [DOI] [PubMed] [Google Scholar]
- 82. Sato T., Diehl T. S., Narayanan S., Funamoto S., Ihara Y., De Strooper B., Steiner H., Haass C., Wolfe M. S. (2007) Active γ-secretase complexes contain only one of each component. J. Biol. Chem. 282, 33985–33993 [DOI] [PubMed] [Google Scholar]