

Abstract
Protease-activated receptors (PARs) are G protein-coupled receptors activated by proteolytic cleavage at their amino termini by serine proteases. PAR activation contributes to the inflammatory response in the gastrointestinal (GI) tract and alters GI motility, but little is known about the specific cells within the tunica muscularis that express PARs and the mechanisms leading to contractile responses. Using real time PCR, we found PARs to be expressed in smooth muscle cells (SMCs), interstitial cells of Cajal (ICC) and platelet-derived growth factor receptor α positive (PDGFRα+) cells. The latter cell-type showed dominant expression of F2r (encodes PAR1) and F2rl1 (encodes PAR2). Contractile and intracellular electrical activities were measured to characterize the integrated responses to PAR activation in whole muscles. Cells were isolated and ICC and PDGFRα+ cells were identified by constitutive expression of fluorescent reporters. Thrombin (PAR1 agonist) and trypsin (PAR2 agonist) caused biphasic responses in colonic muscles: transient hyperpolarization and relaxation followed by repolarization and excitation. The inhibitory phase was blocked by apamin, revealing a distinct excitatory component. Patch clamp studies showed that the inhibitory response was mediated by activation of small conductance calcium-activated K+ channels in PDGFRα+ cells, and the excitatory response was mediated by activation of a Cl− conductance in ICC. SMCs contributed little to PAR responses in colonic muscles. In summary, PARs regulate the excitability of colonic muscles; different conductances are activated in each cell type of the SMC–ICC–PDGFRα+ cell (SIP) syncytium. Motor responses to PAR agonists are integrated responses of the SIP syncytium.
Key points
Activation of protease-activated receptors (PAR) regulates gastrointestinal (GI) motility but little is known about the cells and mechanisms in GI muscles responsible for PAR responses.
Using mouse cells, we found high levels of F2r and F2rl1 PAR-encoding genes expressed in purified platelet-derived growth factor α-positive (PDGFRα+) cells in comparison to other cells in colonic muscles.
PAR1 and PAR2 agonists caused transient hyperpolarization and relaxation of colonic muscles, with relaxation responses followed by excitation.
The inhibitory phase was inhibited by apamin and mediated by activation of small conductance calcium-activated potassium channels in PDGFRα+ cells.
The excitatory response resulted largely from activation of a chloride conductance in interstitial cells of Cajal; small amplitude inward currents were generated in smooth muscle cells by PAR activation, but these responses were too small to be resolved in intact muscles.
PAR receptor responses are integrated responses generated by cells of the smooth muscle, interstitial cells of Cajal and PDGFRα+ cells (SIP syncytium).
Introduction
Protease-activated receptors (PARs) are G protein-coupled receptors activated by proteolytic cleavage of N termini by serine proteases. The peptides liberated are ligands that activate the receptors and initiate intracellular signalling events (Macfarlane et al. 2001; Traynelis & Trejo, 2007). Four PARs (PAR1–4) have been cloned (Vu et al. 1991b; Nystedt et al. 1994; Ishihara et al. 1997; Xu et al. 1998). Many studies have linked PAR activation to second messengers through their association with G proteins (Coughlin, 2000, 2005; Macfarlane et al. 2001; Bunnett, 2006; Traynelis & Trejo, 2007). For example, activation of PAR1 by thrombin and other proteases increases 1,4,5-trisphosphate (IP3) production via Gq/11-mediated activation of phospholipase C (PLCβ) and reduces cAMP due to Gi/Go-mediated inhibition of adenynyl cyclase (Vu et al. 1991a; Hung et al. 1992). Activation of PAR2 by trypsin also increases IP3 production via Gq/11 coupling (Bohm et al. 1996; Ossovskaya & Bunnett, 2004).
PARs are widely distributed in the gastrointestinal (GI) tract and involved in regulating salivary gland secretion, mucus and pepsin production, pancreatic secretions, small intestinal ion transport and motility (Kawabata, 2003). PARs may participate in regulation of motility under physiological and pathological conditions. PARs affect motility by regulating sensory neurons (Mule et al. 2003; Zhao & Shea-Donohue, 2003; Sekiguchi et al. 2006) and directly regulating the excitability of GI smooth muscles. PAR1 and PAR2 agonists induced contraction of gastric smooth muscles but these agonists can also cause relaxation of pre-contracted muscles (Cocks et al. 1997; Sekiguchi et al. 2006). In rat duodenal muscles, PAR2 agonists cause slowly developing, persistent contraction, while PAR1 agonists cause initial relaxation followed by strong contraction (Kawabata et al. 1999). In colon PAR1 and PAR2 agonists elicit relaxation and/or contractions (Corvera et al. 1999; Mule et al. 2002a; Sato et al. 2006). Relaxation induced by PAR agonists can be inhibited by apamin, a blocker of small conductance Ca2+-activated K+ channels (SK channels) (Cocks et al. 1997; Kawabata et al. 1999; Mule et al. 2002a; Sekiguchi et al. 2006). The complexity of responses suggests that multiple receptors may be linked to different mechanisms that are not in temporal alignment. The cells mediating non-neural PAR responses of the tunica muscularis have not been clarified.
GI muscles are not homogenous tissues containing only SMCs and enteric neurons. Interstitial cells are present and affect the excitability of the smooth muscle syncytium. At a minimum, interstitial cells of Cajal (ICC) provide pacemaker activity, transduce inputs from motor neurotransmitters, and mediate responses to stretch (Sanders et al. 2012). Platelet-derived growth factor receptor α-positive (PDGFRα+) cells are also abundant and participate in transduction of neural inputs (Kurahashi et al. 2011). ICC and PDGFRα+ cells are electrically coupled to SMCs, and together SMCs, ICC and PDGFRα+ form an integrated structure called the SIP syncytium (Sanders et al. 2012). Changes in ionic conductances in any of the SIP cells affect smooth muscle excitability and responses to bioagonists regulating motor function. In this study we explored expression of PARs in SIP cells and their specific responses to activation of PARs.
Methods
Animals
C57BL/6 mice, Pdgfratm11(EGFP)Sor/J heterozygote mice (Jackson Laboratory, Bar Harbor, ME, USA), KitcopGFP/+ (as described previously; Zhu et al. 2009), and smMHC/Cre/eGFP mice (donated by Dr Michael Kotlikoff, Cornell University) were used for molecular and electromechanical experiments. Mice (5–8 weeks) were anaesthetized with isoflurane (Baxter Healthcare, Deerfield, IL, USA) and killed humanely by cervical dislocation. Animals were maintained in accordance with the NIH Guide for the Care and Use of Laboratory Animals. All procedures were approved by the Institutional Animal Use and Care Committee at the University of Nevada, Reno.
Isometric force recording
Colons were removed and washed with Krebs–Ringer bicarbonate solution (KRB). Colon segments from 1 cm below the caecocolic junction were opened along the mesenteric border and mucosae were removed. Strips of muscle (1 cm length × 0.2 cm width) were cut parallel to the circular muscle layer, attached to an isometric force transducer (Fort 10, WPI, FL, USA), and suspended in a 5 ml organ bath containing oxygenated (97% O2 and 3% CO2) KRB. Temperature through experiments was maintained at 37 ± 0.5°C, and KRB was changed every 15 min. A resting force of 1–3 mN was applied to set muscles at optimum lengths. Mechanical responses were recorded with a computer running Axoscope (Axon Instruments, Union City, CA, USA). The amplitude, frequency and the area under the curve (AUC) during 2 min recordings were measured, and these parameters were compared before and after drugs. Muscles were pre-treated with tetrodotoxin (TTX, 1 μm) for 10 min before adding PAR agonists to decrease contamination of responses from neural effects.
Transmembrane potential recording
Intracellular microelectrode recordings were used to measure membrane potentials of colonic smooth muscle cells (SMCs). Smooth muscle sheets (0.5 cm length × 0.5 cm width) were pinned to the floor of a recording chamber perfused continuously with oxygenated, pre-warmed (37 ± 0.5°C) KRB. Cells within the circular smooth muscle layer were impaled with microelectrodes with tip resistances of 80–100 MΩ. Transmembrane potential was measured with a high input impedance amplifier (WPI Duo 773, FL, USA) and recorded with a computer running AxoScope. Data were analysed by Clampfit (v9.02, Axon Instruments) and Graphpad Prism (version 5.0, Graphpad Software Inc., CA, USA). Experiments were performed in the presence of TTX (1 μm).
Cell preparation and patch clamp experiment
Whole-cell patch clamp studies were performed on PDGFRα+ cells and ICC identified by fluorescence proteins expressed by these cells, and SMCs were identified by morphology. Cells were obtained by digestion of colonic smooth muscles, first equilibrated for 30 min in Ca2+-free Hanks’ solution at 4°C (containing in mm: 125 NaCl, 5.36 KCl, 15.5 NaHCO3, 0.336 Na2HPO4, 0.44 KH2PO4, 10 glucose, 2.9 sucrose and 11 Hepes and adjusted to pH 7.2 with NaOH) and then exposed for 30 ± 2 min at 37°C to a solution containing (per ml) 3.5 mg collagenase (Worthington Type II; Worthington Biochemical, NJ, USA), 8.0 mg bovine serum albumin (Sigma, St Louis, MO, USA), 8.0 mg trypsin inhibitor (Sigma), as described previously (Kurahashi et al. 2011; Zheng et al. 2014). After washing and trituration, cells were placed on collagen-coated (2.5 mg ml-1, BD Falcon, NJ, USA) glass coverslips. Cells were used for experiments after 1 h at 37°C in a 95% O2–5% CO2 incubator.
After obtaining gigaseals, cells were voltage clamped using an Axopatch 200 B amplifier (Axon Instruments) and digitized with a 12-bit A/D converter (Digidata 1320 A, Axon Instruments) to record membrane currents. Membrane potentials were measured under current clamp (I = 0), as described previously (Kurahashi et al. 2011; Zheng et al. 2014). All data were acquired and digitized using pClamp software (Clampex 10.0.0.61, Axon Instruments) and analysed by Clampfit (v9.02, Axon Instruments) and Graphpad Prism (version 5.0, Graphpad Software Inc.) software. Experiments were performed at 30°C using a CL-100 bath heater (Warner Instruments, CT, USA).
Molecular studies
SMCs, ICC and PDGFRα+ cells were purified by fluorescence-activated cell sorting (Becton Dickinson FACSAria using the blue laser (488 nm) and the GFP emission detector; 530/30 nm). Total RNA isolation, cDNA preparation and amplification of murine colonic muscle strips (mucosa and submucosa removed) were performed as previously reported (Zhu et al. 2009). To investigate the expression of PARs, the following PCR primers designed against murine sequences were used (genebank accession number is given in parenthesis for the reference nucleotide sequence used): F2r (NM_010169), F2rl1 (NM_007974), F2rl2 (NM_010170), F2rl3 (NM_007975). The relative expression levels of PARs was determined by real-time quantitative PCR performed on a ABI PrismM 7000 sequence detector using SYBR Green chemistry (Applied Biosystems, CA, USA). Standard curves were generated for each receptor and constitutively expressed Gapdh from regression analysis of the mean values of RT-PCRs for the log10 diluted cDNA. Each cDNA sample was tested in triplicate and cDNAs were obtained from four murine colons. The reproducibility of the assay was tested by analysis of variance, comparing repeat runs of samples, and the mean values generated at individual time points were compared by Student's t test.
Solutions and drugs
In mechanical and electrical recordings, the muscles were equilibrated for 1–2 h before experiments began in oxygenated KRB (in mm): 120 NaCl; 5.9 KCl; 1.2 MgCl2; 15.5 NaHCO3; 1.2 NaH2PO4; 11.5 dextrose; and 2.5 CaCl2. The pH of KRB was 7.3–7.4 when bubbled with 97% O2–3% CO2 at 37.0 ± 0.5°C. External solution for whole-cell recordings was a Ca2+-containing physiological salt solution (CaPSS) consisting of (in mm): 5 KCl, 135 NaCl, 2 CaCl2, 10 glucose, 1.2 MgCl2, and 10 Hepes, adjusted to pH 7.4 with Tris. K+-rich internal solution solution contained (in mm): 135 KCl, 3 MgATP, 0.1 NaGTP, 2.5 creatine phosphate disodium, 0.1 EGTA, 0.01 CaCl2, 10 Hepes, 10 glucose, adjusted to pH 7.2 with Tris. Cs+-rich internal solution contained (in mm): 30 CsCl, 110 caesium aspartate, 3 MgATP, 0.1 NaGTP, 0.1 EGTA, 0.01 CaCl2, 10 Hepes, 10 glucose, adjusted to pH 7.2 with Tris. The calculated junction potentials in K+-rich solution and Cs+-rich solutions were 5.3 mV and 14.6 mV, respectively. The holding potentials given in the text are ‘command potentials’ and uncorrected for junction potentials. Thrombin, trypsin, TTX, tetraethylammonium (TEA), and 1-[(2-chlorophenyl)diphenylmethyl]-1H-pyrazole (TRAM-34) were purchased from Sigma and apamin was purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA).
Statistical analysis
Data shown are means ± SEM. The Student's t test between two groups and ANOVA followed by a post hoc test among three groups or more were used where appropriate to evaluate significance. P values less than 0.05 were taken as statistically significant and n values refer to the number of recordings from muscle strips in electro-mechanical experiments and isolated cells in whole-cell patch experiments.
Results
Transcriptional expression of protease-activated receptors in colon
Expression of PAR isoforms (F2r-F2rl3) in unsorted cells and sorted SMCs, ICC and PDGFRα+ cells was determined by RT-PCR. Detectable amplicons for F2r (PAR1), F2rl1 (PAR2) and F2rl2 (PAR3) were found in all cell extracts (Fig.1A). However, quantitative analysis showed that F2r and F2rl1 were highly expressed in PDGFRα+ cells, and F2r and F2rl1 were expressed in ICC (Fig.1B). Transcript levels were very low in SMCs.
Figure 1.
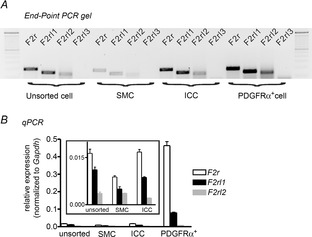
Transcriptional expression of protease-activated receptors (PARs) in murine colonic smooth muscles
A, representative agarose end-point gel of RT-PCR products revealed F2r (195 bp), F2rl1 (151 bp) and F2rl2 (139 bp) expression in unsorted cells after enzymatic dispersion of the tunica muscularis of the colon, sorted smooth muscle cells (SMC), sorted interstitial cells of Cajal (ICC) and sorted platelet-derived growth factor receptor α (PDGFRα+) cells. B, summary data from real-time quantitative PCR analysis of PARs from unsorted and sorted cells (n = 4) showing relative expression of PAR isoforms. Inset graph has expanded y-axis to show the levels of the relatively low expression of PAR genes in SMCs and ICC.
Inhibitory responses of thrombin and trypsin on spontaneous contractions
The amplitude, frequency and AUC of contractions were analysed before and after addition of PAR agonists (i.e. thrombin for PAR1, PAR3 and PAR4 and trypsin for PAR2). Thrombin (50 U ml-1) reduced spontaneous contractions initially to 20.2 ± 10.6% (P < 0.01) in amplitude, from 2.1 ± 0.4 contractions per minute (cpm) to 0.8 ± 0.6 cpm (P < 0.05) in frequency, and to 12.9 ± 5.4% (P < 0.01) in AUC within 2 min of treatment (Fig.2, n = 6). Trypsin (1 μm) also decreased contractions to 35.1 ± 11.9% (P < 0.01) in amplitude, from 2.1 ± 0.2 cpm to 0.9 ± 0.5 cpm (P < 0.05) in frequency, and to 25.7 ± 6.8% (P < 0.01) in AUC within 2 min of treatment (Fig.2, n = 5). Inhibitory responses to thrombin and trypsin were transient (Fig.2A and B). Contractions recovered gradually in the continued presence of thrombin and trypsin, such that the amplitudes of contractions were 68.1 ± 4.7% at 5 min and 105.8 ± 4.2% at 10 min in the presence of thrombin (n = 6), and 83.4 ± 7.9% after 5 min and 101.5 ± 4.8% after 10 min in the presence of trypsin (n = 5). The frequencies of contractions recovered fully within 5 min after thrombin and trypsin were added (i.e. to 2.3 ± 0.3 cpm and 1.9 ± 0.3 cpm, respectively; both P > 0.05 vs. control). AUC had not recovered fully within 5–10 min after addition of thrombin (n = 6; P < 0.01) or trypsin (n = 6; P < 0.01; Fig.2C). These results show that thrombin and trypsin have transient inhibitory effects on colonic contractions. It is possible that the integrated responses resulted from summation of dual effects with different kinetics: an initial rapid inhibitory phase followed by a more slowly developing contractile response.
Figure 2.
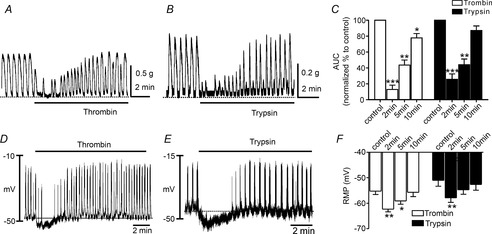
The effects of thrombin and trypsin on contractions and transmembrane potentials of murine colonic smooth muscle
A and B, representative mechanical traces showing spontaneous contractile activity of murine proximal colon and the effects of thrombin (50 U ml−1; A) and trypsin (1 μm; B). PAR agonists caused an initial relaxation followed by slow recovery of contractions to approximately the control level of contractility. C, summary data of the area under the curve (AUC) at 2 min, 5 min and 10 min. The data were normalized to control value (before application of drugs). D and E, representative traces illustrating that thrombin (50 U ml−1; D) and trypsin (1 μm; E) induced hyperpolarization followed by recovery to approximately control levels of electrical rhythmicity and membrane potential. Traces shown in A and B were recorded from different muscles to traces in D and E; however, the time courses of the electrical and mechanical responses are similar. F, summarized data showing average effects of thrombin and trypsin on resting membrane potentials at 2 min, 5 min and 10 min. *P < 0.05, **P < 0.01, ***P < 0.001.
The effects of thrombin and trypsin on resting membrane potential
Intracellular microelectrode recordings were used to study the effects of thrombin and trypsin on resting membrane potentials (RMP) in murine colon because the inhibitory effects were potentially the result of hyperpolarization. Thrombin (50 U ml−1) and trypsin (1 μm) caused transient hyperpolarization of cells (i.e. from −55 ± 1.3 mV to 62 ± 1.0 mV and from −51 ± 2.3 mV to −58 ± 1.8 mV, respectively; P < 0.01 for each drug; Fig.2F, both n = 5). Membrane potential recovered to the control level of RMP after the initial hyperpolarization. Rhythmical electrical activity was inhibited during the period of hyperpolarization (from 2.8 ± 0.4 to 0.3 ± 0.2 cpm (P < 0.01 with thrombin) and from 2.5 ± 0.6 to 0.3 ± 0.2 cpm (P < 0.01 with trypsin; Fig.2D and E, n = 6 for each drug) and recovered after restoration of RMP (i.e. to 2.7 ± 0.3 cpm with thrombin, and 2.3 ± 0.5 cpm with trypsin; n = 6 for each drug).
The effect of apamin on thrombin- and trypsin-induced initial inhibitory responses
PAR1 and PAR2 are typically coupled to Gq/11 (Kawabata et al. 2002; Mule et al. 2002b; Hollenberg, 2005) that can activate phospholipase C-mediated signalling mechanisms, including 1,4,5-trisphosphate (IP3) production and release of Ca2+ from intracellular stores. Previous studies have shown that the inhibitory effects of thrombin and trypsin can be reduced or abolished by apamin, indicating that PAR activation may be coupled to activation of SK channels in rat colon (Mule et al. 2002a). Therefore, we characterized the effects of apamin on inhibitory responses caused by thrombin and trypsin.
Apamin (300 nm) caused an increase in spontaneous contractions due to reduction in the tonic inhibitory influence of purines in the colon (data not shown). The initial inhibitory phases of the responses to thrombin or trypsin (Fig.2) were blocked by apamin, and muscles developed either sustained contraction or increased phasic contractions after addition of thrombin or trypsin in presence of apamin (Fig.3A and B). AUC of responses to thrombin increased from 124.0 ± 4.9 to 141.7 ± 2.8%, and responses to trypsin increased from 114.5 ± 6.0 to 133.9 ± 7.0% (n = 5 each) in the presence of apamin. TEA (1 mm) and TRAM-34 (10 μm), blockers of large and intermediate conductance Ca2+-activated K+ channels, did not affect responses to PAR agonists (data not shown).
Figure 3.
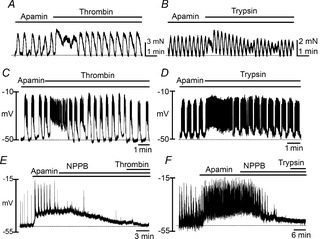
Effects of apamin on the relaxation and hyperpolarization phases of PAR responses
A and B, representative traces showing that apamin (300 nm) pretreatment abolished thrombin (50 U ml−1; A) and trypsin (1 μm; B)-induced relaxation phase and caused development of tone in these phasic muscles. C and D, intracellular microelectrode recordings of transmembrane potentials showing that thrombin (50 U ml−1; C) and trypsin (1 μm; D) induced depolarization and sustained firing of action potentials when the hyperpolarization phase of PAR responses was blocked by apamin (300 nm). E and F, thrombin (50 U ml−1; E) and trypsin (1 μm; F)-induced depolarization were completely abolished by pretreatment of NPPB (10 μm).
We also examined the effects of apamin on electri-cal responses to thrombin and trypsin (Fig.3C and D). Apamin caused depolarization of impaled cells from −57 ± 2.7 to −52 ± 2.1 mV. In the presence of apamin, cells were further depolarized by thrombin to −41 ± 2.7 mV (n = 6; P < 0.01 compared to the membrane potential with apamin alone). In experiments with trypsin, apamin pretreatment depolarized colonic smooth muscles from −59 ± 4.3 to −55 ± 2.4 mV, and trypsin, in the continued presence of apamin, caused further depolarization to −41 ± 3.0 mV (n = 5; P < 0.01 compared to the membrane potential with apamin alone). Action potential discharges, often sustained for several minutes, were noted during the responses to thrombin and trypsin. These observations indicate that tissue responses to PAR agonists are dual in nature, and excitatory responses are masked initially by activation of an apamin-sensitive conductance.
ICC express Ca2+-activated Cl− channels (CaCCs) that are responsible for pacemaker activity (Zhu et al. 2009). We tested whether a CaCC blocker, NPPB (10 μm), blocked the depolarization responses induced by thrombin and trypsin in the presence of apamin (Fig.3E and F). NPPB abolished slow waves and action potentials and caused hyperpolarization to −51 ± 3.5 mV (n = 6, P < 0.01 compared with electrical activity in the presence of apamin alone). Thrombin and trypsin failed to induce depolarization (i.e. −52 ± 3.8 mV and −51 ± 2.9 mV, respectively) in the presence of NPPB. These observations suggest that the depolarization responses to thrombin and trypsin may be mediated by activation of CaCCs.
The effects of thrombin and trypsin on membrane currents and potentials in PDGFRα+ cells
We previously reported that SK current density is much greater in PDGFRα+ cells than in SMCs (Kurahashi et al. 2011). Thus, PDGFRα+ cells may mediate the hyperpolarization and inhibitory contractile responses induced by thrombin and trypsin. We tested the effects of thrombin and trypsin on isolated and identified PDGFRα+ cells under whole-cell patch clamp conditions. The average cell capacitance of freshly isolated PDGFRα+ cells was 6.6 ± 0.8 pF (n = 33), and the average holding current at a holding potential (HP) −50 mV was −4.6 ± 0.6 pA (n = 36) using a K+-rich pipette solution. Thrombin and trypsin evoked large-amplitude outward currents in PDGFRα+ cells, averaging 108 ± 19.4 pA pF–1 and 71 ± 6.0 pA pF–1 (after 1 min exposure) and 34 ± 7.1 and 24 ± 2.1 pA pF–1 (after 2 min exposure), respectively (Fig.4A and D, n = 6 for each drug). The current responses to the PAR agonists were transient in nature, possibly due to desensitization (Ferguson, 2001). Figure 4B and E show current–voltage relationships before and after application of PAR agonists. Thrombin and trypsin shifted the reversal potential of the whole-cell currents close to EK (which was −86 mV under the conditions of these experiments), and the shape of the I–V curves in the presence of PAR agonists is indicative of activation of an SK-type conductance (i.e. voltage independence and inward rectification at positive potentials; Barfod et al. 2001). Apamin, added 1 min after application of thrombin and trypsin, inhibited the outward currents. The average current densities were reduced significantly by apamin to 3.9 ± 1.0 pA pF–1 and 3.6 ± 0.8 pA pF–1 (at 2 min), respectively (Figs4A and D and 5C).
Figure 4.
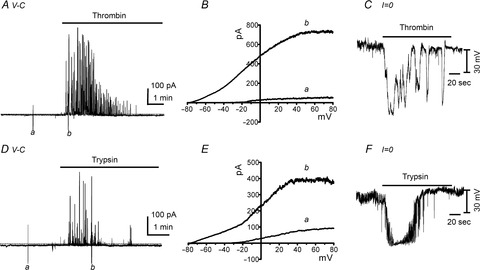
Effects of thrombin and trypsin on membrane currents and transmembrane potentials of isolated, single PDGFRα+ cells
A and D, under voltage-clamp (V-C) conditions, thrombin (50 U ml−1; A) and trypsin (1 μm; D) activated outward currents in PDGFRα+ cells (holding potential was −50 mV in these traces). B and E, expanded time scale showing responses to ramp depolarization (from −80 mV to +80 mV; 1 s ramps) before (a) and during (b) thrombin (B) and trypsin (E) application (current responses are uncorrected for junction potential). C and F, under current clamp conditions (I = 0), thrombin (50 U ml−1; C) and trypsin (1 μm; F) induced hyperpolarization of PDGFRα+ cells.
Figure 5.
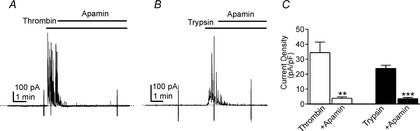
Effects of apamin on thrombin and trypsin-activated outward currents in PDGFRα+ cells
A and B, apamin (300 nm; added 1 min after addition of thrombin or trypsin) blocked the outward currents induced by thrombin (50 U ml-1; A, and trypsin (1 μm; B, The holding potential in these experiments was −50 mV. C, summarized data showing the average current densities evoked by thrombin (open bars; n = 5) or trypsin (filled bars; n = 5) 2 min after the responses were initiated in cells without apamin present (these time controls were tabulated from the experiments depicted in Fig.4). In different cells, the outward currents activated by thrombin (n = 5) or trypsin (n = 5) were blocked by apamin. The ‘+Apamin bars’ show the current density 2 min after addition of thrombin or trypsin and 1 min after addition of apamin. **P < 0.01, ***P < 0.001.
Under current clamp conditions (I = 0), thrombin and trypsin elicited rapid, transient hyperpolarization responses from −12 ± 2.3 mV to −79 ± 3.8 mV and from −14 ± 1.5 mV to −78 ± 3.5 mV, respectively (n = 5 for each drug; Fig.4C and F). Taken together, these results suggest that thrombin and trypsin induce hyperpolarization by activation of the SK conductance that is expressed robustly by PDGFRα+ cells.
The effects of thrombin and trypsin on membrane currents in ICC
Thrombin and trypsin induced depolarization and contractions when the inhibitory mechanism of the SIP syncytium was inhibited by apamin (see Fig.3). Inward currents were never induced in PDGFRα+ cells when the outward current was blocked by apamin. Therefore, we evaluated conductances activated by PAR agonists in ICC and SMCs to determine whether these cells might mediate the excitatory components of PAR responses.
Freshly isolated and positively identified ICC were studied under patch clamp conditions. The average cell capacitance of ICC was 3.0 ± 0.1 pF (n = 10). Spontaneous transient inward currents (STICs) were typically observed in ICC held at −50 mV (K+-rich pipette solution). Thrombin or trypsin increased the amplitude of STICs from 67 ± 24.2 to 324 ± 51.7 pA pF–1 and from 45 ± 14.2 to 287 ± 72.4 pA pF–1, respectively (Fig.6A–C, n = 5 for each drug). The properties of STICs were evaluated using a Cs+-rich pipette solution and setting ECl = −40 mV. Under these conditions, STICs reversed between −30 and −20 mV (before correction of junction potentials, calculated to be 14.6 mV in these experiments), suggesting that the STICs in colonic ICC were due to activation of a Cl− conductance (Fig.6D and E), as previously shown for STICs in ICC of the small intestine (Zhu et al. 2009).
Figure 6.
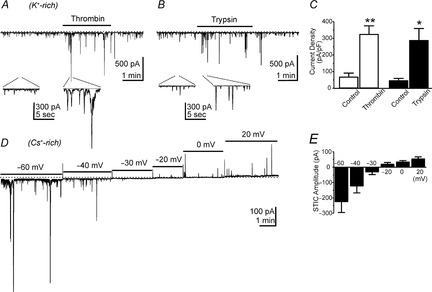
Effects of thrombin and trypsin on interstitial cells of Cajal (ICC)
A and B, representative traces using K+-rich pipette solutions showing enhancement of STICs by thrombin (A) and trypsin (B). Insets under the main traces show selected areas at an expanded sweep speed. Note also that thrombin and trypsin failed to elicit any outward current at −50 mV with K+-rich dialysis of cells. C, summarized data showing increase in average current density in cells after exposure to thrombin (open bars; n = 5) and trypsin (filled bars; n = 5). D shows STICs (using Cs+-rich pipette solutions) at various holding potentials from −60 to +20 mV. E, STICs reversed between −30 and −20 mV (before correction of junction potentials), which approximated the Cl− equilibrium potential in these experiments.
Although expression of gene transcripts for PARs was low in SMCs, we also tested the effects of PAR agonists on isolated colonic SMCs. The average capacitance of SMCs was 38 ± 1.9 pF (n = 22). Thrombin (50 U ml−1) and trypsin (1 μm) evoked small-amplitude, transient inward currents, averaging 1.6 ± 0.3 and 1.3 ± 0.3 pA pF–1 at −50 mV HP, respectively (K+-rich pipette solution; Fig.7A–C; n = 6 for thrombin, n = 5 for trypsin). Similar currents were also activated by PAR agonists at −50 mV HP under conditions with Cs+-rich internal solution (Fig.7D–F, n = 6 for thrombin, n = 5 for trypsin). SMCs have never been shown to manifest resolvable CaCCs, and therefore it is likely that the small inward currents activated in SMCs by thrombin and trypsin were due to activation of non-selective cation channels that are expressed by SMCs (Dwyer et al. 2011). La3+ (10 μm), applied after 1 min exposure to thrombin and trypsin, reduced the inward currents activated by PARs from 1.5 ± 0.3 to 0.5 ± 0.2 pA pF–1 (−50 mV HP; P < 0.05 for currents activated by thrombin) and from 1.3 ± 0.2 to 0.5 ± 0.1 pA pF–1 (−50 mV HP; P < 0.05 for currents activated by trypsin; Fig.8A–C). Pharmacological experiments on intact muscles suggested that a CaCC is responsible for the excitatory responses to PARs. However, we also performed control experiments to test the effects of NPPB on the inward currents activated in SMCs by PARs. The current density of responses to thrombin (1.0 ± 0.1 pA pF–1, n = 4; Fig.8D and F) or trypsin (0.9 ± 0.1 pA pF–1, n = 4; Fig.8E and F) at 2 min exposure after 1 min treatment of NPPB (10 μm) was not different from the effect of a 2 min exposure to thrombin (1.0 ± 0.2 pA pF–1, n = 4; Fig.7D and F) or trypsin (1.0 ± 0.1 pA pF–1, n = 4; Fig.7E and F) alone. Pretreatment of cells with NPPB (10 μm) did not block the inward currents elicited in SMCs by thrombin or trypsin (HP = −50 mV; Fig.8G–I). These data suggest that PAR agonists do not activate CaCCs in SMCs, and NPPB does not block the non-selective cation currents activated in SMCs.
Figure 7.
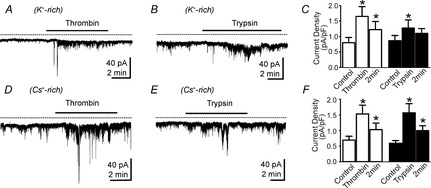
Effects of thrombin and trypsin on smooth muscle cells (SMCs)
A and B, representative traces using K+-rich pipette solutions showing enhancement of noisy inward currents by thrombin (A) and trypsin (B). These responses were of relatively low current density in SMC. Note also that thrombin and trypsin failed to elicit any outward current at −50 mV with K+-rich dialysis of cells. C, summarized data showing increase in average current density in cells after exposure 1 min and 2 min to thrombin (open bars; n = 6) and trypsin (filled bars; n = 5). *P < 0.05 compared with control. D and E show similar responses using Cs+-rich pipette solutions at −50 mV. These experiments demonstrate that the small responses of SMCs are not due to contamination by simultaneous generation of opposing outward currents in these cells. F, summarized data showing increase in average current density in cells after exposure to thrombin (open bars; n = 6) and trypsin (filled bars; n = 5) for 1 min and 2 min. *P < 0.05 compared with control.
Figure 8.
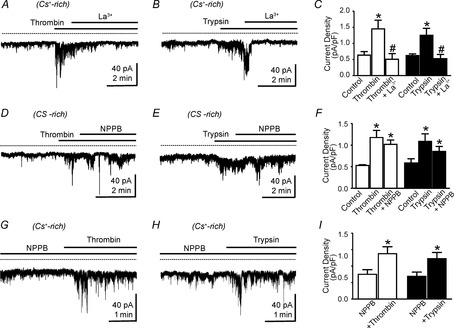
Effects of La3+ and NPPB on thrombin- and trypsin-activated inward currents in smooth muscle cells (SMCs)
A and B, representative traces using Cs+-rich pipette solutions showed that inward currents activated by thrombin (A) and trypsin (B) were blocked by La3+ (10 μm; HP = −50 mV). C, summarized data showing that La3+ decreased the average current density activated by thrombin (n = 4) and trypsin (n = 4). *P < 0.05 compared with control; #P < 0.05 compared with thrombin or trypsin. D and E show effects of NPPB (10 μm) inward currents activated by thrombin (D) and trypsin (E); HP = −50 mV in tests of both PAR agonists. F, summarized data showing increase in average current density after exposure to thrombin (n = 4) and trypsin (n = 4). NPPB had no significant effect on currents activated by either PAR agonist (data were compared with current densities 2 min after addition of thrombin or trypsin in the absence of NPPB; see Fig. 7F). *P < 0.05 compared with control. G and H show thrombin (G) and trypsin (H) effects after pretreatment with NPPB. I, summarized data showing thrombin (n = 4) and trypsin (n = 4) activated inward currents in the presence of NPPB (HP = −50 mV). *P < 0.05 compared with NPPB alone.
Discussion
The findings of this study serve to illustrate an overarching concept that is important in regulation of motor function in the GI tract: unique responses of different cells to bioactive mediators are integrated by the SIP syncytium to modulate smooth muscle excitability and, ultimately, GI motor activity. This is the first study in which the unique responses of SIP cells (SMCs, ICC and PDGFRα+ cells) to any agonist have been compared under the same experimental conditions and related to tissue-level responses. We found that non-neural PAR activation in colonic muscles is complex and mediates responses due to activation of multiple membrane conductances expressed by the different cells of the SIP syncytium. The temporal response to PAR agonists in tissues was biphasic, consisting of an initial inhibitory phase superimposed upon a more extended excitatory phase. Molecular studies of specific classes of SIP cells demonstrated differential expression of PARs in PDGFRα+ cells, ICC and SMCs. F2r (PAR1) and F2r1 (PAR2) were relatively highly expressed in PDGFRα+ cells, and ICC also expressed relatively high levels of F2r (PAR1) in comparison to SMCs. Functional studies measuring contractions of muscle strips, transmembrane potential, and conductances in isolated SIP cells mirrored the expression of PARs: responses were most prominent in PDGFRα+ cells and ICC and less prominent in SMCs. It should be noted, however, that low transcript levels (e.g. F2r1 in ICC and all PAR isoforms in SMCs) were still associated with responses to thrombin and trypsin in these cells. Using thrombin to activate, at a minimum, PAR1 and trypsin to activate PAR2, we found that both classes of receptors coupled to the same types of ionic conductances in SIP cells.
The inhibitory phase of PAR responses in colonic muscles, resulting from stimulation by either PAR1 or PAR2 agonists, is likely to be mediated by Gq/11 signalling (Kawabata et al. 2002; Mule et al. 2002b; Ossovskaya & Bunnett, 2004; Hollenberg, 2005), which typically induces PLCβ activation, increased production of IP3, and Ca2+ release from ER. The inhibitory phase (electrical and mechanical responses) was abolished by apamin, suggesting the involvement of SK channels in these responses. Blockers of large and intermediate conductance Ca2+-activated K+ channels did not affect responses to PAR agonists. Apamin-sensitive SK channels are highly expressed in PDGFRα+ cells in comparison to SMCs in murine colon (Kurahashi et al. 2011, 2012), and SK current density was found to be at least in 100 times greater in PDGFRα+ cells than in SMCs. The outward currents elicited in PDGFRα+ cells by PAR agonists were voltage independent, blocked by apamin, and characterized by current–voltage responses typical of SK currents. Hyperpolarization responses to PAR agonists, leading to muscle relaxation in whole colonic muscles, appeared to be mediated by PDGFRα+ cells, because net outward currents were never activated in ICC or SMCs by PAR agonists. Inward currents were activated in SMCs and ICC by thrombin and trypsin. Thus, we would conclude that the hyperpolarization and relaxation responses induced by PAR activation in colonic muscles are due to activation of SK channels in PDGFRα+ cells.
Depolarization and the excitatory phase of PAR responses appear to be mediated by ICC, because this response was due to activation of a Cl− conductance. ICC express Ano1, which encodes a CaCC that is blocked by NPPB. Ano1 channels are responsible for STICs and the pacemaker activity of ICC (Hwang et al. 2009; Zhu et al. 2011). STICs in ICC are initiated by release of Ca2+ from stores and activation of CaCCs (Zhu et al. 2011). Enhancement in the probability of Ca2+ release, due to an increase in IP3 production by PAR agonists, is likely to be the mechanism by which STICs increased in ICC in response to thrombin and trypsin. Summation of STICs in hundreds of ICC in colonic muscles could produce depolarization responses, as we observed after exposure to PAR agonists in the presence of apamin (Fig.3). The depolarization phase of PAR responses was blocked by NPPB. Colonic SMCs lack Ano1 expression (and other functional CaCCs), but these cells express a variety of non-selective cation channels, some of which are regulated by intracellular Ca2+ (Dwyer et al. 2011). PAR agonists induced small amplitude inward currents in SMCs, but these inward currents were not blocked by NPPB. Thus, contributions from SMCs to the integrated responses of the SIP syncytium to PAR agonists appear to be minor.
At first glance our data may suggest that the inhibitory phase of PAR activation is not a very important regulatory factor in colonic motility because it deactivates or is overcome by an excitatory phase that tends to restore normal contractility. However, in our study PARs were activated in a synchronous manner by adding proteases to the extracellular solution bathing the muscles. The availability of endogenous proteases in vivo may be localized and delivered in a paracrine-like fashion (e.g. such as localized mast-cell degranulation), and therefore PAR responses may lack tissue-wide synchronization. Asymmetrical and asynchronous activation of PARs in PDGFRα+ cells, ICC or SMCs may lead to sustained inhibitory or excitatory influences on the SIP syncytium, and therefore these receptors might modulate basal colonic excitability and responsiveness to normal neural and hormonal inputs. An overlay of such modulation (i.e. a retuning of excitability) could lead to abnormal colonic motility that is not a result of loss or defects in normal neurogenic or myogenic regulatory elements. Thus, an overlay of PAR effects could possibly provide an explanation for some types of ‘functional’ bowel disorders.
This is the first study to examine the specific cellular distribution and responses of non-neural PARs in the tunica muscularis. Thus, we focused our experiments on canonical activation of PARs by thrombin and trypsin. Access to these particular proteases may be rather limited in situ, but a large number of proteases, capable of activating PARs, are widely expressed by cells and tissues (Zhao et al. 2014). Some proteases cleave PARs at different sites than thrombin and trypsin and can couple PAR activation to responses by different second messenger pathways. Whether non-neural PARs of the tunica muscularis are exposed to proteases prone to activate responses different than what we observed with thrombin and trypsin (i.e. biased receptor activation) will be an important question for future investigation. At present little is known about the expression of endogenous proteases by cells of the SIP syncytium or by cells in close proximity to SIP cells. Fibroblasts, to which PDGFRα+ cells have been likened based on morphological criteria, have been shown to produce a variety of matrix metalloproteases (MMPs), and some of these proteases activate PARs (e.g. MMP1, MMP13; Trivedi et al. 2009; Jaffré et al. 2012). The tunica muscularis is rich in resident macrophages, another cell population that expresses proteases (e.g. MMPs, cathepsins; Wynn & Barron, 2010), and macrophages lie in close proximity to SIP cells (Mikkelsen 2010; Rumessen et al. 2011). Activation of macrophages, an important feature of the innate immune response, leads to recruitment and extravasation of leukocytes that also synthesize and secrete proteases (Pham, 2006). Mast cells, which are sparse in muscles of rodents, but present in the tunica muscularis in humans, are also a source of proteases able to activate PARs (Corvera et al. 1999; Molino et al. 1997). At present we are far from understanding the complex milieu that might lead to PAR activation in the SIP syncytium under basal conditions and during responses to pathophysiological conditions and/or tissue regeneration.
Our data suggest that changes in PAR expression in tissues remodelled by pathophysiological processes could impact the excitability of GI muscles. For example, if PAR expression were to decrease in PDGFRα+ cells, then excitatory responses might become dominant. PAR2-mediated relaxation of colonic smooth muscle was reported to be impaired in the dextran sodium sulphate-induced colitis animal model (Sato et al. 2006). It is possible that impairment of PAR-mediated relaxation could be due to remodelling or loss of PDGFRα+ cells. Similarly, ICC are decreased or lost in several motility disorders (He et al. 2000; Nakahara et al. 2002; Farrugia, 2008), and loss of this component might cause dominance of the inhibitory phase of the PAR response. In future studies it will be important to evaluate the expression of PARs in specific classes of SIP cells in disease models.
In conclusion, our results demonstrate how integrated responses of the SIP syncytium can influence the excitability and motor activity of GI muscles. Similar to neurotransmission (Sanders et al. 2010), responses to inflammatory mediators, such as PAR agonists, result in cell specific responses that are integrated by the electrical connectivity between cells of the SIP syncytium to yield tissue and organ level effects. This is the first report demonstrating contributions of PDGFRα+ cells and ICC to the net responses to inflammatory mediators in GI muscles. Our data show that the inhibitory effects of non-neural PAR activation are mediated through activation of SK channels in PDGFRα+ cells, and the excitatory phase of PAR responses is mediated largely by ICC. Responses of different cell types in the SIP syncytium are integrated via electrical coupling between interstitial cells and SMCs. Conductance changes in any of the SIP cells can modulate the gain on smooth muscle excitability, and this retuning of excitability influences myogenic motor activity and responses to other regulatory bioagonists.
Glossary
Abbreviations
- AUC
area under the curve
- CaCC
Ca2+-activated Cl− conductance
- GI
gastrointestinal
- ICC
interstitial cells of Cajal
- KRB
Krebs–Ringer bicarbonate
- PDGFRα
platelet-derived growth factor receptor α
- PAR
protease-activated receptor
- RMP
resting membrane potential
- SIP
SMC–ICC–PDGFRα+ cell (syncytium)
- SK
small conductance Ca2+-activated K+ channel
- SMC
smooth muscle cell
- STICs
spontaneous transient inward currents
- TTX
tetrodotoxin
Additional information
Competing interests
None to declare.
Author contributions
Drs T.S. Sung, H.U. Kim and J.H. Kim equally contributed to this project. Most of the data were collected and analysed by T.S.S., H.U.K. and J.H.K., with the assistance of other authors. Membrane potential studies were performed by H.L. K.M.S. and S.D.K. shared in the design of experiments, interpretation of the data and the writing of the manuscript. All authors approved the final version of the manuscript.
Funding
This study was supported by a program project grant from the NIDDK: P01 DK41315-25.
References
- Barfod ET, Moore AL. Lidofsky SD. Cloning and functional expression of a liver isoform of the small conductance Ca2+-activated K+ channel SK3. Am J Physiol Cell Physiol. 2001;280:C836–C842. doi: 10.1152/ajpcell.2001.280.4.C836. [DOI] [PubMed] [Google Scholar]
- Bohm SK, Kong W, Bromme D, Smeekens SP, Anderson DC, Connolly A, Kahn M, Nelken NA, Coughlin SR, Payan DG. Bunnett NW. Molecular cloning, expression and potential functions of the human proteinase-activated receptor-2. Biochem J. 1996;314:1009–1016. doi: 10.1042/bj3141009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunnett NW. Protease-activated receptors: how proteases signal to cells to cause inflammation and pain. Semin Thromb Hemost. 2006;32:39–48. doi: 10.1055/s-2006-939553. (Suppl 1) [DOI] [PubMed] [Google Scholar]
- Cocks TM, Sozzi V, Moffatt JD. Selemidis S. Protease-activated receptors mediate apamin-sensitive relaxation of mouse and guinea pig gastrointestinal smooth muscle. Gastroenterology. 1999;116:586–592. doi: 10.1016/s0016-5085(99)70180-0. [DOI] [PubMed] [Google Scholar]
- Corvera CU, Déry O, McConalogue K, Böhm SK, Khitin LM, Caughey GH, Payan DG. Bunnett NW. Mast cell tryptase regulates rat colonic myocytes through proteinase-activated receptor2. J Clin Invest. 1997;100:1383–1393. doi: 10.1172/JCI119658. (6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coughlin SR. Thrombin signalling and protease-activated receptors. Nature. 2000;407:258–264. doi: 10.1038/35025229. [DOI] [PubMed] [Google Scholar]
- Coughlin SR. Protease-activated receptors in hemostasis, thrombosis and vascular biology. J Thromb Haemost. 2005;3:1800–1814. doi: 10.1111/j.1538-7836.2005.01377.x. [DOI] [PubMed] [Google Scholar]
- Dwyer L, Rhee PL, Lowe V, Zheng H, Peri L, Ro S, Sanders KM. Koh SD. Basally activated nonselective cation currents regulate the resting membrane potential in human and monkey colonic smooth muscle. Am J Physiol Gastrointest Liver Physiol. 2011;301:G287–G296. doi: 10.1152/ajpgi.00415.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrugia G. Interstitial cells of Cajal in health and disease. Neurogastroenterol Motil. 2008;20(Suppl. 1):54–63. doi: 10.1111/j.1365-2982.2008.01109.x. [DOI] [PubMed] [Google Scholar]
- Ferguson SS. Evolving concepts in G protein-coupled receptor endocytosis: the role in receptor desensitization and signaling. Pharmacol Rev. 2001;53:1–24. [PubMed] [Google Scholar]
- Gomez-Pinilla PJ, Gibbons SJ, Bardsley MR, Lorincz A, Pozo MJ, Pasricha PJ, Van de Rijn M, West RB, Sarr MG, Kendrick ML. Ano1 is a selective marker of interstitial cells of Cajal in the human and mouse gastrointestinal tract. Am J Physiol Gastrointest Liver Physiol. 2009;296:G1370–G1381. doi: 10.1152/ajpgi.00074.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He CL, Burgart L, Wang L, Pemberton J, Young-Fadok T, Szurszewski J. Farrugia G. Decreased interstitial cell of Cajal volume in patients with slow-transit constipation. Gastroenterology. 2000;118:14–21. doi: 10.1016/s0016-5085(00)70409-4. [DOI] [PubMed] [Google Scholar]
- Hollenberg MD. Physiology and pathophysiology of proteinase-activated receptors (PARs): proteinases as hormone-like signal messengers: PARs and more. J Pharmacol Sci. 2005;97:8–13. doi: 10.1254/jphs.fmj04005x2. [DOI] [PubMed] [Google Scholar]
- Hung DT, Wong YH, Vu TK. Coughlin SR. The cloned platelet thrombin receptor couples to at least two distinct effectors to stimulate phosphoinositide hydrolysis and inhibit adenylyl cyclase. J Biol Chem. 1992;267:20831–20834. [PubMed] [Google Scholar]
- Hwang SJ, Blair PJ, Britton FC, O'Driscoll KE, Hennig G, Bayguinov YR, Rock JR, Harfe BD, Sanders KM. Ward SM. Expression of anoctamin 1/TMEM16A by interstitial cells of Cajal is fundamental for slow wave activity in gastrointestinal muscles. J Physiol. 2009;587:4887–4904. doi: 10.1113/jphysiol.2009.176198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara H, Connolly AJ, Zeng D, Kahn ML, Zheng YW, Timmons C, Tram T. Coughlin SR. Protease-activated receptor 3 is a second thrombin receptor in humans. Nature. 1997;386:502–506. doi: 10.1038/386502a0. [DOI] [PubMed] [Google Scholar]
- Jaffré F, Friedman AE, Hu Z, Mackman N. Blaxall BC. ß-Adrenergic receptor stimulation transactivates protease-activated receptor1 via matrix metalloproteinase13 in cardiac cells. Circulation. 2012;125:2993–3003. doi: 10.1161/CIRCULATIONAHA.111.066787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabata A. Gastrointestinal functions of proteinase-activated receptors. Life Sci. 2003;74:247–254. doi: 10.1016/j.lfs.2003.09.011. [DOI] [PubMed] [Google Scholar]
- Kawabata A, Kuroda R, Nishida M, Nagata N, Sakaguchi Y, Kawao N, Nishikawa H, Arizono N. Kawai K. Protease-activated receptor-2 (PAR-2) in the pancreas and parotid gland: Immunolocalization and involvement of nitric oxide in the evoked amylase secretion. Life Sci. 2002;71:2435–2446. doi: 10.1016/s0024-3205(02)02044-1. [DOI] [PubMed] [Google Scholar]
- Kawabata A, Saifeddine M, Al-Ani B, Leblond L. Hollenberg MD. Evaluation of proteinase-activated receptor-1 (PAR1) agonists and antagonists using a cultured cell receptor desensitization assay: activation of PAR2 by PAR1-targeted ligands. J Pharmacol Exp Ther. 1999;288:358–370. [PubMed] [Google Scholar]
- Kurahashi M, Nakano Y, Hennig GW, Ward SM. Sanders KM. Platelet-derived growth factor receptor alpha-positive cells in the tunica muscularis of human colon. J Cell Mol Med. 2012;16:1397–1404. doi: 10.1111/j.1582-4934.2011.01510.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurahashi M, Zheng H, Dwyer L, Ward SM, Koh SD. Sanders KM. A functional role for the ‘fibroblast-like cells’ in gastrointestinal smooth muscles. J Physiol. 2011;589:697–710. doi: 10.1113/jphysiol.2010.201129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macfarlane SR, Seatter MJ, Kanke T, Hunter GD. Plevin R. Proteinase-activated receptors. Pharmacol Rev. 2001;53:245–282. [PubMed] [Google Scholar]
- Mikkelsen HB. Interstitial cells of Cajal, macrophages and mast cells in the gut musculature: morphology, distribution, spatial and possible functional interactions. J Cell Mol Med. 2010;14:818–832. doi: 10.1111/j.1582-4934.2010.01025.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molino M, Barnathan ES, Numerof R, Clark J, Dreyer M, Cumashi A, Hoxie JA, Schechter N, Woolkalis M. Brass LF. Interactions of mast cell tryptase with thrombin receptors and PAR-2. J Biol Chem. 1997;272:4043–4049. doi: 10.1074/jbc.272.7.4043. [DOI] [PubMed] [Google Scholar]
- Mule F, Baffi MC, Capparelli A. Pizzuti R. Involvement of nitric oxide and tachykinins in the effects induced by protease-activated receptors in rat colon longitudinal muscle. Br J Pharmacol. 2003;139:598–604. doi: 10.1038/sj.bjp.0705273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mule F, Baffi MC. Cerra MC. Dual effect mediated by protease-activated receptors on the mechanical activity of rat colon. Br J Pharmacol. 2002a;136:367–374. doi: 10.1038/sj.bjp.0704746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mule F, Baffi MC, Falzone M. Cerra MC. Signal transduction pathways involved in the mechanical responses to protease-activated receptors in rat colon. J Pharmacol Exp Ther. 2002b;303:1265–1272. doi: 10.1124/jpet.102.041301. [DOI] [PubMed] [Google Scholar]
- Nakahara M, Isozaki K, Hirota S, Vanderwinden JM, Takakura R, Kinoshita K, Miyagawa J, Chen H, Miyazaki Y, Kiyohara T, et al. Deficiency of KIT-positive cells in the colon of patients with diabetes mellitus. J Gastroenterol Hepatol. 2002;17:666–670. doi: 10.1046/j.1440-1746.2002.02756.x. [DOI] [PubMed] [Google Scholar]
- Nystedt S, Emilsson K, Wahlestedt C. Sundelin J. Molecular cloning of a potential proteinase activated receptor. Proc Natl Acad Sci U S A. 1994;91:9208–9212. doi: 10.1073/pnas.91.20.9208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossovskaya VS. Bunnett NW. Protease-activated receptors: contribution to physiology and disease. Physiol Rev. 2004;84:579–621. doi: 10.1152/physrev.00028.2003. [DOI] [PubMed] [Google Scholar]
- Pham CT. Neutrophil serine proteases: specific regulators of inflammation. Nat Rev Immunol. 2006;6:541–550. doi: 10.1038/nri1841. [DOI] [PubMed] [Google Scholar]
- Rumessen JJ, Vanderwinden JM. Horn T. Crohn's disease: ultrastructure of interstitial cells in colonic myenteric plexus. Cell Tissue Res. 2011;344:471–479. doi: 10.1007/s00441-011-1175-9. [DOI] [PubMed] [Google Scholar]
- Sanders KM, Hwang SJ. Ward SM. Neuroeffector apparatus in gastrointestinal smooth muscle organs. J Physiol. 2010;588:4621–4639. doi: 10.1113/jphysiol.2010.196030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders KM, Koh SD, Ro S. Ward SM. Regulation of gastrointestinal motility – insights from smooth muscle biology. Nat Rev Gastroenterol Hepatol. 2012;9:633–645. doi: 10.1038/nrgastro.2012.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K, Ninomiya H, Ohkura S, Ozaki H. Nasu T. Impairment of PAR-2-mediated relaxation system in colonic smooth muscle after intestinal inflammation. Br J Pharmacol. 2006;148:200–207. doi: 10.1038/sj.bjp.0706717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiguchi F, Hasegawa N, Inoshita K, Yonezawa D, Inoi N, Kanke T, Saito N. Kawabata A. Mechanisms for modulation of mouse gastrointestinal motility by proteinase-activated receptor (PAR)-1 and -2 in vitro. Life Sci. 2006;78:950–957. doi: 10.1016/j.lfs.2005.06.035. [DOI] [PubMed] [Google Scholar]
- Traynelis SF. Trejo J. Protease-activated receptor signaling: new roles and regulatory mechanisms. Curr Opin Hematol. 2007;14:230–235. doi: 10.1097/MOH.0b013e3280dce568. [DOI] [PubMed] [Google Scholar]
- Trivedi V, Boire A, Tchernychev B, Kaneider NC, Leger AJ, O'Callaghan K, Covic L. Kuliopulos A. Platelet matrix metalloprotease-1 mediates thrombogenesis by activating PAR1 at a cryptic ligand site. Cell. 2009;137:332–343. doi: 10.1016/j.cell.2009.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu TK, Hung DT, Wheaton VI. Coughlin SR. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell. 1991a;64:1057–1068. doi: 10.1016/0092-8674(91)90261-v. [DOI] [PubMed] [Google Scholar]
- Vu TK, Wheaton VI, Hung DT, Charo I. Coughlin SR. Domains specifying thrombin-receptor interaction. Nature. 1991b;353:674–677. doi: 10.1038/353674a0. [DOI] [PubMed] [Google Scholar]
- Wynn TA. Barron L. Macrophages: master regulators of inflammation and fibrosis. Semin Liver Dis. 2010;30:245–257. doi: 10.1055/s-0030-1255354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu WF, Andersen H, Whitmore TE, Presnell SR, Yee DP, Ching A, Gilbert T, Davie EW. Foster DC. Cloning and characterization of human protease-activated receptor 4. Proc Natl Acad Sci U S A. 1998;95:6642–6646. doi: 10.1073/pnas.95.12.6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao P, Metcalf M. Bunnett NW. Biased signaling of protease-activated receptors. Front Endocrinol (Lausanne) 2014;5:1–16. doi: 10.3389/fendo.2014.00067. (67) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao A. Shea-Donohue T. PAR-2 agonists induce contraction of murine small intestine through neurokinin receptors. Am J Physiol Gastrointest Liver Physiol. 2003;285:G696–703. doi: 10.1152/ajpgi.00064.2003. [DOI] [PubMed] [Google Scholar]
- Zheng H, Park KS, Koh SD. Sanders KM. Expression and function of a T-type Ca2+ conductance in interstitial cells of Cajal of the murine small intestine. Am J Physiol Cell Physiol. 2014;306:C705–C713. doi: 10.1152/ajpcell.00390.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu MH, Kim TW, Ro S, Yan W, Ward SM, Koh SD. Sanders KM. A Ca2+-activated Cl– conductance in interstitial cells of Cajal linked to slow wave currents and pacemaker activity. J Physiol. 2009;587:4905–4918. doi: 10.1113/jphysiol.2009.176206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu MH, Sung IK, Zheng H, Sung TS, Britton FC, O'Driscoll K, Koh SD. Sanders KM. Muscarinic activation of Ca2+-activated Cl– current in interstitial cells of Cajal. J Physiol. 2011;589:4565–4582. doi: 10.1113/jphysiol.2011.211094. [DOI] [PMC free article] [PubMed] [Google Scholar]