

Abstract
Influenza viruses present a significant health challenge each year, as in the H3N2 epidemic of 2012-2013. Here, we describe an antibody, F045-092, that possesses broadly neutralizing activity against the entire H3 subtype and accommodates the natural variation and additional glycosylation in all strains tested from 1963 to 2011. Crystal structures of F045-092 in complex with HAs from 1975 and 2011 H3N2 viruses reveal the structural basis for its neutralization breadth through insertion of its 23-residue HCDR3 into the receptor-binding site that involves striking receptor mimicry. F045-092 extends its recognition to divergent subtypes, including H1, H2, and H13, using the enhanced avidity of its IgG to overcome lower affinity Fab binding, as observed with other receptor-binding site antibodies. This unprecedented level of antibody cross-reactivity against the H3 subtype can potentially inform on development of a pan-H3 vaccine or small molecule therapeutics.
Influenza virus infects millions annually and causes severe illness and economic distress. The constant threat of a novel pandemic influenza virus looms large as with the emergence in 2009 of the H1N1 swine flu that antigenically closely resembled the 1918 H1N1 Spanish flu, which resulted in death of around 50 million people worldwide. Fortunately, the novel 2009 H1N1 virus was much less severe and subsequently became the circulating seasonal H1 strain. The H3N2 virus in 2012-2013 was more virulent than usual and caused epidemics in the northeastern United States1. The unpredictable epidemic and pandemic potential of these viruses underscores the need for broad-spectrum therapy. To combat flu infections, small molecules are available that target the neuraminidase surface glycoprotein on influenza virus, but the effectiveness of these drugs is diminishing2,3 and underscores the need for new classes of drugs. Vaccine therapy remains the best means as a preventative countermeasure from flu infections. However, vaccine strains must be reformulated almost every year to stay ahead of the ever-changing virus and may not provide protection if they are an inadequate match to the dominant circulating virus.
Much of the challenge in raising a broadly neutralizing antibody response against influenza by a universal flu vaccine arises from the diversity and hypervariability of the hemagglutinin (HA) surface glycoprotein. There are currently 18 identified HA subtypes (H1 to H18) of influenza A viruses, where aquatic birds act as the primary reservoir. Among these, only the H1 and H3 subtypes currently circulate in humans although sporadic cases of H5, H7, including the most recent H7N9 outbreak4,5, and H9 have caused deaths and severe illness. Thus, emerging viruses are carefully monitored for any propensity to acquire human-to-human transmission that would increase the potential to cause a human pandemic. Despite extensive sequence diversity across HA subtypes, viral attachment to terminal sialic acid glycans on host cells is a conserved function of the HA that initiates viral infection6,7, although the bat H17 and H18 HAs appear to be exceptions8-10. The HA interaction with sialosides is very weak (Kd ~mM) and the virus is thus reliant on multivalency to achieve high enough affinity for productive receptor binding and subsequent receptor-mediated endocytosis.
HA is synthesized as a single polypeptide precursor (HA0) that assembles into trimers, which mature after proteolytic cleavage by host proteases between the HA1 and HA2 subunits to produce a pH-dependent metastable intermediate. In structural and functional terms, HA can be divided into a membrane-proximal “stem”, which constitutes the core fusion machinery that is triggered by low pH in the endosomes11, and a membrane-distal “head”, which contains the receptor-binding site (three per HA trimer). The receptor-binding site is a broad and shallow pocket located at the apex of the HA head and its outer ridges are formed primarily by four loops named after their locations in the HA sequence: the 130 loop, 150 loop, 190 helix, and 220 loop.
Blocking HA-receptor interactions can prevent infection by influenza virus. However, despite the functional conservation of receptor binding, the design of entry inhibitors to the HA still remains a significant challenge due to the shallowness of the receptor-binding pocket and sequence divergence within and across HA subtypes. Nevertheless, a number of HA receptor-binding site targeted antibodies have been structurally characterized that have broader-spectrum activity against a single subtype12-15 or across multiple subtypes16,17, thus suggesting that the receptor-binding pocket is indeed a site of vulnerability on HA18. The breadth of recognition of these antibodies is surprising as it was popularly believed until recently that the HA head could only elicit strain-specific antibodies. All of these antibodies commonly insert a single complementarity-determining region (CDR) loop (HCDR2 or HCDR3) into the receptor-binding site to prevent viral-host recognition; however, due to the larger footprints of antibodies in comparison to the sialic acid receptor, they invariably contact variable residues on the outer edges of the receptor-binding pocket and, hence, have limited breadth.
Here, we describe crystal structures of antibody F045-092 bound to divergent HAs from the 1975 and 2011 H3 subtype. The antibody was selected from a phage-displayed library, derived from human B lymphocytes, and was reported to have activity against a panel of H3 strains from 1968 to 2004 and possesses neutralizing activity against all H3N2 viruses that were tested within this time span as well as certain H1N1, H2N2, and H5N1 viruses19. The antibody contacts a minimal epitope in the HA receptor-binding site primarily by insertion of its 23-residue HCDR3 and achieves exceptional receptor mimicry. Full-coverage of an entire subtype by interaction with the receptor-binding site is of great significance and the structural information from the complexes may potentially advance the design of novel small molecule inhibitors as well as inform on the formulation of immunogens as vaccine candidates against H3 viruses.
Results
Binding affinity of F045-092 against HAs
To further profile its breadth, binding of F045-092 Fab and IgG was measured against a panel of HAs that encompass representative strains from nearly all HA subtypes by bio-layer interferometry (Table 1 and Supplementary Table 1). F045-092 has heterosubtypic activity and binds HAs from from the H1, H2, H3, and H13 subtypes. Most notably, F045-092 Fab binds all 13 representative HAs from strains from the H3 subtype that span five decades from 1963 to 2011, including the 2012-2013 H3 vaccine strain A/Victoria/361/2011 (Vic2011/H3). Although the HAs of some strains are strongly bound by the Fab (Kd ~5 nM to 50 nM), others have only moderate to weak binding (Kd ~50 nM to 5 μM), some of which are outside the threshold thought to be necessary for effective neutralization (Kd <250 nM)17. However, the bivalent F045-092 IgG shows increased affinity through avidity (Kd <20 nM) that significantly improves F045-092 binding to all strains compared to the monovalent Fab (Table 1). These IgG binding data are consistent with the reported neutralization data19. Avidity has also been observed, and now appears to be a common feature, for other receptor-binding site targeted antibodies16,17 as the close packing and high density of HA trimers on viruses20 likely promotes and facilitates bivalent binding by IgG. Moreover, binding of F045-092 was tested against strains beyond the years that were tested for neutralization (1968 to 2004)19. Despite further antigenic variation, the more recent strains (2007-2011) are bound with high affinity by IgG, indicating that F045-092 may truly be a pan-H3 antibody.
Table 1.
Binding breadth of F045-092 Fab and IgG to influenza HA subtypes and strains.
| Subtype | Strain | Binding | Neutralization* | |
|---|---|---|---|---|
| F045-092 Fab | F045-092 IgG | F045-092 IgG | ||
| H1N1 | A/Beijing/262/1995 | + | +++ | NT |
| H1N1 | A/New Caledonia/20/1999 | + | +++ | +++ |
| H2N2 | A/Adachi/2/1957 | + | +++ | NT |
| H2N2 | A/Okuda/1957 | NT | NT | +++ |
| H3N8 | A/duck/Ukraine/1/1963 | + | +++ | NT |
| H3N2 | A/Aichi/2/1968 | NT | NT | ++++ |
| H3N2 | A/Hong Kong/1/1968 | +++ | ++++ | NT |
| H3N2 | A/Victoria/3/1975 | +++ | ++++ | NT |
| H3N2 | A/Bangkok/1/1979 | ++ | ++++ | NT |
| H3N2 | A/Leningrad/360/1986 | +++ | ++++ | NT |
| H3N2 | A/Beijing/353/1989 | ++ | ++++ | NT |
| H3N2 | A/Kitakyushu/159/1993 | NT | NT | ++++ |
| H3N2 | A/Shangdong/9/1993 | ++++ | ++++ | NT |
| H3N2 | A/Panama/2007/1999 | +++ | ++++ | NT |
| H3N2 | A/Moscow/10/1999 | ++++ | ++++ | NT |
| H3N2 | A/Wyoming/3/2003 | + | +++ | NT |
| H3N2 | A/Brisbane/10/2007 | ++ | ++++ | NT |
| H3N2 | A/Perth/16/2009 | ++ | ++++ | NT |
| H3N2 | A/Victoria/361/2011 | ++ | ++++ | NT |
| H13N6 | A/gull/Maryland/704/1977 | + | +++ | NT |
Virus neutralizing activities adapted from Ohshima et al.19
Dissociation constants:
NT, not tested
+ Kd = 500–5000 nM
++ Kd = 50–500 nM
+++ Kd = 5–50 nM or IC50 < 5 μg/mL
++++ Kd ≤ 5 nM or IC50 < 0.05 μg/mL
Crystal structures of the F045-092-HA complexes
To decipher the structural basis for this pan-H3 recognition by F045-092, crystal structures of the F045-092 Fab in complex with the HAs from the A/Victoria/3/1975 (Vic1975/H3) and A/Victoria/361/2011 (Vic2011/H3) viruses were determined at 2.75 Å and 6.50 Å resolution (Table 2). In addition, crystal structures of the F045-092 Fab and the unliganded Vic2011/H3 HA were determined at 1.50 Å and 1.75 Å resolution (Table 2), respectively, and served as high-resolution starting models for refinement of the Fab-HA complexes. In the crystal asymmetric unit, one copy of the Fab is bound to one HA protomer of the HA trimer in the F045-092-Vic1975/H3 complex. The HA protomer and the Fab variable domain are well ordered, as judged from the electron density, whereas the Fab constant domain appears to be disordered, as frequently found in other Fab-HA complexes14,17,21-23 due to flexibility of the elbow region connecting the Fab variable and constant domains24. The asymmetric unit of the F045-092-Vic2011/H3 complex contains two HA trimers bound by six copies of the Fab. Despite the moderate resolution, the HA and Fab molecules can be clearly observed in the electron density maps (Supplementary Fig. 1). All six Fab variable domains similarly recognize HA, but have a ~14° rotation in approach angle on average in comparison to the F045-092-Vic1975/H3 complex. No major conformational changes were observed between the apo and bound Fab in either complex. There is also good structural agreement between the HAs from the Vic1975/H3 and Vic2011/H3 complexes (Cα RMSD of ~0.4 Å) despite their divergence over 36 years, which amounted to 62 amino-acid mutations in the HA ectodomain.
Table 2.
Data collection and refinement statistics.
| F045-092- Vic1975/H3 complex |
F045-092- Vic2011/H3 complex |
F045-092 Fab | Vic2011/H3 HA | |
|---|---|---|---|---|
| Data collection | ||||
| Space group | P321 | C2 | P212121 | H32 |
| Cell dimensions □ □ | ||||
| a, b, c (Å) | 99.1, 99.1 336.7 | 318.1, 187.2, 353.6 | 69.5, 77.3 81.5 | 100.7, 100.7, 383.3 |
| □□□□ α, β, γ (°) | 90, 90, 120 | 90, 90.5, 90 | 90, 90, 90 | 90, 90, 120 |
| Resolution (Å) | 50–2.75 | 50–6.50 | 50–1.50 | 50–1.75 |
| (2.92–2.75)* | (6.89–6.50) | (1.60–1.50) | (1.85–1.75) | |
| R merge | 0.10 (0.71) | 0.07 (0.50) | 0.04 (0.51) | 0.07 (0.89) |
| I / σI | 13.3 (2.3) | 10.8 (2.1) | 22.7 (2.3) | 21.2 (2.4) |
| Completeness (%) | 99.5 (97.7) | 92.8 (91.7) | 96.8 (83.4) | 99.8 (99.0) |
| Redundancy | 6.1 (6.1) | 2.0 (2.0) | 5.6 (4.4) | 8.9 (8.6) |
| Refinement | ||||
| Resolution (Å) | 49.0–2.75 | 49.0–6.50 | 43.6–1.50 | 46.8–1.75 |
| No. of reflections | 50,811 | 38,102 | 67,979 | 75,930 |
| Rwork / Rfree | 17.7 / 22.4 | 20.1 / 24.8 | 17.5 / 20.7 | 16.5 / 18.5 |
| No. of atoms | ||||
| Protein | 7,078 | 42,816 | 3,436 | 3,929 |
| Glycan/ligand/ion | 236 | 1,489 | 5 | 178 |
| Water | 76 | 0 | 457 | 426 |
| B-factors | ||||
| Protein | 57 | 145 | 26 | 34 |
| Glycan/ligand/ion | 86 | 168 | 73 | 69 |
| Water | 37 | – | 34 | 37 |
| R.m.s. deviations | ||||
| Bond lengths (Å) | 0.010 | 0.006 | 0.010 | 0.009 |
| Bond angles (°) | 1.25 | 1.22 | 1.26 | 1.18 |
| PDB code | 4O58 | 4O5I | 4O5L | 4O5N |
Values in parentheses are for highest-resolution shell.
The F045-092 Fab binds near the apex of HA to a single protomer of the HA trimer (Fig. 1) and contacts portions of antigenic sites A, B, and D25. The interactions are mediated only through the heavy chain using all three of its CDR loops, whereas the light chain is positioned ~14 Å away from the HA main chain and only comes within ~10 Å of a conserved glycan at Asn165 from a neighboring HA protomer in the trimer (Fig. 1c). Indeed, the heavy chain CDRs form a ridge and protrude much further from the Fab core than the CDRs from the light chain (Supplementary Fig. 2). F045-092 reaches into the receptor-binding site through insertion of its long 23-residue HCDR3, which notably contains an intrachain disulfide separated by only two amino acids (Fig. 2a). This unusual disulfide in HCDR3 has only been seen in only a few other antibody structures26-28. As the HCDR3 and sialoglycan receptor bind to the same pocket (Fig. 2b), the F045-092 antibody is able to block viral-host interactions. Only ~970 Å2 of total molecular surface is buried in the F045-092-Vic1975/H3 interaction, divided evenly between HA and the Fab, and is the smallest footprint for any HA-antibody complex to date (Supplementary Table 2).
Figure 1. Antibody F045-092 binds the influenza HA receptor-binding site.

Crystal structures of F045-092 Fab in complex with (a) Vic1975/H3 HA and (b) Vic2011/H3 HA. One Fab-HA protomer is colored with HA1 in light blue, HA2 in green, the Fab heavy chain in purple, and the Fab light chain in pink. Glycans are shown as spheres with carbon in yellow, oxygen in red, and nitrogen in blue. (c) F045-092 binds HA using only its heavy chain and inserts its HCDR3 into the HA receptor-binding site. The HCDR3 contains a disulfide bridge, which is shown in yellow sticks.
Figure 2. Receptor-binding site recognition by F045-092 to Vic1975/H3 in comparison to the α2,6 sialoglycan receptor.
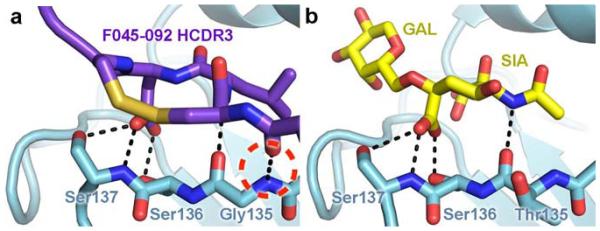
(a) F045-092 inserts its HCDR3 into the HA receptor-binding site. The carboxylate side chain of F045-092 Asp100e overlaps with the (b) carboxylate moiety of the sialic acid of the α2,6 sialoglycan receptor (PDB 2YP4) and uses identical hydrogen bonding interactions, which are shown as black dashed lines. An additional hydrogen bond between the main chains of F045-092 and HA is circled in red dashed lines. The Leu100d side chain of F045-092 overlaps with the acetamide moiety of sialic acid.
The VH1-69 germline is well known for producing heterosubtypic HA antibodies, which use a hydrophobic HCDR2 loop to target the stem of group 1 HAs21,26. However, other VH1-69 germline-encoded antibodies have been shown to insert their HCDR2 into the receptor-binding site14. F045-092 is also a VH1-69 germline-encoded antibody but primarily uses its HCDR3, which comprises 58% of the total surface area contributed by the Fab. The hydrophobic residues at the tip of HCDR2 (somatic Phe53 and germline Phe54) that are the hallmark of the VH1-69 family surprisingly only make minimal contacts with the outer ridges of the 150 loop and the 190 helix of HA.
Sequence analysis of the F045-092 epitope
To delineate the F045-092 binding determinants, the sequence of the HA epitope was analyzed. F045-092 contacts a minimal epitope in and around the conserved receptor-binding site (Fig. 3a). Although F045-092 does indeed contact absolutely conserved residues involved in receptor recognition, the antibody also contacts some variable residues outside the receptor-binding site (Fig. 3b). The F045-092 epitope on the HA is quite variable across all human H3 strains and, at first glance, broad-spectrum recognition to a site with this relatively poor sequence conservation, apart from a handful of key residues that bind sialic acid, would not have been predicted as compared with the near absolute conservation of the HA stem-targeted epitopes21,22,26,29-32 (Fig. 3c and Supplementary Table 3). Yet, F045-092 can tolerate natural variation in H3 strains that span from 1963 to 2011 (Table 1). Furthermore, the current 2013-2014 H3 vaccine strain (A/Texas/50/2012) is antigenically similar to Vic2011/H3 and has an identical F045-092 epitope.
Figure 3. Sequence conservation of the F045-092 epitope across human H3 viruses.
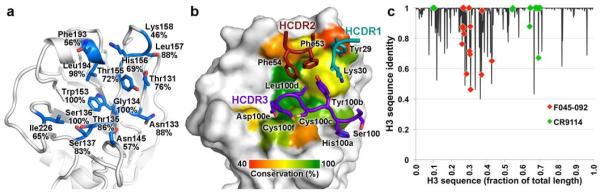
(a) The F045-092 contact residues on HA are depicted as sticks. The percent conservation for the most common residue found in the H3 subtype is labeled, which is not always identical to the residue at that position in the Vic1975/H3 sequence. (b) Interaction of F045-092 with Vic1975/H3 illustrating the sequence conservation of neutralizing epitope on the HA surface. For identification of the HA residues and positions that are color coded on the surface, see (a). The F045-092 contact residues are labeled, colored by HCDR, and shown as sticks. (c) Percent identity of the most common residue among human H3 isolates plotted against the linear sequence of HA. Residues contacted by F045-092 or CR9114 are indicated by red or green diamonds, respectively. Although F045-092 contacts more hypervariable residues than CR9114, it can still broadly neutralize the H3 subtype.
Among the four loops that form the receptor-binding pocket, only the 150 loop does not contact the sialoglycan receptor and, therefore, it possesses high sequence variability as it is not functionally constrained for receptor recognition. Notwithstanding, the 150 loop contributes 40% of the total HA buried surface area in the F045-092-HA complex. However, the antibody appears to make non-specific interactions with the side chains of variable residues of HA such as with residues 156 and 158 that have a sequence conservation of 69% and 46% across all human H3 viruses, respectively (Supplementary Fig. 3). Interactions with these and other variable residues appear to be permissive and do not create destabilizing clashes with F045-092, as the antibody is able to tolerate natural variation across decades of antigenic drift (Table 1).
In addition to the sequence variability in and around the receptor-binding site, insertions and deletions at or near the receptor-binding site loops can alter the conformations of the loops to evade immune recognition. In particular, the conformation of the 130 loop is important for F045-092 recognition as the antibody contacts this loop primarily through main-chain interactions and, thus, is not restricted by the natural variation of the residues in this loop in the H3 subtype. For instance, the 133a insertion (between residues 133 and 134) observed in ~78% of human H1 strains (observed in the 1918 and 2009 pandemic viruses) and in ~78% of human H5 strains, produces a bulge in the 130 loop that protrudes from the receptor-binding site and would clash with F045-092 and other antibodies12,16,17. Notably, the H1 and H2 isolates recognized by F045-092 do not contain the 133a insertion. Also, a conserved two amino-acid insertion is present in the 150 loop of all H7 strains that likely will sterically prevent F045-092 binding. Although these idiosyncratic structural features of the receptor-binding loops limit the recognition of these particular strains and subtypes by F045-092 (Supplementary Table 1), no insertions or deletions exist for any H3 strains suggesting that sequence variation alone dictates immune evasion in the H3 subtype.
HA glycan evasion
In addition to continual antigenic drift, influenza HA can evade immune detection by masking neutralizing sites with glycans33-37. Within the H3 subtype, five conserved glycans exist at HA positions 22, 38, 165, and 285 on the HA1 subunit as well as 154 on HA2. Over time, H3 strains have incorporated and generally have retained several additional, potential N-linked glycosylation sites (PNGs) on the HA head (Fig. 4a). For example, Vic1975/H3 has seven PNGs per HA protomer, whereas Vic2011/H3 has these same seven and five additional PNGs in the HA head. This finding suggests that addition of glycans to H3 HAs is accretive, similar to the continual selection of variants in the HA amino-acid sequence by antigenic drift38. The added PNGs do not appear to occlude receptor binding but decorate the regions surrounding the receptor-binding site. Nevertheless, F045-092 evades these glycans to contact the HA protein surface. Most notably, a PNG in the 130 loop at Asn133 appeared in H3 viruses in 1996 and has been retained in successive viruses. As seen in the Vic2011/H3 complex, the glycan at this site is directed away from the receptor-binding site and does not interfere with F045-092 binding as the antibody slides in between the glycan and the HA 140 loop (Fig. 4b). Accordingly, the A/Panama/2007/1999 (H3N2) and A/Moscow/10/1999 (H3N2) strains, which have a PNG at Asn133, are both bound with very high affinity by the F045-092 Fab (Kd <20 nM). In addition, a PNG at Asn144 began to emerge in 1996, but no corresponding density for carbohydrate was observed in the Vic2011/H3 complex; however, the Asn side chain is directed away from the combining site (Fig. 4b) and suggests that potential glycosylation at this site would not obstruct F045-092 recognition, which is substantiated by the binding data.
Figure 4. Emergence of predicted N-linked glycans (PNGs) across human H3 viruses and evasion by F045-092.

(a) Stacked graph showing the normalized percentage of strains that contain an additional PNG on HA1 at each indicated position. Seven PNGs have been incorporated in the HA since 1968 in addition to the five absolutely conserved PNGs in H3 strains (HA1 Asn22, Asn38, Asn165, Asn285; HA2 Asn154), except for the PNG at Asn81 (1968-1977). (b) F045-092 evades the glycan at Asn133 proximal to the receptor-binding site in the Vic2011/H3 complex. The glycan is shown as spheres, with carbon in yellow, oxygen in red, and nitrogen in blue. No density corresponding to the PNG at Asn144 (shown as sticks) was observed.
F045-092 receptor mimicry
The F045-092 HCDR3 adopts a protruding, blunt–ended conformation, which inserts into the receptor-binding site. The inserted residues include a disulfide loop consisting of residues Cys100c, Leu100d, Asp100e, and Cys100f, which form a type I-like β turn. Remarkably, the side chains of the two central residues of the disulfide loop overlap with and chemically resemble sialic acid of the sialoglycan receptor (Fig. 2). In particular, the isobutyl side chain of Leu100d aligns with the acetamide of sialic acid and similarly enters the hydrophobic pocket formed by the nearly universally conserved HA residues Trp153 and Leu194. Moreover, the carboxylate side chain of Asp100e closely aligns with the carboxylate of sialic acid and uses the same network of hydrogen bonds, including Ser136, which was predicted to contribute to antibody recognition19. Furthermore, the F045-092 HCDR3 curls over the 130 loop and creates an additional hydrogen bond between the Fab main chain at Tyr100b and HA1 Gly135 (Fig. 2).
The importance of the HCDR3 disulfide loop was investigated by alanine scanning mutagenesis. Individually mutating either Leu100d or Asp100e to alanine or eliminating the disulfide loop abolishes binding by F045-092 Fab to nearly every H3 strain tested (Supplementary Table 4). These results suggest the identities of these four residues are the key determinants for F045-092 recognition of the H3 HA.
Alanine mutagenesis of His100a, which is positioned in the groove between the 130 and 140 loops, does not have any major effect on the affinity of F045-092 to HA (Supplementary Table 4). Its side chain is directed away from the HA and evades the glycan at Asn133 in the Vic2011/H3 complex (Fig. 4b). The residues that form and surround the vicinity of this groove, which is adjacent to the receptor-binding pocket, are highly conserved among H3 isolates (Supplementary Fig. 4). This cleft may then be a potential secondary hotspot for the expansion of receptor-binding site directed small molecules or engineered proteins.
Discussion
Influenza H3N2 viruses have circulated in humans ever since the 1968 Hong Kong pandemic. In the span of nearly five decades, the H3 HAs have diverged by antigenic drift so that the current H3 strain has only ~80% sequence identity to the 1968 H3 strain, with the majority of that variation focused in the HA head. In addition, glycans are periodically incorporated and retained as an immune evasion strategy employed by H3 viruses to shield antigenic sites33,34. The extent of the additional masking of the HA surface by glycans from 1968 to present can be clearly visualized in the Vic2011/H3 HA structure (Supplementary Fig. 5). The crystal structures of heterosubtypic, pan-H3 antibody F045-092 in complex with the HAs from 1975 and 2011 H3N2 viruses reveal how the antibody can broadly neutralize the H3 subtype through insertion of a long HCDR3 loop into the HA receptor-binding site. Although F045-092 does not make specific contacts with the HA glycans around the receptor-binding site but rather avoids them, its mode of binding to HA resembles to some extent the exceptionally long HCDR3 loops of many anti-HIV antibodies, especially those that use their HCDR3 loops to reach into recessed sites or penetrate through the glycan shield of HIV-1 Env to contact the protein surface below39-42.
The principal contacts of F045-092 to the HA are via HCDR3, which contains a disulfide loop. The antibody binds a minimal HA epitope that is located in and around the receptor-binding site and has the smallest footprint on HA to date (Supplementary Table 2). The HCDR3 Asp100e and Leu100d, which are between the Cys residues (Cys100c, Cys100f) that form the disulfide loop, mimic the hydrogen-bonding network of the carboxylate and the hydrophobic acetamide moiety of the endogenous sialoglycan receptor, respectively, and are critical for antibody recognition to the HA. Furthermore, an additional hydrogen bond is formed between the main chains of F045-092 and the HA 130 loop, which represent an interaction that sialoglycan receptors do not utilize.
Yet, due to the larger footprint of the antibody on HA (~490 Å2) compared to sialic acid (~190 Å2), it is inevitable that F045-092 will contact some non-conserved residues outside the receptor-binding site. However, F045-092 is able to overcome the antigenic variation of the H3 subtype, as well as the addition and accumulation of glycans, surrounding the receptor-binding site and binds some HA strains with a Kd ~nM as compared to a Kd ~mM for the sialoglycan receptor, a difference of six orders of magnitude. Studies43 indicate that H3 strains with increased glycosylation have significantly decreased affinity for receptor, particularly in strains that appeared around and after 2003 and, in accord, the F045-092 Fab exhibits weaker binding for these more recent H3 strains (Table 1). However, the antibody binding appears to be tuned such that weaker Fab binding can be enhanced and accommodated by avidity of the bivalent IgG so as to extend the antibody’s breadth of recognition, as observed with some other receptor-binding site targeted antibodies15-17. In addition, F045-092 uses different approach angles to bind the Vic1975/H3 and Vic2011/H3 HAs, pivoting around the receptor-binding site likely to accommodate variable epitope residues. Altogether, these binding features allow F045-092 to have exceptional binding breadth against the entire H3 subtype as well as strains from the H1, H2, and H13 subtypes.
Common trends of recognition, as seen with F045-092 and other antibodies, are now emerging that show recurring modes of binding to attain broad recognition of the receptor-binding site. For instance, the use of receptor mimicry has been observed in other antibodies that similarly insert their HCDR3 into the HA receptor-binding site, which all use an Asp carboxylate at the tip of the loop to mimic the carboxylate of sialic acid12,13,15,44,45. These findings suggest that receptor mimicry of the carboxylate may be important for broad recognition of HA, which is utilized by half of the receptor-binding site targeted antibodies solved in complex with HA to date (Fig. 5a), although other antibodies recognize this portion of the receptor-binding site by other means14,16,17,23. In addition, nearly all receptor-binding site targeted antibodies commonly insert a hydrophobic residue into the universally conserved hydrophobic pocket formed by Trp153 and Leu194 that would be occupied by the acetamide moiety of sialic acid. The identity of this hydrophobic residue can vary from a smaller residue such as proline or valine, to a bulky aromatic residue such as tyrosine, phenylalanine, or tryptophan (Fig. 5b). Among the structurally characterized receptor-binding site targeted antibodies, F045-092 most closely resembles C05 as both antibodies contact a very small HA footprint primarily through their long HCDR3 loops16. In contrast, these antibodies differ in their angle of approach by ~113° (Supplementary Fig. 6) and the interaction of C05 with HA does not resemble those of the receptor, suggesting that there is considerable versatility in targeting the receptor-binding site.
Figure 5. Common trends in recognition of the HA by receptor-binding site targeted antibodies.

(a) Antibodies HC19 (red, PDB 2VIR), HC63 (light blue, PDB 1KEN), CH65 (green, PDB 3SM5), and 5J8 (orange, PDB 4M5Z) use an Asp residue to insert a carboxylate in the receptor-binding site that would be occupied by the carboxylate of sialic acid of the α2,6 sialoglycan receptor (dashed red circle). (b) Antibodies HC19 (red, PDB 2VIR), C05 (yellow, PDB 4FP8), 8F8 (blue, PDB 4HF5), 8M2 (pink, PDB 4HFU), and 2G1 (green, PDB 4HG4) use hydrophobic residues to target the receptor-binding site that would be occupied by the acetamide moiety of sialic acid (dashed blue circle). The disulfide loop of the F045-092 HCDR3 is shown and colored purple.
The crystal structures of F045-092 in complex with divergent HAs reveal that the receptor-binding site is a hotspot for antibody recognition and is a key site of vulnerability against influenza HA. The challenge now is how the information gained from these structures can be applied to inform the design of improved vaccines or broad-spectrum small molecule therapeutics, which are currently unavailable as a countermeasure against influenza HA. For instance, multimerization of the HA heads onto a self-assembling platform in combination with epitope focusing of the receptor-binding site may elicit a greater antibody response as an alternative vaccination strategy46,47. Since F045-092 only uses its VH domain to contact a very minimal epitope on HA, it may be a promising candidate as a nanobody therapeutic or a diagnostic against H3 viruses48,49. Due to its extremely small footprint on HA, another possibility may be to computationally design small proteins to bind the receptor-binding site, as previously achieved for the HA stem50,51. However, the synthesized F045-092 HCDR3 peptide does not compete with Fab binding nor could it be co-crystallized with HA, suggesting that the antibody stabilizes the proper conformation of the antibody HCDR3 loop by utilizing contacts from the other HCDRs, as also seen for the C05 antibody16.
Although the ultimate goal is for universal therapy against all flu viruses, it may be more practical to also focus some attention on the H1, H3, and flu B viruses that currently circulate in humans and have been responsible for all human pandemics, epidemics and seasonal flu in the last 46 years. No previous antibody has been able to achieve complete HA subtype recognition at the receptor-binding site as described and characterized here for F045-092, and the structural information has great potential to serve as a template for the design of next-generation therapeutics against all H3 influenza viruses.
Methods
Fab and IgG expression and purification
F045-092 Fab and Fab mutants (HisH100aAla; CysH100cAla, CysH100fAla; LeuH100dAla; AspH100eAla) were individually cloned into the pFastBac Dual vector (Invitrogen) with N-terminal gp67 signal peptides fused to the light and heavy chains and a C-terminal His6 tag fused to the heavy chain. Recombinant bacmid DNA was generated using the Bac-to-Bac system (Invitrogen) and baculovirus was generated by transfecting purified bacmid DNA into Sf9 cells using Cellfectin II (Invitrogen). F045-092 Fab and mutants were expressed by infecting suspension cultures of High Five cells (Invitrogen) with baculovirus shaking at 110 rpm for 72 hours at 28 °C. The proteins were purified by Ni-NTA (Qiagen), protein G, Mono S chromatography (GE Healthcare), followed by a final step of gel filtration (GE Healthcare).
F045-092 Fab was also prepared in E. coli DH12S cells (Invitrogen). C-terminal His6 and FLAG tags were fused to the heavy and light chains, respectively. DH12S cells harboring the F045-092 Fab expression plasmids were cultured in 2x YT medium at 30 °C and induced with 1 mM IPTG at an optical density of 0.7 overnight. The culture supernatant was recovered and subjected to 60% ammonium sulfate precipitation. The precipitant was dissolved in PBS and the Fab was purified by Ni-NTA (Qiagen), anti-FLAG antibody M2 (Sigma), Mono S chromatography (GE Healthcare), followed by a final step of gel filtration (GE Healthcare).
To generate F045-092 IgG, the antibody light and heavy chains were each cloned into a modified pCAGGS vector52 containing a dihydrofolate reductase (DHFR) gene. CHO (DHFR–) cells were co-transfected with the resultant plasmids and cultured in serum-free media. F045-092 IgG was purified from the culture supernatant by Capto Q chromatography (GE Healthcare) and Protein A affinity chromatography (ProteNova).
HA expression and purification
Baculovirus-expressed HA was prepared for binding studies and crystallization. Briefly, each HA was fused with an N-terminal gp67 signal peptide and a C-terminal BirA biotinylation site, thrombin cleavage site, trimerization domain, and a His6 tag. The HAs were expressed, as described above for the F045-092 Fabs, as either HA0 (human HAs) or mature HA (HA1/HA2; due to the polybasic linker in avian HAs between HA1 and HA2) and purified by Ni-NTA (Qiagen). For crystallization, the HA0 of Vic1975/H3 was treated with trypsin (NEB) to remove the C-terminal foldon and His6 tags and to produce mature HA. The HA was then purified by a final step of gel filtration (GE Healthcare). For binding studies, each HA0 or mature HA was biotinylated with BirA and purified by gel filtration (GE Healthcare). The biotinylation reactions contained 100 mM Tris pH 8.0, 10 mM ATP, 10 mM magnesium acetate, 50 μM biotin, and less than 50 mM NaCl, and were incubated at 37 °C for 1-2 hours.
Kd determination
Kd values were determined by bio-layer interferometry using an Octet RED instrument (ForteBio, Inc.). Biotinylated HAs at ~10-50 μg/mL in 1x kinetics buffer (1x PBS pH 7.4, 0.01% BSA, and 0.002% Tween 20) were immobilized onto streptavidin-coated biosensors and incubated with varying concentrations of baculovirus-expressed F045-092 Fab or IgG. All binding data were collected at 30 °C. The kon and koff values of each Fab and IgG were measured in real-time to determine Kd values for each Fab and IgG for each HA tested. The sequences of the HA proteins used in the binding studies and the experimental binding curves for each Fab and IgG for fitting kon and koff are reported in Supplementary Fig. 7.
Crystallization and structural determination of Fab F045-092
Bacterially expressed F045-092 Fab crystals were grown by sitting drop vapor diffusion at 20 °C by mixing 0.75 μL of concentrated protein sample (7.3 mg/mL) with 0.75 μL of mother liquor (30% PEG 600, 0.1 M phosphate-citrate pH 5.0). Crystals appeared in 7 days and were cryoprotected with mother liquor supplemented with 20% ethylene glycol. X-ray diffraction data were collected to 1.50 Å resolution at beamline 11-1 (wavelength 0.97945 Å) at the Stanford Synchrotron Radiation Lightsource (SSRL) and were processed in space group P212121 using XDS53. The structure was determined by molecular replacement with Phaser54 using the variable and constant domains of Fab New (PDB 7FAB55) as search models and one Fab was found in the asymmetric unit. The model was iteratively rebuilt using Coot56 and refined in Phenix57. Refinement parameters include initial rigid body refinement (each Ig domain) and simulated annealing followed by restrained refinement including TLS refinement (each Ig domain as a TLS group). In the final F045-092 Fab structure, 97.9% of the residues are in favored regions of the Ramachandran plot with 0% outliers, as calculated by MolProbity58.
Crystallization and structural determination of the Vic2011/H3 HA
EndoH-treated baculovirus-expressed Vic2011/H3 HA was crystallized by sitting drop vapor diffusion at 4 °C by mixing 0.75 μL of concentrated protein sample (3 mg/mL) with 0.75 μL of mother liquor (5% PEG 8,000, 20% PEG 300, 10% glycerol, 0.1 M Tris pH 8.7). Crystals appeared in 3 days. X-ray diffraction data were collected to 1.75 Å resolution at SSRL beamline 11-1 (wavelength 0.97945 Å) and were processed in space group H32 using XDS53. The structure was determined by molecular replacement with Phaser54 using a protomer of the apo 2005 Hong Kong (H3N2) HA (PDB 2YP743) as the search model. One HA protomer (HA1/HA2) of the HA trimer was found in the asymmetric unit. The model was iteratively rebuilt using Coot56 and refined in Phenix57. Refinement parameters include initial rigid body refinement and simulated annealing followed by restrained refinement including TLS refinement (each HA1 and HA2 chain). In the final Vic2011/H3 HA structure, 96.6% of the residues are in favored regions of the Ramachandran plot with 0% outliers58.
Crystallization and structural determination of F045-092 Fab-HA complexes
For Fab-HA complex formation, baculovirus-expressed Fab F045-092 was added to Vic1975/H3 or Vic2011/H3 HA in a molar ratio of 3.2:1 to achieve 3 Fabs per HA trimer, incubated at room temperature for 1 hour, and the complex was then purified from unbound Fab by gel filtration. F045-092-Vic1975/H3 crystals were grown by sitting drop vapor diffusion at 4 °C by mixing 0.75 μL of concentrated protein sample (5.7 mg/mL) with 0.75 μL of mother liquor (1.7 M ammonium sulfate, 0.1 M Tris pH 8.1, 4% PEG 400). Crystals appeared in 7 days and were cryoprotected with mother liquor supplemented with increasing concentrations of glycerol (5% steps, 10 minutes per step) to a final concentration of 25%. X-ray diffraction data were collected to 2.75 Å resolution at the GM/CA CAT 23ID-B beamline (wavelength 1.03322 Å) at the Advanced Photon Source (APS) and were processed in space group P321 using XDS53. The structure was determined by molecular replacement with Phaser54, using the variable domain of the high-resolution Fab structure and the HA protomer of Vic1975/H3 (PDB 4GMS17, chains A and B) as models. After rigid body refinement of the HA and Fab variable domain, density was observed for the constant domain, which was docked in by hand and refined using rigid body refinement. The model was iteratively rebuilt using Coot56 and refined in Phenix57. Refinement parameters include rigid body refinement (each Ig domain and HA protomer) and simulated annealing followed by restrained refinement including TLS refinement (each Ig domain and HA chain). In the final F045-092-Vic1975/H3 structure, 96.2% of the residues are in favored regions of the Ramachandran plot with 0.3% outliers58.
F045-092-Vic2011/H3 crystals were grown by sitting drop vapor diffusion at 20 °C by mixing 0.75 μL of concentrated protein sample (9.1 mg/mL) with 0.75 μL of mother liquor (20% PEG 400, 30% 1,2-propanediol, 0.1 M HEPES pH 7.5). Crystals appeared in 7 days. X-ray diffraction data were collected to 6.50 Å resolution at the GM/CA CAT 23ID-B beamline (wavelength 1.03318 Å) at APS and were processed in space group C2 using XDS53. The structure was determined by molecular replacement with Phaser54 using the high-resolution apo A/Victoria/361/2011 (H3N2) HA trimer as a search model to locate two HA trimers in the asymmetric unit. Next, six copies of the variable domain of the high-resolution F045-092 Fab structure were used as search models after fixing the position and orientation of the two HA trimers but only five variable domain copies were found. After rigid body refinement, density was observed for the remaining Fab variable and constant domains, which were then docked in by hand and refined with rigid body refinement. The model was iteratively rebuilt using Coot56 and refined in Phenix57 using the high-resolution Fab and HA structures to generate reference model restraints and with similar parameters as in the F045-092-Vic1975/H3 complex. In the final F045-092-Vic2011/H3 structure, 97.4% of the residues are in favored regions of the Ramachandran plot with 0% outliers58.
Structural analyses
Hydrogen bonds and van der Waals contacts were calculated using HBPLUS59 and CONTACSYM60, respectively. Molecular surface area buried upon Fab binding was calculated using MS61. MacPyMol (DeLano Scientific) was used to render structure figures. Kabat numbering was applied to the variable domains of the Fabs using the AbNum server62. The final coordinates were validated using the JCSG quality control server (v2.8), which includes MolProbity58.
Sequence analysis of F045-092 epitope conservation
All full-length and non-redundant human influenza A HA sequences were downloaded on December 12, 2013 from the Influenza Virus Resource at the NCBI database63. The sequences were aligned using MUSCLE64 and analyzed using GCG (Accelrys) and custom shell scripts (available from the authors upon request).
Supplementary Material
Acknowledgements
We thank H. Tien of the Robotics Core at the Joint Center for Structural Genomics for automated crystal screening, the staff of the Advanced Photon Source (APS) GM/CA-CAT beamline 23ID-B and the Stanford Synchrotron Radiation Lightsource (SSRL) beamline 11-1 for support, M. Elsliger (The Scripps Research Institute) and the instructors from the 2012 CCP4 School held at APS for crystallographic and computational support, and J.-P. Julien, X. Zhu (The Scripps Research Institute), and D.C. Ekiert (University of California, San Francisco) for helpful discussions. The work was funded in part by NIH grants R56 AI099275 (I.A.W.) and GM080209 for the NIH Molecular Evolution Training Program at TSRI (P.S.L.), and the Skaggs Institute for Chemical Biology. SSRL is a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the U.S. Department of Energy Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393) and the National Center for Research Resources (P41RR001209). The GM/CA CAT has been funded in whole or in part with Federal funds from National Cancer Institute (Y1-CO-1020) and National Institute of General Medical Sciences (Y1-GM-1104). Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Science, under contract No. DE-AC02-06CH11357. The content is solely the responsibility of the authors and does not necessarily represent the official views of NIGMS or the NIH. This is The Scripps Research Institute manuscript number 25075.
Competing financial interests: The authors declare no competing financial interests.
Footnotes
Additional information
Accession codes: Coordinates and structure factors have been deposited in the RCSB Protein Data Bank under accession codes 4O58 (F045-092-Vic1975/H3 complex), 4O5I (F045-092-Vic2011/H3 complex), 4O5L (apo F045-092 Fab), and 4O5N (apo Vic2011/H3 HA).
Supplementary Information accompanies this paper.
References
- 1.CDC Update: Influenza activity - United States, September 30, 2012-February 9, 2013. MMWR Morb. Mortal. Wkly. Rep. 2013;62:124–130. [PMC free article] [PubMed] [Google Scholar]
- 2.Le QM, et al. Avian flu: isolation of drug-resistant H5N1 virus. Nature. 2005;437:1108. doi: 10.1038/4371108a. [DOI] [PubMed] [Google Scholar]
- 3.Stephenson I, et al. Neuraminidase inhibitor resistance after oseltamivir treatment of acute influenza A and B in children. Clin Infect Dis. 2009;48:389–396. doi: 10.1086/596311. [DOI] [PubMed] [Google Scholar]
- 4.Gao R, et al. Human infection with a novel avian-origin influenza A (H7N9) virus. N. Engl. J. Med. 2013;368:1888–1897. doi: 10.1056/NEJMoa1304459. [DOI] [PubMed] [Google Scholar]
- 5.Influenza Cohen, J. New flu virus in China worries and confuses. Science. 2013;340:129–130. doi: 10.1126/science.340.6129.129. [DOI] [PubMed] [Google Scholar]
- 6.Wilson IA, Skehel JJ, Wiley DC. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 Å resolution. Nature. 1981;289:366–373. doi: 10.1038/289366a0. [DOI] [PubMed] [Google Scholar]
- 7.Weis W, et al. Structure of the influenza virus haemagglutinin complexed with its receptor, sialic acid. Nature. 1988;333:426–431. doi: 10.1038/333426a0. [DOI] [PubMed] [Google Scholar]
- 8.Zhu X, et al. Hemagglutinin homologue from H17N10 bat influenza virus exhibits divergent receptor-binding and pH-dependent fusion activities. Proc. Natl. Acad. Sci. USA. 2013;110:1458–1463. doi: 10.1073/pnas.1218509110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun X, et al. Bat-derived influenza hemagglutinin H17 does not bind canonical avian or human receptors and most likely uses a unique entry mechanism. Cell Rep. 2013;3:769–778. doi: 10.1016/j.celrep.2013.01.025. [DOI] [PubMed] [Google Scholar]
- 10.Tong S, et al. New world bats harbor diverse influenza A viruses. PLoS Pathog. 2013;9:e1003657. doi: 10.1371/journal.ppat.1003657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bullough PA, Hughson FM, Skehel JJ, Wiley DC. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature. 1994;371:37–43. doi: 10.1038/371037a0. [DOI] [PubMed] [Google Scholar]
- 12.Whittle JR, et al. Broadly neutralizing human antibody that recognizes the receptor-binding pocket of influenza virus hemagglutinin. Proc. Natl. Acad. Sci. USA. 2011;108:14216–14221. doi: 10.1073/pnas.1111497108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schmidt AG, et al. Preconfiguration of the antigen-binding site during affinity maturation of a broadly neutralizing influenza virus antibody. Proc. Natl. Acad. Sci. USA. 2013;110:264–269. doi: 10.1073/pnas.1218256109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu R, et al. A recurring motif for antibody recognition of the receptor-binding site of influenza hemagglutinin. Nat. Struct. Mol. Biol. 2013;20:363–370. doi: 10.1038/nsmb.2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hong M, et al. Antibody recognition of the pandemic H1N1 influenza hemagglutinin receptor binding site. J. Virol. 2013;87:12471–12480. doi: 10.1128/JVI.01388-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ekiert DC, et al. Cross-neutralization of influenza A viruses mediated by a single antibody loop. Nature. 2012;489:526–532. doi: 10.1038/nature11414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee PS, et al. Heterosubtypic antibody recognition of the influenza virus hemagglutinin receptor binding site enhanced by avidity. Proc. Natl. Acad. Sci. USA. 2012;109:17040–17045. doi: 10.1073/pnas.1212371109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Julien JP, Lee PS, Wilson IA. Structural insights into key sites of vulnerability on HIV-1 Env and influenza HA. Immunol. Rev. 2012;250:180–198. doi: 10.1111/imr.12005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ohshima N, et al. Naturally occurring antibodies in humans can neutralize a variety of influenza virus strains, including H3, H1, H2, and H5. J. Virol. 2011;85:11048–11057. doi: 10.1128/JVI.05397-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harris A, et al. Influenza virus pleiomorphy characterized by cryoelectron tomography. Proc. Natl. Acad. Sci. USA. 2006;103:19123–19127. doi: 10.1073/pnas.0607614103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ekiert DC, et al. Antibody recognition of a highly conserved influenza virus epitope. Science. 2009;324:246–251. doi: 10.1126/science.1171491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Corti D, et al. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science. 2011;333:850–856. doi: 10.1126/science.1205669. [DOI] [PubMed] [Google Scholar]
- 23.Tsibane T, et al. Influenza human monoclonal antibody 1F1 interacts with three major antigenic sites and residues mediating human receptor specificity in H1N1 viruses. PLoS Pathog. 2012;8:e1003067. doi: 10.1371/journal.ppat.1003067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stanfield RL, Zemla A, Wilson IA, Rupp B. Antibody elbow angles are influenced by their light chain class. J. Mol. Biol. 2006;357:1566–1574. doi: 10.1016/j.jmb.2006.01.023. [DOI] [PubMed] [Google Scholar]
- 25.Wiley DC, Wilson IA, Skehel JJ. Structural identification of the antibody-binding sites of Hong Kong influenza haemagglutinin and their involvement in antigenic variation. Nature. 1981;289:373–378. doi: 10.1038/289373a0. [DOI] [PubMed] [Google Scholar]
- 26.Sui J, et al. Structural and functional bases for broad-spectrum neutralization of avian and human influenza A viruses. Nat. Struct. Mol. Biol. 2009;16:265–273. doi: 10.1038/nsmb.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Almagro JC, et al. Characterization of a high-affinity human antibody with a disulfide bridge in the third complementarity-determining region of the heavy chain. J. Mol. Recognit. 2012;25:125–135. doi: 10.1002/jmr.1168. [DOI] [PubMed] [Google Scholar]
- 28.Kong L, et al. Hepatitis C virus E2 envelope glycoprotein core structure. Science. 2013;342:1090–1094. doi: 10.1126/science.1243876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ekiert DC, et al. A highly conserved neutralizing epitope on group 2 influenza A viruses. Science. 2011;333:843–850. doi: 10.1126/science.1204839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dreyfus C, et al. Highly conserved protective epitopes on influenza B viruses. Science. 2012;337:1343–1348. doi: 10.1126/science.1222908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dreyfus C, Ekiert DC, Wilson IA. Structure of a classical broadly neutralizing stem antibody in complex with a pandemic H2 hemagglutinin. J. Virol. 2013;87:7149–7154. doi: 10.1128/JVI.02975-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Friesen RH, et al. A common solution to group 2 influenza virus neutralization. Proc. Natl. Acad. Sci. USA. 2014;111:450–455. doi: 10.1073/pnas.1319058110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Skehel JJ, et al. A carbohydrate side chain on hemagglutinins of Hong Kong influenza viruses inhibits recognition by a monoclonal antibody. Proc. Natl. Acad. Sci. USA. 1984;81:1779–1783. doi: 10.1073/pnas.81.6.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abe Y, et al. Effect of the addition of oligosaccharides on the biological activities and antigenicity of influenza A/H3N2 virus hemagglutinin. J. Virol. 2004;78:9605–9611. doi: 10.1128/JVI.78.18.9605-9611.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang CC, et al. Glycans on influenza hemagglutinin affect receptor binding and immune response. Proc. Natl. Acad. Sci. USA. 2009;106:18137–18142. doi: 10.1073/pnas.0909696106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu R, et al. Structural basis of preexisting immunity to the 2009 H1N1 pandemic influenza virus. Science. 2010;328:357–360. doi: 10.1126/science.1186430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wei CJ, et al. Cross-neutralization of 1918 and 2009 influenza viruses: role of glycans in viral evolution and vaccine design. Sci Transl Med. 2010;2 doi: 10.1126/scitranslmed.3000799. 24ra21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Karlsson Hedestam GB, et al. The challenges of eliciting neutralizing antibodies to HIV-1 and to influenza virus. Nat. Rev. Microbiol. 2008;6:143–155. doi: 10.1038/nrmicro1819. [DOI] [PubMed] [Google Scholar]
- 39.Pejchal R, et al. A potent and broad neutralizing antibody recognizes and penetrates the HIV glycan shield. Science. 2011;334:1097–1103. doi: 10.1126/science.1213256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McLellan JS, et al. Structure of HIV-1 gp120 V1/V2 domain with broadly neutralizing antibody PG9. Nature. 2011;480:336–343. doi: 10.1038/nature10696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Julien JP, et al. Asymmetric recognition of the HIV-1 trimer by broadly neutralizing antibody PG9. Proc. Natl. Acad. Sci. USA. 2013;110:4351–4356. doi: 10.1073/pnas.1217537110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pancera M, et al. Structural basis for diverse N-glycan recognition by HIV-1-neutralizing V1-V2-directed antibody PG16. Nat. Struct. Mol. Biol. 2013;20:804–813. doi: 10.1038/nsmb.2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin YP, et al. Evolution of the receptor binding properties of the influenza A (H3N2) hemagglutinin. Proc. Natl. Acad. Sci. USA. 2012;109:21474–21479. doi: 10.1073/pnas.1218841110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bizebard T, et al. Structure of influenza virus haemagglutinin complexed with a neutralizing antibody. Nature. 1995;376:92–94. doi: 10.1038/376092a0. [DOI] [PubMed] [Google Scholar]
- 45.Barbey-Martin C, et al. An antibody that prevents the hemagglutinin low pH fusogenic transition. Virology. 2002;294:70–74. doi: 10.1006/viro.2001.1320. [DOI] [PubMed] [Google Scholar]
- 46.Jardine J, et al. Rational HIV immunogen design to target specific germline B cell receptors. Science. 2013;340:711–716. doi: 10.1126/science.1234150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kanekiyo M, et al. Self-assembling influenza nanoparticle vaccines elicit broadly neutralizing H1N1 antibodies. Nature. 2013;499:102–106. doi: 10.1038/nature12202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang J, et al. Pentamerization of single-domain antibodies from phage libraries: a novel strategy for the rapid generation of high-avidity antibody reagents. J. Mol. Biol. 2004;335:49–56. doi: 10.1016/j.jmb.2003.09.034. [DOI] [PubMed] [Google Scholar]
- 49.Wesolowski J, et al. Single domain antibodies: promising experimental and therapeutic tools in infection and immunity. Med. Microbiol. Immunol. 2009;198:157–174. doi: 10.1007/s00430-009-0116-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fleishman SJ, et al. Computational design of proteins targeting the conserved stem region of influenza hemagglutinin. Science. 2011;332:816–821. doi: 10.1126/science.1202617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Whitehead TA, et al. Optimization of affinity, specificity and function of designed influenza inhibitors using deep sequencing. Nat. Biotechnol. 2012;30:543–548. doi: 10.1038/nbt.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- 53.Kabsch W. Acta Crystallogr. D Biol. Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. XDS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McCoy AJ, et al. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Saul FA, Poljak RJ. Crystal structure of human immunoglobulin fragment Fab New refined at 2.0 Å resolution. Proteins. 1992;14:363–371. doi: 10.1002/prot.340140305. [DOI] [PubMed] [Google Scholar]
- 56.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen VB, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McDonald IK, Thornton JM. Satisfying hydrogen bonding potential in proteins. J. Mol. Biol. 1994;238:777–793. doi: 10.1006/jmbi.1994.1334. [DOI] [PubMed] [Google Scholar]
- 60.Sheriff S, Hendrickson WA, Smith JL. Structure of myohemerythrin in the azidomet state at 1.7/1.3 Å resolution. J. Mol. Biol. 1987;197:273–296. doi: 10.1016/0022-2836(87)90124-0. [DOI] [PubMed] [Google Scholar]
- 61.Connolly ML. Analytical molecular surface calculation. J. Appl. Crystallogr. 1983;16:548–558. [Google Scholar]
- 62.Abhinandan KR, Martin AC. Analysis and improvements to Kabat and structurally correct numbering of antibody variable domains. Mol. Immunol. 2008;45:3832–3839. doi: 10.1016/j.molimm.2008.05.022. [DOI] [PubMed] [Google Scholar]
- 63.Bao Y, et al. The influenza virus resource at the National Center for Biotechnology Information. J. Virol. 2008;82:596–601. doi: 10.1128/JVI.02005-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.