

Highlights
-
•
Atg8 lipidation can be efficiently reconstituted in vitro.
-
•
Lipidation and de-lipidation of Atg8 by Atg4 can be analyzed.
-
•
Reconstitution of Atg8-lipidation using giant unilamellar vesicles offers spatial insights.
-
•
These assays allow determining the effect of modifications on Atg8 lipidation/de-lipidation.
Keywords: Autophagy, Protein conjugation, Autophagosome, Atg8, Atg4, Atg12
Abstract
Macroautophagy is a major bulk degradation pathway for cytoplasmic material in eukaryotic cells. During macroautophagy, double membrane-bound organelles called autophagosomes are formed in a de novo manner. In the course of their formation autophagosomes capture cytoplasmic material, which is subsequently degraded upon fusion with the lysosomal system in complex eukaryotes or the vacuole in yeast. Several proteins are required for autophagosome formation. Among these are the components of two ubiquitin-like conjugation reactions that collectively mediate the conjugation of the ubiquitin-like Atg12 to the Atg5 protein and of the ubiquitin-like protein Atg8 to the headgroup of the membrane lipid phosphatidylethanolamine. The lipidated form of Atg8 is membrane-bound and marks the growing autophagosomal membrane as well as the completed autophagosome. Here we describe assays for the in vitro reconstitution of the Atg8 lipidation reaction using recombinantly expressed and purified proteins derived from Saccharomycescerevisiae in combination with small and giant unilamellar vesicles. The assays enable the study of the biochemical mechanisms of action of the Atg8 lipidation machinery and to analyze the impact of mutations and post-translational modifications of the conjugation machinery on Atg8 lipidation.
1. Introduction
Macroautophagy (hereafter autophagy) is a major intracellular bulk degradation system that enables cells to survive during starvation and that mediates quality control by the removal of harmful intracellular structures [1]. In addition, autophagy functions in the clearance of intracellular pathogens [2].
Upon induction of autophagy cells respond with the formation of double membrane-bound organelles called autophagosomes. Autophagosomes are formed in a de novo manner. Initially small membrane structures called isolation membranes (or phagophores) are observed. As they grow isolation membranes gradually enclose cellular cargo and when they close to give rise to autophagosomes the cargo is completely isolated from the cytoplasm. Eventually, autophagosomes fuse with the lysosomal system wherein the inner membrane and the cargo are degraded [3].
A number of genes that are essential for autophagosome formation have been identified, mainly in genetic screen in Saccharomyces cerevisiae [4,5]. These genes are conserved from yeast to human and are referred to as autophagy-related genes (ATGs) [6]. A surprising number of these genes functions during the unusual conjugation of the ubiquitin-like Atg8 protein to the headgroup of the membrane lipid phosphatidylethanolamine (PE) on the growing isolation membrane (Fig. 1) [7,8].
Fig. 1.

The Atg8 and Atg12 conjugation machinery. S. cerevisiae Atg8 is initially synthesized with a C-terminal arginine residue (R117). The cysteine protease Atg4 mediates the cleavage of this C-terminal arginine exposing the penultimate glycine, which becomes activated by the E1-like enzyme Atg7 under consumption of ATP. From Atg7 Atg8 becomes transferred to a cysteine in the E2-like Atg3. From there Atg8 is transferred to the headgroup of the membrane lipid phosphatidylethanolamine. Alternatively, phosphatidylserine can serve as an acceptor of Atg8. The transfer of Atg8 to the lipid headgroup is massively stimulated by a complex composed of the Atg5, Atg12 and Atg16 proteins. This complex is itself the result of an ubiquitin-like conjugation reaction. Here the C-terminus of the ubiquitin-like Atg12 is covalently attached to a lysine residue (K149) of the Atg5 protein. Atg7 and the E2-like Atg10 protein mediate this conjugation. The resulting Atg12–Atg5 conjugate forms a complex with the Atg16 protein giving rise to the Atg12–Atg5-Atg16 complex. Atg4, the same enzyme that mediates the removal of R117 from Atg8, is able to remove Atg8 from the membrane by hydrolyzing the bond between the C-terminal glycine and the lipid headgroup.
In S. cerevisiae only a single Atg8 protein exists while there are 7 isoforms in humans [9]. Atg8 is initially synthesized with a C-terminal arginine. This arginine is removed by the cysteine protease Atg4 exposing the penultimate glycine residue [10]. The C-terminal glycine is then activated by the E1-like enzyme Atg7 under consumption of ATP and formation of a thioester bond. From there Atg8 is transferred to a cysteine of the E2-like Atg3 and finally to the headgroup of PE [8,11–13]. A protein complex composed of the Atg5, Atg12 and Atg16 proteins, which acts analogous to E3 enzymes during ubiquitination reactions, vastly stimulates this last step [14–17].
The complex composed of Atg5, Atg12 and Atg16 is itself the product of an ubiquitin-like conjugation reaction [18]. Here the ubiquitin-like Atg12 is activated by the E1-like Atg7, transferred to the E2-like Atg10 and from there to a lysine residue of Atg5 [18–20]. The resulting Atg12–Atg5 conjugate binds to Atg16 giving rise to the Atg12–Atg5-Atg16 complex [21].
The cysteine protease Atg4 is able to release lipidated Atg8 from the membrane by catalyzing the deconjugation of Atg8 [10,22–24].
All components of the Atg8 and Atg12 conjugation systems are required for autophagosome formation [4,5,8,18,24]. The function of membrane localized Atg8–PE for cargo recruitment during selective autophagy has been well established (reviewed in [25]) but the role of the conjugation systems for autophagosome formation in general is still obscure. In selective autophagy cargo material is linked to the growing isolation membrane. Cargo receptor proteins that simultaneously bind the cargo and Atg8 on the isolation membrane mediate this selectivity [25–28]. Atg8 therefore serves as an identifier of the future autophagosomal membrane. However, the Atg8 and Atg12 conjugation systems are also essential for autophagosome formation during non-selective autophagy. Electron microscopy studies in Atg3 and Atg5 knockout mouse cells and in rodent cells expressing dominant negative versions of ATG4 showed that autophagosome formation is blocked at rather late stages. In particular, in these cells extended isolation membranes are formed but these fail to close [29–33].
While in vivo experiments are essential to uncover the function of a given gene product, in vitro systems are able to reveal whether a given protein is not only essential but also sufficient for the process of interest. In addition, in vitro systems allow one to gain insights into the biochemical mechanism of action of the factors under investigation. Indeed, many key insights into the molecular mechanism of action of the conjugation machinery have been gained from biochemical reconstitution systems. Most studies have focused on the Atg8 conjugation system derived from S. cerevisiae. The reason is that, as opposed to Atg12, recombinant Atg8 can be produced in its non-conjugated form. Furthermore, Atg12 appears to be constitutively conjugated to Atg5 and so far no studies implicate the conjugation of Atg12–Atg5 as regulatory mechanism, while lipidation of Atg8 correlates with autophagosome formation [7,8,10,18,24]. For the Atg8 system all intermediates including the Atg8–AMP, Atg8–Atg7, the Atg8–Atg3 and the final Atg8–PE product have been identified and the contribution of each component of the conjugation machinery to their formation can thus be tested [11–13,15,34]. One of the key findings was that the yeast Atg12–Atg5-Atg16 complex acts in an E3-like manner facilitating the transfer of Atg8 from Atg3 to PE or PS [15,16,35]. This is consistent with the requirement of Atg5, Atg12, Atg10 and Atg16 for Atg8–PE formation in vivo. In vitro, using small unilamellar vesicles (SUVs) the Atg12–Atg5 conjugate is sufficient to promote Atg8 lipidation [14–16]. It likely acts by direct activation of the catalytic activity of Atg3 [17]. However, under more stringent conditions using giant unilamellar vesicles (GUVs) that are much less curved than SUVs, the presence of Atg16 also facilitates Atg8 lipidation possibly via activation of membrane binding by the Atg12–Atg5-Atg16 complex [14]. Whether the mammalian ATG12–ATG5-ATG16L complex promotes lipidation of ATG8-family proteins in a similar manner remains to be tested. However, given the conservation of the components and the same requirements of the respective genes for ATG8 family protein conjugation in mammalian cells likely possesses similar biochemical properties.
Reconstitution experiments have also revealed extensive crosstalk between the conjugation machinery and the membrane. Thus a high content of PE facilitates Atg8 conjugation [16,34,35]. The positive correlation between the PE content of the membrane and the efficiency of Atg8–PE formation may not only reflect the higher availability of the PE substrate. Since PE has a small headgroup it aids the insertion of proteins into the lipid bilayer and will therefore promote membrane binding by the Atg8 conjugation machinery [14,34,36]. A high degree of membrane curvature has a similar effect since the lipid headgroups are further apart aiding membrane insertion by proteins [37–39]. Indeed, a recent study showed that ATG3 contains a membrane inserting, curvature sensitive domain and that at least in the absence of the mammalian ATG12–ATG5-ATG16L complex, the conjugation of the ATG8-family proteins LC3B, GABARAPL1 and GABARAPL2 occurs preferentially on highly curved membranes [34]. In vivo, Atg8 conjugation may therefore occur on the highly curved edge of the growing isolation membrane.
In vitro studies have not only revealed an impact of the membrane on the efficiency of Atg8 lipidation but also an effect of the conjugation machinery and the final product on the properties of the membrane. Yeast Atg8 and the mammalian ATG8-family protein, are able to efficiently tether membranes in form of SUVs or GUVs [14,40–42]. Moreover, it was reported that lipidated yeast Atg8 mediates membrane hemifusion [14,41], which is an intermediate step toward full membrane fusion [43] and that the lipidated forms of the mammalian ATG8 proteins LC3B and GATE-16/GABARAPL2 mediate full membrane fusion [42]. A subsequent study showed a strong influence of the lipid composition on the fusion activity of yeast Atg8 [40].
Employing GUVs more recent reconstitution studies have shown that the yeast Atg12–Atg5-Atg16 complex is also able to tether membranes and that it forms patch-like assemblies on the membrane [14]. It was further suggested that the Atg12–Atg5-Atg16 complex binds Atg8 on the membrane of the GUVs resulting in the formation of a scaffold-like assembly that can be disassembled by Atg4-mediated deconjugation of Atg8–PE [44].
As evident from the studies outlined above in vitro reconstitution systems have provided numerous insights into the biochemical properties of the autophagic conjugation machinery.
Here we will describe how to set up in vitro systems for reconstituting the lipidation and de-lipidation of yeast Atg8. We will outline the requirements and highlight the steps where special caution is needed. The system should allow researchers to directly test the effect of further factors in the system [45], to study mutants or to study the impact of post-translational modifications on the activity of the conjugation machinery [46].
2. Materials and methods
2.1. Accession numbers
All proteins used were from S. cerevisiae. Atg3: NP_014404; Atg4: NP_014176.2; Atg5: NP_015176.1; Atg7: NP_012041.1; Atg8: NP_009475.1; Atg10: NP_013058.1; Atg12: NP_009776.1; Atg16: NP_013882.1.
2.2. Protein expression and purification
2.2.1. Expression and purification of S. cerevisiae Atg3 (NP_014404)
Full length Atg3 was expressed as an N-terminal GST fusion protein from pGEX4T1 in Escherichia coli. Rosetta pLySS cells were grown at 37 °C to an OD600 of 0.8, induced with 50 μM IPTG and grown for further 16 h at 18 °C. Cells were pelleted and resuspended in a buffer containing 50 mM HEPES pH 7.5, 300 mM NaCl, 2 mM MgCl2, 1 mM DTT, complete protease inhibitors (Roche) and DNAse I (Sigma). Cells were lysed by freeze thawing followed by brief sonication and the lysate was centrifuged at 40,000 rpm (Beckman Ti45 rotor) for 40 min at 4 °C. The supernatant was incubated with glutathione-beads (GE Healthcare) for 2 h at 4 °C. Beads were washed 5× with 50 mM HEPES pH 7.5, 300 mM NaCl, 1 mM DTT followed by 2 washes with 50 mM HEPES pH 7.5, 1 M NaCl, 1 mM DTT and two washes with 50 mM HEPES pH 7.5, 300 mM NaCl, 1 mM DTT. The protein was cleaved off from the GST tag by incubation with thrombin protease (Serva) overnight at 4 °C. The supernatant containing the cleaved off Atg3 protein was diluted to reach a final salt concentration of 150 mM NaCl and further purified using a 16/60 Q-Sepharose column. The protein was eluted on a gradient ranging from 150 mM to 1 M NaCl. Fractions containing Atg3 were pooled, concentrated and run on a 16/60 Superdex 75 size exclusion column in 50 mM HEPES pH 7.5, 150 mM NaCl, 1 mM DTT. Fractions containing pure Atg3 were pooled, concentrated, diluted 1:1 with glycerol, and stored at −20 °C.
2.2.2. Expression and purification of S. cerevisiae Atg4 (NP_014176.2)
Atg4 was purified and expressed as an N-terminal GST fusion protein from pGEX4T3 in E. coli Rosetta pLysS cells. Cells were grown at 37 °C until of OD600 of 0.8. Protein expression was induced by addition of IPTG to a final concentration of 0.1 mM and the protein was expressed at 18 °C over night. The cell pellets were resuspended in 50 mM HEPES pH 7.5, 300 mM NaCl, 1 mM DTT, 1 mM MgCl2, DNAse I, complete protease inhibitors, PEFABLOC. Cells were lysed by freezing in liquid N2 and thawing followed by brief sonication. The lysate was cleared by centrifugation at 40,000 rpm (Beckman Ti45 rotor) for 40 min at 4 °C and the supernatant was incubated with equilibrated glutathione beads (GE Healthcare) for 2 h at 4 °C. The beads were washed 5 times with 50 mM HEPES pH 7.5, 300 mM NaCl, 1 mM DTT, 2 times with 50 mM HEPES pH 7.5, 700 mM NaCl, 1 mM DTT and finally 2 times with 50 mM HEPES pH 7.5, 300 mM NaCl, 1 mM DTT. The protein was eluted with 50 mM HEPES pH 7.5, 300 mM NaCl, 1 mM DTT supplemented with 20 mM l-glutathione and cleaved with thrombin. The cleaved protein was run on a Superdex 200 16/60 column (GE Healthcare) and the peak fractions containing Atg4 were pooled, concentrated and flash frozen in liquid N2.
2.2.3. Expression and purification of S. cerevisiae Atg7 (NP_012041.1)
Full length Atg7 was expressed as an N-terminal His-tagged protein from pOPTHrsTEV in E. coli. Rosetta pLySS cells were grown at 37 °C to an OD600 of 0.8, induced with 50 μM IPTG and grown for further 16 h at 18 °C. Cells were pelleted and resuspended in a buffer containing 50 mM HEPES pH 7.5, 300 mM NaCl, 10 mM Imidazole, 1 mM MgCl2, 2 mM β-mercaptoethanol, complete protease inhibitors (Roche) and DNAse I (Sigma). Cells were lysed by freeze thawing followed by brief sonication and the lysate was centrifuged at 40,000 rpm (Beckman Ti45 rotor) for 40 min at 4 °C. The supernatant was incubated with nickel beads (5 Prime) for 2 h at 4 °C. Beads were washed with 50 mM HEPES pH 7.5, 300 mM NaCl, 10 mM Imidazole, 2 mM β-mercaptoethanol and the His-tag was cleaved off with TEV protease at room temperature. The Atg7 protein was diluted to reach a final salt concentration of 150 mM and the protein was further purified on a 16/60 Q-Sepharose column. The protein was eluted using a gradient reaching from 150 mM to 1 M NaCl. Fractions containing Atg7 were pooled, concentrated and run on a 16/60 Superdex 200 size exclusion column in 50 mM HEPES pH 7.5, 150 mM NaCl, 1 mM DTT. Fractions containing pure Atg7 were pooled, concentrated, diluted 1:1 with glycerol, and stored at −20 °C.
2.2.4. Expression and purification of S. cerevisiae Atg8 (NP_009475.1)
Atg8 lacking the C-terminal arginine was expressed as N-terminal His-tagged protein from pOPC-His-TEV-Atg8 in E. coli. Rosetta pLySS cells were grown at 37 °C to an OD600 of 0.8, induced with 0.5 mM IPTG and grown for further 3 h at 37 °C. Cells were pelleted and resuspended in a buffer containing 50 mM HEPES pH 7.5, 300 mM NaCl, 10 mM Imidazole, 1 mM MgCl2, 2 mM β-mercaptoethanol, complete protease inhibitors (Roche) and DNAse I (Sigma). Cells were lysed by freeze thawing followed by brief sonication and the lysate was centrifuged at 40,000 rpm (Beckman Ti45 rotor) for 40 min at 4 °C. The supernatant was incubated with nickel beads (5 Prime) for 2 h at 4 °C. Beads were washed with 50 mM HEPES pH 7.5, 300 mM NaCl, 10 mM Imidazole, 2 mM β-mercaptoethanol and the His-tag was cleaved off for several hours with TEV protease at room temperature. The Atg8 protein was diluted to reach final salt concentration of 150 mM and the protein was further purified on 16/60 SP-Sepharose column. The protein was eluted using a gradient reaching from 150 mM to 1 M NaCl. Fractions containing Atg8 were pooled, concentrated and run on a 16/60 Superdex 75 size exclusion column in 50 mM HEPES pH 7.5, 150 mM NaCl, 1 mM DTT. Fractions containing pure Atg8 were pooled, concentrated, diluted 1:1 with glycerol, and stored at −20 °C.
2.2.5. Expression of N-terminal monomeric eGFP fusion of S. cerevisiae Atg8 (NP_009475.1)
The fusion gene coded for an N-terminal His-tag followed by a TEV protease recognition site and the Atg8 open reading frame (lacking the C-terminal R117) was expressed from pETDuet-1 in E. coli. Rosetta pLySS cells were grown at 37 °C until an OD600 of 0.8, induced with 50 μM IPTG and grown for further 16 h at 18 °C. Cells were pelleted and resuspended in a buffer containing 50 mM HEPES pH 7.5, 300 mM NaCl, 10 mM Imidazole, 1 mM MgCl2, 2 mM β-mercaptoethanol, complete protease inhibitors (Roche) and DNAse I. Cells were lysed by freeze thawing followed by brief sonication and the lysate was centrifuged at 40,000 rpm (Beckman Ti45 rotor) for 40 min at 4 °C. The supernatant was passed over a nickel column and the protein was eluted in a step gradient ranging from 50 to 300 mM Imidazole. The His-tag was cleaved off with TEV protease at room temperature. The Atg8 protein was diluted to reach final imidazole concentration of 40 mM and the TEV protease containing an N-terminal His-tag was removed by incubation with nickel beads. The supernatant containing meGFP-Atg8 was concentrated and run on a 16/60 Superdex 75 size exclusion column in 50 mM HEPES pH 7.5, 150 mM NaCl, 1 mM DTT. Fractions containing pure Atg8 fusion proteins were pooled, concentrated and stored at −80 °C.
2.2.6. Expression and purification of the Atg12–Atg5 conjugate and the Atg12–Atg5-Atg16 complex (Atg5: NP_015176.1, Atg12: NP_009776.1, Atg16: NP_013882.1)
The Atg12–Atg5 conjugate and the Atg12–Atg5-Atg16 complex were produced in E. coli Rosetta pLySS by co-expression of Atg12, hexahistidine-tagged Atg5, Atg7, Atg10 and, if applicable, Atg16. Atg5 and Atg12 were co-expressed from pETDuet-1, Atg7 and Atg10 were co-expressed from pCOLADuet-1 and Atg16 was expressed from pCDFDuet-1.
Cells were grown at 37 °C to an OD600 of 0.8, induced with 1 mM IPTG and grown for another 4 h at 37 °C. Harvested cells were resuspended in the resuspension buffer (300 mM NaCl, 50 mM HEPES, pH 7.5, 10 mM Imidazole, 2.5 mM PEFABLOC (Roth), 1 mM MgCl2, 2 mM β-mercaptoethanol, 10 μg/ml DNAse), and disrupted by freeze–thaw method and sonication. The cleared lysate was applied to a HisTrap column (GE Healthcare) and the proteins were eluted by a step-wise imidazole gradient at 150 mM imidazole. For Atg12–Atg5, the eluate was concentrated using Amicon Ultra centrifugal filter (MW cut-off 30 kDa) and further purified using a 16/60 Superdex 200 size exclusion column (GE Healthcare). The protein was eluted from the column with 150 mM NaCl, 50 mM HEPES pH 7.5, 1 mM DTT. For Atg12–Atg5-Atg16 the HisTrap fractions containing the protein were concentrated and run on a Q-Sepharose column (GE Healthcare). The protein eluted in a continuous NaCl gradient at 530 mM NaCl. The eluate was concentrated and applied to a 16/60 Superdex 200 size exclusion column (GE Healthcare). The protein was eluted with 150 mM NaCl, 50 mM HEPES pH 7.5, 1 mM DTT.
2.3. Preparation of small unilamellar vesicles (SUVs)
SUVs were prepared using an E. coli total extract (Avanti Polar Lipids, Inc., 100500C), which contains between 55% and 60% PE (weight/weight) for the assays shown in Figs. 2 and 3A. For the Atg4 deconjugation assay shown in Fig. 3B the SUVs were composed of 40% POPC, 35% POPS, 20% POPE, 5% PI3P. 100 μl of the lipid stock (10 mg/ml) were transferred into a glass vial and dried under an argon stream. The dried lipids were further dried for an additional hour in a desiccator. Subsequently, the dried lipids were incubated with 1 ml of 50 mM HEPES, pH 7.5, 0.2 mM DTT (or 1 mM DTT for Fig. 3B) for 15 min. The lipids were resuspended by tapping and gently sonicated for 2 min in a water bath sonicator. The resuspended SUVs were then extruded 21 times through 0.4 μm membrane followed by extrusion through a 0.1 μm membrane (Whatman, Nucleopore) using the Mini Extruder from Avanti Polar Lipids Inc. The final SUVs suspension has a concentration of 1 mg lipids/ml buffer. SUVs are stable for 1–2 days when stored at 4 °C.
Fig. 2.

In vitro Atg8 lipidation reaction using SUVs. The figure shows a 16% urea SDS page gel of an overnight Atg8 lipidation reaction. The conversion of Atg8 to Atg8–PE is detected by a characteristic size shift towards a faster migration behavior in this gel system. The addition of 6 M urea to the separating gel greatly facilitates the detection of the lipidated form of yeast Atg8. Lipidation of Atg8 occurs only in the presence of Atg3, Atg7, SUVs (derived from the E. coli lipids in the reaction shown) and ATP plus MgCl2 (all components). The Atg8–Atg3 intermediate can also be detected on this gel. The faster migrating Atg8 band in the absence of liposomes presumably represents the Atg8–AMP product formed by Atg7 [34]. The numbers on the left of the gel indicate the molecular weights of the marker bands in kDa.
Fig. 3.

In vitro Atg8 lipidation/de-lipidation reaction using SUVs. (A) Two Coomassie-stained gels of two time series of Atg8 conjugation reactions using SUVs derived from the E. coli lipids. The first reaction was conducted in the absence (left) and the second reaction was conducted in the presence (right) of the Atg12–Atg5-Atg16 complex. The lower gels shown in the bottom were run at lower polyacrylamide concentrations (12% as opposed to 16% for the upper two gels) in order to separate the Atg7 protein from the Atg12–Atg5 conjugate. Note the massive stimulation of Atg8 lipidation in the presence of the Atg12–Atg5-Atg16 complex. The numbers between the gels indicate the molecular weights of the marker bands in kDa. (B) Coomassie-stained gel of an Atg8 lipidation/de-lipidation reaction. The SUVs were composed of 40% POPC, 35% POPS, 20% POPE and 5% PI3P. Atg8 was first completely lipidated in the presence of Atg3, Atg7, the Atg12–Atg5 conjugate and ATP and MgCl2. After 30 min Atg4 and EDTA were added to final concentrations of 0.07 μM and 2 mM, respectively. After 2 min Atg8 was completely de-lipidated. The numbers to the left of the gel indicate the molecular weights of the marker bands in kDa. Note that the 35 kDa and 15 kDa bands separate into two bands in the presence of 6 M urea. (M: marker).
2.4. Atg8 conjugation and deconjugation assay using SUVs
The conjugation and deconjugation reactions were performed at 30 °C and all buffers, solutions and the SUVs with the exception of the proteins were pre-warmed at this temperature. Atg3, Atg7, Atg12–Atg5 and Atg12–Atg5-Atg16 were used at a final concentration of 1 μM whereas Atg8 (lacking the C-terminal arginine) was used at a final concentration of 5 μM. Atg4 was used at a concentration of 0.07 μM. ATP and EDTA were used at a final concentration of 2 mM while MgCl2 was used at a final concentration of 1 mM. The reactions were stopped by the addition of SDS loading buffer. The reactions were run on 13.5% SDS–polyacrylamide gels containing 6 M urea in the separating gels.
2.5. Preparation of giant unilamellar vesicles (GUVs)
GUVs were prepared by electroformation. 3 μl of a lipid mixture (10 mg/ml in chloroform/methanol (3:1)) were applied onto the surface of indium-tin-oxide coated glass slides in a drop wise manner and desiccated for at least 3 h under vacuum. Subsequently, electroformation chambers were assembled using silicon gaskets and the chambers were filled with a solution containing 300 mM sucrose. The electroformation protocol contained 3 phases. Phase 1: 30 min. Sine wave with frequency f = 10 Hz. Peak-to-peak amplitude ramps up linearly from 0.05 V to 1.41 V. Phase 2: 120 min. Sine wave with frequency f = 10 Hz. Peak-to-peak amplitude held constant at 1.41 V. Phase 3: 30 min. Square wave with frequency f = 4.5 Hz. Peak-to-peak amplitude held constant at 2.12 V. The electroformation was conducted at 24 °C. The GUVs were diluted in 15 mM HEPES, pH 7.5, 135 mM NaCl.
The following lipid mix was used: 40% DOPC, 35% DOPS, 20% DOPE, 5% PI3P. 2% Marina Blue (Invitrogen) at the expense of DOPC was added to the membrane labeled GUVs shown in Fig. 4. All lipids were purchased from Avanti Polar Lipids Inc. Marina blue PE was from Invitrogen.
Fig. 4.
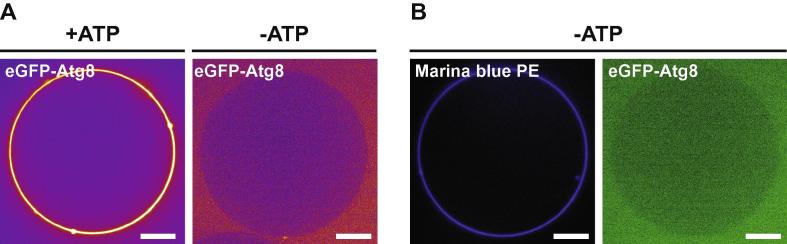
Reconstitution of eGFP-Atg8 conjugation using GUVs. (A) GUVs incubated with monomeric eGFP-Atg8 in the presence of Atg3, Atg7 and the Atg12–Atg5-Atg16 complex. On the left ATP was added to the reaction. On the right ATP was omitted. The fluorescence signal of eGFP-Atg8 is shown in false color (spectrum). (B) Same reaction as in (A) right panel but here the membrane of the GUV is labeled by incorporation of Marina blue PE. For the conjugation reaction the proteins were used at the final concentrations of 80 nM (Atg3 and Atg7) and 400 nM (eGFP-Atg8 and Atg12–Atg5-Atg16). The reaction was performed in the presence or absence of 1 mM ATP. Scale bars: 5 μm.
2.6. Atg8 conjugation on GUVs
Electroformed GUVs were diluted 1:2 or 1:4 in 15 mM HEPES, pH 7.5, 135 mM NaCl buffer and gently transferred to a 96-well glass-bottom microplate (Greiner Bio-One). Prior to transferring GUVs, the wells were incubated with 5 mg/ml BSA in 50 mM Tris–HCl pH 7.4, 150 mM NaCl buffer for 1 h and subsequently washed once with 15 mM HEPES, pH 7.5, 135 mM NaCl buffer. To conjugate Atg8 to PE containing membrane the conjugation reaction was performed at the following final proteins concentrations: 400 nm mGFP-Atg8 and Atg12–Atg5-Atg16, 80 nM Atg3 and Atg7. ATP and MgCl2 were added to final concentrations of 1 mM and 0.5 mM, respectively. All components required for Atg8 lipidation were pipetted directly to the 96-well glass-bottom microplate (Greiner Bio-One) containing GUVs. The final volume of the Atg8 lipidation mix and GUVs was 40 μl per well. The reaction was carried for 30 min in room temperature in the dark. Microscopic images were acquired using confocal spinning disc microscope (Visitron) and processed with ImageJ software.
3. Results and discussion
In order to successfully reconstitute the Atg8–PE conjugation reaction and obtain reproducible results two factors are particularly important. First, the proteins used have to be of sufficient purity and should be purified via a gel filtration column to remove potentially aggregated and chaperone bound proteins. Secondly, the lipids used to prepare the SUVs or GUVs should be of high quality and stored at −80 °C under argon or nitrogen to prevent hydrolysis and oxidation. The lipids used in this study for the SUVs were from E. coli total extracts or composed of synthetic lipids. In principle SUVs can be of any lipid composition given that the mixture is able to form bilayers. When preparing the SUVs or GUVs the phase transition temperatures of the lipids in the mix should be considered and if applicable the temperature at which the vesicles are prepared should be increased to a temperature above this value.
Fig. 2 shows a Coomassie-stained urea SDS–PAGE gel of an Atg8 conjugation reaction using Atg7 (1 μM), Atg3 (1 μM), Atg8 (5 μM) and E. coli lipid derived SUVs. In addition, ATP and MgCl2 were added (all components). The reaction was allowed to proceed over night at 30 °C. Atg8-lipidation is generally inferred from the conversion into a faster migrating band. The faster running behavior of lipidated Atg8 is more apparent when 6 M urea is added to the separating gel. In order to convert Atg8 to a faster migrating species the E1-like enzyme Atg7, the E2-like Atg3, the substrate in form of PE or PS containing SUVs and ATP are required (Fig. 2 and [35]). A number of reaction intermediates can be detected using this system. One of these is the Atg8–Atg3 substrate-E2 intermediate shown in Fig. 1 [15,35]. Another intermediate is the Atg8–AMP produced by the E1-like Atg7 appearing between the free and the lipidated Atg8 [34]. In addition, the Atg8–Atg7 intermediate is detectable in this system [11,12].
As apparent from Fig. 2 the machinery including Atg7, Atg3 and high PE containing SUVs is sufficient for Atg8 lipidation. However, the reaction is vastly stimulated by the presence of the Atg12–Atg5 conjugate or the Atg12–Atg5-Atg16 complex (Fig. 3A and [14,15]). This stimulating activity offers an explanation for the absolute requirement of these components for Atg8 lipidation in vivo. Fig. 3A shows a direct side-by-side comparison of two Atg8 lipidation reactions in the absence (left) and presence (right) of the Atg12–Atg5-Atg16 complex. The reactions contained 1 μM Atg7, 1 μM Atg3, 1 μM Atg12–Atg5-Atg16, 5 μM Atg8 and E. coli derived SUVs. Whereas in the presence of the Atg12–Atg5-Atg16 complex conjugation is readily detectable after 15 min and almost complete after 60 min, in the absence of the Atg12–Atg5-Atg16 complex the first sign of Atg8 lipidation is detectable only after 60 min and the reaction is not even complete after 4 h. It is therefore advantageous to include the Atg12–Atg5-Atg16 complex or the Atg12–Atg5 conjugate in the assay if effects of the lipid composition, buffer compositions or mutations/modifications in the conjugating proteins are studied. Otherwise there is the risk that reaction intermediates are studied that are not rate limiting in vivo.
A number of in vitro assays to analyze the catalytic cleavage of the C-terminus of ATG8 family proteins by ATG4 isoforms have been devised [23,46–51]. Here we present an assay to determine the de-lipidation activity of S. cerevisiae (Fig. 3B). This assay is similar to the LC3B and GABARAP de-lipidation assay described for human ATG4B [51] and exploits a shift towards slower migration behavior on a urea-containing SDS–PAGE gel (Fig. 3B). In the assay shown Atg8 lacking the C-terminal arginine is lipidated by the conjugation machinery containing Atg7, Atg3 and the Atg12–Atg5 conjugate in the presence of SUVs, ATP and MgCl2. After 30 min recombinant Atg4 and EDTA to chelate the MgCl2 are added to the reaction. As apparent from the gel the deconjugation reaction is complete after only 2 min showing that Atg4 has a high catalytic activity towards the lipidated form of Atg8. The assay is therefore well suited to analyze the effect of the lipid composition, posttranslational modifications or other factors on the activity of Atg4. We have noticed that at very high concentrations of Atg4 relative to its substrate Atg8 tended to non-specifically degrade proteins. This effect should be controlled for as it may result in misinterpretation of the de-lipidation experiments.
Reconstitution experiments using SUVs have yielded numerous crucial insights into the Atg8 conjugation machinery. However, due to their small size (usually around 100 nm diameter) they do not yield spatial information when imaged by light microscopy. This drawback can be overcome using GUVs that are much larger (>1 μm diameter) than SUVs and can thus be imaged using wide-field, spinning disk or confocal microscopy. Reconstitution systems employing GUVs have been successfully used for the reconstitution of Atg8 conjugation [14,44,45]. Spinning disk and confocal imaging can be combined with fluorescence recovery after photo bleaching (FRAP) experiments to obtain information about the turnover rate of the tagged or labeled proteins [44]. Atg8 itself or the other factors of the conjugation machinery can be fluorescently tagged and their membrane recruitment and localization pattern on the GUV membrane observed. Fluorescent labeling can be achieved by fusion of the protein to fluorescent proteins such as monomeric eGFP or mCherry [14,45] or by coupling of the protein to a fluorophore [44]. In case fluorescent fusion proteins are used their functionality can be tested in vivo by expressing the receptive fusion protein in a deletion background followed by assays for autophagic activity. Fig. 4 shows a conjugation reaction using GUVs. In our experience almost no detectable Atg8 conjugation as inferred from the localization of eGFP-Atg8 conjugation to the GUV membrane can be observed in the absence of Atg12–Atg5 conjugate or the Atg12–Atg5-Atg16 complex [14]. It is therefore mandatory to include either the conjugate or the complex in the assay. For the GUV reconstitution system the protein concentration can be lower than for the bulk assay employing SUVs as fluorescent imaging is able to detect relatively small concentrations of the fluorescent protein on the two-dimensional surface of the GUVs. The membrane of the GUVs can be labeled by incorporation of small amounts (usually 0.1–2% mol/mol) of fluorescently labeled lipids. Most commonly these are derivatives of PE coupled fluorophores such as rhodamine, Oregon-green or Marina-blue.
4. Conclusions
In vitro systems reconstituting the conjugation of Atg8 family proteins have yielded crucial insights into their mechanisms of action. Notably, these systems are the only means of defining whether a given factor is not only necessary but also sufficient for the activity of interest. This information can then be used to introduce targeted mutations, the effect of which can then be tested on autophagosome formation in vivo. It is expected that in vitro reconstitution systems will continue to yield valuable information on the mechanism of action of further autophagy proteins and/or their post-translational modifications.
Acknowledgements
The research leading to these results has received funding from the European Research Council under the European Community’s Seventh Framework Programme (FP7/2007-2013)/ERC grant agreement No. 260304, from the FWF Austrian Science Fund (grant number P25546-B20) and by the EMBO Young Investigator Program to S.M. We also acknowledge funding by the VIPS Program of the Austrian Federal Ministry of Science and Research and the City of Vienna to J.S.-M. The authors declare no conflict of interests.
References
- 1.Mizushima N., Komatsu M. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 2.Randow F., MacMicking J.D., James L.C. Science. 2013;340:701–706. doi: 10.1126/science.1233028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kraft C., Martens S. Curr. Opin. Cell Biol. 2012;24:496–501. doi: 10.1016/j.ceb.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 4.Tsukada M., Ohsumi Y. FEBS Lett. 1993;333:169–174. doi: 10.1016/0014-5793(93)80398-e. [DOI] [PubMed] [Google Scholar]
- 5.Harding T.M., Morano K.A., Scott S.V., Klionsky D.J. J. Cell Biol. 1995;131:591–602. doi: 10.1083/jcb.131.3.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klionsky D.J., Cregg J.M., Dunn W.A., Jr., Emr S.D., Sakai Y., Sandoval I.V., Sibirny A., Subramani S., Thumm M., Veenhuis M., Ohsumi Y. Dev. Cell. 2003;5:539–545. doi: 10.1016/s1534-5807(03)00296-x. [DOI] [PubMed] [Google Scholar]
- 7.Kirisako T., Baba M., Ishihara N., Miyazawa K., Ohsumi M., Yoshimori T., Noda T., Ohsumi Y. J. Cell Biol. 1999;147:435–446. doi: 10.1083/jcb.147.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ichimura Y., Kirisako T., Takao T., Satomi Y., Shimonishi Y., Ishihara N., Mizushima N., Tanida I., Kominami E., Ohsumi M., Noda T., Ohsumi Y. Nature. 2000;408:488–492. doi: 10.1038/35044114. [DOI] [PubMed] [Google Scholar]
- 9.Shpilka T., Weidberg H., Pietrokovski S., Elazar Z. Genome Biol. 2011;12:226. doi: 10.1186/gb-2011-12-7-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kirisako T., Ichimura Y., Okada H., Kabeya Y., Mizushima N., Yoshimori T., Ohsumi M., Takao T., Noda T., Ohsumi Y. J. Cell Biol. 2000;151:263–276. doi: 10.1083/jcb.151.2.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hong S.B., Kim B.W., Lee K.E., Kim S.W., Jeon H., Kim J., Song H.K. Nat. Struct. Mol. Biol. 2011;18:1323–1330. doi: 10.1038/nsmb.2165. [DOI] [PubMed] [Google Scholar]
- 12.Noda N.N., Satoo K., Fujioka Y., Kumeta H., Ogura K., Nakatogawa H., Ohsumi Y., Inagaki F. Mol. Cell. 2011;44:462–475. doi: 10.1016/j.molcel.2011.08.035. [DOI] [PubMed] [Google Scholar]
- 13.Taherbhoy A.M., Tait S.W., Kaiser S.E., Williams A.H., Deng A., Nourse A., Hammel M., Kurinov I., Rock C.O., Green D.R., Schulman B.A. Mol. Cell. 2011;44:451–461. doi: 10.1016/j.molcel.2011.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Romanov J., Walczak M., Ibiricu I., Schuchner S., Ogris E., Kraft C., Martens S. EMBO J. 2012;31:4304–4317. doi: 10.1038/emboj.2012.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hanada T., Noda N.N., Satomi Y., Ichimura Y., Fujioka Y., Takao T., Inagaki F., Ohsumi Y. J. Biol. Chem. 2007;282:37298–37302. doi: 10.1074/jbc.C700195200. [DOI] [PubMed] [Google Scholar]
- 16.Oh-oka K., Nakatogawa H., Ohsumi Y. J. Biol. Chem. 1852;283(2008):21847–21852. doi: 10.1074/jbc.M801836200. [DOI] [PubMed] [Google Scholar]
- 17.Sakoh-Nakatogawa M., Matoba K., Asai E., Kirisako H., Ishii J., Noda N.N., Inagaki F., Nakatogawa H., Ohsumi Y. Nat. Struct. Mol. Biol. 2013;20:433–439. doi: 10.1038/nsmb.2527. [DOI] [PubMed] [Google Scholar]
- 18.Mizushima N., Noda T., Yoshimori T., Tanaka Y., Ishii T., George M.D., Klionsky D.J., Ohsumi M., Ohsumi Y. Nature. 1998;395:395–398. doi: 10.1038/26506. [DOI] [PubMed] [Google Scholar]
- 19.Mizushima N., Sugita H., Yoshimori T., Ohsumi Y. J. Biol. Chem. 1998;273:33889–33892. doi: 10.1074/jbc.273.51.33889. [DOI] [PubMed] [Google Scholar]
- 20.Shintani T., Mizushima N., Ogawa Y., Matsuura A., Noda T., Ohsumi Y. EMBO J. 1999;18:5234–5241. doi: 10.1093/emboj/18.19.5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mizushima N., Noda T., Ohsumi Y. EMBO J. 1999;18:3888–3896. doi: 10.1093/emboj/18.14.3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakatogawa H., Ishii J., Asai E., Ohsumi Y. Autophagy. 2012;8:177–186. doi: 10.4161/auto.8.2.18373. [DOI] [PubMed] [Google Scholar]
- 23.Satoo K., Noda N.N., Kumeta H., Fujioka Y., Mizushima N., Ohsumi Y., Inagaki F. EMBO J. 2009;28:1341–1350. doi: 10.1038/emboj.2009.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim J., Huang W.-P., Klionsky D.J. J. Cell Biol. 2001;152:51–64. doi: 10.1083/jcb.152.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rogov V., Dötsch V., Johansen T., Kirkin V. Mol. Cell. 2014;53:167–178. doi: 10.1016/j.molcel.2013.12.014. [DOI] [PubMed] [Google Scholar]
- 26.Noda N.N., Kumeta H., Nakatogawa H., Satoo K., Adachi W., Ishii J., Fujioka Y., Ohsumi Y., Inagaki F. Genes Cells. 2008;13:1211–1218. doi: 10.1111/j.1365-2443.2008.01238.x. [DOI] [PubMed] [Google Scholar]
- 27.Ichimura Y., Kumanomidou T., Sou Y.-S., Mizushima T., Ezaki J., Ueno T., Kominami E., Yamane T., Tanaka K., Komatsu M. J. Biol. Chem. 2008;283:22847–22857. doi: 10.1074/jbc.M802182200. [DOI] [PubMed] [Google Scholar]
- 28.Pankiv S., Clausen T.H., Lamark T., Brech A., Bruun J.-A., Outzen H., Øvervatn A., Bjørkøy G., Johansen T. J. Biol. Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 29.Ylä-Anttila P., Vihinen H., Jokitalo E., Eskelinen E.-L. Autophagy. 2009;5:1180–1185. doi: 10.4161/auto.5.8.10274. [DOI] [PubMed] [Google Scholar]
- 30.Hayashi-Nishino M., Fujita N., Noda T., Yamaguchi A., Yoshimori T., Yamamoto A. Nat. Cell Biol. 2009 doi: 10.1038/ncb1991. [DOI] [PubMed] [Google Scholar]
- 31.Kishi-Itakura C., Koyama-Honda I., Itakura E., Mizushima N. J. Cell Sci. 2014;127:4089–4102. doi: 10.1242/jcs.156034. [DOI] [PubMed] [Google Scholar]
- 32.Sou Y.S., Waguri S., Iwata J., Ueno T., Fujimura T., Hara T., Sawada N., Yamada A., Mizushima N., Uchiyama Y., Kominami E., Tanaka K., Komatsu M. Mol. Biol. Cell. 2008;19:4762–4775. doi: 10.1091/mbc.E08-03-0309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Uemura T., Yamamoto M., Kametaka A., Sou Y.S., Yabashi A., Yamada A., Annoh H., Kametaka S., Komatsu M., Waguri S. Mol. Cell. Biol. 2014;34:1695–1706. doi: 10.1128/MCB.01327-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nath S., Dancourt J., Shteyn V., Puente G., Fong W.M., Nag S., Bewersdorf J., Yamamoto A., Antonny B., Melia T.J. Nat. Cell Biol. 2014;16:415–424. doi: 10.1038/ncb2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ichimura Y., Imamura Y., Emoto K., Umeda M., Noda T., Ohsumi Y. J. Biol. Chem. 2004;279:40584–40592. doi: 10.1074/jbc.M405860200. [DOI] [PubMed] [Google Scholar]
- 36.Hanada T., Satomi Y., Takao T., Ohsumi Y. FEBS Lett. 2009;583:1078–1083. doi: 10.1016/j.febslet.2009.03.009. [DOI] [PubMed] [Google Scholar]
- 37.McMahon H.T., Kozlov M.M., Martens S. Cell. 2010;140:601–605. doi: 10.1016/j.cell.2010.02.017. [DOI] [PubMed] [Google Scholar]
- 38.Gerlach H., Laumann V., Martens S., Becker C.F.W., Goody R.S., Geyer M. Nat. Chem. Biol. 2010;6:46–53. doi: 10.1038/nchembio.268. [DOI] [PubMed] [Google Scholar]
- 39.Vanni S., Hirose H., Barelli H., Antonny B., Gautier R. Nat. Commun. 2014;5:4916. doi: 10.1038/ncomms5916. [DOI] [PubMed] [Google Scholar]
- 40.Nair U., Jotwani A., Geng J., Gammoh N., Richerson D., Yen W.L., Griffith J., Nag S., Wang K., Moss T., Baba M., McNew J.A., Jiang X., Reggiori F., Melia T.J., Klionsky D.J. Cell. 2011;146:290–302. doi: 10.1016/j.cell.2011.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakatogawa H., Ichimura Y., Ohsumi Y. Cell. 2007;130:165–178. doi: 10.1016/j.cell.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 42.Weidberg H., Shpilka T., Shvets E., Abada A., Shimron F., Elazar Z. Dev. Cell. 2011;20:444–454. doi: 10.1016/j.devcel.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 43.Martens S., Mcmahon H. Nat. Rev. Mol. Cell Biol. 2008;9:543–556. doi: 10.1038/nrm2417. [DOI] [PubMed] [Google Scholar]
- 44.Kaufmann A., Beier V., Franquelim H.G., Wollert T. Cell. 2014;156:469–481. doi: 10.1016/j.cell.2013.12.022. [DOI] [PubMed] [Google Scholar]
- 45.Sawa-Makarska J., Abert C., Romanov J., Zens B., Ibiricu I., Martens S. Nat. Cell Biol. 2014;16:425–433. doi: 10.1038/ncb2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scherz-Shouval R., Shvets E., Fass E., Shorer H., Gil L., Elazar Z. EMBO J. 2007;26:1749–1760. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li M., Chen X., Ye Q.Z., Vogt A., Yin X.M. Autophagy. 2012;8:401–412. doi: 10.4161/auto.18777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nguyen T.G., Honson N.S., Arns S., Davis T.L., Dhe-Paganon S., Kovacic S., Kumar N.S., Pfeifer T.A., Young R.N. Assay Drug Dev. Technol. 2014;12:176–189. doi: 10.1089/adt.2013.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li M., Hou Y., Wang J., Chen X., Shao Z.-M., Yin X.-M. J. Biol. Chem. 2011;286:7327–7338. doi: 10.1074/jbc.M110.199059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shu C.W., Drag M., Bekes M., Zhai D., Salvesen G.S., Reed J.C. Autophagy. 2010;6:936–947. doi: 10.4161/auto.6.7.13075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kumanomidou T., Mizushima T., Komatsu M., Suzuki A., Tanida I., Sou Y.S., Ueno T., Kominami E., Tanaka K., Yamane T. J. Mol. Biol. 2006;355:612–618. doi: 10.1016/j.jmb.2005.11.018. [DOI] [PubMed] [Google Scholar]