

Abstract
Morphogenesis is crucial during development to generate organs and tissues of the correct size and shape. During Drosophila late eye development, interommatidial cells (IOCs) rearrange to generate the highly organized pupal lattice, in which hexagonal ommatidial units pack tightly. This process involves the fine regulation of adherens junctions (AJs) and of adhesive E-Cadherin (E-Cad) complexes. Localized accumulation of Bazooka (Baz), the Drosophila PAR3 homolog, has emerged as a critical step to specify where new E-Cad complexes should be deposited during junction remodeling. However, the mechanisms controlling the correct localization of Baz are still only partly understood. We show here that Drosophila Magi, the sole fly homolog of the mammalian MAGI scaffolds, is an upstream regulator of E-Cad-based AJs during cell rearrangements, and that Magi mutant IOCs fail to reach their correct position. We uncover a direct physical interaction between Magi and the Ras association domain protein RASSF8 through a WW domain-PPxY motif binding, and show that apical Magi recruits the RASSF8-ASPP complex during AJ remodeling in IOCs. We further show that this Magi complex is required for the cortical recruitment of Baz and of the E-Cad-associated proteins α- and β-catenin. We propose that, by controlling the proper localization of Baz to remodeling junctions, Magi and the RASSF8-ASPP complex promote the recruitment or stabilization of E-Cad complexes at junction sites.
Keywords: Adherens junctions, MAGI scaffolds, Morphogenesis
Highlighted article: The apical scaffold protein Magi regulates localization of the polarity protein Bazooka and hence E-cadherin deposition and adherens junction remodelling in Drosophila eye epithelial cells.
INTRODUCTION
During development, tissues undergo morphogenesis to adopt their specific sizes and shapes, a process driven by the behaviors of individual cells (Heisenberg and Bellaïche, 2013). For instance, the stereotypical arrangement of hexagonal ommatidia in the Drosophila adult compound eye is achieved during earlier pupal stages through cell rearrangements. Each ommatidium is composed of eight photoreceptors beneath four cone cells, and is surrounded by two primary pigment cells (Bao et al., 2010). Ommatidia are separated by secondary and tertiary pigment cells [interommatidial cells (IOCs)] arranged in a repetitive hexagonal lattice. Initially, an excess of unpatterned interommatidial precursor cells (IPCs) lies between the ommatidia. Between 18 h and 42 h after puparium formation (APF), the unpatterned IPCs first rearrange into a single row between neighboring ommatidia and the excess are removed by apoptosis, giving rise to the stereotypical repetitive hexagonal units of the Drosophila eye (Cagan, 2009).
Regulated cell-cell adhesion is crucial for correct formation of the interommatidial lattice. For example, IPC rearrangement requires the careful modulation of adherens junctions (AJs) (Tepass and Harris, 2007). This remodeling of AJs involves the destruction of old E-Cadherin (E-Cad)-based contacts and the creation of new ones (E-Cad is also known as Shotgun – FlyBase). Studies in Drosophila embryos and pupae have highlighted crucial roles for Bazooka (Baz), the Drosophila PAR3 homolog, in specifying de novo E-Cad deposition during remodeling (Desai et al., 2013; McGill et al., 2009; McKinley et al., 2012; Walther and Pichaud, 2010) and for the actin cytoskeleton in controlling the dynamics of individual junctions (Levayer et al., 2011; Rauzi et al., 2010). In the Drosophila pupal eye, IOC rearrangements and hence junction remodeling are also regulated by heterophilic interactions between Irre family adhesion molecules, such as Hibris and Roughest [homologs of vertebrate Nephrin (NPHS1) and NEPH1 (KIRREL), respectively] (Bao and Cagan, 2005; Bao et al., 2010; Reiter et al., 1996). Loss of any of these adhesion molecules leads to defects in IOC rearrangement as well as in apoptosis, suggesting that IOC rearrangement is necessary for apoptosis.
Recently, the N-terminal Ras association (RA) domain-containing protein RASSF8 (Ras association domain family 8) was shown to regulate Drosophila pupal eye morphogenesis and the AJ integrity of IOCs (Langton et al., 2009). In Drosophila epithelial cells, RASSF8 is localized apically and binds to ASPP (Ankyrin-repeat, SH3-domain, and proline-rich-region containing protein), the fly homolog of the p53 activating partners ASPP1 (PPP1R13B) and ASPP2 (TRP53BP2). The RASSF8-ASPP complex regulates the activity of Src kinases, which promote AJ remodeling (Langton et al., 2007, 2009; Vidal et al., 2006). How this complex interacts with other key players of AJ regulation to control AJ remodeling during eye morphogenesis remains unexplored.
MAGI (membrane-associated guanylate kinase inverted) proteins are molecular scaffolds with several protein-protein interaction (WW and PDZ) domains. MAGI proteins localize at apical junctions and bind β-catenin as well as other apical proteins (Dobrosotskaya and James, 2000; Ide et al., 1999; Kawajiri et al., 2000). For example, MAGI1 binds and activates the guanine nucleotide exchange factor for the junction remodeling small GTPase RAP1, which in turn regulates maturation of the AJs (Sakurai et al., 2006). MAGIs also bind to and stabilize the tumor suppressor PTEN (phosphatase and tensin homolog) at the membrane, leading to increased PTEN activity (Kotelevets et al., 2005; Subauste et al., 2005; Wu et al., 2000a,b; Zmajkovicova et al., 2013). Taken together, these studies suggest that MAGIs may modulate epithelial cell adhesion and, through PTEN, restrict proliferation. MAGI2 and MAGI3 mutations have been identified in prostate, colorectal and breast cancer genomes (Banerji et al., 2012; Berger et al., 2011; Pleasance et al., 2010), suggesting that MAGI proteins might be involved in cancers.
Here, we investigate the function of Drosophila Magi, the sole fly homolog of the three human MAGI genes, and demonstrate that it regulates AJ dynamics and IOC number. In Magi mutants, IOCs of the pupal lattice fail to rearrange, resulting in eyes with mislocalized supernumerary cells. We have identified RASSF8 as a Magi binding partner and show that these proteins interact physically and functionally during pupal eye development. Magi localizes at the AJs in Drosophila pupal eye epithelial cells, where it recruits the RASSF8-ASPP complex. During IOC rearrangement, Magi is required for E-Cad-based junction integrity, a function dependent on its WW domains and on its interaction with RASSF8. We show that Magi, RASSF8 and ASPP are required for the recruitment of Baz at the membrane. We propose that, by recruiting the RASSF8-ASPP complex at the AJs during remodeling in Drosophila eye IOCs, Magi helps specify domains of the membrane where Baz promotes the accumulation of E-Cad to generate new junctions.
RESULTS
Magi controls cell numbers in the Drosophila pupal eye
To study Magi function in Drosophila we generated mutations by mobilizing a transposon (P element P{GSV6}GS6093) located just upstream of the Magi locus, between Magi and CG9406. Among the local genomic deletions generated by the imprecise excision of this transposon, excision ex214 affected only Magi. Genomic PCR mapped the deficiency to the first three exons of Magi, deleting the transcription start site, the initiating methionine, and the first half of the first WW domain. The rest of the Magi gene and the neighboring genes mago, CG9406 and Xbp1 are unaffected (supplementary material Fig. S1A and Fig. S2A). In Magiex214 mutant cells, no Magi protein expression could be detected using a polyclonal anti-Magi antibody that we generated directed against the PDZ3 and 4 domains (supplementary material Fig. S1B). Finally, western blot analysis using the anti-Magi antibody revealed the absence of a band at the expected size for the protein (∼130 kDa) in Magiex214 mutants and did not reveal any shorter products (supplementary material Fig. S1C), suggesting that no truncated protein is expressed. Together, these results suggest that Magiex214 is a molecular null allele.
Magiex214 homozygous flies are viable and fertile, but exhibit slightly enlarged wings (supplementary material Table S1) and mildly rough eyes (Fig. 1B,B′). The latter arises because the ommatidia are not surrounded by the proper numbers of neighbors and interommatidial bristles (six and three, respectively, in wild type) resulting in packing defects (Fig. 1D). In Magi mutants, ∼13% of ommatidia have abnormal numbers of neighbors compared with 0.4% in wild-type controls (Fig. 1D). Similarly, ∼35% of Magi mutant ommatidia have abnormal numbers of interommatidial bristles compared with 9.5% in wild type (Fig. 1E). This phenotype is not enhanced when Magiex214 is transheterozygous with a mapped deficiency that uncovers the Magi locus [Df(2R)Exel6072], arguing that Magiex214 behaves as a genetic null mutation. The rough eye phenotype and cell packing defects are completely rescued by low-level ubiquitous expression of full-length Magi [using the ubiquitin promoter, which drives expression of Magi transgenes at levels similar to (∼1.5-fold) the endogenous protein; supplementary material Fig. S1D,E], proving that the defects observed are specific to the Magi mutation (Fig. 1C-E).
Fig. 1.

Magi controls morphogenesis of the Drosophila pupal eye lattice. (A-C) Scanning electron micrographs showing the effect of Magi mutation on the external architecture of the adult eye. Magi mutant adults have a mild rough eye phenotype (B) compared with wild type (A), which is rescued by providing a ubiquitously expressed Magi transgene (C). (A′-C′) High magnification of the adult compound eye corresponding to A-C. The regular hexagonal packing of ommatidial units of the wild type (A′) is affected in Magi mutants (B′) and rescued by providing wild-type Magi (C′). (A″-C″) Pupal eye at 44 h APF stained for E-Cad, showing the ommatidial organization corresponding to A-C. Magi mutant eyes have extra IOCs (B″, arrowheads) compared with wild-type controls (A″). Total rescue is seen when a wild-type Magi transgene is provided (C″). (D,E) Quantification of the effects observed in A′-C′. The total number of ommatidia scored ranged from 433 to 541 depending on genotype. (F) Quantification of the number of IOCs per ommatidium (see Materials and Methods). (D-F) Error bars indicate s.e.m.; ***P<0.001, **P<0.01, n.s., not significant (unpaired t-test). Scale bars: 100 µm in A; 50 µm in A′; 10 µm in A″.
The mild rough eye in Magiex214 can be explained by the presence of additional mislocalized IOCs as well as extra interommatidial bristles in pupal retinas (Fig. 1A″,B″). There were on average 12.25±1.00 IOCs per ommatidium in the Magi mutant, as compared with 9.04±0.06 in wild type (see supplementary Materials and Methods for quantification procedures; Fig. 1F). Magiex214 pupal eyes also exhibited sorting defects, whereby secondary pigment cells contacted other secondary pigment cells instead of being surrounded by primary pigment cells (Fig. 1B″, arrowheads). The number of photoreceptors, cone cells and primary pigment cells was unaffected. These defects in IOC numbers and sorting were completely rescued by a wild-type Magi transgene (average of 8.97±0.14 IOCs per ommatidium; Fig. 1C″,F). Conversely, overexpression of Magi in all pupal eye cells (using GMR-Gal4) led to a loss of IOCs (average of 7.40±0.65 IOCs per ommatidium; supplementary material Fig. S2B, arrowheads), leading to packing defects and an externally rough eye (see Fig. 3K). These results show that Magi is required for the control of IOC number and for the proper sorting of IOCs during Drosophila pupal eye development.
Fig. 3.

Magi acts through RASSF8. (A-F) Pupal eye at 44 h APF stained for E-Cad showing ommatidial organization. Arrowheads highlight extra IOCs compared with wild type. Whereas the supernumerary IOC phenotype in Magi mutants (A) is completely rescued by introducing a wild-type Magi transgene (B), only partial rescue is seen with a MagiΔWW transgene (C). Similarly, only a wild-type RASSF8 transgene (E), and not a RASSF8YA transgene mutated in its Magi-interacting motif (F), is able to fully rescue the extra IOC phenotype in RASSF8 mutants (D). (G) Quantification of the number of IOCs per ommatidium in A-F (see Materials and Methods). s.e.m. is shown; ***P<0.001, **P<0.01, *P<0.05 (unpaired t-test). (H) Strength of rescue obtained after normalization. Normalization was obtained by dividing the number of IOCs in the different rescue experiments by the number of IOCs in the corresponding Magi or RASSF8 mutant background. (I-M) Scanning electron microscopy of Drosophila adult eyes following GMR-Gal4-driven overexpression. Full-length Magi overexpression produces a mild rough eye (K) compared with wild type (I), while MagiΔWW has a weaker effect (J) (all transgenes are inserted at the same locus ensuring equivalent expression levels). This effect of Magi is strongly suppressed by removing one copy of RASSF8 (RASSF86/+; L) but is enhanced by co-expressing RASSF8 (M). (I′-M′) High magnification of adult eyes corresponding to I-M. Note the almost wild-type packing after MagiΔWW (J′) overexpression, and the very disorganized eye structure after Magi and RASSF8 co-overexpression (M′). (N) Quantification of the effects observed in I′-M′, highlighting the suppression of the effects of Magi overexpression by RASSF8 mutants. n, total number of ommatidia scored per genotype. Scale bars: 100 µm in I; 50 µm in I′.
We then tested whether the extra IOCs in Magi mutants could be due to a defect in the developmental apoptosis of IOCs occurring between 18 and 42 h APF (Cagan, 2009). Assessing the number of apoptotic cells in wild-type and Magi mutant tissues within the same retinas, we did not detect any significant difference (6.12±2.20 and 5.64±1.89 apoptotic cells per arbitrary surface unit, respectively; P=0.395, paired t-test; supplementary material Fig. S2C,D). We note, however, that the small number of extra IOCs in Magi mutants suggests that Magi would only affect a small proportion of the cells undergoing developmental apoptosis in the pupal eye, making it difficult to distinguish the Magi-regulated effects from the bulk of apoptotic events.
RASSF8 is a binding partner of Magi
To elucidate Magi function during eye development, we sought to identify its molecular partners. One candidate was ASPP, as ASPP loss-of-function alleles cause similar phenotypes to Magi mutations in the developing Drosophila eye (Langton et al., 2007), and because human ASPP2 binds to human MAGI1 (Pirozzi et al., 1997). Furthermore, several clones of Drosophila Magi were isolated in a yeast two-hybrid screen using RASSF8, a partner of ASPP, as bait. RASSF8 is a strong binding partner of ASPP, and RASSF8 mutant pupal eyes exhibit a phenotype similar to that of Magi mutants (Langton et al., 2009). We therefore first tested the Magi-RASSF8 interaction by co-immunoprecipitation (co-IP), and confirmed that Myc-tagged RASSF8 could be co-purified with FLAG-tagged Magi (Fig. 2B). Although ASPP alone did not co-immunoprecipitate with Magi, when ASPP was co-expressed with RASSF8 both proteins co-purified with FLAG-Magi (Fig. 2B). This suggests that ASPP might not interact directly with Magi in Drosophila, but forms a ternary complex through RASSF8.
Fig. 2.

Magi binds to RASSF8. (A) Schematics of Drosophila RASSF8 and Magi structure. Highlighted are the RASSF8 residues 294-303 with a PPxY motif, the Magi residues 285-378 (green line) representing the common fragment of the Magi clones identified by two-hybrid analysis using RASSF8 as bait, and the Magi residues 790-1202 (red line) representing the fragment used to raise the anti-Magi antibody. The PDZ (PSD95/Dlg1/Zo1), WW, Ras association (RA) and coiled-coil (C) domains are shown. (B) RASSF8 co-immunoprecipitates with Magi. (Left) Western blot of cleared lysates from cells expressing either FLAG-tagged GFP (FLAG-GFP, negative control) or FLAG-Magi with Myc-ASPP or Myc-RASSF8, or both. (Right) Western blot following immunoprecipitation with anti-FLAG. Myc-RASSF8 co-immunoprecipitates only with FLAG-Magi (lanes 5 and 6) and not with FLAG-GFP (lanes 2 and 3). Myc-ASPP co-immunoprecipitates with FLAG-Magi only in the presence of RASSF8 (lane 6). (C) The PPxY motif in RASSF8 is required for the interaction with Magi. (Left) Western blot of cleared lysates from cells expressing Myc-Magi with either FLAG-GFP (negative control) or FLAG-RASSF8 (WT), or FLAG-RASSF8Y297A in which Y297 is substituted for alanine (Y297A). (Right) Western blot following immunoprecipitation with anti-FLAG. Myc-Magi co-immunoprecipitates only with FLAG-RASSF8 (lane 2) and not with FLAG-GFP (lane 1) or FLAG-RASSF8Y297A (lane 3). (D) The WW domains in Magi are required for the interaction with RASSF8. (Left) Western blot of cleared lysates from cells expressing HA-RASSF8 with different FLAG-Magi mutant constructs: wild-type Magi (WT), Magi deleted of the two WW domains (ΔWW), Magi in which P232 in the first WW is mutated (P232A), and Magi in which P370 in the second WW is mutated (P370A). (Right) Western blot following immunoprecipitation with anti-FLAG. HA-RASSF8 co-immunoprecipitates only with HA-Magi (WT, lane 1) and not with MagiΔWW (ΔWW, lane 2) or Magi with point mutations in the WW domains (lanes 3 and 4).
The Magi fragments isolated in the two-hybrid screen with RASSF8 spanned the two WW domains of Magi (Fig. 2A; supplementary material Table S2). Since RASSF8 contains a PPxY motif, which is a known ligand for WW domains, we tested whether Magi could be interacting, via its WW domains, with RASSF8 through this motif. We generated a mutant RASSF8 protein in which tyrosine residue 297 of the PPxY motif was mutated to an alanine. This RASSF8Y297A mutant could no longer interact with Magi in co-IP experiments (Fig. 2C). To confirm that RASSF8 binds to the WW domains of Magi we generated three different mutant versions by (1) deleting the two WW domains (ΔWW), (2) substituting proline 323 in the first WW domain with an alanine (P323A), and (3) substituting proline 370 in the second WW domain with an alanine (P370A). In co-IP experiments, only the wild-type form of Magi pulled down RASSF8 (Fig. 2D). Mutating either WW domain was sufficient to abrogate the binding. These results show that binding of RASSF8 and Magi is mediated by a PPxY motif-WW domain interaction.
The physical interaction between RASSF8 and Magi is required for their full function
As Magi and RASSF8 form a complex, we investigated the consequences of disrupting the Magi-RASSF8 physical interaction on their activities.
Whereas the ubiquitous expression of full-length Magi fully rescued the adult external rough eye phenotype and packing defects of the Magi mutant (Fig. 1C-F), a mutant form of Magi lacking the WW RASSF8-interacting domains gave only a partial rescue (2% of ommatidia with the improper number of neighbors and 23% with bristle abnormalities). Furthermore, whereas the extra IOC phenotype in Magi mutants (12.25±1.00 IOCs per ommatidium compared with 9.04±0.06 in wild type, a 36% increase) was completely rescued by wild-type Magi (Fig. 3B,G), it was only partially rescued by MagiΔWW (10.69±0.92 IOCs per ommatidium, an 18% increase; Fig. 3C,G). The MagiΔWW mutant is expressed at very similar levels to wild-type Magi and ∼1.5-fold more than endogenous Magi, showing that the weaker effects of MagiΔWW are not due to reduced protein levels (supplementary material Fig. S1D,E). This demonstrates that the WW domains of Magi are required for full function.
Similarly, the extra IOC phenotype observed in RASSF8 mutants (10.91±0.36, a 21% increase compared with wild type) was completely rescued by expression of a wild-type form of RASSF8 (Fig. 3E,G), but only partially rescued by expression of the RASSF8Y297A variant in which the Magi-binding PPxY motif was mutated (9.48±0.28, a 5% increase; Fig. 3F,G). We note that when normalized to the extra IOC phenotype observed in the Magi and RASSF8 mutants, the extents of rescue by MagiΔWW and RASSF8Y297A were similar (0.872 and 0.869, respectively; Fig. 3H). Together, these results strongly suggest that the physical interaction between Magi and RASSF8 is required for their functions in controlling IOC numbers and their rearrangement.
RASSF8 mediates the effects of Magi in the Drosophila eye
To further establish the relevance of RASSF8 for Magi function, we tested the functional requirements for RASSF8 under conditions in which Magi overexpression elicits ommatidial packing defects. As expected, MagiΔWW lacking the RASSF8-interacting WW domains gave a far milder phenotype than wild-type Magi when overexpressed (Fig. 3J,K,N), suggesting that the Magi-RASSF8 interaction is important for the effects of excess Magi.
Furthermore, the rough eye phenotype induced by Magi overexpression (Fig. 3K) was efficiently suppressed by removing one copy of RASSF8 and was strongly enhanced by co-expressing RASSF8 (Fig. 3L-N; note that overexpressed RASSF8 alone had no effect, Fig. 3N). In eyes with overexpressed Magi, 58.5% of ommatidia had defects. The proportion of defective ommatidia was reduced to 34.9% in a RASSF8 heterozygous background, but increased to 79.7% when RASSF8 was co-expressed (Fig. 3N). These results argue that RASSF8 mediates the effect of Magi (see also supplementary material Table S3) and, taken with the other data, suggest that the control of IOC numbers, cell rearrangements and packing during Drosophila pupal eye development requires a complex containing Magi and RASSF8.
Magi recruits RASSF8 and ASPP at AJs in the pupal eye
The RASSF8-ASPP complex is localized at AJs and has been proposed to regulate AJ remodeling that occurs in the eye around 24 h APF (Langton et al., 2007, 2009). Similar to RASSF8, in wing imaginal discs (Fig. 4A and supplementary material Fig. S1B) and in 24-h APF pupal eye discs (Fig. 4D) Magi colocalized with E-Cad at AJs (Fig. 4A,D). Its position was restricted to the AJ: apical to the septate junction protein Discs large (Dlg) (Fig. 4B) and basal to the subapical marker aPKC (Fig. 4A).
Fig. 4.
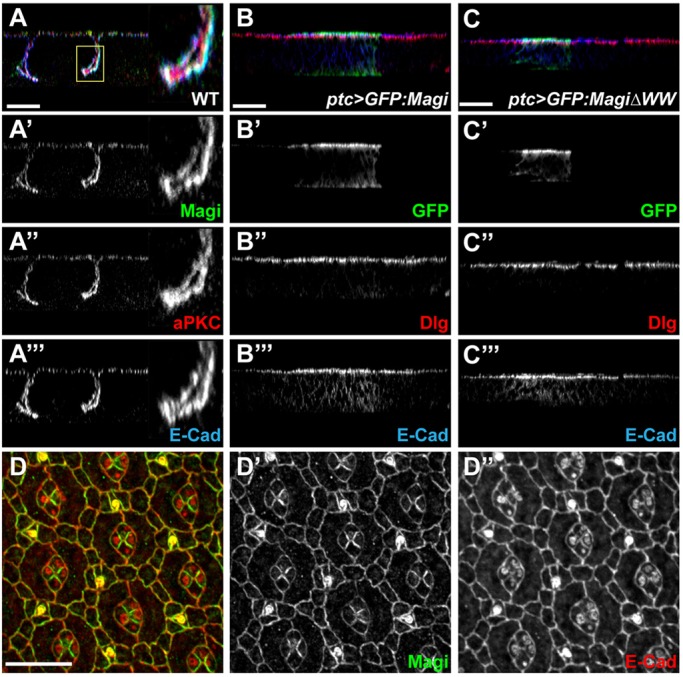
Magi AJ localization is independent of RASSF8 and ASPP. (A-A‴) In larval wing imaginal disc epithelial cells Magi (green, A′) colocalizes with E-Cad (blue, A‴) at the AJ level, basal to aPKC (red, A″). A confocal z-section is shown. The boxed region is shown at higher magnification to the right. (B-C‴) Confocal z-sections of larval wing discs overexpressing GFP-tagged Magi (B) or MagiΔWW (C) using the ptc-Gal4 driver. Both Magi constructs (GFP, green, B′,C′) colocalize with E-Cad (blue, B‴,C‴), apical to the septate junction protein Dlg (red, B″,C″). (D-D″) In 24-h APF pupal eye discs, Magi (green, D′) is localized at cell junctions in IOCs together with E-Cad (red, D″). Scale bars: 20 µm in A-C; 10 µm in D.
The physical interaction between Magi and RASSF8 raises the possibility that they could reciprocally regulate their localization at AJs. However, Magi localization at the AJ was found to be independent of both RASSF8 and ASPP. Focusing on the particular timing when RASSF8 and ASPP are required for AJ remodeling, both wild-type Magi and a mutant form lacking the WW domains localized correctly at AJs (expressed using ubiquitin promoter; Fig. 5A,B), even in the absence of endogenous Magi (Magi null mutant background). We obtained similar results in larval wing discs, where GFP-tagged wild-type and WW-deficient forms of Magi both localized at the AJs (expressed using patched-Gal4; Fig. 4B,C). These results show that the RASSF8-interacting WW domains are dispensable for Magi localization at AJs. Furthermore, the localization of Magi was unaffected in either RASSF8 or ASPP mutant cells, whether in wing imaginal discs (supplementary material Fig. S3A,B,E,F) or in 24-h APF eye discs (Fig. 5C,D). Finally, even in the absence of both RASSF8 and ASPP, Magi was still properly localized at the membrane of IOCs at 24 h APF (Fig. 5E), ruling out any potential redundancy between RASSF8 and ASPP. Taken together, these results show that Magi localizes at AJs independently of either RASSF8 or ASPP.
Fig. 5.
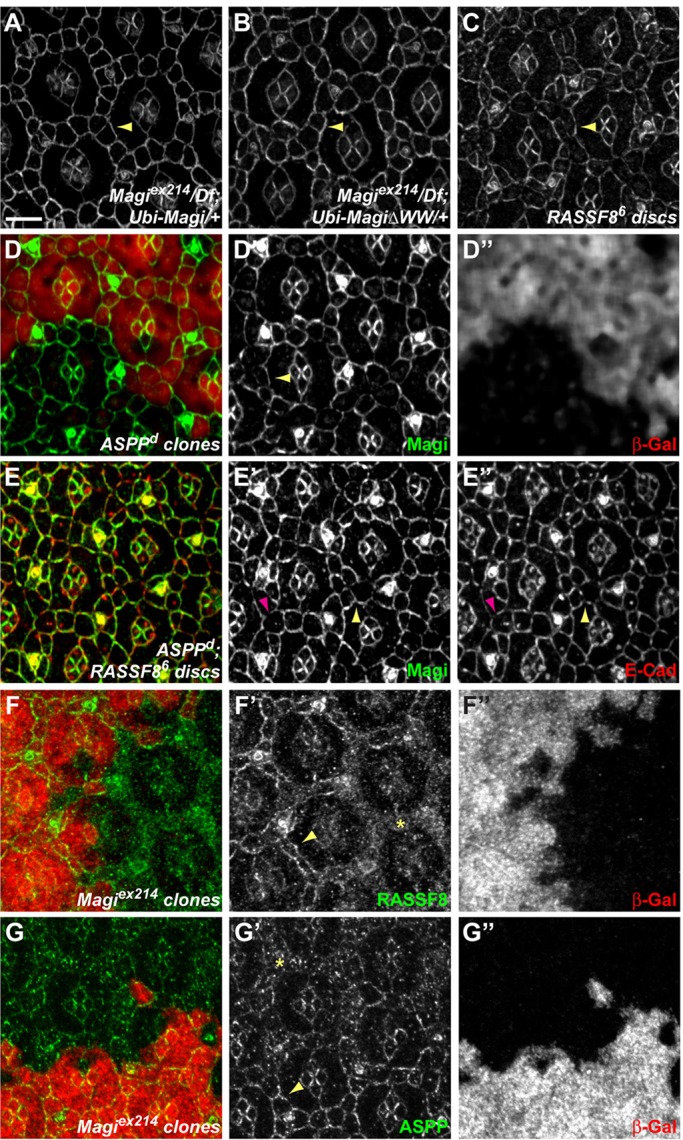
Magi recruits RASSF8 and ASPP. (A-C) 24-h APF pupal eye discs stained for Magi. Arrowheads mark membrane recruitment of Magi. In Magi null mutant discs, both Magi (A) and MagiΔWW (B) are recruited at the membrane. Similarly, in a RASSF8 whole mutant disc, Magi is correctly localized at junctions (C). (D-D″) At 24-h APF, in ASPP mutant clones marked by the absence of β-galactosidase (red, D″), Magi (green, D′) protein levels and apical localization are unaffected (arrowhead). (E-E″) At 24 h APF, in ASPP; RASSF8 double-mutant discs, Magi (green, E′) is properly localized at the cortex, even though a few gaps can be seen. There are many more gaps for E-Cad (red, E″), which overlap (purple arrowhead) or not (yellow arrowhead) with the Magi gaps. (F-G″) 24-h APF Magi mutant clones marked by the absence of β-galactosidase (red, F″,G″). RASSF8 (F, green; F′) and ASPP (G, green; G′) are mislocalized (asterisk) in Magi mutant IOCs compared with their membrane localization in wild-type tissues (arrowheads). Scale bar: 5 µm.
By contrast, RASSF8 and ASPP localization at AJs in pupal eye discs was dependent on Magi. In Magi mutant clones at 24 h APF both RASSF8 and ASPP were absent from the membrane and accumulated in the cytoplasm of IOCs (Fig. 5F,G). We note, however, that a residual amount of ASPP was still cortical in Magi mutant clones (Fig. 5G). This suggests the existence of an in vivo ternary molecular complex between Magi and RASSF8-ASPP and validates in pupal IOCs the direct physical interaction detected between Magi and RASSF8. However, RASSF8 and ASPP were not dependent on Magi for their AJ localization in larval wing disc epithelial cells (supplementary material Fig. S3C,D,G,H). This suggests that either the interaction between Magi and the RASSF8-ASPP complex is context dependent or that, at least in the larval disc, redundant localization mechanisms to Magi exist.
Magi affects AJ integrity in the pupal eye similarly to RASSF8 and ASPP
As ASPP and RASSF8 are required for AJ integrity and IPC rearrangement, we examined these processes in Magi mutants. During the formation of the interommatidial lattice, IPCs undergo extensive rearrangements. This process requires the redistribution of AJs (Tepass and Harris, 2007) and fails in the RASSF8 or ASPP mutants, leading to interrupted AJs at 24 h APF (Langton et al., 2007, 2009). We therefore investigated the morphology of AJs in Magi mutant cells, and observed interruptions in the E-Cad belt at 24 h APF (Fig. 6A). Whereas we could detect on average 0.25±0.10 interruptions in the cortical E-Cad staining per IOC in wild-type clones, this increased to 0.73±0.27 in neighboring Magi mutant clones (Fig. 6B). Using the same quantification method, we could detect 1.49±0.32 E-Cad gaps per IOC in ASPP mutants, confirming the similarity in the early E-Cad disruption phenotypes.
Fig. 6.

Magi mutant has abnormal AJs. (A-A″,C-C″,F-F″) 24-h APF pupal eye discs with Magi mutant clones marked by the absence of β-galactosidase (red, A″,F″) or the absence of Magi (red, C″). Yellow lines (A′,C′,F′) mark the boundaries between wild-type and mutant tissues. (A) In Magi mutant tissue, E-Cad staining (green, A′) is frequently interrupted (yellow arrowheads) compared with wild type (purple arrowheads). (C,F) In Magi mutant tissue, β-catenin (Arm; green, C′) and α-catenin (α-Cat; green, F′) are less cortical and accumulate in cytoplasmic vesicles in IOCs. (B,D,G) Quantification of E-Cad interruptions (B), Arm vesicles (D) and α-Cat vesicles (G) in wild-type and Magi mutant tissues corresponding to A, C and F, respectively. s.e.m. is shown; ***P<0.001 (unpaired t-test). (E,H) Quantification of Arm (E) and α-Cat (H) membrane levels between IOCs in wild-type and Magi mutant tissues corresponding to C and F, respectively. Eight independent pairs of wild-type and Magi mutant tissues are shown and plotted by decreasing arbitrary average pixel intensity. Levels in wild type are always higher than those in Magi. s.e.m. is shown; P<0.001 (paired t-test). Scale bars: 5 µm.
Given the effect of Magi on E-Cad localization during AJ remodeling, we tested whether Magi could also affect α- and β-catenins, which are core components of Cadherin-based junctions. Strikingly, in Magi mutant clones at 24 h APF, the membrane levels of both α-catenin and β-catenin [Armadillo (Arm) – FlyBase] were reduced compared with neighboring wild-type cells (Fig. 6C,E,F,H). This was accompanied by an increase in intracellular puncta containing these proteins. Within Magi mutant clones, there were 6.76±1.58 α-catenin and 5.88±0.64 β-catenin intracellular puncta per IOC, as compared with wild-type cells which had 3.01±0.84 and 3.66±0.93 puncta, respectively (Fig. 6D,G). Taken together, these results suggest that during remodeling Magi mutant cells form abnormal AJs with less localized E-Cad and catenin proteins. Halving the dosage of β-catenin strongly enhances the E-Cad interruptions in IOCs seen in Magi mutants at 24 h APF (supplementary material Fig. S4A,B), suggesting that Magi could be acting redundantly with other β-catenin-localizing mechanisms to regulate E-Cad-based AJs.
We note, however, that such interruptions to AJ structure in Magi mutants occur transiently at the time of IOC sorting, and are not evident at later stages (pupal retina lattice at 44 h APF; Fig. 1B″) or in the larval wing disc epithelia. Magi function might therefore only be crucial in tissues undergoing rapid cell rearrangements.
Magi controls Baz localization during IOC remodeling
The apical scaffold protein Baz/PAR3 has been implicated in AJ stability and remodeling in several developmental processes (Desai et al., 2013; McGill et al., 2009; McKinley et al., 2012; Walther and Pichaud, 2010). It has been proposed to promote the coalescence of small cadherin-catenin clusters to form nascent junctions during cellularization in the embryo (McGill et al., 2009; McKinley et al., 2012), a process mediated at least in part through the binding and recruitment of β-catenin (Wei et al., 2005). Since E-Cad-based junctions and catenin membrane distributions were disrupted in Magi mutant cells, we tested whether Baz could also be affected. Strikingly, Baz was lost from the membrane of IOCs in Magi mutant clones at 24 h APF, and accumulated in the cytoplasm (Fig. 7A). Baz localization was however normal in Magi mutant cells by 44 h APF (supplementary material Fig. S4C).
Fig. 7.
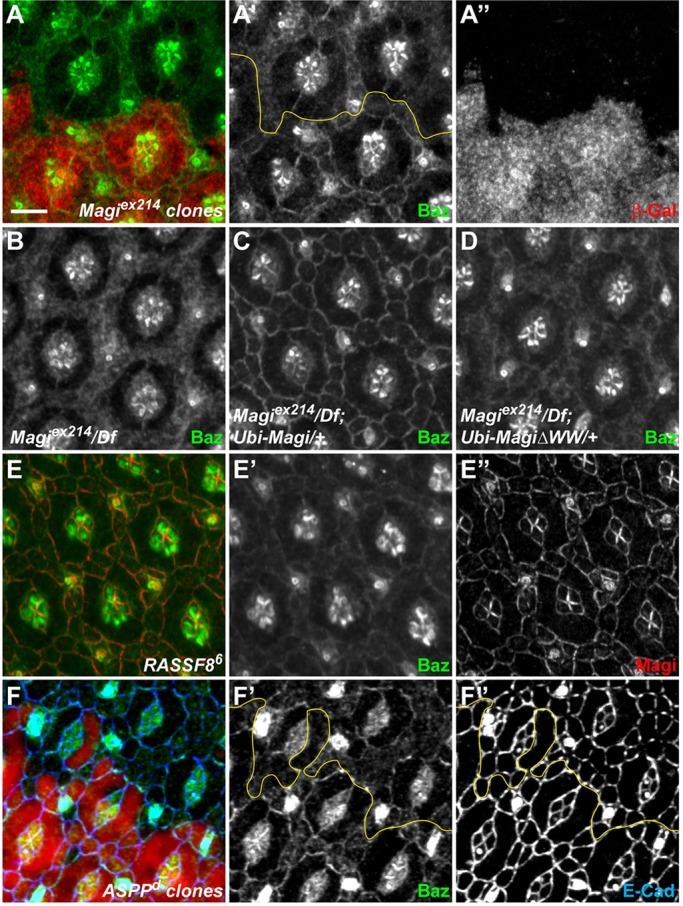
Magi is required for Baz recruitment. (A-F″) 24-h APF pupal eye discs in different mutant backgrounds. The yellow lines (A′,F′,F″) mark the boundaries between wild-type and mutant tissues. (A-A″) In Magi mutant clones marked by the absence of β-galactosidase (red, A″), Baz (green, A′) is less cortical and accumulates in the cytoplasm. (B-D) In a whole Magi mutant background, Baz (white) is absent from the membrane in IOCs (B). Whereas cortical Baz localization is completely rescued by providing a Magi transgene (C), only very faint cortical recruitment of Baz could be observed with a mutant MagiΔWW transgene. (E-E″) In whole RASSF8 mutant discs, Baz (green, E′) is very weakly associated with the cortex, whereas Magi localization (red, E″) is unaffected. (F-F″) In ASPP mutant clones marked by the absence of β-galactosidase (red), Baz (green, F′) is weaker at the membrane, in particular between IOCs [Baz at membrane in ASPP versus wild type=0.86; P=0.005 (paired t-test)], whereas Baz localization in primary pigment cells is less affected. Membranes are highlighted by E-Cad (blue, F″). Scale bar: 5 µm.
We then investigated whether the interaction between Magi and the RASSF8-ASPP complex was required for Baz membrane recruitment. First, we confirmed that the effects were completely rescued by expression of wild-type Magi. By contrast, Baz localization to the membrane was still defective in the presence of MagiΔWW (Fig. 7B-D). This suggests that the binding of Magi to RASSF8 is required for Baz membrane localization in IOCs at 24 h APF. In agreement, Baz was also mislocalized in RASSF8 and ASPP mutant cells, albeit to a lesser extent than in Magi mutants (Fig. 7E,F). Thus, Magi, and its interaction with the RASSF8-ASPP complex, is required for normal Baz localization at AJs in IOCs at 24 h APF during tissue remodeling.
baz mutants show similar IOC defects to Magi mutants
We then tested whether defects in Baz membrane recruitment could be causing interrupted AJs and remodeling defects in IOCs. At 44 h APF, baz mutant clones (using the strong hypomorphic baz815-8 allele) contained an excess of mislocalized IOCs (Fig. 8A′, arrowheads). Furthermore, since baz is required at earlier stages for correct ommatidial development, including correct morphogenesis of photoreceptors (Walther and Pichaud, 2010), many baz mutant ommatidia were severely disrupted; these were excluded from the subsequent analysis of AJs. In the remaining ommatidia, E-Cad-based AJs were disrupted in baz mutant clones at 24 h APF, similar to what was seen with Magi mutants. Whereas there were on average 0.72±0.16 interruptions in the cortical E-Cad staining per IOC in baz heterozygous tissues, this was increased to 1.26±0.30 in neighboring baz homozygous mutant IOCs (Fig. 8B,C). Similar results were obtained using bazXI, an independent hypomorphic allele (0.68±0.21 interruptions per IOC in bazXI heterozygous, rising to 0.97±0.24 in bazXI homozygous; Fig. 8C).
Fig. 8.

Baz controls IOC remodeling. baz mutant clones are marked by the absence of GFP (red, A″,B″,D″). Yellow lines (A′,B′,D′) mark the boundaries between wild-type and mutant tissues. (A-A″) 44-h APF pupal eye discs stained for E-Cad (green, A′). In the baz mutant, supernumerary mislocalized IOCs are present (arrowheads). (B-B″) 24-h APF pupal eye discs. In baz mutant tissue, E-Cad staining (green, B′) is frequently interrupted (yellow arrowheads) compared with baz heterozygous tissue (purple arrowheads). (C) Quantification of E-Cad interruptions, as shown in B, in control and in two independent baz mutant tissues. s.e.m. is shown; **P=0.009, *P=0.021 (paired t-test). (D-D″) 24-h APF pupal eye discs. In baz mutant tissue, Magi (green, D′) is found at the cortex, similarly to wild type. Scale bars: 5 µm.
We note that the defects in baz heterozygous tissue were stronger than in Magi or ASPP heterozygous tissues (where we detected ∼0.25 interruptions per IOC). This might suggest that the E-Cad interruption phenotype is sensitive to the dosage of baz. Nevertheless, this phenotype increased in severity in baz homozygous tissue and was observed in two independent baz alleles, ruling out nonspecific genetic background effects. Finally, the defects in IOC numbers and in AJ interruptions observed in baz mutant tissues were not due to defects in Magi localization, as Magi was localized normally at the AJ in baz mutant clones (Fig. 8D). These observations support a model whereby abnormal and interrupted E-Cad/AJs in Magi mutant IOCs are partly due to a failure in Baz recruitment.
DISCUSSION
As Magi is the sole Drosophila homolog of the three vertebrate MAGI scaffolds, it offers a powerful system with which to investigate the functions of these important proteins. Using newly generated null alleles, we have shown that Magi coordinates the number and packing of IOCs in the developing Drosophila pupal eye by regulating AJ dynamics. Magi is necessary in the IOCs to localize the RASSF8-ASPP complex correctly during their junctional remodeling. This ensures the integrity of E-Cad-based junctions and the correct localization of Baz, α- and β-catenin. Based on these observations and on the growing evidence of a role for Baz in AJ remodeling (Desai et al., 2013; McGill et al., 2009; McKinley et al., 2012; Wei et al., 2005), we propose a model whereby, during AJ remodeling in IOCs, Magi recruits the RASSF8-ASPP complex, which helps to localize Baz at the membrane and regulates the sites of E-Cad accumulation.
Magi and AJ remodeling
Junction remodeling is a key step during morphogenesis, in which cells in a tissue change position and neighbors. For instance, in the developing pupal eye, IOCs found between ommatidia organize as a single row of cells (Cagan, 2009). During this process existing contacts are eliminated and new ones are established by remodeling E-Cad-based junctions. In Magi mutants, we observed rearrangement defects and some incorrect localization of IOCs. At the same time, E-Cad-based AJs were interrupted in Magi mutant cells. We propose that this defect in AJ remodeling leads to IOCs remaining at the wrong place in the lattice. The most parsimonious model is that the defects in AJ remodeling trigger the defects in cell numbers seen in Magi mutants by preventing apoptosis, although we were unable to fully substantiate this as the effect of Magi on apoptosis was not statistically significant. If the model is correct, it still remains unclear how disrupted junctions would lead to a failure in apoptosis. One possibility is that IOCs only receive the correct ‘death signal’ when they have rearranged to contact the correct cells. Thus, in Magi mutants, the defective AJs would lead to apoptosis failure because the IOCs did not attain their position in the ‘death zone’ (Monserrate and Brachmann, 2007) to receive the killing signal.
These junctional defects are reminiscent of those seen for magi-1 mutants in the nematode C. elegans, in which magi-1 loss of function enhanced the defects caused by cadherin and catenin mutations and disrupted cell migration during enclosure (Lynch et al., 2012). MAGI scaffolds are thus implicated in the fine regulation of AJs in both flies and nematodes. A similar role has been suggested for MAGI proteins in mammalian epithelial cells. In overexpression studies, human MAGI1 reduced the Src-induced invasiveness of MDCK cells and stabilized E-Cad-mediated intercellular aggregation (Kotelevets et al., 2005). By analogy, the overexpression phenotype of Drosophila Magi could thus be due to stronger AJs, although this remains to be experimentally tested. The overexpression effects of MAGI-1b were sensitive to PTEN and AKT activities (Kotelevets et al., 2005) and mammalian MAGI scaffolds have also been implicated in PTEN activation through their direct binding to PTEN (Kotelevets et al., 2005; Subauste et al., 2005; Wu et al., 2000a,b; Zmajkovicova et al., 2013). However, we did not detect any physical interaction between Drosophila Magi and Pten, and the overexpression phenotype of Magi, at least in the Drosophila eye, appeared insensitive to Pten (supplementary material Table S3). Although these are negative observations, they suggest that in Drosophila Magi and Pten do not form a complex to regulate AJs.
Despite its effects on eye development, Magi mutants exhibit slightly enlarged wings. Whether this is dependent on E-Cad belt integrity and AJ dynamics remains to be established. The fact that ASPP shows a very similar wing phenotype supports this model (Langton et al., 2007).
Baz membrane recruitment
Rather than binding and modulating the activity of Pten, our analysis supports a model whereby Magi, by binding to the RASSF8-ASPP complex, recruits and stabilizes Baz at the membrane. Accumulation of Baz has been shown to specify and initiate the formation of new AJs both in cellularizing embryos and in photoreceptors (McGill et al., 2009; McKinley et al., 2012; Walther and Pichaud, 2010). We propose that Baz recruited at the membrane of IOCs will in turn promote the stabilization or the proper distribution around the cell cortex of AJ material. Since biochemical and genetic experiments suggest that RASSF8 and Magi act together in a complex, we propose that the effects of Magi on AJs and on Baz membrane recruitment are mediated by RASSF8, and are thus likely to involve ASPP. Indeed, mammalian ASPP2 binds PAR3 (Cong et al., 2010; Hauri et al., 2013; Sottocornola et al., 2010) and is required for PAR3 localization at junctions both in cell culture and in the mouse neuroepithelium. This suggests that Magi might control Baz localization through ASPP. However, Baz membrane recruitment is unlikely to be the only step to form correct AJs downstream of Magi/RASSF8/ASPP. Previous studies have implicated C-terminal Src kinase (Csk) and its action on Src kinase (Langton et al., 2007, 2009), and the relationships between Magi, Baz and Csk should be investigated in the future.
Magi membrane localization
During IOC remodeling, Magi therefore appears to be a crucial upstream regulator of AJs. However, the mechanisms governing Magi membrane localization are still unknown. One hypothesis is that the membrane recruitments of different AJ components and regulators are dependent on each other in stabilization loops. However, this is unlikely to be the case for Magi as it is still perfectly localized at the membrane in ASPP, RASSF8 and baz mutants, and in ASPP; RASSF8 double mutants.
Another possibility is that Magi would require mature AJs with E-Cad to be at the membrane. We found, however, no direct correlation between E-Cad accumulation around the apical membrane and Magi membrane localization. For instance, in ASPP; RASSF8 double-mutant cells, we could detect E-Cad belt interruptions either without (Fig. 5E, purple arrowhead) or with (Fig. 5E, yellow arrowhead) Magi, indicating that Magi localization does not require E-Cad directly. Furthermore, an extensive domain mapping of Magi failed to identify a single domain (WW or PDZ) that would be required for Magi recruitment (our unpublished observations), suggesting that it might be independent of these domains or that several redundant mechanisms may be at play. The nature of the signal required for Magi membrane localization thus remains to be uncovered.
A MAGI-ASPP complex in mammals
Even though Magi binds to RASSF8 directly and both proteins function together during Drosophila eye morphogenesis, their mutant phenotypes are not identical. First, RASSF8 mutants have a wing rounding phenotype (Langton et al., 2009), which is absent in Magi mutants. Second, whereas RASSF8 has a significant role in the global developmental apoptosis rate in the pupal eye (Langton et al., 2009), no significant effect could be detected for Magi and ASPP (Langton et al., 2007). Taken together, this suggests that the assembly of a Magi-RASSF8-ASPP complex might be context dependent or that RASSF8 has Magi-independent functions.
Although the human N-terminal RASSF (RASSF7-10) proteins lack any PPxY motifs, one is present in ASPP2 and has been shown to bind to MAGI1 (Pirozzi et al., 1997). It is therefore possible that MAGI-ASPP complexes are formed in all organisms but the precise mode of interaction differs: mediated by RASSF8 in the fly, but direct in humans.
MAGI scaffolds have been suggested to play a role in tumorigenesis. First, they are bound and inactivated by several viral oncoproteins (Glaunsinger et al., 2000; Thomas et al., 2002). Second, MAGI1 has been shown to exhibit tumor suppressor activity in colorectal cancer cell lines in xenograft models (Zaric et al., 2012). Finally, mutations in MAGI2 and MAGI3 are reported in colon, prostate and breast cancers. Documented alterations include deletion of the second WW motif of MAGI2 (Pleasance et al., 2010), inversion of MAGI2 (Berger et al., 2011) and a MAGI3:AKT3 fusion leading to a disruption of MAGI3 (Banerji et al., 2012). Based on our work, we propose that these are loss-of-function mutations. It would be interesting to investigate whether changes in AJ dynamics are associated with these MAGI mutations in human cancers and whether they contribute to tumorigenesis.
MATERIALS AND METHODS
Drosophila genetics
The viable Magiex214 null allele was generated by imprecise excision of the P element P{GSV3}GS6093. The Magi null phenotype is obtained either as Magiex214 homozygous or by crossing Magiex214 with the Df(2R)Exel6072 spanning the Magi locus. The full-length Magi coding sequence, or Magi lacking the WW domains (MagiΔWW, removing amino acids 292-372), was amplified by PCR from cDNA LD27118 [Drosophila Genomics Resource Center (DGRC), University of Indiana, Bloomington, IN, USA] and cloned either in the pKC26w-pUbiq rescue plasmid that allows expression of the cloned fragments under the ubiquitous ubiquitin-63E promoter (see supplementary Materials and Methods for construction of the pKC26w-pUbiq rescue plasmid) or in frame with an N-terminal GFP in the pUASt vector for overexpression studies. All transgenes (pKC26w-pUbiq and pUASt) were inserted at the same chromosomal location (86F8) using phiC31-mediated integration on the M{3xP3-RFP.attP}ZH-86Fb landing platform.
Overexpression studies were performed using GMR-Gal4 (all cells in the differentiating eye) or patched[559.1]-Gal4 (stripe in the wing disc) to drive expression of the UASt GFP:Magi, UASt GFP:MagiΔWW, UASt RASSF8 (Langton et al., 2009) and UASt ASPP (Langton et al., 2007) transgenes.
Genetic interactions were performed at 25°C driving UASt GFP:Magi under the control of GMR-Gal4 and crossed to flies carrying mutations or overexpression UASt transgenes for the different genes of interest (see supplementary material Table S3 for list and origin).
Mutant clones were generated at high frequency in the wing using abxUbxFLPase or in the eye using eyFLPase in combination with FRT42D Magiex214, FRT42D ASPPd, FRT82B RASSF86, FRT9-2 baz815-8 or FRT9-2 bazXI.
Plasmids for cell culture
RASSF8 and ASPP plasmids have been described before (Langton et al., 2009). All expression plasmids were generated with the Gateway system (Invitrogen). Destination vectors are from the Drosophila Gateway vector collection (http://emb.carnegiescience.edu/labs/murphy/Gateway%20vectors.html). The Magi ORF was amplified from the LD27118 cDNA by PCR and cloned into pDONR/Zeo (Invitrogen). The point mutations in RASSF8 and Magi were introduced in Entry vectors by mutagenesis PCR using PfuTurbo (Agilent). Mutations are TAT→GCT for RASSF8-Y297A, CCC→GCC for Magi-P323A, and CCA→GCA for Magi-P370A. MagiΔWW was subcloned using pUASt MagiΔWW as template (see above).
Yeast two-hybrid screen
The RASSF8 yeast two-hybrid screen was performed by Hybrigenics (Paris, France). Full-length RASSF8 was cloned as an N-terminal LexA fusion in pB29 and used as a bait against a Drosophila whole embryo (0-24 h) cDNA library (RP2) in 100 mM 3-amino-triazole. 2.45×107 clones were screened, from which 380 positive clones were recovered and retested under the same conditions as the screen.
Western blotting and immunoprecipitation
All immunoprecipitations were performed using lysates from S2 cells grown in Schneider's Drosophila medium (Gibco) supplemented with 10% FCS (Sigma-Aldrich), 100 units/ml penicillin and 100 μg/ml streptomycin. 3×106 S2 cells were transfected using Effectene (Qiagen). 72 h after transfection, S2 cells were lysed with HEPES lysis buffer (50 mM HEPES-NaOH pH 7.5, 150 mM NaCl, 0.5% Triton X-100) supplemented with 1 mM DTT and Complete Protease Inhibitor Cocktail (Roche). Half of the lysate was incubated with anti-FLAG M2 beads (Sigma-Aldrich) for 1 h at 4°C. The beads were washed with HEPES lysis buffer four times for 2 min each. Proteins were visualized by immunoblotting using rabbit anti-Myc (sc-789, Santa Cruz; 1:5000), rat anti-HA (3F10, Roche; 1:2000), mouse anti-FLAG (F1804, Sigma-Aldrich; 1:5000) and mouse anti-tubulin (DM1A, Sigma-Aldrich; 1:10,000) antibodies.
Immunocytochemistry
Antibody staining of wing imaginal discs or pupal eyes were performed using standard protocols (see supplementary Materials and Methods for a full list of the antibodies used). Images were acquired with a Leica SP2-405 or a Zeiss LSM780 confocal microscope and processed using Adobe Photoshop or ImageJ (NIH).
Supplementary Material
Acknowledgements
We thank Michael Wehr and Paulo Ribeiro for the pKC26-pUbiq plasmid and Magi Entry clone; Florence Frayssinoux for help with directed two-hybrid tests; Elodie Forest, Lisa Héron-Milhavet and Rémi Logeay for help with ‘blind’ quantifications; Daniel St Johnston and Andreas Wodarz for sharing flies and antibodies; and the Bloomington Stock Center, the DGRC Kyoto Stock Center and the Developmental Studies Hybridoma Bank for flies and antibodies.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceived experiments: S.J.B., N.T. and A.D. Performed experiments: S.Z., Y.Z. and A.D. Analyzed the data: S.Z., Y.Z., N.T. and A.D. Wrote the manuscript: S.Z., S.J.B., N.T. and A.D.
Funding
This work was supported by an Atip/Avenir grant to A.D.; a Wellcome Trust project grant to A.D. and S.J.B. [WT083576MA]; and a Medical Research Council programme grant to S.J.B. [G0800034]. Y.Z. acknowledges support from the Boehringer Ingelheim Fonds and Cancer Research UK (CRUK). The N.T. lab is supported by CRUK. Deposited in PMC for release after 6 months.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.116277/-/DC1
References
- Banerji S., Cibulskis K., Rangel-Escareno C., Brown K. K., Carter S. L., Frederick A. M., Lawrence M. S., Sivachenko A. Y., Sougnez C., Zou L. et al. (2012). Sequence analysis of mutations and translocations across breast cancer subtypes. Nature 486, 405-409 10.1038/nature11154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao S. and Cagan R. (2005). Preferential adhesion mediated by Hibris and Roughest regulates morphogenesis and patterning in the Drosophila eye. Dev. Cell 8, 925-935 10.1016/j.devcel.2005.03.011 [DOI] [PubMed] [Google Scholar]
- Bao S., Fischbach K.-F., Corbin V. and Cagan R. L. (2010). Preferential adhesion maintains separation of ommatidia in the Drosophila eye. Dev. Biol. 344, 948-956 10.1016/j.ydbio.2010.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger M. F., Lawrence M. S., Demichelis F., Drier Y., Cibulskis K., Sivachenko A. Y., Sboner A., Esgueva R., Pflueger D., Sougnez C. et al. (2011). The genomic complexity of primary human prostate cancer. Nature 470, 214-220 10.1038/nature09744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cagan R. (2009). Principles of Drosophila eye differentiation. Curr. Top. Dev. Biol. 89, 115-135 10.1016/S0070-2153(09)89005-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong W., Hirose T., Harita Y., Yamashita A., Mizuno K., Hirano H. and Ohno S. (2010). ASPP2 regulates epithelial cell polarity through the PAR complex. Curr. Biol. 20, 1408-1414 10.1016/j.cub.2010.06.024 [DOI] [PubMed] [Google Scholar]
- Desai R., Sarpal R., Ishiyama N., Pellikka M., Ikura M. and Tepass U. (2013). Monomeric α-catenin links cadherin to the actin cytoskeleton. Nat. Cell Biol. 15, 261-273 10.1038/ncb2685 [DOI] [PubMed] [Google Scholar]
- Dobrosotskaya I. Y. and James G. L. (2000). MAGI-1 interacts with beta-catenin and is associated with cell-cell adhesion structures. Biochem. Biophys. Res. Commun. 270, 903-909 10.1006/bbrc.2000.2471 [DOI] [PubMed] [Google Scholar]
- Glaunsinger B. A., Lee S. S., Thomas M., Banks L. and Javier R. (2000). Interactions of the PDZ-protein MAGI-1 with adenovirus E4-ORF1 and high-risk papillomavirus E6 oncoproteins. Oncogene 19, 5270-5280 10.1038/sj.onc.1203906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauri S., Wepf A., van Drogen A., Varjosalo M., Tapon N., Aebersold R. and Gstaiger M. (2013). Interaction proteome of human Hippo signaling: modular control of the co-activator YAP1. Mol. Syst. Biol. 9, 713 10.1002/msb.201304750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heisenberg C.-P. and Bellaïche Y. (2013). Forces in tissue morphogenesis and patterning. Cell 153, 948-962 10.1016/j.cell.2013.05.008 [DOI] [PubMed] [Google Scholar]
- Ide N., Hata Y., Nishioka H., Hirao K., Yao I., Deguchi M., Mizoguchi A., Nishimori H., Tokino T., Nakamura Y. et al. (1999). Localization of membrane-associated guanylate kinase (MAGI)-1/BAI-associated protein (BAP) 1 at tight junctions of epithelial cells. Oncogene 18, 7810-7815 10.1038/sj.onc.1203153 [DOI] [PubMed] [Google Scholar]
- Kawajiri A., Itoh N., Fukata M., Nakagawa M., Yamaga M., Iwamatsu A. and Kaibuchi K. (2000). Identification of a novel beta-catenin-interacting protein. Biochem. Biophys. Res. Commun. 273, 712-717 10.1006/bbrc.2000.3002 [DOI] [PubMed] [Google Scholar]
- Kotelevets L., van Hengel J., Bruyneel E., Mareel M., van Roy F. and Chastre E. (2005). Implication of the MAGI-1b/PTEN signalosome in stabilization of adherens junctions and suppression of invasiveness. FASEB J. 19, 115-117. [DOI] [PubMed] [Google Scholar]
- Langton P. F., Colombani J., Aerne B. L. and Tapon N. (2007). Drosophila ASPP regulates C-terminal Src kinase activity. Dev. Cell 13, 773-782 10.1016/j.devcel.2007.11.005 [DOI] [PubMed] [Google Scholar]
- Langton P. F., Colombani J., Chan E. H. Y., Wepf A., Gstaiger M. and Tapon N. (2009). The dASPP-dRASSF8 complex regulates cell-cell adhesion during Drosophila retinal morphogenesis. Curr. Biol. 19, 1969-1978 10.1016/j.cub.2009.10.027 [DOI] [PubMed] [Google Scholar]
- Levayer R., Pelissier-Monier A. and Lecuit T. (2011). Spatial regulation of Dia and Myosin-II by RhoGEF2 controls initiation of E-cadherin endocytosis during epithelial morphogenesis. Nat. Cell Biol. 13, 529-540 10.1038/ncb2224 [DOI] [PubMed] [Google Scholar]
- Lynch A. M., Grana T., Cox-Paulson E., Couthier A., Cameron M., Chin-Sang I., Pettitt J. and Hardin J. (2012). A genome-wide functional screen shows MAGI-1 is an L1CAM-dependent stabilizer of apical junctions in C. elegans. Curr. Biol. 22, 1891-1899 10.1016/j.cub.2012.08.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGill M. A., McKinley R. F. A. and Harris T. J. C. (2009). Independent cadherin-catenin and Bazooka clusters interact to assemble adherens junctions. J. Cell Biol. 185, 787-796 10.1083/jcb.200812146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinley R. F. A., Yu C. G. and Harris T. J. C. (2012). Assembly of Bazooka polarity landmarks through a multifaceted membrane-association mechanism. J. Cell Sci. 125, 1177-1190 10.1242/jcs.091884 [DOI] [PubMed] [Google Scholar]
- Monserrate J. P. and Brachmann C. B. (2007). Identification of the death zone: a spatially restricted region for programmed cell death that sculpts the fly eye. Cell Death Differ. 14, 209-217 10.1038/sj.cdd.4401947 [DOI] [PubMed] [Google Scholar]
- Pirozzi G., McConnell S. J., Uveges A. J., Carter J. M., Sparks A. B., Kay B. K. and Fowlkes D. M. (1997). Identification of novel human WW domain-containing proteins by cloning of ligand targets. J. Biol. Chem. 272, 14611-14616 10.1074/jbc.272.23.14611 [DOI] [PubMed] [Google Scholar]
- Pleasance E. D., Cheetham R. K., Stephens P. J., McBride D. J., Humphray S. J., Greenman C. D., Varela I., Lin M.-L., Ordóñez G. R., Bignell G. R. et al. (2010). A comprehensive catalogue of somatic mutations from a human cancer genome. Nature 463, 191-196 10.1038/nature08658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauzi M., Lenne P.-F. and Lecuit T. (2010). Planar polarized actomyosin contractile flows control epithelial junction remodelling. Nature 468, 1110-1114 10.1038/nature09566 [DOI] [PubMed] [Google Scholar]
- Reiter C., Schimansky T., Nie Z. and Fischbach K. F. (1996). Reorganization of membrane contacts prior to apoptosis in the Drosophila retina: the role of the IrreC-rst protein. Development 122, 1931-1940. [DOI] [PubMed] [Google Scholar]
- Sakurai A., Fukuhara S., Yamagishi A., Sako K., Kamioka Y., Masuda M., Nakaoka Y. and Mochizuki N. (2006). MAGI-1 is required for Rap1 activation upon cell-cell contact and for enhancement of vascular endothelial cadherin-mediated cell adhesion. Mol. Biol. Cell 17, 966-976 10.1091/mbc.E05-07-0647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sottocornola R., Royer C., Vives V., Tordella L., Zhong S., Wang Y., Ratnayaka I., Shipman M., Cheung A., Gaston-Massuet C. et al. (2010). ASPP2 binds Par-3 and controls the polarity and proliferation of neural progenitors during CNS development. Dev. Cell 19, 126-137 10.1016/j.devcel.2010.06.003 [DOI] [PubMed] [Google Scholar]
- Subauste M. C., Nalbant P., Adamson E. D. and Hahn K. M. (2005). Vinculin controls PTEN protein level by maintaining the interaction of the adherens junction protein beta-catenin with the scaffolding protein MAGI-2. J. Biol. Chem. 280, 5676-5681 10.1074/jbc.M405561200 [DOI] [PubMed] [Google Scholar]
- Tepass U. and Harris K. P. (2007). Adherens junctions in Drosophila retinal morphogenesis. Trends Cell Biol. 17, 26-35 10.1016/j.tcb.2006.11.006 [DOI] [PubMed] [Google Scholar]
- Thomas M., Laura R., Hepner K., Guccione E., Sawyers C., Lasky L. and Banks L. (2002). Oncogenic human papillomavirus E6 proteins target the MAGI-2 and MAGI-3 proteins for degradation. Oncogene 21, 5088-5096 10.1038/sj.onc.1205668 [DOI] [PubMed] [Google Scholar]
- Vidal M., Larson D. E. and Cagan R. L. (2006). Csk-deficient boundary cells are eliminated from normal Drosophila epithelia by exclusion, migration, and apoptosis. Dev. Cell 10, 33-44 10.1016/j.devcel.2005.11.007 [DOI] [PubMed] [Google Scholar]
- Walther R. F. and Pichaud F. (2010). Crumbs/DaPKC-dependent apical exclusion of Bazooka promotes photoreceptor polarity remodeling. Curr. Biol. 20, 1065-1074 10.1016/j.cub.2010.04.049 [DOI] [PubMed] [Google Scholar]
- Wei S.-Y., Escudero L. M., Yu F., Chang L.-H., Chen L.-Y., Ho Y.-H., Lin C.-M., Chou C.-S., Chia W., Modolell J. et al. (2005). Echinoid is a component of adherens junctions that cooperates with DE-Cadherin to mediate cell adhesion. Dev. Cell 8, 493-504 10.1016/j.devcel.2005.03.015 [DOI] [PubMed] [Google Scholar]
- Wu X., Hepner K., Castelino-Prabhu S., Do D., Kaye M. B., Yuan X.-J., Wood J., Ross C., Sawyers C. L. and Whang Y. E. (2000a). Evidence for regulation of the PTEN tumor suppressor by a membrane-localized multi-PDZ domain containing scaffold protein MAGI-2. Proc. Natl. Acad. Sci. USA 97, 4233-4238 10.1073/pnas.97.8.4233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y., Dowbenko D., Spencer S., Laura R., Lee J., Gu Q. and Lasky L. A. (2000b). Interaction of the tumor suppressor PTEN/MMAC with a PDZ domain of MAGI3, a novel membrane-associated guanylate kinase. J. Biol. Chem. 275, 21477-21485 10.1074/jbc.M909741199 [DOI] [PubMed] [Google Scholar]
- Zaric J., Joseph J.-M., Tercier S., Sengstag T., Ponsonnet L., Delorenzi M. and Rüegg C. (2012). Identification of MAGI1 as a tumor-suppressor protein induced by cyclooxygenase-2 inhibitors in colorectal cancer cells. Oncogene 31, 48-59 10.1038/onc.2011.218 [DOI] [PubMed] [Google Scholar]
- Zmajkovicova K., Jesenberger V., Catalanotti F., Baumgartner C., Reyes G. and Baccarini M. (2013). MEK1 is required for PTEN membrane recruitment, AKT regulation, and the maintenance of peripheral tolerance. Mol. Cell 50, 43-55 10.1016/j.molcel.2013.01.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.