

Abstract
Background
The fungus Pochonia chlamydosporia parasitizes nematode eggs and has become one of the most promising biological control agents (BCAs) for plant-parasitic nematodes, which are major agricultural pests that cause tremendous economic losses worldwide. The complete mitochondrial (mt) genome is expected to open new avenues for understanding the phylogenetic relationships and evolution of the invertebrate-pathogenic fungi in Hypocreales.
Results
The complete mitogenome sequence of P. chlamydosporia is 25,615 bp in size, containing the 14 typical protein-coding genes, two ribosomal RNA genes, an intronic ORF coding for a putative ribosomal protein (rps3) and a set of 23 transfer RNA genes (trn) which recognize codons for all amino acids. Sequence similarity studies and syntenic gene analyses show that 87.02% and 58.72% of P. chlamydosporia mitogenome sequences match 90.50% of Metarhizium anisopliae sequences and 61.33% of Lecanicillium muscarium sequences with 92.38% and 86.04% identities, respectively. A phylogenetic tree inferred from 14 mt proteins in Pezizomycotina fungi supports that P. chlamydosporia is most closely related to the entomopathogenic fungus M. anisopliae. The invertebrate-pathogenic fungi in Hypocreales cluster together and clearly separate from a cluster comprising plant-pathogenic fungi (Fusarium spp.) and Hypocrea jecorina. A comparison of mitogenome sizes shows that the length of the intergenic regions or the intronic regions is the major size contributor in most of mitogenomes in Sordariomycetes. Evolutionary analysis shows that rps3 is under positive selection, leading to the display of unique evolutionary characteristics in Hypocreales. Moreover, the variability of trn distribution has a clear impact on gene order in mitogenomes. Gene rearrangement analysis shows that operation of transposition drives the rearrangement events in Pezizomycotina, and most events involve in trn position changes, but no rearrangement was found in Clavicipitaceae.
Conclusions
We present the complete annotated mitogenome sequence of P. chlamydosporia. Based on evolutionary and phylogenetic analyses, we have determined the relationships between the invertebrate-pathogenic fungi in Hypocreales. The invertebrate-pathogenic fungi in Hypocreales referred to in this paper form a monophyletic group sharing a most recent common ancestor. Our rps3 and trn gene order results also establish a foundation for further exploration of the evolutionary trajectory of the fungi in Hypocreales.
Electronic supplementary material
The online version of this article (doi:10.1186/s12866-015-0341-8) contains supplementary material, which is available to authorized users.
Keywords: Nematode egg-parasite fungus, Pochonia chlamydosporia, Biological control agent, Mitochondrial genome, rps3, Rearrangement, Evolution, Phylogeny
Background
The nematophagous fungus Pochonia chlamydosporia (Goddard) Zare & Gams (previously named Verticillium chlamydosporium, teleomorph Metacordyceps chlamydosporia) is a widespread soil fungus distributed worldwide in nematode suppressive soils. Pochonia chlamydosporia can infect the females and eggs of plant endoparasitic nematodes as a facultative parasite [1,2]. It has already demonstrated its efficacy as a BCA against both root-knot nematodes (RKNs; Meloidogyne spp.) and cyst nematodes (Heterodera spp. and Globodera spp.) [2-5], major agricultural pests that cause tremendous economic losses worldwide estimated at 100 billion dollars annually [6].
The fungus P. chlamydosporia belongs to the family Clavicipitaceae (Ascomycota: Pezizomycotina: Sordariomycetes: Hypocreales). Based on spore morphology and polymorphisms of the nuclear ribosomal internal transcribed spacer (ITS) region, this species has been recognized as having at least two distinct varieties, P. chlamydosporia var. chlamydosporia and P. chlamydosporia var. catenulate [7,8]. Many members of Clavicipitaceae are invertebrate-pathogenic fungi. It was previously suggested that the host relatedness and host habitat hypotheses could explain the evolution of parasite-host relationships [9]. Therefore, exploring the phylogenetic relationships and evolutionary trajectories of the invertebrate-pathogenic fungi in Clavicipitaceae is a topic of interest. Multi-gene phylogenetic analyses showed that Clavicipitaceae is paraphyletic and consists of three well-defined clades, at least one of which is shared with the members of another fungal family (Hypocreaceae) [10]. The phylogenetic placement of the genus Pochonia in the family Clavicipitaceae was inferred from six nuclear genes (nrSSU, nrLSU, beta-tubulin, EF-1alpha, RPB1, RPB2) and one mt gene (atp6) using phylogenetic analysis, and the phylogenetic trees inferred from different genes produced different topological structures [10]. Therefore, a comparison of larger scale genome sequences is necessary because it can provide deep insights into the phylogenetic relationships and evolution of the fungi in Clavicipitaceae. Recently, the complete nuclear genome sequence of P. chlamydosporia was published. Based on a phylogenetic analysis inferred from nuclear genome sequences, P. chlamydosporia appeared to be most closely related to the Metarhizium species [11].
Mitochondrial markers can be successfully applied in evolutionary biology and systematics because mt genomes often evolve faster than nuclear genomes and allow for robust phylogenetic analyses based on the conserved proteins of the oxidative phosphorylation system [12]. The continuously increasing number of recent fungal mt genome studies – more than 165 complete fungal mt genomes are available today at NCBI - provide powerful tools for comparative studies to reveal the patterns and mechanisms of mitogenome evolution [13,14]. Thus far, fungal mitogenomes vary considerably in size, the largest being 147,264 bp long in Rhizoctonia solani AG1 IA (Basidiomycota) [15] and the smallest being 18,844 bp long in Hanseniaspora uvarum (Ascomycota) [16]. Researchers have demonstrated that different lengths of intronic sequences and intergenic regions result in different sizes of mitogenomes [17-19], particularly for the mitogenome of Moniliophthora perniciosa in Basidiomycota, which has a length of 109,103 bp and encodes several hypothetical ORFs in the larger sized intergenic regions and introns [19]. However, a depiction of the size variation and related mechanisms in Ascomycota has not yet been reported. Moreover, the nearly ubiquitous presence of rps3 genes, which are encoded in the group I introns within rnl genes, has been attributed to a vertical mode of inheritance rather than horizontal inheritance during evolution [20]. In one study, the molecular evolution of the rps3 gene in filamentous Ascomycetes fungi (especially Ophiostomatoid fungi) was analysed, and the group I intron-encoded version of rps3 appeared to have a rather complex evolutionary history in ascomycetes fungi [21]. However, only a few fungi in Hypocreales were included in that study. To better understand the evolution of the fungal mitogenome in Hypocreales, a phylogenetic analysis of the rps3 genes is required. Furthermore, it was reported that trn genes are able to change their location in genomes and participate in horizontal gene transfer (HGT) events, as they have editing, excision and integration capabilities [22,23]. Because changes in trn location are relatively rare events, the locations of trn genes in fungal mt genomes have been used to study fungal evolution and phylogenetic signals [22]. To date, 20 different types of aminoacyl-tRNAs (aa-tRNAs) have been identified in fungal mitogenomes, and conserved trn clusters are found in Pezizomycotina by employing comparative genomic approaches [12,24-26]. The distribution of trn genes may contribute to gene order variation in fungal mitogenomes [22]. Six operations have been used to explain rearrangements in mt gene order: inversion, transposition, reverse transposition, tandem-duplication-random-loss [27], deletion and replication slippage [28]. However, the evolution of gene orders in Pezizomycotina mitogenomes is suggested to be mainly driven by transpositions [12]. As in animal mt genomes, trn in fungal mitogenomes might have played a role in gene shuffling as they intervene between genes and can act like mobile elements [29,30]. Taking advantage of published fungal mitogenomes and the novel P. chlamydosporia mitogenome, we investigated the trn rearrangements in Pezizomycotina, and addressed the role of rearrangements in evolution.
In this paper, the P. chlamydosporia strain 170 isolated from a RKN in China was sequenced using Illumina sequencing, and a complete mt genome of 25,615 bp was obtained. We aim to (i) present a complete and annotated mt genome sequence of P. chlamydosporia, (ii) compare the mt genome of P. chlamydosporia with the genomes of other fungi in Pezizomycotina to identify the common and specific characteristics of the mt genomes of invertebrate-pathogenic fungi, and (iii) provide powerful insights into the evolution and phylogenetic relationships of the invertebrate-pathogenic fungi in Hypocreales.
Results
General characteristics of the mt genome in P. chlamydosporia
The complete mt genome of P. chlamydosporia is a circular DNA molecule with a length of 25,615 bp. The low G + C content (28.3%) is similar to other fungal mitogenomes in Clavicipitaceae (Table 1). The mitogenome encodes an essential set of conserved genes including three cytochrome c oxidase subunits (cox1, cox2, cox3), apocytochrome b (cob), three ATP synthase subunits (atp6, atp8, atp9), seven subunits of NADH dehydrogenase (nad1, nad2, nad3, nad4, nad4L, nad5, nad6), the small and large ribosomal RNA subunits (rns, rnl), an intronic ORF coding for a putative rps3, and 23 trn. All of the 40 genes are encoded on the same DNA strand (Figure 1). The set of trn genes found (including two trnA genes and three trnM genes) is the smallest set currently known for Pezizomycotina fungi (Table 1), but the trn genes can recognize the codons for all 20 amino acids. In total, the coding regions (CRs), of which the 14 typical protein-coding genes related to oxidative phosphorylation account for 13,068 bp, and together with the rps3 (1,317 bp) within the rnl group I intron (1,652 bp, in total), cover 56.16% of the total mt genome, and the rest are trn, rRNA genes and intergenic regions.
Table 1.
General features of the mitogenomes
| Species | Length (bp) | GC (%) | Coding genes | trn | rRNAs | Accession |
|---|---|---|---|---|---|---|
| Pochonia chlamydosporia | 25,615 | 28.3 | 15 | 23 | 2 | KF479445 |
| Metarhizium anisopliae | 24,673 | 28.4 | 15 | 24 | 2 | AY884128 |
| Beauveria bassiana | 29,961 | 27.2 | 15 | 25 | 2 | EU371503 |
| Beauveria pseudobassiana | 28,006 | 27.5 | 15 | 25 | 2 | NC_022708 |
| Cordyceps brongniartii | 33,926 | 27.3 | 15 | 25 | 2 | NC_011194 |
| Lecanicillium muscarium | 24,499 | 27.1 | 15 | 25 | 2 | AF487277 |
| Cordyceps militaris | 33,277 | 26.8 | 15 | 26 | 2 | NC_022834 |
| Hypocrea jecorina | 42,130 | 27.2 | 19 | 26 | 2 | AF447590 |
| Fusarium solani | 62,978 | 28.9 | 30 | 25 | 2 | NC_016680 |
| Fusarium graminearum | 95,676 | 31.8 | 50 | 28 | 2 | NC_009493 |
| Fusarium verticillioides | 53,735 | 32.6 | 21 | 27 | 2 | NC_016687 |
| Fusarium oxysporum | 34,477 | 31.0 | 16 | 25 | 2 | AY945289 |
| Verticillium dahliae | 27,184 | 27.3 | 15 | 25 | 2 | DQ351941 |
| Neurospora crassa | 64,840 | 36.1 | 28 | 28 | 2 | Broad Institute |
| Aspergillus fumigatus | 31,762 | 25.4 | 20 | 31 | 2 | JQ346809 |
| Aspergillus niger | 31,103 | 26.9 | 16 | 25 | 2 | NC_007445 |
| Aspergillus tubingensis | 33,656 | 26.8 | 16 | 25 | 2 | NC_007597 |
| Trichophyton rubrum | 26,985 | 23.5 | 17 | 25 | 2 | NC_012824 |
| Epidermophyton floccosum | 30,910 | 23.4 | 24 | 25 | 2 | NC_007394 |
| Paracoccidioides brasiliensis | 71,335 | 21.1 | 17 | 25 | 2 | NC_007935 |
| Candida parapsilosis | 32,745 | 23.8 | 20 | 24 | 2 | NC_005253 |
Figure 1.
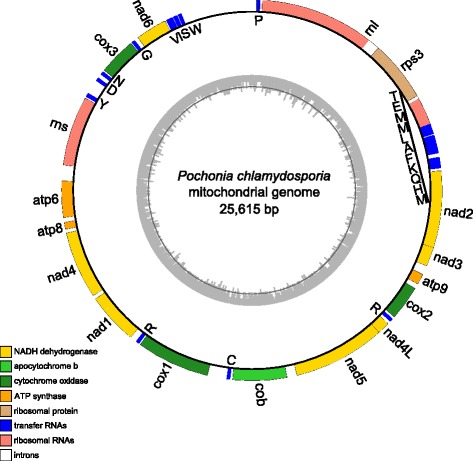
Functional map of the complete mitogenome of P. chlamydosporia . All genes are transcribed on the same strand. There are 15 encoding genes, 23 transfer RNA genes and 2 ribosomal RNA genes (rns and rnl). The circular map was generated using OrganellarGenomeDRAW [31].
Using the abundant sequenced resources available (the average coverage per base is 1,058 ×, see the Methods section), the sequence variance of the P. chlamydosporia mitogenome was investigated, but no single nucleotide polymorphism (SNP) was found. This indicates that a high quality mitogenome was assembled. Moreover, we compared the published mt gene sequences of P. chlamydosporia with our genome sequence. In total, 12 mt gene sequences from NCBI are available, including six coding segments in the IMI 113169 strain (nad3-atp9, atp6, rns, nad1, nad3, cox3), four genes in the IMI 156157 strain (rns, nad1, nad3, cox3), one gene in the CBS 101244 strain (atp6) and one gene in the CBS 504.66 strain (atp6). Our analysis showed that 11 sequences were matched to our genome sequence with identities greater than or equal to 95% (Additional file 1: Table S1), suggesting that these gene sequences are highly conserved in P. chlamydosporia strains.
Phylogenetic relationships of P. chlamydosporia to other fungi in Pezizomycotina
Utilizing published fungal mitogenomes in addition to the novel P. chlamydosporia mitogenome, the phylogenetic relationships in 20 species in Pezizomycotina were inferred, using Candida parapsilosis as an outgroup (Figure 2A). Phylogenetic trees were constructed based on the 14 conserved protein-coding genes associated with the oxidative phosphorylation system (cox1-3, cob, atp6, atp8-9, nad1-6 and nad4L) using a Maximum Likelihood (ML) approach. The topological structures based on both nucleotide and amino acid sequences were identical, and a clear genealogical relationship was shown (Figure 2A). P. chlamydosporia is most closely related to the entomopathogenic fungus M. anisopliae, with a bootstrap value of 100%, which is similar to the result of phylogenetic analysis based on genome-encoded orthologous proteins [11]. The two fungi form a cluster with five additional entomopathogenic fungi, namely, Cordyceps brongniartii, Beauveria pseudobassiana, B. bassiana, Lecanicillium muscarium and C. militaris. Interestingly, although both P. chlamydosporia and L. muscarium were once put into the same genus (Verticillium), the two species are assigned to different sub-clades in the phylogenetic tree. However, all of the invertebrate-pathogenic fungi cluster into a clade (clade A) clearly separated from the plant-parasitic fungi, including four Fusarium plant pathogens (F. solani, F. graminearum, F. oxysporum and F. verticillioides), which form clade B with the fungus Hypocrea jecorina that specializes in colonizing pre-degraded wood. Both invertebrate-pathogenic and plant-pathogenic fungi shared a common ancestor in clade C represented by a soil born plant pathogen, Verticillium dahliae, suggesting that Hypocreales is a monophyletic group (Figure 2A). The phylogeny shows that plant pathogenic and invertebrate-pathogenic fungi in Hypocreales form independent clusters and have evolved separately. A third group comprises fungi that infect animals or humans, including several members of Eurotiomycetes, i.e., Aspergillus fumigates, Paracoccidioides brasiliensis, Epidermophyton floccosum and Trichophyton rubrum; this group is distant from P. chlamydosporia and the entomopathogenic fungi. The phylogenetic relationships inferred from mt proteins are principally consistent with the current Pezizomycotina taxonomic system (Figure 2A).
Figure 2.
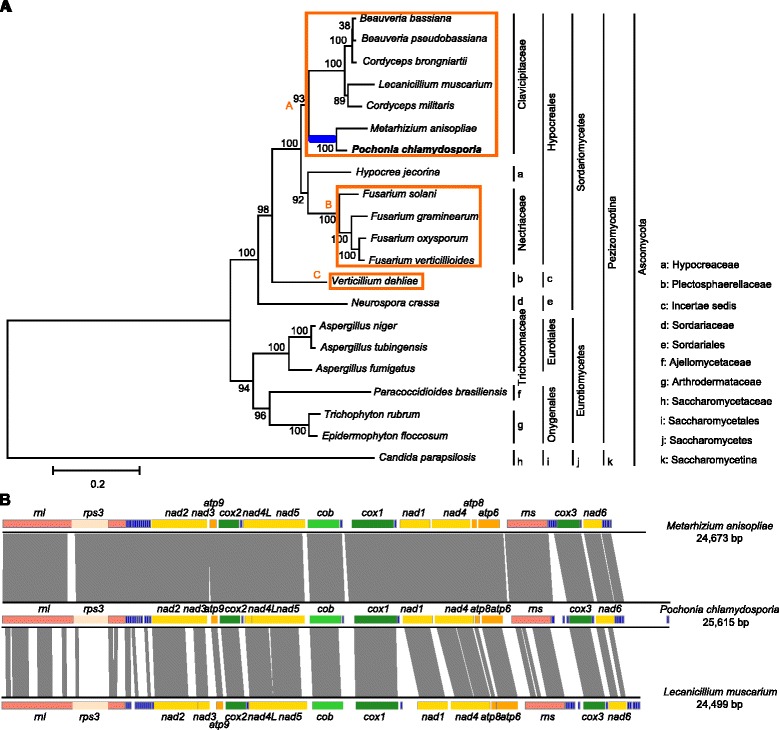
The mitogenome phylogenetic analysis and comparative analysis. A). Phylogenetic placement of P. chlamydosporia using 21 fungal mitogenomes. The phylogeny was generated based on 14 conserved encoded proteins (cox1-3, cob, nad1-6, nad4L, atp6, atp8-9) using a ML approach. C. parapsilosis was used as an outgroup. P. chlamydosporia is closely related to M. anisopliae with a bootstrap support value of 100%. B). Synteny comparisons between P. chlamydosporia and M. anisopliae, P. chlamydosporia and L. muscarium. Syntenic regions were identified by performing BLASTN with a threshold of 1e-5, and alignment blocks of less than 100 bp are not shown.
Comparison of mitogenome organization in P. chlamydosporia and related species
We first compared the mitogenomes of P. chlamydosporia, M. anisopliae and L. muscarium (previously widely known as Verticillium lecanii), and found that they are highly similar in both size (25,615 bp, 24,673 bp and 24,499 bp, respectively) and G + C content (28.3%, 28.4% and 27.1%, respectively) (Table 1). The total length of the gene regions (including protein-coding genes, trn and rRNA genes) in P. chlamydosporia mt genome (20,997 bp) is 142 bp less than that of L. muscarium and 208 bp less than that of M. anisopliae. However, the entire length of the P. chlamydosporia mitogenome is 1,116 bp and 942 bp larger than L. muscarium and M. anisopliae respectively, mainly due to longer intergenic regions in P. chlamydosporia. We also compared the contents of the simple repeat motifs in each of the genomes, including low complexity sequences (such as AT-rich sequences), simple repeats (such as (TAA) n), inverted repeats and palindromes, which have been previously suggested to be putative elements for recombination or regulation [24]. It was found that ~4.14% (1,061 bp, including 948 bp tandem repeats, 64 bp inverted repeats and 172 bp palindromes with 123 bp overlaps between tandem repeats and palindromes) of the P. chlamydosporia mitogenome consists of repeat sequences compared with only ~2.71% (664 bp) of the L. muscarium mitogenome and ~3.99% (985 bp) of the M. anisopliae mitogenome. The distributions of repeat elements are also different (Additional file 1: Figure S1). In L. muscarium, there are no repeat sequences located in the regions between nad6 and rnl, nad5 and cob, rns and cox3. However, in M. anisopliae and P. chlamydosporia, there are no repeat sequences in the regions between rnl and nad2, cox3 and nad6. There are repeat sequences residing in the regions between rns and cox3 in P. chlamydosporia, but only one AT rich motif is found in this region in M. anisopliae (Additional file 1: Figure S1). Repeat sequences in this region display the unique mitogenomic features of P. chlamydosporia.
In addition, sequence similarity studies and syntenic gene analyses were performed comparing P. chlamydosporia with five other mitogenomes, including M. anisopliae, L. muscarium, B. bassiana, V. dahliae and A. fumigatus. The analyses show identical gene order in M. anisopliae, L. muscarium, B. bassiana and P. chlamydosporia mt genomes. V. dahliae, as shown in previous work [32], displayed differences at the regions nad6-rnl, nad3-atp9 and cox1-nad1 with a gene order of nad6-atp9, nad3-nad1 and cox1-rnl instead (Additional file 1: Figure S2). A larger difference of gene order is observed between the mitogenomes of P. chlamydosporia and A. fumigatus (Additional file 1: Figure S2). Comparing the two mitogenomes of P. chlamydosporia and M. anisopliae, 22,331 bp (90.50%) of M. anisopliae mitogenome sequences match to 22,290 bp (87.02%) of P. chlamydosporia mitogenome sequences with 92.38% identity determined by BLASTN (Figure 2B). A long-range synteny between the P. chlamydosporia and M. anisopliae mt genomes is observed. The majority of the unmatched sequences reside within the intergenic regions between nad6 and rnl. However, comparing the P. chlamydosporia and L. muscarium mitogenomes, 15,026 bp (61.33%) of the L. muscarium mitogenome sequences match to 58.72% of the P. chlamydosporia mitogenome sequences with 86.04% identity (Figure 2B). Comparisons of P. chlamydosporia mt genomes with the other three species (B. bassiana and V. dahliae in Sordariomycetes, and A. fumigatus in Eurotiomycetes) showed that only 15,624 bp (52.15%) of B. bassiana, 13,412 bp (49.34%) of V. dahliae and 8,147 bp (25.65%) of A. fumigatus mitogenomes matched to P. chlamydosporia mt sequences with 87.52%, 84.62% and 82.82% identities, respectively (Additional file 1: Figure S2). Although the sequences of most of the mt genes in V. dahliae matched to homologous genes in P. chlamydosporia mitogenome, the alignments of some genes (such as rns, nad5 and nad6) between the two species present multiple blocks, and no alignments are found in the intergenic regions in the P. chlamydosporia and V. dahliae mitogenomes. Based on the above syntenic analysis results, an evolutionary relationship between P. chlamydosporia and the other fungi is also indicated, that is, that P. chlamydosporia is most closely related to M. anisopliae. The result is similar to that obtained from the genomic DNA analysis [11].
Analysis of mitogenome size variation among the fungi in Sordariomycetes
The sizes of the mitogenome in the fungi in Sordariomycetes vary significantly, from 24,673 bp in M. anisopliae to 95,676 bp in F. graminearum (Table 1). To identify the causes of this mitogenome size variation in Sordariomycetes, five mitogenomes (P. chlamydosporia, M. anisopliae, F. oxysporum, H. jecorina and Neurospora crassa) of different sizes were used for a comparative analysis (Figure 3). Despite the size variance ranging from 24,673 bp in M. anisopliae to 64,840 bp in N. crassa, 15 common mt protein-coding gene sets, including cox1-3, cob, nad1-6, nad4L, atp6, atp8-9 and rsp3, are encoded in all of the five genomes. We calculated the size of the CRs, trn regions, rRNA regions, introns and intergenic regions in each mitogenome separately (Additional file 1: Table S2). In P. chlamydosporia mitogenome, the lengths of the CRs, trn regions, rRNA regions, introns and intergenic regions are 14,385 bp (56.16%), 1,697 bp (6.63%), 6,232 bp (24.33%), 335 bp (1.31%) and 4,618 bp (18.03%) of the genome sequences, respectively. A comparison of the five mitogenomes showed that, in general, the length of each region (CRs, trn regions, rRNA regions, introns and intergenic regions) increases with genome size, as well as the proportions of introns and intergenic regions, with the exception of H. jecorina, which has the smallest length of rRNA genes but the largest length of introns of the five mitogenomes. In addition, the length of its intergenic regions is less than that of F. oxysporum, although the genome size of H. jecorina is larger than that of F. oxysporum (Additional file 1: Table S2). The results indicate that, in Sordariomycetes, CRs, intergenic regions and introns are the three main contributors to mitogenome size variation, with the length of intergenic regions or introns being the primary contributor in most of the mitogenomes. The length of the CRs in N. crassa and H. jecorina is the second highest contributor because several intron-encoded ORFs and unidentified reading frames exist in their intergenic regions (Additional file 1: Table S3).
Figure 3.

A comparison of the mitogenomes of five Pezizomycotina fungi with considerable size differences. There are 15, 15, 16, 19, and 28 protein-coding genes in P. chlamydosporia, M. anisopliae, F. oxysporum, H. jecorina, and N. crassa, respectively. Size comparisons indicate that the larger the mitogenomes are primarily due to larger intergenic regions, secondarily to introns, and to a lesser extent to longer coding regions.
Codon usage in the fungal mitogenomes of Pezizomycotina
We analysed codon usage bias in the mitogenome of P. chlamydosporia, and compared it with other fungal mitogenomes in Pezizomycotina. Genetic code four [33] was used to transcribe the coding gene sequences in the P. chlamydosporia mt genome. A total of 58 codons were found, including the high-frequency codons TTA (Leu), ATA (Ile), GTA (Val) and TTC (Phe) (Additional file 1: Table S4), which are amino acids with hydrophobic side chains. Six codons (CTC, CTG, AGG, CGC, CGG and TGG) were absent in P. chlamydosporia but present in the closely related species M. anisopliae. However, two codons (CGA, TAG) were absent in M. anisopliae but present in P. chlamydosporia. Clearly, nearly in all of the fungal mitogenomes, codon usage is biased strongly towards codons ending in A or T, and more than 84% of the codons in P. chlamydosporia mitogenomes end in A or T (Additional file 1: Table S5), possibly due to the high AT content found in fungal mitogenomes. Similar results were obtained from M. anisopliae and L. muscarium mitogenomes [24,30].
Molecular evolution of rps3 in Pezizomycotina
We identified and annotated the rps3 genes in the 20 fungal mitogenomes. The length of the rps3 sequences ranged from 1,140 bp in the T. rubrum and E. floccosum mitogenomes to 1,545 bp in the F. verticillioides mitogenome (Additional file 1: Table S6). When the 20 Rps proteins were aligned by BLASTP the similarity levels were greater than or equal to 30%. However, the C-terminal domains of these proteins obtained from the Pfam database indicated a common function (Additional file 1: Figure S3A). The addition of 19 more fungal rps3 gene sequences showed matches with E-value cutoffs of less than 2e-29, hence they were omitted from comparisons. E-value cutoffs of 0 were obtained for the rps3 genes of F. oxysporum, M. anisopliae, H. jecorina, F. solani, F. graminearum and F. verticillioides (Additional file 1: Figure S3B). The unique evolutionary relationships among these species can be elucidated from phylogenetic analysis based on the sequences of the rps3 genes (Additional file 1: Figure S3C). A phylogenetic tree shows that P. chlamydosporia rps3 is more closely related to F. oxysporum and M. anisopliae rps3. They then group together with H. jecorina and other three Fusarium species to form clade A. Clade A separates distinctly from clade B, which contains the other five entomopathogenic fungi (L. muscarium, C. militaris, B. pseudobassiana, C. brongniartii and B. bassiana) (Additional file 1: Figure S3C). Evidently, the phylogenetic relationships among the species of Hypocreales inferred from the rps3 genes are different from those inferred from the other 14 protein-coding genes (Figure 2A).
To measure the rate and selective pressure of the rps3 genes, we calculated nonsynonymous/synonymous substitution rate ratios (ω = dN/dS) by adopting different models (see the Methods section). The dN/dS value for the 20 rps3 genes is 0.090 using the CODEML program with a JTT model in PAML [34]. Because every functional protein contains amino acid sites under selective constraints, averaging the evolutionary rates across sites leads to a lower capacity to detect positive selection. Thus, we adopted the site models M1 (neutral), M2 (selection), M7 (beta) and M8 (beta & ω) for the ω ratio analysis [35]. Because the M7-M8 comparison is a very stringent test of positive selection [36], we calculated the value of the likelihood ratio test (LRT) statistic (2Δ = 25.854, P-value = 2.432e-06, the Chi-square test with degrees of freedom, i.e. d.f. = 2), and the significant results of this comparison clearly indicates signals of positive selection in the rps3 genes. To further confirm whether the Hypocreales rps3 genes are evolving under positive selection or not, we took the rps3 sequence of T. rubrum as a reference to calculate the dN and dS of each rps3 sequence with the reference sequence using DnaSP [37] and CODEML [34]. Similar results were obtained, that is, dN/dS > 1 in Hypocreales (Additional file 1: Figure S3C and Table S7). The above results indicate that the fungal rps3 protein is not only under functional constraints but also under positive selection in Hypocreales.
trn gene distribution and putative gene rearrangements in Pezizomycotina
The distribution of trn genes on mt genomes is considered as a factor that potentially contributes to the gene order variation in Pezizomycotina [22]. Firstly, we analyzed the distribution of 23 trn genes in the P. chlamydosporia mitogenome and found that they cluster into three groups (YDN, VISWP and TEMMLAFKQHM), with the exception of four trn genes (trnG, trnR, trnC, trnR) that scatter as a single gene throughout the mt genome (Figure 1). Then, we compared the 13 trn genes (trnD, trnE, trnF, trnH, trnI, trnK, trnL, trnM, trnN, trnR, trnS, trnW, and trnY) in the 20 fungal mitogenomes of Pezizomycotina and found that they are presented in all mitogenomes whereas some of the other seven trn genes (trnA, trnC, trnG, trnP, trnQ, trnT and trnV) are often missing from a fungal mitogenome (Figure 4A) as reported in previous studies [24,25,30,32,38,39]. Some trn genes have multiple copies, such as trnM, which has at least two copies in each mitogenome. A sequence alignment showed that the trn gene sequences display high similarities among these fungal species, such as for trnN, where 43 out of the 71/72 bp (61%) are in consensus in the 20 mitogenomes, and completely identical (100%) in B. bassiana, B. pseudobassiana and C. brongniartii (Figure 4B). A comparative analysis showed that the distribution of trn genes is variable in Pezizomycotina, but identical in the Order Hypocreales (Figure 4A). The trn clusters are conspicuously observed in these mitogenomes, as reports in previous studies [24-26]. Excepting the largest trn cluster (TEMMLAFKLQHM) conserved in all of the Pezizomycotina mitogenomes, the others are generally conserved at the Order level, such as (YDSN) and (VISWP) in Hypocreales, (KGDSW) in Sordariales, (KGDSWISP) in Eurotiales and (KGDSIWSP) in Onygenales, although minor differences exist owing to the lack of a trn or a transposition event (Figure 4A). The clustering of trn is suggested to be a unique characteristic of all Pezizomycotina [24].
Figure 4.
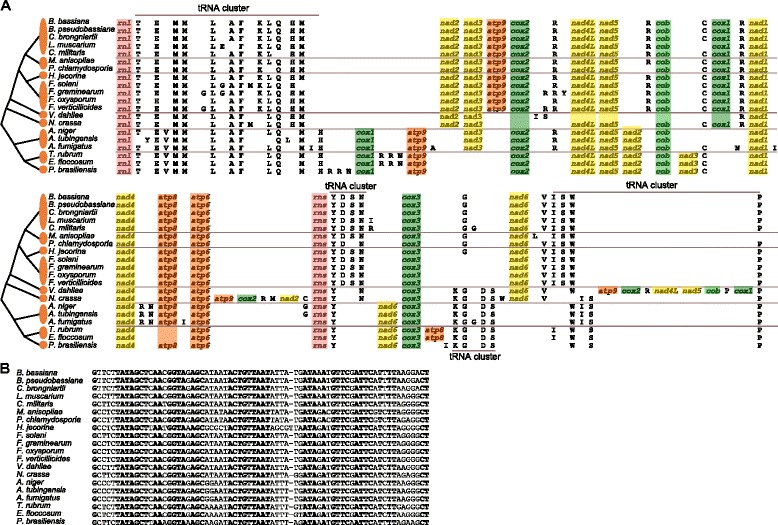
The schematic representation of trn clusters. A). Analysis of the trn genes residing in the complete mitogenomes of 20 fungi that belong to nine families. The nine families are Clavicipitaceae, Hypocreaceae, Nectriaceae, Plectosphaerellaceae, Sordariaceae, Lasiosphaeriaceae, Trichocomaceae, Ajellomycetaceae and Arthrodermataceae. B). An example of high sequence similarities in trn genes (trnN). The consensus bases are marked in boldface.
Based on a display of the orthologous protein-coding genes, trn genes and rRNAs in the 20 mitogenomes (Figure 4A), a similar gene order can be observed in the Hypocreales mitogenomes, with the exception of trnG, which changed position in Fusarium species. The gene order is essentially identical in the seven invertebrate-parasitic fungi (B. bassiana, B. pseudobassiana, C. brongniartii, L. muscarium, C. militaris, M. anisopliae and P. chlamydosporia) and H. jecorina, as well as the four Fusarium fungi, as found in previous studies [24-26]. The same is also observed in Eurotiales and Onygenales. However, marked differences in gene order are observed between Sordariomycetes and Eurotiomycetes (Figure 4A). This indicates that in Pezizomycotina, which has closer phylogenetic relationships among its fungi, a greater number of identical gene orders are displayed, supporting the conclusion that the greater the identical gene orders the closer relationship they share [25].
Using CREx analysis [40], possible gene rearrangements in the 20 mitogenomes were predicted. A total of 21 transposition events were identified, 15 of which involve changes in trn position (Additional file 1: Figure S4). Among these, there are some rearrangement events that only occur in certain fungi (such as trnL in A. tubingensis from ‘LQ’ to ‘QL’ ), but no transposition event was found in Clavicipitaceae. Several events occur at the Order level, for example, the trnG location in Hypocreales involves at least three events (‘G nad6 V I S W P rnl T E M M L’ vs ‘nad6 V I S W P rnl T E M M L G’; ‘G nad6 V I S W P rnl T E M M’ vs ‘nad6 V I S W P rnl T E M M G’; ‘L G’ vs ‘G L’). Most of the rearrangements occur in Pezizomycotina, such as trnH (‘H M’ vs ‘M H’), trnI and trnS (‘I S W’ vs ‘W I S’, ‘S W’ vs ‘W S’), between Sordariomycetes and Eurotiomycetes (Figure 4A). Notably, many transposition events (13/21) occur in the region near nad6 (Additional file 1: Figure S4), perhaps due to historical changes in gene order in this region. Although four types of rearrangement operations (i.e. transpositions, reverse transpositions, reversals and tandem-duplication-random-loss) were considered in the program CREx [40], only transposition events were predicted in our study, supporting the suggestion that the evolution of gene orders within Pezizomycotina is mostly driven by transpositions [12]. Even though the mechanisms of rearrangements are not fully understood, these rearrangement events may provide clues about the phylogeny and evolution of the fungal species in Pezizomycotina.
Discussion
The mt genome represents a major model system in studies on evolutionary genomics, such as in animals [29] and fungi [12]. Here, the organization of the complete mt genome of P. chlamydosporia that is a promising BCA parasitizing nematode eggs is provided and its phylogenetic relationships to other invertebrate-pathogenic fungi are investigated. Synteny and phylogenetic analyses show that P. chlamydosporia is more closely related to M. anisopliae than to L. muscarium and other entomopathogenic fungi (Figure 2), even though both P. chlamydosporia and L. muscarium were once assigned to the same genus, Verticillium. Our results are consistent with previous results on the nuclear genome, in which the genome of P. chlamydosporia was found to be most closely related to entomopathogenic fungi Metarhizium spp. [11], as well as on the phylogenetic analyses of the P. chlamydosporia serine proteases [41,42]. Our phylogenetic results also show that fungi attributed to the same genus group together at first, such as the three species of Beauveria (B. bassiana, B. pseudobassiana and C. brongniartii, the latter is the teleomorph of B. brongniartii [43]), and the two species of Lecanicillium (L. muscarium and C. militaris, the teleomorph of L. militaris [44]). These five species then cluster into a clade because all of their teleomorphic forms are attributed to the same genus, Cordyceps [44,45]. Similarly, the four Fusarium species and the three Aspergillus species form two separate clades. According to our results, P. chlamydosporia and M. anisopliae also group together with a bootstrap value of 100%. The teleomorph of P. chlamydosporia is Metacordyceps chlamydosporia, but the teleomorph of M. anisopliae is unknown, therefore, we hypothesize that the teleomorphic form of M. anisopliae may also belong to the genus Metacordyceps.
Our phylogenetic tree showed that, in Hypocreales, all of the invertebrate-pathogenic fungi cluster together to form a monophyletic group, which is noticeably distinguished from a cluster comprising plant pathogens. This suggests that these invertebrate-pathogenic fungi have a most recent common ancestor (MRCA). Notably, in our phylogenetic tree, the fungus H. jecorina (the anamorphic name Trichoderma reesei [46]) groups together with the four Fusarium species, and is obviously outside the group entomopathogens. This is in agreement with some previous studies [47,48], but distinctly different from that in others [11,12,24,30,32], in which H. jecorina was within entomophagous group and more closely related to M. anisopliae. Because the results were obtained from both the total genomic data [11,48] and the mitogenomic data [12,24,30,32,47], we believe this may reflect to the different evolutionary rates of the nuclear and mt genomes or the much bigger data (number of genes) provided by the nuclear genome in comparison with mt genes. Considering H. jecorina that is attributed to the Family Hypocreaceae in fungal taxonomy, we think it is more reasonable that H. jecorina is outside the group entomopathogens, which belong to the Family Clavicipitaceae. Based on the phylogenetic relationship in our study, the evolution of fungi in Hypocreales may be speculated to have evolved from being parasitic on plants towards being parasitic on invertebrates. We think that a host habitat hypothesis can be used to explain the host shift in Hypocreales fungi, which suggests that host shifts tend to follow the host’s microhabitat or feeding habitat, resulting in a group of related endoparasites that exploit distantly related organisms at higher taxonomic levels [9]. Nikoh and Fukatsu reported that the entomoparasitic fungi of Cordyceps (Hypocreales) have an interkingdom host jumping from Animalia to Fungi by overlapping the ecological niches of the unrelated hosts [9]. Because most Hypocreales fungi grow naturally in soil, interkingdom host jumping events in Hypocreales might occur in an underground environment. It is thus worthwhile to study the evolution of Hypocreales fungi in depth in the future.
Among the ascomycete fungi, it was reported that the group I intron encoded version of rps3 appears to have a rather complex evolutionary history [21]. However, the evolution of rps3 in Hypocreales fungi has not been reported so far. In this study, our phylogenetic analysis shows that mitogenome rps3 genes display an evolutionary pattern distinctly different from those inferred from the 14 mt protein-coding genes in Hypocreales fungi (Figure 2A). A JTT model showed a dN/dS value less than 1, indicating that the encoded protein is under functional constraints, i.e., natural selection is operating to minimize the number of amino acid changes, thereby maintaining the activity of the protein; a result similar to that found in a study with Ophiostomatoid fungi [21]. However, a site model analysis showed a clear signal of positive selection in these rps3 sequences. Moreover, the dN/dS values obtained for all of the rps3 sequences in Hypocreales - in relation to the reference sequence of T. rubrum - are also greater than 1, suggesting that positive selection acts on rps3 in Hypocreales fungi. Such a positive selection on rps3 was also observed previously in gymnosperms [49]. Although the special function of rps3 in ascomycetes fungi is unclear, it is known that rps3 plays a critical role in ribosome biogenesis and DNA repair in other eukaryotes [50]. Therefore, further investigation is needed to fully understand the evolution of fungal rps3 genes.
As shown recently by the comparison of 38 complete fungal mt genomes from all the major phyla, remarkable variation is observed in genome size, gene order, composition of intergenic regions, repeats and introns [22]. In our study, we compared the sizes of the mt genomes from fungal genera belonging to Sordariomycetes and showed that, CRs, intronic and intergenic regions all contributed to genome size variation. However, the major contributor was the length of the intergenic regions or the intronic regions in the majority of the fungal mitogenomes (Additional file 1: Table S3). Our result indicates that, in Sordariomycetes, the length of intergenic regions is also an important contributor to fungal mitogenome size variation, the same as the length of introns.
A remarkable characteristic of fungal mitogenomes is that, similar to plant mitogenomes, fungal mitogenomes show signals of recombination. A comparison of 38 complete fungal mitogenomes (including the major fungal group) showed that the patterns of rearrangements may be explained by the combined influences of recombination, accumulated repeats, especially at intergenic regions, and to a lesser extent, mobile element dynamics [22]. In this study, we compared the trn in the 20 mitogenomes of Pezizomycotina fungi, and found that the gene order for all the protein- and rRNA-coding genes were principally conserved in Sordariomycetes, but trn clusters were conserved at the Order level (Figure 4A). Obvious differences of gene order and trn clusters were observed between Sordariomycetes and Eurotiomycetes. Our results support the previous reports that the Sordariomycetes and the Eurotiomycetes have highly conserved gene arrangements [22]. Moreover, several gene order rearrangement events in the 20 Pezizomycotina fungi mitogenomes were estimated by CREx analysis, and 21 transposition events in the mitogenomes were identified (Additional file 1: Figure S4). Out of these, 15 events displayed changes in trn order. Our results support the view that the evolution of gene order in Pezizomycotina is mostly driven by transpositions [12], although four operations of inversion, transposition, reverse-transposition and tandem-duplication-random-loss were considered in CREx program [27]. The observed transpositions also support the previous suggestion that trn genes should be considered as mobile elements involved in gene rearrangement [29,30]. Our results may provide an illustration of trn genes location changes in gene rearrangement in Pezizomycotina fungi mitogenomes.
Conclusions
In this study, the complete and annotated mt genome sequence of P. chlamydosporia is provided. The relationships among the invertebrate pathogenic fungi in Hypocreales are determined. According to different model predictions, our results indicate that the rps3 gene has experienced positive selection leading to a unique evolutionary pattern in Hypocreales. A comparison of the mitogenome sizes in Sordariomycetes shows that intergenic regions are as important as introns contributing to mitogenome size variation in Sordariomycete. Gene rearrangement analysis shows that the operation of transposition drives the rearrangement events of gene order in Pezizomycotina, and most of them display changes in trn order.
Methods
Fungal isolate
The fungal isolate of P. chlamydosporia stain 170 used in this study was originally isolated from RKN Meloidogyne incognita eggs and confirmed using ITS sequences. This isolate was deposited into the China General Microbiological Culture Collection Center (CGMCC, number 8860). The fungus was grown on potato dextrose agar at 28°C.
DNA preparation, sequencing and assembly
Total DNA of P. chlamydosporia strain 170 was isolated from freeze-dried mycelium from liquid cultures following Fountaine’s description [51]. Three libraries with average insert sizes of 165 bp, 760 bp and 4,261 bp were constructed and sequenced using an Illumina Hiseq 2000 at BGI-Shenzhen (China). A total of 57,274,568 paired-end reads of the three libraries were produced (Additional file 1: Table S8). For these reads, ALLPATHS-LG revision 42305 [52] was used for assembly. From the assembled sequences, one 25,710 bp contig encoding several mt genes was discovered after an alignment to the NCBI NT database using BLASTN. The contig was then selected to amplify both ends of the DNA sequence with the primers (5′-GTACCTATTAACGGTACGGCTA-3′ and 5′-TTAGCCGAGGCAGAATCTGAGT-3′) using PCR technology and sequencing. A single 661 bp PCR product was generated. Using the obtained DNA sequence, a complete 25,615 bp mt genome of P. chlamydosporia was confirmed, and the sequence was submitted to NCBI (KF479445). The sequencing coverage of the contig was estimated by aligning the reads from a short insert library (165 bp) to the genome using BWA [53], and an average coverage of 1,058 × per base was identified. SNPs were investigated using mpileup and bcftools in the samtools package [54].
Gene prediction and genome annotation
The mt genes were predicted by the method described in [15]. All the ORFs were obtained by searching for the mitogenome sequence in the Swiss-Prot [55] and NCBI NR databases using exonerate [56]. From the displayed high sequence similarities identified in the Swiss-Prot/ NR databases, 15 conserved mt genes (cox1-3, cob, nad1-6, nad4L, atp6, atp8-9, rps3) were discovered and annotated. The prediction software tRNAscan-SE v1.3.1 [57] was used to discover the trn. Additionally, the rfam_scan.pl program available in the Rfam database was used for trn and rRNA identification by searching for the mitogenome sequences in Rfam v11.0 [58]. The annotated trn genes from published mt genomes, such as those from Hypocrea jecorina mitogenome, were used as reference genes for prediction via BLASTN. A total of 23 trn genes were confirmed. Moreover, RepeatMasker with default parameters [59], EMBOSS einverted and palindrome [60] were used to identify the repeats in the mitogenomes, and repeat content was discovered. We also used 12 published P. chlamydosporia mt gene sequences to investigate the variance in the P. chlamydosporia mitogenome, including six coding segments in strain IMI 113169 (nad3-atp9, atp6, rns, nad1, nad3, cox3) [61,62], four genes in strain IMI 156157 (rns, nad1, nad3, cox3) [62], one atp6 gene in strain CBS 101244 and one atp6 gene in strain CBS 504.66 [10].
Comparative analysis
A total of 21 fungal mitogenomes were used for the comparative analysis (Table 1). The genome sequences and annotations of 19 mitogenomes were obtained from NCBI genbank files, with the exception of the N. crassa sequence, which was downloaded from the Broad Institute [63]. The N. crassa trn were predicted using a method similar to that used for the P. chlamydosporia trn identification. The trn analyses were performed based on comparing the trn arrangements in the mitogenomes of 20 Pezizomycotina fungi, including 12 Hypocreales fungi (P. chlamydosporia, M. anisopliae [24], B. bassiana [26], B. pseudobassiana, C. brongniartii [61], L. muscarium [30], C. militaris [38], H. jecorina [64], F. solani [65], F. graminearum [65], F. verticillioides [65] and F. oxysporum [25] ), one Phyllachorales fungus (V. dahliae [32] ), one Sordariales fungus (N. crassa), three Eurotiales fungi (A. fumigatus [66], A. niger [39] and A. tubingensis [67] ), and three Onygenales fungi (T. rubrum [68], E. floccosum [69] and P. brasiliensis [70] ). The proteins of seven fungi were selected for codon usage analysis, including two Clavicipitaceae species (M. anisopliae, P. chlamydosporia), and one from each of the families Hypocreaceae (H. jecorina), Nectriaceae (F. oxysporum), Plectosphaerellaceae (V. dahliae), Sordariaceae (N. crassa) and Trichocomaceae (A. fumigatus) and using C. parapsilosis of Saccharomycotina as the outgroup for phylogeny comparisons. Five genomes displaying sizes with different variances were used for the size comparison. The rearrangement events in the mitogenomes were identified with CREx [40]. The gene orders of the 20 mitogenomes were uploaded to the CREx web server [71] which covers inversions, transpositions, reverse transpositions, and tandem-duplication-random-losses. In this study, only transposition operations were identified.
Phylogenetic analysis
The 21 fungal mitogenomes were used for inferring a phylogenetic tree. In total, 14 mt genes (cox1-3, cob, nad1-6, nad4L, atp6, and atp8-9) were identified in the 21 organisms, however, two nad2 (NCU16001, 235 aa; NCU16006, 584 aa) were encoded in N. crassa mitochondria, and both nad2 genes had stop codons. Both of these nad2 proteins showed a high level of sequence similarity to nad2 in other fungi using BLASTP with a threshold of 1e-50. NCU16001 is shorter than NCU16006. A block (1–195 aa) of the sequence NCU16001 aligned to the NCU16006 block (1–233 aa) with an E-value of 2e-66. However, the size of NCU16006 is similar to the nad2 of other Pezizomycotina species (such as H. jecorina), and NCU16006 is adjacent to nad3, which is a conserved unit in Pezizomycotina. To infer the true phylogeny, two groups of proteins (group I used 14 proteins in 21 species, including NCU16006 but not NCU16001 in N. crassa, and group II used 14 proteins in 20 species except N. crassa) were investigated through phylogenetic analysis, and the results displayed similar topologies.
The phylogenetic trees were constructed from both the nucleotide and amino acid sequences of the mt genes (Additional file 1: Table S9) using a ML method. A multiple sequence alignment of each gene was analysed using MUSCLE version 3.8.31 [72]. Certain poor alignment positions may have been saturated by multiple substitutions, and these regions may be obstacles to obtaining a reliable molecular phylogeny. The amino acid sequences of these core genes were concatenated, and the regions of poor alignment, including gaps, were removed using Gblocks version 0.91 with default parameters [73]. The unambiguously aligned portions of the amino acid sequences were obtained, and the corresponding nucleotide sequences were generated by running an in-house perl script. The best models for phylogenetic analysis of the nucleotide sequences were evaluated using jModelTest version 2.1.4 [74], which calculated 20 models. The evaluations generated by the Akaike Information Criterion (AIC) [75] and the Bayesian Information Criterion (BIC) [76] suggested that the General Time Reversible (GTR) [77] model coupled with rate variation among sites (+G) was the best model. Therefore, the GTR + G model was used for phylogenetic analysis. The nucleotide sequences were used to generate an ML tree with bootstrapping (1000 replicates) using MEGA version 6.06 [78] and PhyML version 3.1 [79]. Additionally, ProtTest version 3.4 [80] indicated that cpREV [81] combined with rate variation among sites (+G) and empirical frequencies (+F) was the best-fit model out of 120 models for the amino acid sequences, according to both AIC [75] and BIC [76]. The aligned amino acid sequences were used to construct the ML trees with 1000 bootstrap replicates using MEGA [78] and PhyML [79]. The topologies generated by both the nucleotide sequences and amino acids were similar to each other.
The rps3 genes were identified and annotated in 20 fungi, and used to infer phylogeny. jModelTest [74] and ProtTest [80] were performed to select the best-fit models for the sequences of rps3. For the nucleotide sequences, the GTR + G model was used. For the amino acid sequences, the Jones-Taylor-Thornton (JTT) [82] model combining rate variation among sites (+G) and empirical frequencies (+F) was chosen. The two models (GTR + G, JTT + G + F) were evaluated with AIC [75] and BIC [76]. The ML trees of the sequences were constructed using MEGA [78] and PhyML [79]. The dN/dS ratio of rps3 was analysed using CODEML with a JTT [82] model in PAML v4.7a [34], and site models M1 (neutral), M2 (selection), M7 (beta) and M8 (beta & ω) were used to detect the positive selection pressure on the genes. Although the values of dN/dS are less than or equal to 0.158, the LRT statistic for comparing M7 (lnL (log likelihood value) = −10,186.296) and M8 (lnL = −10,173.369) is 25.854 (2Δ = 2* (10,186.296-10,173.369) = 25.854), with a P-value of 2.432e-06 using the Chi-square test (with d.f. = 2). Furthermore, the dN/dS values of each rps3 sequence with the rps3 sequence in T. rubrum were calculated using DnaSP 5.10.1 [37] and CODEML [34].
Data depositions
The mt genome sequence of P. chlamydosporia has been submitted to NCBI’s GenBank under the accession number: KF479445. And the phylogenetic data in this study has been submitted to TreeBASE [83].
Acknowledgements
We thank all the members of the laboratories involved in this project: Guokun Liu at Fujian Agriculture and Forestry University for his kind gift of the strain, Aiping Zheng at Sichuan Agricultural University and Jianfeng Ren at Shanghai Ocean University for their helpful comments on the article, and BGI-Shenzhen for sequencing. This work was supported by the National Basic Research Program of China (No. 2009CB11900); The Special Fund for Agro-scientific Research in the Public Interest (No. 201003018); and the earmarked fund for China Agriculture Research System (No. CARS-25-B-01).
Abbreviations
- BCA
Biological control agent
- ITS
Internal transcribed spacer
- mt
Mitochondrial
- RKN
Root-knot nematode
- cox
Cytochrome c oxidase subunit
- cob
Apocytochrome b
- atp
ATP synthase subunit
- nad
Subunit of NADH dehydrogenase
- rps3
Ribosomal protein S3
- rns
Small ribosomal RNA
- rnl
Large ribosomal RNA
- trn
Transfer RNA genes
- SNP
Single nucleotide polymorphism
- CDS
Coding DNA sequence
- CR
Coding region
- LRT
Likelihood ratio test
- d.f.
Degrees of freedom
- ML
Maximum likelihood
- MRCA
Most recent common ancestor
Additional file
Comparison of the assembled mitogenome sequences with the published P. chlamydosporia mt genes. Table S2. Gene and intergenic regions sizes from the complete mitogenomes of five Pezizomycotina fungal species. Table S3. The three primary contributors to mitogenome size variation. Table S4. Distinct codon usage for fungal mitogenome genes. Table S5. Codon usage bias statistics. Table S6. Nucleotide sequence lengths (bp) of the rps3 gene and group I introns. Table S7. The dN/dS values of the rps3 sequences. Table S8. Summary of the sequenced data of P. chlamydosporia. Table S9. Proteins used for phylogenetic analyses are shown. Figure S1. Distribution of the genes and repeats in the mitogenomes of P. chlamydosporia, M. anisopliae and L. muscarium. Figure S2. Synteny between P. chlamydosporia and B. bassiana, V. dahliae, A. fumigatus. Figure S3. Evolutionary characteristics of rps3. Figure S4. Putative transposition events in the mitogenomes of 20 fungi detected by the use of CREx algorithm.
Footnotes
Runmao Lin and Chichuan Liu contributed equally to this work.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
LR assembled the genome. LR, LC, and BM annotated the genome. SB and BM conducted the molecular biology experiments. LR, LC, SB, BM, LJ, CG, and MZ analysed the data. LR, LC, SB, BM and CX prepared the graphs and tables. LR, CX and XB wrote the manuscript. CX and XB participated in the design of the study. LR and LC contributed equally to this work. All authors have read and approved the final manuscript.
Contributor Information
Runmao Lin, Email: linrm2010@gmail.com.
Chichuan Liu, Email: chichuanliu@hotmail.com.
Baoming Shen, Email: baobao8759@163.com.
Miao Bai, Email: baimiao2010@gmail.com.
Jian Ling, Email: lingjian2005@126.com.
Guohua Chen, Email: chenguohua01@caas.cn.
Zhenchuan Mao, Email: maozhenchuan@caas.cn.
Xinyue Cheng, Email: chengxy@bnu.edu.cn.
Bingyan Xie, Email: xiebingyan@caas.cn.
References
- 1.Escudero N, Lopez-Llorca LV. Effects on plant growth and root-knot nematode infection of an endophytic GFP transformant of the nematophagous fungus Pochonia chlamydosporia. Symbiosis. 2012;57(1):33–42. doi: 10.1007/s13199-012-0173-3. [DOI] [Google Scholar]
- 2.Manzanilla-López RH, Esteves I, Finetti-Sialer MM, Hirsch PR, Ward E, Devonshire J, et al. Pochonia chlamydosporia: advances and challenges to improve its performance as a biological control agent of sedentary endo-parasitic nematodes. Journal of Nematology. 2013;45(1):1. [PMC free article] [PubMed] [Google Scholar]
- 3.Kerry BR. Rhizosphere interactions and the exploitation of microbial agents for the biological control of plant-parasitic nematodes. Annu Rev Phytopathol. 2000;38:423–41. doi: 10.1146/annurev.phyto.38.1.423. [DOI] [PubMed] [Google Scholar]
- 4.Kerry BR, Bourne JM. The importance of rhizosphere interactions in the biological control of plant parasitic nematodes—a case study using Verticillium chlamydosporium. Pesticide Science. 1996;47(1):69–75. doi: 10.1002/(SICI)1096-9063(199605)47:1<69::AID-PS386>3.0.CO;2-6. [DOI] [Google Scholar]
- 5.Casas-Flores S, Herrera-Estrella A. Antagonism of plant parasitic nematodes by fungi. In: Kubicek C, Druzhinina I, editors. The mycota IV: environmental and microbial relationships. 2. Berlin, Germany: Springer; 2007. pp. 147–57. [Google Scholar]
- 6.Sasser J, Freckman D. A world perspective on nematology: the role of the society. In: Veech JA, Dickson DW, editors. Vistas on nematology. Hyatsville, MD USA: Society of Nematologists; 1987. pp. 7–14. [Google Scholar]
- 7.Zare R, Gams W, Evans H. A revision of Verticillium section Prostrata. V. The genus Pochonia, with notes on Rotiferophthora. Nova Hedwigia. 2001;73(1):51–86. [Google Scholar]
- 8.Morton CO, Mauchline TH, Kerry R, Hirsch PR. PCR-based DNA fingerprinting indicates host-related genetic variation in the nematophagous fungus Pochonia chlamydosporia. Mycol Res. 2003;107(2):198–205. doi: 10.1017/S0953756203007251. [DOI] [PubMed] [Google Scholar]
- 9.Nikoh N, Fukatsu T. Interkingdom host jumping underground: phylogenetic analysis of entomoparasitic fungi of the genus cordyceps. Mol Biol Evol. 2000;17(4):629–38. doi: 10.1093/oxfordjournals.molbev.a026341. [DOI] [PubMed] [Google Scholar]
- 10.Sung G-H, Sung J-M, Hywel-Jones NL, Spatafora JW. A multi-gene phylogeny of Clavicipitaceae (Ascomycota, Fungi): Identification of localized incongruence using a combinational bootstrap approach. Mol Phylogenet Evol. 2007;44(3):1204–23. doi: 10.1016/j.ympev.2007.03.011. [DOI] [PubMed] [Google Scholar]
- 11.Larriba E, Jaime MD, Carbonell-Caballero J, Conesa A, Dopazo J, Nislow C, et al. Sequencing and functional analysis of the genome of a nematode egg-parasitic fungus, Pochonia chlamydosporia. Fungal Genet Biol. 2014;65:69–80. doi: 10.1016/j.fgb.2014.02.002. [DOI] [PubMed] [Google Scholar]
- 12.Duò A, Bruggmann R, Zoller S, Bernt M, Grünig CR. Mitochondrial genome evolution in species belonging to the Phialocephala fortinii s.l. - Acephala applanata species complex. BMC Genomics. 2012;13(1):166. doi: 10.1186/1471-2164-13-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paquin B, Laforest M-J, Forget L, Roewer I, Wang Z, Longcore J, et al. The fungal mitochondrial genome project: evolution of fungal mitochondrial genomes and their gene expression. Curr Genet. 1997;31(5):380–95. doi: 10.1007/s002940050220. [DOI] [PubMed] [Google Scholar]
- 14.Gray MW. Mitochondrial evolution. Cold Spring Harbor Perspectives in Biology. 2012; 4(9):a011403 [DOI] [PMC free article] [PubMed]
- 15.Zheng A, Lin R, Zhang D, Qin P, Xu L, Ai P, et al. The evolution and pathogenic mechanisms of the rice sheath blight pathogen. Nat Commun. 2013;4:1424. doi: 10.1038/ncomms2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pramateftaki PV, Kouvelis VN, Lanaridis P, Typas MA. The mitochondrial genome of the wine yeast Hanseniaspora uvarum: a unique genome organization among yeast/fungal counterparts. FEMS Yeast Res. 2006;6(1):77–90. doi: 10.1111/j.1567-1364.2005.00018.x. [DOI] [PubMed] [Google Scholar]
- 17.Belcour L, Rossignol M, Koll F, Sellem CH, Oldani C. Plasticity of the mitochondrial genome in Podospora. Polymorphism for 15 optional sequences: group-I, group-II introns, intronic ORFs and an intergenic region. Curr Genet. 1997;31(4):308–17. doi: 10.1007/s002940050210. [DOI] [PubMed] [Google Scholar]
- 18.Hausner G. Fungal mitochondrial genomes, plasmids and introns. Applied Mycology and Biotechnology. 2003;3:101–31. doi: 10.1016/S1874-5334(03)80009-6. [DOI] [Google Scholar]
- 19.Formighieri EF, Tiburcio RA, Armas ED, Medrano FJ, Shimo H, Carels N, et al. The mitochondrial genome of the phytopathogenic basidiomycete Moniliophthora perniciosa is 109 kb in size and contains a stable integrated plasmid. Mycol Res. 2008;112(10):1136–52. doi: 10.1016/j.mycres.2008.04.014. [DOI] [PubMed] [Google Scholar]
- 20.Sethuraman J, Majer A, Friedrich N, Edgell D, Hausner G. Genes within genes: multiple LAGLIDADG homing endonucleases target the ribosomal protein S3 gene encoded within an rnl group I intron of Ophiostoma and related taxa. Mol Biol Evol. 2009;26(10):2299–315. doi: 10.1093/molbev/msp145. [DOI] [PubMed] [Google Scholar]
- 21.Sethuraman J, Majer A, Iranpour M, Hausner G. Molecular evolution of the mtDNA encoded rps3 gene among filamentous ascomycetes fungi with an emphasis on the ophiostomatoid fungi. J Mol Evol. 2009;69(4):372–85. doi: 10.1007/s00239-009-9291-9. [DOI] [PubMed] [Google Scholar]
- 22.Aguileta G, de Vienne DM, Ross ON, Hood ME, Giraud T, Petit E, et al. High variability of mitochondrial gene order among fungi. Genome Biol Evol. 2014;6(2):451–65. doi: 10.1093/gbe/evu028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tuller T, Girshovich Y, Sella Y, Kreimer A, Freilich S, Kupiec M, et al. Association between translation efficiency and horizontal gene transfer within microbial communities. Nucleic Acids Res. 2011;39(11):4743–55. doi: 10.1093/nar/gkr054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghikas DV, Kouvelis VN, Typas MA. The complete mitochondrial genome of the entomopathogenic fungus Metarhizium anisopliae var. anisopliae: gene order and trn gene clusters reveal a common evolutionary course for all Sordariomycetes, while intergenic regions show variation. Arch Microbiol. 2006;185(5):393–401. doi: 10.1007/s00203-006-0104-x. [DOI] [PubMed] [Google Scholar]
- 25.Pantou MP, Kouvelis VN, Typas MA. The complete mitochondrial genome of Fusarium oxysporum: Insights into fungal mitochondrial evolution. Gene. 2008;419(1):7–15. doi: 10.1016/j.gene.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 26.Xu J, Huang B, Qin C, Li Z-Z. Sequence and phylogenetic analysis of Beauveria bassiana with mitochondrial genome. Mycosystema. 2009;28(5):718–23. [Google Scholar]
- 27.Perseke M, Fritzsch G, Ramsch K, Bernt M, Merkle D, Middendorf M, et al. Evolution of mitochondrial gene orders in echinoderms. Mol Phylogenet Evol. 2008;47(2):855–64. doi: 10.1016/j.ympev.2007.11.034. [DOI] [PubMed] [Google Scholar]
- 28.Cameron SL, Yoshizawa K, Mizukoshi A, Whiting MF, Johnson KP. Mitochondrial genome deletions and minicircles are common in lice (Insecta: Phthiraptera) BMC Genomics. 2011;12:394. doi: 10.1186/1471-2164-12-394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saccone C, Gissi C, Reyes A, Larizza A, Sbisa E, Pesole G. Mitochondrial DNA in metazoa: degree of freedom in a frozen event. Gene. 2002;286(1):3–12. doi: 10.1016/S0378-1119(01)00807-1. [DOI] [PubMed] [Google Scholar]
- 30.Kouvelis VN, Ghikas DV, Typas MA. The analysis of the complete mitochondrial genome of Lecanicillium muscarium (synonym Verticillium lecanii) suggests a minimum common gene organization in mtDNAs of Sordariomycetes: phylogenetic implications. Fungal Genet Biol. 2004;41(10):930–40. doi: 10.1016/j.fgb.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 31.Lohse M, Drechsel O, Kahlau S, Bock R. OrganellarGenomeDRAW--a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013;41(Web Server issue):W575–81. [DOI] [PMC free article] [PubMed]
- 32.Pantou MP, Kouvelis VN, Typas MA. The complete mitochondrial genome of the vascular wilt fungus Verticillium dahliae: a novel gene order for Verticillium and a diagnostic tool for species identification. Curr Genet. 2006;50(2):125–36. doi: 10.1007/s00294-006-0079-9. [DOI] [PubMed] [Google Scholar]
- 33.Fox TD. Natural variation in the genetic code. Annu Rev Genet. 1987;21(1):67–91. doi: 10.1146/annurev.ge.21.120187.000435. [DOI] [PubMed] [Google Scholar]
- 34.Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24(8):1586–91. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 35.Yang Z, Nielsen R. Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol Biol Evol. 2002;19(6):908–17. doi: 10.1093/oxfordjournals.molbev.a004148. [DOI] [PubMed] [Google Scholar]
- 36.Anisimova M, Bielawski JP, Yang Z. Accuracy and power of the likelihood ratio test in detecting adaptive molecular evolution. Mol Biol Evol. 2001;18(8):1585–92. doi: 10.1093/oxfordjournals.molbev.a003945. [DOI] [PubMed] [Google Scholar]
- 37.Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25(11):1451–2. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- 38.Sung G-H. Complete mitochondrial DNA genome of the medicinal mushroom Cordyceps militaris (Ascomycota, Cordycipitaceae). Mitochondrial DNA. 2013 10.3109/19401736.2013.855754 (in press). [DOI] [PubMed]
- 39.Juhász Á, Pfeiffer I, Keszthelyi A, Kucsera J, Vágvölgyi C, Hamari Z. Comparative analysis of the complete mitochondrial genomes of Aspergillus niger mtDNA type 1a and Aspergillus tubingensis mtDNA type 2b. FEMS Microbiol Lett. 2008;281(1):51–7. doi: 10.1111/j.1574-6968.2008.01077.x. [DOI] [PubMed] [Google Scholar]
- 40.Bernt M, Merkle D, Ramsch K, Fritzsch G, Perseke M, Bernhard D, et al. CREx: inferring genomic rearrangements based on common intervals. Bioinformatics. 2007;23(21):2957–8. doi: 10.1093/bioinformatics/btm468. [DOI] [PubMed] [Google Scholar]
- 41.Morton CO, Hirsch PR, Peberdy JP, Kerry BR. Cloning of and genetic variation in protease VCP1 from the nematophagous fungus Pochonia chlamydosporia. Mycol Res. 2003;107(1):38–46. doi: 10.1017/S0953756202007050. [DOI] [PubMed] [Google Scholar]
- 42.Larriba E, Martín-Nieto J, Lopez-Llorca LV. Gene cloning, molecular modeling, and phylogenetics of serine protease P32 and serine carboxypeptidase SCP1 from nematophagous fungi Pochonia rubescens and Pochonia chlamydosporia. Can J Microbiol. 2012;58(7):815–27. doi: 10.1139/w2012-054. [DOI] [PubMed] [Google Scholar]
- 43.Shimazu M, Mitsuhashi W, Hashimoto H. Cordyceps brongniartii sp. nov., the teleomorph of Beauveria brongniartii. Transactions of the Mycological Society of Japan. 1988;29:323–30. [Google Scholar]
- 44.Zare R, Gams W. A revision of Verticillium section Prostrata. IV. The genera Lecanicillium and Simplicillium gen. nov. . Nova Hedwigia. 2001;73(1–2):1–50. [Google Scholar]
- 45.Li Z, Li C, Huang B, Fan M. Discovery and demonstration of the teleomorph of Beauveria bassiana (Bals.) Vuill., an important entomogenous fungus. Chinese Science Bulletin. 2001;46(9):751–3. doi: 10.1007/BF03187215. [DOI] [Google Scholar]
- 46.Kuhls K, Lieckfeldt E, Samuels GJ, Kovacs W, Meyer W, Petrini O, et al. Molecular evidence that the asexual industrial fungus Trichoderma reesei is a clonal derivative of the ascomycete Hypocrea jecorina. Proc Natl Acad Sci U S A. 1996;93(15):7755–60. doi: 10.1073/pnas.93.15.7755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee J, Young JPW. The mitochondrial genome sequence of the arbuscular mycorrhizal fungus Glomus intraradices isolate 494 and implications for the phylogenetic placement of Glomus. New Phytologist. 2009;183(1):200–11. doi: 10.1111/j.1469-8137.2009.02834.x. [DOI] [PubMed] [Google Scholar]
- 48.Xiao G, Ying SH, Zheng P, Wang ZL, Zhang S, Xie XQ, et al. Genomic perspectives on the evolution of fungal entomopathogenicity in Beauveria bassiana. Sci Rep. 2012;2:483. doi: 10.1038/srep00483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ran JH, Gao H, Wang XQ. Fast evolution of the retroprocessed mitochondrial rps3 gene in Conifer II and further evidence for the phylogeny of gymnosperms. Mol Phylogenet Evol. 2010;54(1):136–49. doi: 10.1016/j.ympev.2009.09.011. [DOI] [PubMed] [Google Scholar]
- 50.Kim Y, Kim HD, Kim J. Cytoplasmic ribosomal protein S3 (rpS3) plays a pivotal role in mitochondrial DNA damage surveillance. Biochim Biophys Acta. 2013;1833(12):2943–52. doi: 10.1016/j.bbamcr.2013.07.015. [DOI] [PubMed] [Google Scholar]
- 51.Fountaine J, Shaw M, Napier B, Ward E, Fraaije B. Application of real-time and multiplex polymerase chain reaction assays to study leaf blotch epidemics in barley. Phytopathology. 2007;97(3):297–303. doi: 10.1094/PHYTO-97-3-0297. [DOI] [PubMed] [Google Scholar]
- 52.Gnerre S, MacCallum I, Przybylski D, Ribeiro FJ, Burton JN, Walker BJ, et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci U S A. 2011;108(4):1513–8. doi: 10.1073/pnas.1017351108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25(14):1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Consortium U. Update on activities at the universal protein resource (UniProt) in 2013. Nucleic Acids Res. 2013;41(D1):D43–7. doi: 10.1093/nar/gks1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Slater GS, Birney E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics. 2005;6(1):31. doi: 10.1186/1471-2105-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25(5):0955–64. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Burge SW, Daub J, Eberhardt R, Tate J, Barquist L, Nawrocki EP, et al. Rfam 11.0: 10 years of RNA families. Nucleic Acids Res. 2013;41(D1):D226–32. doi: 10.1093/nar/gks1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.RepeatMasker. [http://www.repeatmasker.org/]
- 60.EMBOSS einverted and palindrome. [http://emboss.bioinformatics.nl/cgi-bin/emboss/]
- 61.Ghikas DV, Kouvelis VN, Typas MA. Phylogenetic and biogeographic implications inferred by mitochondrial intergenic region analyses and ITS1-5.8 S-ITS2 of the entomopathogenic fungi Beauveria bassiana and B. brongniartii. BMC Microbiol. 2010;10(1):174. doi: 10.1186/1471-2180-10-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pantou MP, Strunnikova OK, Shakhnazarova VY, Vishnevskaya NA, Papalouka VG, Typas MA. Molecular and immunochemical phylogeny of Verticillium species. Mycol Res. 2005;109(8):889–902. doi: 10.1017/S0953756205003345. [DOI] [PubMed] [Google Scholar]
- 63.Neurospora crassa database. [http://www.broadinstitute.org/annotation/genome/neurospora/MultiHome.html]
- 64.Chambergo FS, Bonaccorsi ED, Ferreira AJ, Ramos AS, Ferreira JR, Abrahao-Neto J, et al. Elucidation of the metabolic fate of glucose in the filamentous fungus Trichoderma reesei using expressed sequence tag (EST) analysis and cDNA microarrays. Journal of Biological Chemistry. 2002;277(16):13983–8. doi: 10.1074/jbc.M107651200. [DOI] [PubMed] [Google Scholar]
- 65.Al-Reedy RM, Malireddy R, Dillman CB, Kennell JC. Comparative analysis of Fusarium mitochondrial genomes reveals a highly variable region that encodes an exceptionally large open reading frame. Fungal Genet Biol. 2012;49(1):2–14. doi: 10.1016/j.fgb.2011.11.008. [DOI] [PubMed] [Google Scholar]
- 66.Joardar V, Abrams NF, Hostetler J, Paukstelis PJ, Pakala S, Pakala SB, et al. Sequencing of mitochondrial genomes of nine Aspergillus and Penicillium species identifies mobile introns and accessory genes as main sources of genome size variability. BMC Genomics. 2012;13(1):698. doi: 10.1186/1471-2164-13-698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Juhász Á, Engi H, Pfeiffer I, Kucsera J, Vágvölgyi C, Hamari Z. Interpretation of mtDNA RFLP variability among Aspergillus tubingensis isolates. Antonie Van Leeuwenhoek. 2007;91(3):209–16. doi: 10.1007/s10482-006-9110-x. [DOI] [PubMed] [Google Scholar]
- 68.Wu Y, Yang J, Yang F, Liu T, Leng W, Chu Y, et al. Recent dermatophyte divergence revealed by comparative and phylogenetic analysis of mitochondrial genomes. BMC Genomics. 2009;10(1):238. doi: 10.1186/1471-2164-10-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tambor JHM, Guedes RF, Nobrega MP, Nobrega FG. The complete DNA sequence of the mitochondrial genome of the dermatophyte fungus Epidermophyton floccosum. Curr Genet. 2006;49(5):302–8. doi: 10.1007/s00294-006-0057-2. [DOI] [PubMed] [Google Scholar]
- 70.Cardoso MAG, Tambor JHM, Nobrega FG. The mitochondrial genome from the thermal dimorphic fungus Paracoccidioides brasiliensis. Yeast. 2007;24(7):607–16. doi: 10.1002/yea.1500. [DOI] [PubMed] [Google Scholar]
- 71.CREx web server. [http://pacosy.informatik.uni-leipzig.de/crex]
- 72.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–7. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, et al. Phylogeny. fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008;36(2):W465–9. doi: 10.1093/nar/gkn180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Darriba D, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9(8):772. doi: 10.1038/nmeth.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Akaike H. Information theory and an extension of the maximum likelihood principle. In: Second international symposium on information theory. Akademinai Kiado: 1973; 267–281.
- 76.Schwarz G. Estimating the dimension of a model. The Annals of Statistics. 1978;6(2):461–4. doi: 10.1214/aos/1176344136. [DOI] [Google Scholar]
- 77.Tavaré S. Some probabilistic and statistical problems in the analysis of DNA sequences. Lectures on Mathematics in the Life Sciences. 1986;17:57–86. [Google Scholar]
- 78.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–9. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59(3):307–21. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 80.Darriba D, Taboada GL, Doallo R, Posada D. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics. 2011;27(8):1164–5. doi: 10.1093/bioinformatics/btr088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Adachi J, Waddell PJ, Martin W, Hasegawa M. Plastid genome phylogeny and a model of amino acid substitution for proteins encoded by chloroplast DNA. J Mol Evol. 2000;50(4):348–58. doi: 10.1007/s002399910038. [DOI] [PubMed] [Google Scholar]
- 82.Jones DT, Taylor WR, Thornton JM. The rapid generation of mutation data matrices from protein sequences. Computer Applications in the Biosciences: CABIOS. 1992;8(3):275–82. doi: 10.1093/bioinformatics/8.3.275. [DOI] [PubMed] [Google Scholar]
- 83.Phylogenetic data. [http://purl.org/phylo/treebase/phylows/study/TB2:S16935]