

Abstract
Nonviral plasmid DNA gene therapy represents a promising approach for the treatment of many diseases including cancer. Intracellular delivery of DNA can be achieved with the application of electroporation, which facilitates the initial transport of exogenous DNA across the cell membrane into the cytoplasm. However, it does not guarantee further transport of the DNA from the cytoplasm to the nucleus for subsequent mRNA expression, resulting in varying degrees of exogenous gene translation and a major limitation in comparison to viral approaches. To overcome these expression difficulties, we developed a proof-of-concept vector enhanced expression vector (EEV), which incorporates elements from viral systems including nuclear localization sequences and a viral replicase from the Semliki Forest virus. The replicase allows for cytoplasmic mRNA expression and bypasses the need for nuclear localization to generate high levels of gene expression. We have demonstrated that our EEV is capable of achieving high levels of expression in a variety of tissue types. Antitumor effects of pEEV were demonstrated by the delayed growth and increased survival of the nontherapeutic pEEV-treated CT26 tumor model. Using a novel endoscopic electroporation system, EndoVe, we demonstrate and compare, for the first time, both standard cytomegalovirus (CMV) promoter-driven plasmid and EEV gene expression in intraluminal porcine tissues. Our EEV plasmid displays reliable and superior expression capability, and due to its inherent induced oncolytic activity in transfected cells, it may enhance the efficacy and safety of several cancer immunogene therapy approaches.
Introduction
Gene therapy approaches for cancer have been gaining increasing clinical adoption and impact.1 Delivery of therapeutic genes has typically been approached through utilizing viral vectors with the vast majority of clinical trials, to date, involving the use of viral vectors.2,3 Generally, viral vectors can provide efficient gene transfer, but there are still some disadvantages, such as the potential for toxicity associated with chronic overexpression or insertional mutagenesis and the possibility of nonspecific inflammatory response and antivector cellular immunity.4–6 Other approaches such as using plasmid DNA have received less attention due in part to the weaker levels of gene expression achieved relative to viral systems.1,7 There are nevertheless several limitations, which must be considered including: toxicity, long-term uncontrolled expression, chromosomal integration, immune response generation, and subsequent efficacy of repeat treatment.8 Nonviral approaches with plasmid DNA offer potential advantages for clinical application particularly as they offer a reduced risk profile and a simplified preparation process.
DNA plasmid vectors can allow the transfer of significantly larger genetic material, than is possible using a viral system; are less expensive to manufacture; are considered safe, nontoxic, and less immunogenic than viral vectors; and allow repeat dosing, if required.7,9–11 However, plasmid DNA vectors can often have lower expression profiles than viruses and therefore lack potency in human clinical trials.12 Ultimately, for efficacy in gene therapy, the plasmid must reliably express the gene of interest at adequate levels in the target cell.11,12
The technology of electroporation has been employed effectively in both preclinical and clinical settings for the delivery of plasmid DNA.13–15 It has also been used to enable the uptake by passive diffusion of specific chemotherapeutic drugs, with very high reported antitumor efficacy and negligible side effects.15–18 The process of electroporation involves the delivery of a microsecond pulse directly to the targeted tissue, which increases the local porosity of the tissue to macromolecules.19 Electroporation has been established as a safe and effective method clinically with excellent responses observed in several cancer gene therapy studies. Therapies such as the electroporation delivery of plasmid DNA encoding for interleukin-12 and antiangiogenic metargidin peptide have advanced to clinical trials.20,21 While electroporation facilitates the cytoplasmic absorption of plasmid DNA, it still lags behind viral methods for inducing a high degree of exogenous mRNA expression in the target cells. Therefore, it is important to establish and refine methods to improve electroporation-based gene delivery for clinical use. Optimizing strategies include modulation of electric field strength and pulse duration to enhance plasmid delivery, alteration of the extracellular matrix with enzymatic and chemical methods, and the introduction of reactive oxygen species inhibitors to reduce plasmid DNA destruction by reactive oxygen species generation postelectroporation.22–26 All have an impact on increased transfer and gene expression efficiency via electroporation.
Viruses ensure expression of their genome within an infected cell by expressing a copy of their own replicase, which can transcribe copies of its viral genome within the cytoplasm. In general, the major concern for using virus as an expression vector is their infective nature, which involves biological risks.8 However, the utilization of replication-deficient viral vectors such as Semliki Forest virus (SFV) avoids this problem while allowing rapid and high-level gene delivery.8 The SFV is a positive-stranded RNA virus of the genus Alphavirus of the family Togaviridae.27,28 It is a relatively simple virus, encoding only nine functional proteins of unique sequence. The SFV replicon is a eukaryotic expression vector that includes the cis-acting sequence for 5′ and 3′ ends of the virus genome and all the nonstructural proteins, with the virus structural proteins replaced by the foreign gene.29,30 By utilizing a DNA replicase copy from the SFV and incorporating nuclear localization sequences to facilitate initial nuclear localization of the pEEV and replicase expression, we have been able to overcome some of the drawbacks that have hindered the application of nonviral approaches to cancer gene therapy.
In this study, we sought to deliver a nonviral vector system that has the potential for clinical application in electroporation-based gene therapy. We report a proof-of-concept enhanced expression vector (pEEV), which encompasses a viral replicase from the SFV. We aimed to improve upon nuclear entry by the incorporation of a nuclear localization sequence. We demonstrated cytoplasmic expression of the vector. We also investigated the oncolytic capabilities of the EEV. Furthermore, we compared expression levels of the pEEV with standard available plasmids in murine and porcine animal models.
Results
Construction of pEEV
The EEV construct consists of the cytomegalovirus (CMV) IE/T7 promoter and SV40 polyadenylation (pA) (Figure 1). It includes the entire SFV capsid gene, which functions as a self-cleaving translation enhancer, an Ori colE1 (Ori) and capsid (CAP) gene. It also includes the SFV 1–4 nonstructural components and an ampicillin resistance cassette. To this, an expanded multiple cloning site was incorporated for the inclusion of foreign transcripts. For our study, the lacZ transcript was incorporated. A nuclear localization sequence: CACATAACGGGAGGGCCGGCGGTTACCAGGTCGACGGATATGACGGCAGG was inserted to allow for better transport in the nucleus.
Figure 1.

Plasmid construct. Schematic representation of the circular enhanced expression vector (EEV) plasmid constructed for this study. The circular pEEV construct incorporates a CMV IE/T7 promoter, an Semliki Forest virus (SFV) replicase (nonstructural proteins 1–4), nuclear localization sequence, the entire SFV capsid gene, which functions as a self-cleaving translation enhancer, an Ori colE1 (Ori), 26S subgenomic promoter, capsid (CAP) gene and an ampicillin resistance cassette (AmpR).
Comparison of pEEV-lacZ to standard pCMV-lacz
The EEV plasmid (15.462 kb) is approximately three times larger, than the standard pCMV plasmid (6.233 kb), which could impact on transfection efficacy and expression. To establish this, we compared the transfection efficiency in vivo of pEEV with the pCMV plasmid. In order to determine efficiency, copy numbers of each plasmid expressing the lacZ transgene delivered by electroporation were measured. Both plasmids were detected in all the tissues tested. In general, they were significantly more (P < 0.001) copies of pCMV than pEEV (Figure 2a), with the spleen and tumor having highest plasmid transfection.
Figure 2.
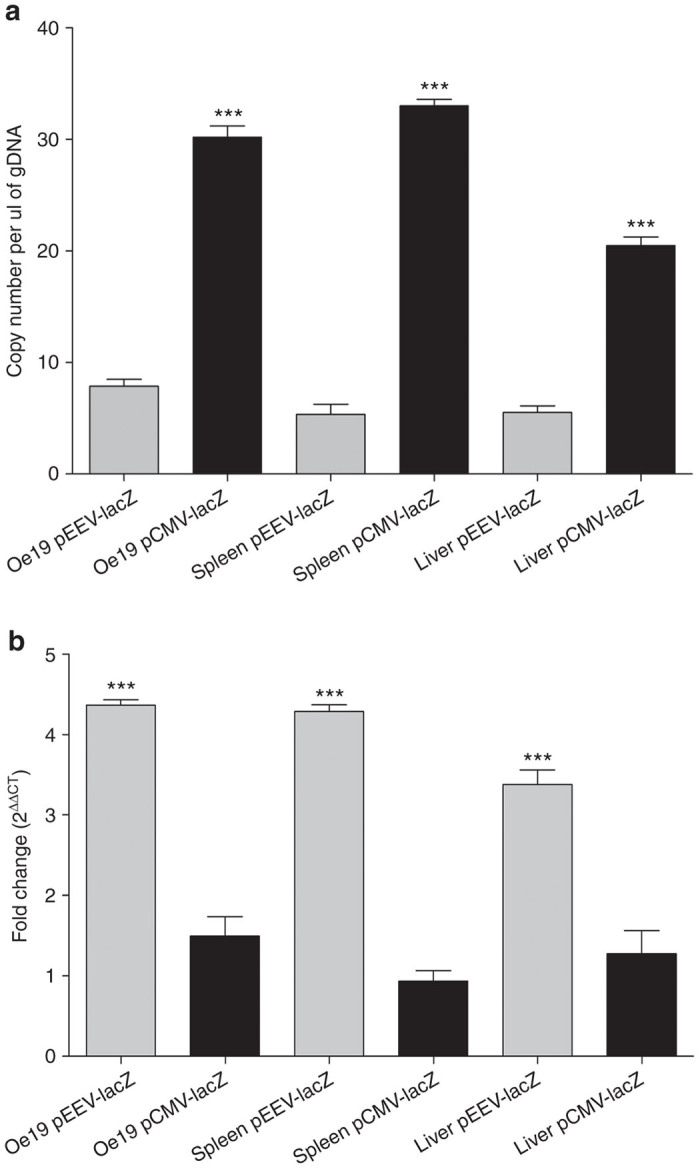
β-galactosidase expression in murine tissue. (a) Copy numbers of lacZ transgene ascertained by quantitative polymerase chain reaction (PCR) in murine tissue. Bar graph showing absolute copy number of the lacz transgene per nanogram of genomic DNA 2 days after electroporation obtained from MF1-nu/nu mouse tissue. All gDNA samples were normalized to 100 ng of DNA prior to PCR. Each individual sample was analyzed in triplicate for each quantiative PCR (qPCR). qPCR values are means ± standard error of the mean (SEM) of triplicate measurements. A comparison of enhanced expression vector (pEEV) (light gray bars) and pCMV (dark gray bars) samples were performed. ***P < 0.0001. (b) Evaluation of lacZ mRNA transcript expression by qRT-PCR in murine tissue. Bar graph presenting the relative expression of the lacZ mRNA 2 days after electroporation obtained from MF1-nu/nu mouse tissue. All qPCR data were normalized using 18S RNA as reference gene. Relative expression levels are plotted as means ± SEM of triplicate measurements. pEEV (light gray bars) and pCMV (dark gray bars). pEEV lacZ expressed was significantly higher than pCMV lacZ. ***P < 0.0001.
To evaluate expression, quantitative polymerase chain reaction (qPCR) was used to detect lacZ transgene expression (as a reporter system (Figure 2b). Transgene expression from the pEEV was significantly higher (P < 0.0001), and on average fourfold higher, than the respective pCMV plasmid. Taken together, these data indicate that high expression is achieved via an active vector, where, although present in fewer copies, pEEV allows an enhanced expression profile over the standard pCMV.
Demonstration of cytoplasmic expression of luciferase
The process for transcription of exogenous DNA material requires its translocation from the cytoplasm to the nucleus. The transcribed mRNA is then transported out of the nucleus for protein synthesis. Given that pEEV contains its own replicase, translation of the EEV-reported gene could occur within the cytoplasm. To assess the functionality of pEEV in a cell-free assay, we used the functional activity of mRNA encoding T7 RNA polymerase, combined with the luciferase reporter gene. Luciferase expression from two plasmid constructs, the standard pCMV plasmid and pEEV, were compared (Figure 3). We could only detect pEEV-driven luciferase expression during this assay. These data confirmed the ability of pEEV to self-express within the cell cytoplasm.
Figure 3.
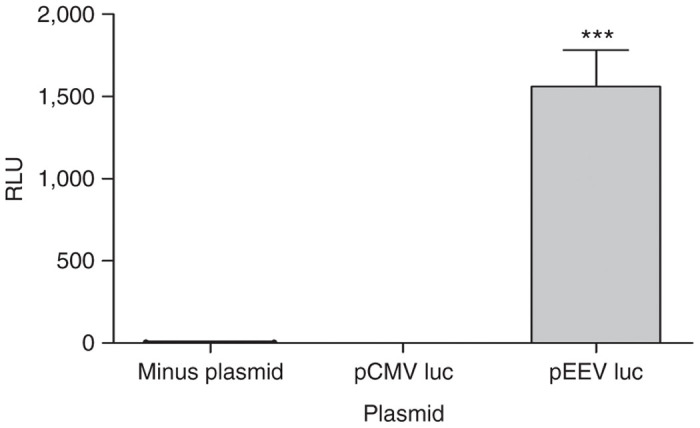
Cytoplasmic expression of enhanced expression vector (pEEV) luciferase. Luciferase reporter assay to verify activity of plasmid in a nuclear free preparation. Relative luciferase expression was used as an indicator of expression and values are plotted as means ± standard error of the mean of triplicate measurements. Only pEEV luciferase expression was detected.
Cytolytic activity of pEEV
In our construction of pEEV, we utilized components of the SFV, and we therefore hypothesized that the nonviral pEEV may delay tumor growth due to excessive gene expression inducing oncolytic activity and leading to cell lysis of transfected cells. We transfected pEEV in an established tumor model to determine antitumor activity in vivo. In growing CT26 tumors, pEEV lacZ, pEEV backbone, pCMV lacZ, and pMG backbone were transfected by electroporation, and growth and survival rates were examined and compared to its respective untreated CT26 tumor. The nontherapeutic bearing pEEV significantly (P < 0.05) reduced tumor volume when compared to the untreated tumor (Figure 4a). Furthermore, we also observed improved survival of animal’s transfected with nontherapeutic pEEV. Interestingly, these collective data indicate that nontherapeutic pEEV can improve survival rates, which may be due to the oncolytic effect of pEEV lacZ (Figure 4b).
Figure 4.

Antitumor effects of nontherapeutic plasmid. (a) Tumor growth curve of CT26 tumors treated with pCMV backbone, pCMV lacZ, enhanced expression vector (pEEV) backbone, pEEV lacZ, and an untreated tumor growing on Balb/c mice (n = 6 per group) is presented as tumor diameter measured. Tumor volume was calculated as previously described. Data presented are means ± SEM of six individual tumors. *Compared to the untreated group and pEEV lacZ, *P < 0.05. (b) Representative Kaplan–Meier survival curve. The respective survival cure of the CT26-treated tumors growing on Balb/c mice (n = 6 per group). The pEEV lacZ-treated mice survived longer than any other group P < 0.05 versus the untreated tumor. The pEEV lacZ group survived 4 days longer than the pEEV backbone. (c) Oncolytic activity of pEEV lacZ in murine muscle. In vivo intramuscular plasmid delivery. In vivo muscle transfection by electroporation was demonstrated by luciferase activity, analyzed daily postintramuscular pCMVluc and pEEVluc, plasmid transfection via electroporation and subsequent gene expression was assessed using whole body imaging of luciferase expression. pCMV expression peaked day 2 in both C57BL/6J (C) and MF1-nu/nu mice (D) and pEEV peaked day 7 in both MF1-nu/nu (A) and C57BL/6J mice (B). No detection of pEEVluc was detected by day 9. (E) Bioluminesence data plotted over time for pEEVluc-treated MF1-nu/nu mice.
To further demonstrate the oncolytic effect of nonviral pEEV, we transfected muscle of MF1-nu/nu mice and C57BL/6 with pCMV luciferase and pEEV luciferase (Figure 4c). Bioluminescent imaging revealed peak luciferase expression for pCMV at day 2 and day 6 for pEEV. pEEV expression was no longer detected by day 9, whereas pCMV expression continued to be detected at low levels up until day 100. This quenching in luciferase expression of pEEV suggests cell lysis due to the oncolytic effect of pEEV lacZ.
Quantification of apoptosis
To assess the ability of pEEV vector in inducing apoptosis, we analyzed in vivo electroporated CT26 tumors for terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick-end labeling (TUNEL) staining. TUNEL staining shown in Figure 5 shows that pEEV lacZ electroporation tumors were abundant in apoptotic nuclei, with double-strand DNA breaks, which are hallmarks of apoptosis. Notably, pCMV-treated tumors did not have any TUNEL staining (i.e., apoptosis). These data further confirm the oncolytic effect of pEEV.
Figure 5.
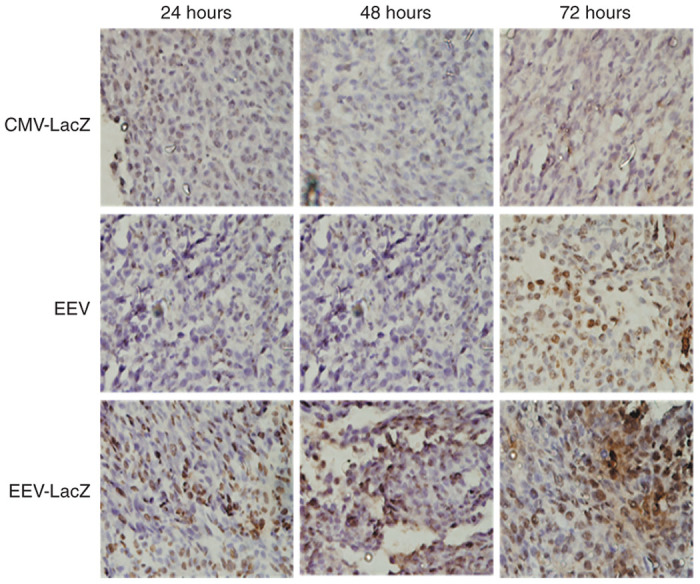
Apoptosis in tumors. Apoptotic dead was evaluated using terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick-end labeling (TUNEL) staining on CT26 tumor model grown on Balb/c mice. Tumors were removed from mice at various time points following treatment; paraffin-embedded and stained sections were examined microscopically. TUNEL stained for evidence of apoptotic cells indicate significant apoptosis in the tumor treated with pEEV lacZ and pEEV backbone but not from untreated groups.
β-Galactosidase expression in a large animal model
Previous studies have indicated that DNA vaccines are often less effective in large animals when compared to responses induced in mice. Translation of plasmid expression into patients is essential for any therapeutic potential to be established. To determine if pEEV had this potential, we tested the plasmid expression in a porcine model. To test transgene expression after electroporation, we examined LacZ transgene-driven β-galactosidase expression. Histological analyses of transfected tissues (Figure 6a) demonstrated positive β-galactosidase expression in all test tissues with all controls negative for β-galactosidase expression. Importantly, the visual expression profile of pEEV lacZ was more abundant in comparison to the standard pCMV plasmid.
Figure 6.

β-Galactosidase expression in porcine tissue. (a) Representative image of β-galactosidase staining in porcine tissues. β-Galactosidase expression significantly increased in enhanced expression vector (pEEV) lacZ in all tissues been examined. (b) Evaluation of lacZ mRNA transcript expression by qRT-PCR in porcine tissue. Bar graph presenting the relative expression of the lacZ mRNA 2 days after electroporation. All qPCR data were normalized using 18S RNA as reference gene. Relative expression levels are plotted as means ± standard error of the mean (SEM) of triplicate measurements. pEEV (light gray bars) and pCMV (dark gray bars). pEEV lacZ expressed was significantly higher than pCMV lacZ. (c) Copy numbers of lacZ transgene ascertained by quantitative PCR in porcine tissue. Bar graph showing absolute copy number of the lacz transgene per nanogram of genomic DNAs 2 days after electroporation. All gDNA samples were normalized to 100 ng of DNA prior to PCR. Each individual sample was analyzed in triplicate for each qPCR. qPCR values are means ± SEM of triplicate measurements. A comparison of pEEV (light gray bars) and pCMV (dark gray bars) samples were performed.
We again utilized the LacZ as a reporter transgene to compare the amount of gene expression induced from plasmids after electroporation, and to determine if the transgene expression was consistent with β-galactosidase staining (Figure 6b). As observed in the mouse models, pEEV lacZ was expressed at a significantly higher level when compared to the standard pCMV lacZ. To investigate the efficiency of transfection in our large animal model, we determined overall copy number (Figure 6c). While we observed that overall copy number was significantly lower for pEEV when compared to pCMV (similar to murine studies), we again observed that pEEV gave a significantly stronger expression profile when compared to pCMV. Taken together, these results demonstrate that pEEV can be used to effectively express exogenous genes within multiple tissues in a large animal model.
Discussion
The current study provides evidence for significant enhancements in the potential for nonviral DNA vector systems. Clinically, non–virus-mediated transgene expression has received increased attention because it is relatively safe, simple, and inexpensive compared to the administration of viral vectors, which are associated with the induction of neutralizing antibodies and cytotoxicity.7,8 Electroporation is considered as a very useful tool for gene therapies and has received a lot of attention in the development and improvement of electrogene transfer protocols. Studies have included modulating the electric pulse, electrodes, extracellular matrix, which has an effect on transfection efficiency, and expression of the delivered gene.22,24,25,31,32 Others have modulated the size of the plasmid being delivered and used plasmid types such as minicircle plasmid.33 We have incorporated elements of a virus and nuclear localization sequence to allow for an enhanced expression of the gene of interest.
We have shown that a DNA-based pEEV has improved expression capabilities across a range of tissue histologies over standard nonviral DNA vectors. pEEV was developed utilizing the SFV replicase, which allows for high levels of cytoplasmic expression and thereby overcoming the requirement and rate-limiting step for all plasmid copies to enter the nucleus in order for mRNA transcription to occur. In the present study, we evaluated our pEEV construct and demonstrated its superior efficacy using reporter gene analysis while comparing findings to a standard plasmid. pEEV delivered via electroporation led to rapid mRNA expression in cancer cells. This approach was also found to have several advantages specifically for cancer therapy including: greatly improved and consistent gene expression and eventual direct oncolytic effect (cellular exhaustion) from continued mRNA production, which obviates the risk of long-term exogenous gene expression.
The EEV construct contains a DNA sequence of 15 kb size. There was initial concern that the large size of pEEV would impact negatively on its transfection efficacy after electroporation. Our data indicate that transfection levels for pEEV, i.e., copies of plasmid DNA within the cell postelectroporation, were indeed lower in comparison to the standard pCMV plasmid, which was only 6.2 kb in size. However, previous studies have indicated that self-amplifying vectors only need a single RNA copy to reach the cytoplasm, whereby they can generate high expression levels almost independent of initial transfection and transcription efficiency. Indeed, although we observed overall lower transfection rates of pEEV, when compared to the standard pCMV plasmid, this did not, however, result in a proportional decrease in mRNA expression levels within a nuclear free lysate. Significantly, mRNA expression levels were in fact four- to fivefold higher in pEEV-transfected cells, when compared to the more efficiently transfected standard commercial pCMV.
Targeting tumors with oncolytic viruses has demonstrated a promising approach for cancer treatment. These are modified or naturally oncotropic viruses, which selectively replicate in malignant tumor cells and finally destroy them via oncolysis, but this approach also encounters similar disadvantages as other viral therapies. Alphaviruses, like SFV, have also been successfully used as oncolytic agents in several preclinical models of cancer. They are enveloped viruses containing a single positive strand RNA genome which, after, can replicate in the cytoplasm. The SFV system is suicidal and are characterized by their high expression levels. This process induces a strong cytopathic effect that leads to cell death by apoptosis. Taking these characteristics into account, we incorporated these elements from the SFV to develop the pEEV. We demonstrated cytoplasmic expression of pEEV and wanted to further characterize if pEEV had oncolytic effects like its viral parent. The stable presence and expression of plasmid DNA vectors in muscle have been demonstrated in several studies. We observed that in vivo muscle expression peaked on day 6 for pEEV transfection with expression levels declining and undetectable by day 9. This cellular oncolysis feature of pEEV could benefit cancer gene therapy applications such as by acting as a safety feature for over or continuous expression. Antitumor effects of pEEV were demonstrated by the delay in growth and increased survival of a nontherapeutic pEEV-treated CT26 tumor model. This oncolytic characteristic and the high expression level trait of pEEV highlight the potential of this vector as an anticancer vector.
The electroporation gene transfer protocol appears to be safe and nontoxic. All mice remained healthy throughout the course of the experiments, and there were no treatment-related deaths. This suggests that electroporation of solid tumours is not traumatic and that the levels produced by the pEEV did not induce systemic toxicity. Other studies have observed similar safe and nontoxic effects of electrogene transfer.34,35 The ultimate aim of any preclinical gene therapy study is to translate findings to the clinic. The safety of electroporation as part of treatment in human patients has been validated in several studies of electrochemotherapy and electrogene transfer.21,36
We wanted to assess the expression levels of pEEV; however, efficacy in mouse models does not necessarily translate into patients. While our pEEV preclinical studies in mice revealed very promising results, we proceeded to validate the approach in the larger pig in vivo model. Pigs are an excellent larger animal model to study human physiology/disease due to many similar attributes including anatomy, genetics, and physiology. Indeed, pig models have been used to evaluate the safety and efficacy of gene therapy; however, studies have yielded varying results with regards to detectable levels of gene expression. Employing the EndoVe, a novel endoscopic electroporation device, we were able to demonstrate the reliability of the pEEV to express the β-galactosidase reporter gene across a range of intraluminal porcine tissue types. The high levels of pEEV reporter gene expression in this pig study indicates the potential utility of pEEV in the clinic and as a therapeutic tool in gene therapy. The data also clearly reveal the lack of consistent expression for the pCMV vector when translated from murine to porcine tissue; in contrast, consistent levels of reporter gene expression were detected across all tissue types for the pEEV plasmid.
A growing number of clinical studies have established that electroporation is safe and effective in delivering plasmid DNA and chemotherapeutic drugs efficiently.20,21 In this study, we used a previously reported safe and nontoxic parameters of electroporation-mediated transfer of the DNA plasmid to deliver pEEV in vivo. We found that the combination of an already established electroporation methodology and pEEV allows for an extremely reliable means of gene delivery and expression.
Taken together, our results clearly demonstrate the potential of pEEV as a safe DNA vector with superior expression capabilities over standard available vectors. This vector, in combination with electroporation, allows for a safe and minimally invasive method of delivery. This study suggests that the pEEV expressing a therapeutic gene may have potential for clinical development with high detectable levels of expression.
Materials and Methods
Animals
Female Balb/c, C57BL/6, and MF1-nu/nu mice (6–8 weeks) were obtained from Harlan Laboratories (Oxfordshire, England). Female pigs (Landrace) were obtained from Teagasc, Moorepark, Ireland, and the pigs used were 25–30 kg. Standard laboratory food and water were provided ad libitum.
Ethics statement
All animal husbandry and experimental procedures were approved by the University College Cork Animal Experimentation Ethics Committee and carried out under licenses issued by the Department of Health, Ireland as directed by the Cruelty to Animals Act Ireland and EU Statutory Instructions.
Cell tissue culture
Tumor cell lines were obtained from the American Type Culture Collection (Manassas, VA). The murine colon adenocarcinoma, CT26 cell line was cultured with Dulbecco’s modified Eagle’s medium (Sigma, Wicklow, Ireland), supplemented with 10% v/v fetal calf serum and 300 µg/ml L-glutamine. The human adenocarcinoma, OE19 cell line was cultured in RPMI-1640 (Sigma) supplemented with 10% v/v fetal calf serum and 300 µg/ml L-glutamine. Cells were maintained in logarithmic phase growth at 37 °C in a humidified atmosphere supplemented with 5% CO2.
Tumor induction
For routine tumor induction, 1 × 106 CT26 or OE19 tumor cells, suspended in 200 µl of serum-free Dulbecco’s modified Eagle’s media were injected subcutaneously into the flank Balb/C (CT26) or MF1-nu/nu mice (OE19). Tumors were allowed to develop and size was monitored by measurements in two dimensions using vernier calipers. Tumor volume was calculated according to the formula V = ab2π/6, where a is the longest diameter of the tumor and b is the longest diameter perpendicular to diameter a. From these volumes, tumor growth curves were constructed. Mice were euthanized when tumor volume reached 1.7 cm3 as approved by the University College Cork Animal Experimentation Ethics Committee.
Plasmids
For plasmid transfection, a pCMV-lacz plasmid (Plasmid factory) was used encoding a β-galactosidase protein (LacZ) gene under the control of a CMV promoter. The EEV plasmid was created by incorporating a SFV DNA replicase sequence (kindly donated by Prof Greg Atkins, Virus Group, Department of Microbiology, School of Genetics and Microbiology, Trinity College Dublin). A nuclear localization sequence was also incorporated to allow for more efficient nuclear targeting of the pEEV plasmid (Figure 1). The pMG plasmid was purchased from InvivoGen (Toulouse, France) (Supplementary Figure S1). Plasmids were propagated in Escherichia coli strain Top10 and purified on endotoxin-free Qiagen-tip 500 columns (Qiagen, Manchester, UK).
DNA transfection and in vivo electroporation
A range of tissues were transfected including a subcutaneous tumor (Oe19) and healthy tissue (muscle, liver, and spleen). Tissues were injected with 50 µg of the respective plasmid and surgically removed 48 hours following in vivo transfection by electroporation. The number of plasmid DNA and LacZ expression was then determined. Procedures were carried out under general anesthesia, using intraperitoneal administration of 200 µg xylazine and 2 mg ketamine. The skin overlying the flank muscle and tumor was shaved and a midline laparotomy was used to expose the liver and spleen. Fifty micrograms of plasmid DNA in 50 µl sterile injectable phosphate-buffered saline (PBS) was injected into the tumor, muscle, liver, and spleen. Parallel plate electrodes,37 with an adjustable separation setting, were inserted on either side of the marked DNA injection point and electroporation pulses delivered within 1 minute of DNA injection. For this study, plate electrodes were fixed at 1 cm apart.
Pigs were sedated with 400 mg of ketamine and 80 mg of xylazine injected into the cervical musculature followed by 150 mg/3 ml intravenous administration of the cardioprotective agent amiodarone hydrochloride. Anesthesia was maintained using a 200 mg ketamine and 10 mg midazolam solution. Surgical access to muscle tissue was obtained by making a 6 cm linear incision over the appropriate flank using bipolar diathermy. Open surgical access to the liver and spleen was obtained through a midline laparotomy incision. Access to the gastrointestinal tissues—esophagus and rectum was achieved using the EndoVe device (Cork Cancer Research Centre, Cork, Ireland). Fifty micrograms of plasmid DNA in 50 µl sterile injectable PBS was injected directly into the target tissues. Electric pulses were delivered using the EndoVe. In vivo electroporation parameters were a single 1,200 V/cm, 100 μsec pulse followed after 1 second by a train of eight 120 V/cm, 20 msec pulses at 1 Hz administered in sequence using the E.Pore Gx (Cork Cancer Research Centre) electroporation generator.
Histological evaluation
Animals were sacrificed; tissues extracted and washed three times in PBS. β-galactosidase staining was performed on tissue using the β-galactosidase reporter gene staining kit (Sigma). Tissues were fixed in a 1X fixation solution for 30 minutes. After fixation, the tissues were rinsed three times in PBS and incubated for 24 hours in X-gal staining solution. Tissues from each animal were processed separately. At the end of the incubation period, tissues were rinsed with PBS. Tissues were embedded in paraffin and serial sections were cut at 5 µm and mounted on slides. Hematoxylin and eosin staining was performed for both murine and porcine samples to assess for tissue damage.
RNA isolation and reverse transcriptase reaction
Frozen tissue samples preserved in RNAlater were homogenized in the presence of Trizol (Sigma), and total RNA was isolated according to manufacturer’s instructions. Each sample was treated with DNase I (Ambion, Cambridgeshire, UK) to eliminate any possible DNA contamination and subsequently tested for presence of DNA using PCR amplification without the reverse transcription step. Total RNA concentration was determined from spectrophotometric optical density measurement (260 and 280 nm). For each sample tested, the ratio between the spectrophotometric readings at 260 and 280 nm (OD260/OD280) was used to provide an estimate of the purity of the nucleic acid, and the ratio in all samples ranged between 1.7 and 2.0. Reverse transcriptase reactions were carried out using the omniscript reverse transcription kit (Qiagen) and Oligo(dt)15 primer (Promega, Hampshire, UK). Each reaction tube contained 1 µg of DNAse-treated total RNA in a volume of 20 µl.
Genomic DNA extraction
Genomic DNA was prepared according to manufacturer’s protocol using the genElute mammalian genomic DNA miniprep kit (Sigma-Aldrich, Wicklow, Ireland). Final DNA concentration was determined with the use of spectrophotometric measurement.
Real-time PCR
Real-time PCR amplification was performed with the lightCycler fastStart DNA master SYBR green I (Roche, Hertfordshire, UK) at final concentrations of 3 mmol/l magnesium, 0.5 µmol/l of each primer, and 1X LightCycler FastStart DNA master SYBR green I. The final volume of each reaction was 20 µl, containing 50 ng of reverse transcription product as template. The amplification program started with 1 cycle at 95 °C for 10 minutes, followed by 38 cycles at 95 °C for 15 seconds, 60 °C for 20 seconds, and 72 °C for 20 seconds. The program ended with 1 cycle at 72 °C for 15 minutes. The following primers for lacZf and lacZr were designed to amplify 185 bp, and their sequences are as follows: lacZf: 5′-GCG TGG ATG AAG ACC AGC-3′, lacZr: 5′-CGA AGC CGC CCT GTA AAC-3′. The housekeeping 18s rRNA was used as an internal control;38 18srRNAf: 5′-TTG ACG GAA GGG CAC CAC CAG-3′, 18srRNAr: 5′-GCA CCA CCA CCC ACG GAA TCG-3′. For genomic PCR, all samples were normalized to 100 ng/ μl of gDNA. Hundred nanograms of DNA were used with similar PCR mixture contents and cycling parameters (40 cycles). LacZgf: 5′-GAC GTC TCG TTG CTG CAT AA-3′, LacZgr: 5′-CAG CAG CAG ACC ATT TTC AA-3′.
Quantification of apoptosis by TUNEL
To assess whether pEEV vector could also induce apoptotic death in vivo, pEEV, pEEV lacZ, and pCMV lacZ was delivered into CT26 tumors via electroporation. Evaluation of the tumors for TUNEL (Roche) staining (which marks apoptotic cells) was utilized. Balb/C mice bearing CT26 tumors were subcutaneously injected with 50 μg of plasmid DNA followed by electroporation. Mice were culled on 24, 48, and 72 hours posttreatment and tumors were removed for detecting in situ apoptosis by TUNEL assay. The TUNEL-positive cells were stained brown. TUNEL staining of tissues was performed according to the manufacturer’s protocol with minor modifications. In brief, processed tissues were embedded in paraffin, cut into 5 µm thick sections, placed on superfrost plus slides, and adhered by heating at 37 °C for 12 hours. Slides were deparaffinized and rehydrated by successive incubations in xylene, absolute ethanol, and 70% ethanol. Nuclei in tissue sections were stripped of protein by treating with proteinase k (20 µg/ml) for 20 minutes followed by four successive washes with PBS. Slides were incubated with TUNEL reaction mixture at 37 °C for 60 minutes in a humidified atmosphere followed by three successive washes with PBS. Sections were incubated with horse-radish peroxidise (Roche) for 30 minutes, rinsed in PBS, and stained with DAB substrate kit (Dako, Dublin, Ireland) according to manufactures’ recommendations. A positive control was established by incubating tissue section with DNase (1 µg/ml) after deproteination and peroxidase inactivation. Intense staining was observed in cells from the Dnase-treated sections. A negative control was established by incubating tissue sample with DAB after deproteination and peroxidase inactivation. No staining was observed.
Bioluminescence imaging
Bioluminescence imaging was performed as previously described,14 with an IVIS 100 charge-coupled device imaging system (Xenogen, Alameda, CA). Bioluminescent signals were quantified by the creation of regions of interest. To standardize the data, light emission from the same surface area (regions of interest) was quantified for each transfected animal. In addition, background light emission, taken from regions of interest created on untransfected control animals, was subtracted from transfected test animals. Imaging data were analyzed and quantified with Living Image Software 2.50 (Xenogen) and expressed as photons/second/cm2.
Cytoplasmic expression assay
Cytoplasmic expression was assessed for both plasmid systems using a nuclear free cytoplasmic preparation. The standard commercial rabbit reticulocyte lysate cell-free system (Promega) was used to characterize translation products from mRNA encoding T7 RNA polymerase. This preparation consisted of a cytoplasmic free of nuclear material lysate and mRNA encoding T7 RNA polymerase. Each plasmid was incubated at 30 °C for a period of 2 hours. Luciferase expression was detected using the IVIS 100 charge-coupled device imaging system (Xenogen). pCMV was added as a control plasmid to demonstrate the efficiency of pEEV expression in cytoplasm lystae when there is a mRNA encoding T7 RNA polymerase present and the requirement of the T7 promoter.
Statistical analysis
Experimental results were plotted and analyzed for significance with Prism 4 software (GraphPad software, La Jolla, CA). P < 0.05 was considered significant.
Acknowledgments
The authors thank Stephen D Robinson, School of Biological Sciences, University of East Anglia, for his appraisal of this manuscript. This project was funded through the Health Research Board Ireland (HRA/2009/91).
References
- Kay MA. State-of-the-art gene-based therapies: the road ahead. Nat Rev Genet. 2011;12:316–328. doi: 10.1038/nrg2971. [DOI] [PubMed] [Google Scholar]
- Miest TS, Cattaneo R. New viruses for cancer therapy: meeting clinical needs. Nat Rev Microbiol. 2014;12:23–34. doi: 10.1038/nrmicro3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A, Hacein-Bey-Abina S, Lagresle C, Garrigue A, Cavazana-Calvo M. Gene therapy of severe combined immunodeficiency disease: proof of principle of efficiency and safety issues. Gene therapy, primary immunodeficiencies, retrovirus, lentivirus, genome. Bull Acad Natl Med. 2005;189:779–85; discussion 786. [PubMed] [Google Scholar]
- Tomanin R, Scarpa M. Why do we need new gene therapy viral vectors? Characteristics, limitations and future perspectives of viral vector transduction. Curr Gene Ther. 2004;4:357–372. doi: 10.2174/1566523043346011. [DOI] [PubMed] [Google Scholar]
- Nair V. Retrovirus-induced oncogenesis and safety of retroviral vectors. Curr Opin Mol Ther. 2008;10:431–438. [PubMed] [Google Scholar]
- Davidson BL, Stein CS, Heth JA, Martins I, Kotin RM, Derksen TA. Recombinant adeno-associated virus type 2, 4, and 5 vectors: transduction of variant cell types and regions in the mammalian central nervous system. Proc Natl Acad Sci USA. 2000;97:3428–3432. doi: 10.1073/pnas.050581197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niidome T, Huang L. Gene therapy progress and prospects: nonviral vectors. Gene Ther. 2002;9:1647–1652. doi: 10.1038/sj.gt.3301923. [DOI] [PubMed] [Google Scholar]
- Thomas CE, Ehrhardt A, Kay MA. Progress and problems with the use of viral vectors for gene therapy. Nat Rev Genet. 2003;4:346–358. doi: 10.1038/nrg1066. [DOI] [PubMed] [Google Scholar]
- Pérez-Martínez FC, Guerra J, Posadas I, Ceña V. Barriers to non-viral vector-mediated gene delivery in the nervous system. Pharm Res. 2011;28:1843–1858. doi: 10.1007/s11095-010-0364-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morille M, Passirani C, Vonarbourg A, Clavreul A, Benoit JP. Progress in developing cationic vectors for non-viral systemic gene therapy against cancer. Biomaterials. 2008;29:3477–3496. doi: 10.1016/j.biomaterials.2008.04.036. [DOI] [PubMed] [Google Scholar]
- Gill DR, Pringle IA, Hyde SC. Progress and prospects: the design and production of plasmid vectors. Gene Ther. 2009;16:165–171. doi: 10.1038/gt.2008.183. [DOI] [PubMed] [Google Scholar]
- Sheridan C. Gene therapy finds its niche. Nat Biotechnol. 2011;29:121–128. doi: 10.1038/nbt.1769. [DOI] [PubMed] [Google Scholar]
- Caracò C, Mozzillo N, Marone U, Simeone E, Benedetto L, Di Monta G. Long-lasting response to electrochemotherapy in melanoma patients with cutaneous metastasis. BMC Cancer. 2013;13:564. doi: 10.1186/1471-2407-13-564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spugnini EP, Di Tosto G, Salemme S, Pecchia L, Fanciulli M, Baldi A. Electrochemotherapy for the treatment of recurring aponeurotic fibromatosis in a dog. Can Vet J. 2013;54:606–609. [PMC free article] [PubMed] [Google Scholar]
- Campana LG, Valpione S, Falci C, Mocellin S, Basso M, Corti L. The activity and safety of electrochemotherapy in persistent chest wall recurrence from breast cancer after mastectomy: a phase-II study. Breast Cancer Res Treat. 2012;134:1169–1178. doi: 10.1007/s10549-012-2095-4. [DOI] [PubMed] [Google Scholar]
- Salwa SP, Bourke MG, Forde PF, O’Shaughnessy M, O’Sullivan ST, Kelly EJ. Electrochemotherapy for the treatment of ocular basal cell carcinoma; a novel adjunct in the disease management. J Plast Reconstr Aesthet Surg. 2014;67:403–406. doi: 10.1016/j.bjps.2013.07.019. [DOI] [PubMed] [Google Scholar]
- Jahangeer S, Forde P, Soden D, Hinchion J. Review of current thermal ablation treatment for lung cancer and the potential of electrochemotherapy as a means for treatment of lung tumours. Cancer Treat Rev. 2013;39:862–871. doi: 10.1016/j.ctrv.2013.03.007. [DOI] [PubMed] [Google Scholar]
- Escoffre JM, Rols MP. Electrochemotherapy: progress and prospects. Curr Pharm Des. 2012;18:3406–3415. [PubMed] [Google Scholar]
- Weaver JC. Electroporation: a general phenomenon for manipulating cells and tissues. J Cell Biochem. 1993;51:426–435. doi: 10.1002/jcb.2400510407. [DOI] [PubMed] [Google Scholar]
- Daud AI, DeConti RC, Andrews S, Urbas P, Riker AI, Sondak VK. Phase I trial of interleukin-12 plasmid electroporation in patients with metastatic melanoma. J Clin Oncol. 2008;26:5896–5903. doi: 10.1200/JCO.2007.15.6794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanggaard I, Snoj M, Cavalcanti A, Bouquet C, Sersa G, Robert C. Gene electrotransfer of plasmid antiangiogenic metargidin peptide (AMEP) in disseminated melanoma: safety and efficacy results of a phase I first-in-man study. Hum Gene Ther Clin Dev. 2013;24:99–107. doi: 10.1089/humc.2012.240. [DOI] [PubMed] [Google Scholar]
- Cemazar M, Golzio M, Sersa G, Rols MP, Teissié J. Electrically-assisted nucleic acids delivery to tissues in vivo: where do we stand? Curr Pharm Des. 2006;12:3817–3825. doi: 10.2174/138161206778559740. [DOI] [PubMed] [Google Scholar]
- Miklavcic D, Beravs K, Semrov D, Cemazar M, Demsar F, Sersa G. The importance of electric field distribution for effective in vivo electroporation of tissues. Biophys J. 1998;74:2152–2158. doi: 10.1016/S0006-3495(98)77924-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cemazar M, Golzio M, Sersa G, Escoffre JM, Coer A, Vidic S. Hyaluronidase and collagenase increase the transfection efficiency of gene electrotransfer in various murine tumors. Hum Gene Ther. 2012;23:128–137. doi: 10.1089/hum.2011.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markelc B, Tevz G, Cemazar M, Kranjc S, Lavrencak J, Zegura B. Muscle gene electrotransfer is increased by the antioxidant tempol in mice. Gene Ther. 2012;19:312–320. doi: 10.1038/gt.2011.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabri N, Pelissier B, Teissie J. Ascorbate increases electrotransformation efficiency of intact maize cells. Anal Biochem. 1998;264:284–286. doi: 10.1006/abio.1998.2878. [DOI] [PubMed] [Google Scholar]
- Atkins GJ, Sheahan BJ, Liljeström P. The molecular pathogenesis of Semliki Forest virus: a model virus made useful? J Gen Virol. 1999;80 (Pt 9):2287–2297. doi: 10.1099/0022-1317-80-9-2287. [DOI] [PubMed] [Google Scholar]
- Rayner JO, Dryga SA, Kamrud KI. Alphavirus vectors and vaccination. Rev Med Virol. 2002;12:279–296. doi: 10.1002/rmv.360. [DOI] [PubMed] [Google Scholar]
- Kääriäinen L, Ahola T. Functions of alphavirus nonstructural proteins in RNA replication. Prog Nucleic Acid Res Mol Biol. 2002;71:187–222. doi: 10.1016/S0079-6603(02)71044-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrengruber MU. Alphaviral gene transfer in neurobiology. Brain Res Bull. 2002;59:13–22. doi: 10.1016/s0361-9230(02)00858-4. [DOI] [PubMed] [Google Scholar]
- Miklavcic D, Snoj M, Zupanic A, Kos B, Cemazar M, Kropivnik M. Towards treatment planning and treatment of deep-seated solid tumors by electrochemotherapy. Biomed Eng Online. 2010;9:10. doi: 10.1186/1475-925X-9-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miklavčič D, Serša G, Brecelj E, Gehl J, Soden D, Bianchi G. Electrochemotherapy: technological advancements for efficient electroporation-based treatment of internal tumors. Med Biol Eng Comput. 2012;50:1213–1225. doi: 10.1007/s11517-012-0991-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Jiang W, Chen Y, Liu P, Sheng C, Chen S. In vivo electroporation of minicircle DNA as a novel method of vaccine delivery to enhance HIV-1-specific immune responses. J Virol. 2014;88:1924–1934. doi: 10.1128/JVI.02757-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller R, Shirley S, Guo S, Donate A, Heller L. Electroporation based gene therapy–from the bench to the bedside. Conf Proc IEEE Eng Med Biol Soc. 2011;2011:736–738. doi: 10.1109/IEMBS.2011.6090167. [DOI] [PubMed] [Google Scholar]
- Heller L, Pottinger C, Jaroszeski MJ, Gilbert R, Heller R. In vivo electroporation of plasmids encoding GM-CSF or interleukin-2 into existing B16 melanomas combined with electrochemotherapy induces long-term antitumour immunity. Melanoma Res. 2000;10:577–583. doi: 10.1097/00008390-200012000-00010. [DOI] [PubMed] [Google Scholar]
- Heller LC, Heller R. Electroporation gene therapy preclinical and clinical trials for melanoma. Curr Gene Ther. 2010;10:312–317. doi: 10.2174/156652310791823489. [DOI] [PubMed] [Google Scholar]
- Soden DM, Larkin JO, Collins CG, Tangney M, Aarons S, Piggott J. Successful application of targeted electrochemotherapy using novel flexible electrodes and low dose bleomycin to solid tumours. Cancer Lett. 2006;232:300–310. doi: 10.1016/j.canlet.2005.03.057. [DOI] [PubMed] [Google Scholar]
- Goidin D, Mamessier A, Staquet MJ, Schmitt D, Berthier-Vergnes O. Ribosomal 18S RNA prevails over glyceraldehyde-3-phosphate dehydrogenase and beta-actin genes as internal standard for quantitative comparison of mRNA levels in invasive and noninvasive human melanoma cell subpopulations. Anal Biochem. 2001;295:17–21. doi: 10.1006/abio.2001.5171. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.