

Summary
Aims
The pathology of stroke consists of multiple pro‐death processes, and CD36 has been suggested as a multimodal target to reduce oxidative stress and inflammation in ischemic stroke. Using CD36‐deficient mice and SS‐31, a cell permeable tetrapeptide known to down‐regulate CD36 pathways, the current study investigated whether targeting CD36 is effective in transient and permanent ischemic stroke.
Methods
Wild‐type or CD36‐deficient mice were subjected to either 30‐min transient or permanent focal ischemic stroke. In parallel, a cohort of mice subjected to either transient or permanent stroke received either vehicle or 5 mg/kg of SS‐31. Monocyte chemoattractant protein‐1 (MCP‐1) and its receptor CCR2, mRNA levels, and infarct volume and percent hemispheric swelling were measured in the postischemic brain.
Results
CD36 deficiency or SS‐31 treatment significantly attenuated MCP‐1 or CCR2 mRNA up‐regulation and injury size in the transient ischemic stroke. However, the approaches failed to show the protective effect in permanent ischemic stroke.
Conclusion
The study revealed that targeting CD36 has a beneficial effect on transient but not permanent focal ischemic stroke. The study thus precludes a generalized strategy targeting CD36 in ischemic stroke and suggests careful consideration of types of stroke and associated pathology in developing stroke therapies.
Keywords: CD36, Inflammation, Permanent focal ischemia, SS‐31, Transient focal ischemia
Introduction
Stroke is a leading cause of disability and death worldwide. More than 80% of stroke patients suffer from ischemic stroke, with or without spontaneous reperfusion. Tissue plasminogen activator (tPA) is an endogenous thrombolytic agent that aids in recanalization when given within 3–6 h after stroke onset 1. While early reperfusion limits brain injury, it also introduces excessive oxidative stress and inflammation, leading to ischemic/reperfusion injury 2, 3, 4. Published reports have shown that levels of reactive oxygen species (ROS) and inflammatory chemokines and cytokines are significantly increased in the ischemic brain, and they are positively correlated with injury size 5, 6, 7, 8, 9. Thus, targeting ROS production and/or inflammation might be a useful strategy to minimize the ischemic/reperfusion injury.
CD36 is a class B scavenger receptor that is expressed in microglia, astrocytes, microvascular endothelial cells, peripheral monocytes/macrophages, and platelets 10, 11, 12, 13, 14. By binding a host of ligands, CD36 plays a role in inflammation, innate immunity, and vascular dysfunction 11, 15, 16, 17, 18, 19. Previously, we reported that CD36 contributes to acute ischemic injury by increasing oxidative stress and inflammation, and the genetic deficiency of CD36 resulted in neuroprotection 8, 9, 20. CD36 is up‐regulated in ischemic brain and regulates the expression of monocyte chemoattractant protein‐1 (MCP‐1) and CCR2, an important chemokine/receptor axis for mononuclear cell infiltration in stroke 9, 20, 21. The involvement of CD36 in postinjury inflammatory responses suggests CD36 as a potential target against focal ischemic stroke. SS‐31 is the first compound shown to attenuate the up‐regulation of CD36 and reduce brain injury in transient focal ischemic stroke 22. In a model of renal ischemia–reperfusion injury, SS‐31 has also been shown to reduce oxidative stress and ameliorate the up‐regulation of MCP‐1 and TNF‐α 23.
Permanent occlusion has been shown to be refractory to the treatments that were effective in transient ischemic stroke. For instance, hyperbaric oxygen preconditioning, dextromethorphan treatment, an antiintercellular adhesion molecule‐1 antibody, and peroxisome proliferator‐activated receptor‐γ (PPAR‐γ) showed benefits in tMCAO but not in pMCAO 24, 25, 26. Although CD36‐induced inflammation is a known component of ischemia–reperfusion injury, it is not clear whether CD36 is up‐regulated in permanent ischemic stroke, and whether prevention of CD36 up‐regulation, either genetically or pharmacologically, impacts on inflammation and outcome in permanent focal ischemic stroke. Here, we report that inflammatory responses are largely blunted in permanent ischemic stroke compared to transient ischemic stroke. While targeting CD36 was effective against transient ischemic stroke, the strategy has little efficacy in permanent occlusion, suggesting alternative strategies for focal ischemic stroke without reperfusion.
Materials and methods
Animals
The use of animals and procedures was approved by the Institutional Animal Care and Use Committee (IACUC) of Weill Medical College of Cornell University and in accordance with the IACUC, National Institutes of Health, and ARRIVE guidelines. Experiments were performed in 10‐ to 11‐week‐old male C57BL/6, wild‐type (WT), or CD36 knock‐out (CD36 KO) mice. C57BL/6 mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA). Breeding pairs of each WT and CD36 KO strain (seven times backcrossed with C57BL/6, 99.7% C57BL/6 background) were derived at the time of the heterozygote cross. From these breeding pairs, only F1 generation was used for the study without further sister–brother mating. The procedures for genotyping have been described previously 16, 27. The mice were housed at the institute's animal facility, which monitors and maintains temperature, humidity, and 12‐h light/dark cycle. Maximum five mice were housed in a cage with an individual ventilating system and irradiated bedding (1/8″ Bed O's Cobs; The Anderson, Maumee, OH, USA). Sterilized food (PicoLab Rodent diet 5053; LabDiet, St. Louise, MO, USA) and water were freely accessible in their cage.
Transient or Permanent Middle Cerebral Artery Occlusion
Mice were randomly selected and subjected to middle cerebral artery occlusion (MCAO) as previously described 20, 28. A fiber optic probe was glued to the parietal bone (2 mm posterior and 5 mm lateral to the bregma) and connected to a Laser Doppler Flowmeter (Periflux System 5010; Perimed, Järfälla, Sweden) for continuous monitoring of cerebral blood flow (CBF) in the ischemic territory. For MCAO, a 6‐0 Teflon‐coated black monofilament surgical suture (Doccol Co., Redland, CA, USA) was inserted into the exposed external carotid artery, advanced into the internal carotid artery, and wedged into the cerebral arterial circle to obstruct the origin of the MCA. For transient MCAO (tMCAO), the filament was withdrawn to allow reperfusion after 30‐min MCAO. Only animals that exhibited greater than 80% reduction in CBF during MCAO and greater than 80% reperfusion 10 min following reperfusion were included in the study. For permanent MCAO (pMCAO), the CBF reduction was monitored for 10 min and the incision was closed by wound clip without the filament withdrawing. Buprenorphine, lidocaine, and bupivacaine were administered during postischemia as analgesics. Mice were then placed in a recovery cage until the animal regained consciousness and resumed activity. Using a rectal probe controlled by a Masterflex pump and thermistor temperature controller (Cole‐Parmer, Vernon Hills, IL, USA), the animals' body temperature was maintained at 37 ± 0.5°C during MCAO and recovery after surgery. The mice were then returned to their home cages where they were previously housed together. For hydrating animals after surgery, hydrogel (ClearH2O, Portland, ME, USA) was provided with food.
SS‐31 Treatment
SS‐31 (D‐Arg‐Dmt‐Lys‐Phe‐NH2; Dmt‐2′,6′‐dimethyltyrosine) was prepared by Dr. Peter W. Schiller (Clinical Research Institute of Montreal, Montreal, QC, Canada) using solid‐phase synthesis as described previously 29. Stroked C57BL/6 male mice were randomized to receive saline (veh) or SS‐31. The treatment and dosages were followed according to a previous report 22. Briefly, animals were treated intraperitoneally with veh or 5 mg/kg of SS‐31 immediately after the reperfusion for tMCAO or immediately before the mice were released from anesthesia for pMCAO, and at 6, 24, and 48 h postischemia.
Tissue Collection for Infarct Volume and Gene Assessment
To obtain tissue that contains the entire infarct territory in an unbiased manner, an unbiased stereological sampling strategy was used according to the method described in the previous study 20, 28. One or three days after tMCAO or pMCAO, brains were excised, frozen, and serial sections spanning about 6 mm rostrocaudal (roughly +2.8 mm and extending to −3.8 mm from bregma) were collected. The entire infarct region was cryosectioned for infarct volume measurement (20 μm thickness) and collected serially at 600‐micron intervals. Infarct volume and % hemispheric swelling were measured using Axiovision software (Zeiss, Jena, Germany). Infarct volume was corrected for swelling by a method described previously 30. Tissues between sections for infarct volume were serially cryosectioned, cut in half, and collected from each hemisphere for gene assessment.
Measurement of Gene Expression
Gene expression levels were quantified by real‐time quantitative RT‐PCR (qPCR) using fluorescent TaqMan technology as described previously 20, 28. Briefly, total RNA was extracted from brain tissues using Tri reagent (MRC, Cincinnati, OH, USA). RNAs were reverse transcribed using the QuantiTech reverse transcription kit (QIAGEN, Valencia, CA, USA). PCR primers and probes specific for CD36, MCP‐1, CCR2, and β‐actin (an internal control) were obtained as TaqMan predeveloped optimized assay reagents for gene expression (Life Technologies, Grand Island, NY, USA). The PCR reaction was performed using FastStart Universal Probe Master Mix (Roche, Indianapolis, IN, USA), according to the manufacturer's instructions. Reactions were performed in 20 μL total volume and incubated at 95°C for 10 min followed by 40 cycles of 15 seconds at 95°C and 1 min at 60°C. The results were analyzed using 7500 Fast Real‐Time PCR System software (Life Technologies).
Data Analysis
Sample size for infarct measurement (minimum n = 8/group) was calculated based on predicting detectable differences to reach power of 0.80 at a significance level of <0.05, assuming a 30% difference in mean and a 20% SD at the 95% confidence level. Infarct volume and percent hemispheric swelling were expressed as mean ± 95% confidence interval (CI). Gene expression levels were presented as the β‐actin normalized value according to the formula, Value = 2(Ct of β‐actin − Ct of target gene). Comparison between two groups was statistically evaluated using Student's t‐test. Multiple comparisons were made using ANOVA followed by a post hoc Newman–Keuls test. Differences were considered significant at P < 0.05.
Results
The Absence of CD36 Reduces Inflammation and Brain Injury in Transient Focal Ischemia
To investigate the relevance of CD36 to inflammatory response following transient focal ischemic stroke, MCP‐1 and CCR2 gene expressions in the brain were determined in WT and CD36 knock‐out (CD36 KO) mice at 1 day after tMCAO. Compared to WT mice, the absence of CD36 attenuated stroke‐induced MCP‐1 and CCR2 expressions (Figure 1A and B). As we previously reported 8, CD36 deficiency resulted in reduced brain injury size and swelling assessed at 3 days poststroke (Figure 1C and D). The association between attenuated MCP‐1 and CCR2 expressions and improved outcomes in CD36 KO mice confirms the involvement of CD36 in inflammation and injury in transient focal ischemia.
Figure 1.
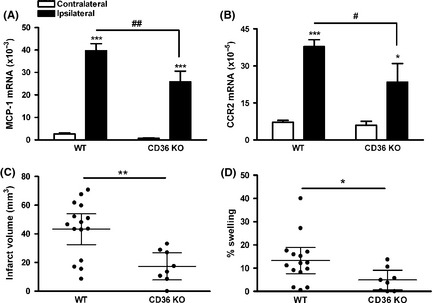
Absence of CD36 decreases inflammation and injury size in transient focal ischemia. (A and B) MCP‐1 (A) and CCR2 (B) mRNA levels in the ischemic brain at 1 day postischemia. n = 5–6/group, *P < 0.05 and ***P < 0.001 versus contralateral, # P < 0.05 and ## P < 0.01 versus wild‐type (WT), two‐way ANOVA. (C and D) Infarct volume (C) and % swelling (D) in WT and CD36 KO mice at 3 days after tMCAO. n = 8–15/group, *P < 0.05 and **P < 0.01 versus WT, Student's t‐test.
CD36 Deficiency does not Reduce Inflammatory Responses and Ischemic Brain Injury in Permanent Focal Ischemia
To investigate the role of CD36 in acute stroke without reperfusion, MCA was permanently occluded (pMCAO) in WT and CD36 KO mice. MCP‐1 gene expression was significantly elevated at 1 day poststroke, and the levels were similar between WT and CD36 KO mice (Figure 2A). Interestingly, the increase in MCP‐1 levels in pMCAO was smaller compared to tMCAO (tMCAO vs. pMCAO [×10−3], 39.7 ± 3.1 vs. 19.7 ± 2.9, P < 0.01) in WT mice, suggesting less inflammation in ischemic stroke without reperfusion. Stroke also increased CCR2 gene expressions in WT mice, and the induction was reduced in CD36 KO mice (Figure 2B). Infarct size and % swelling were significantly larger with pMCAO compared to tMCAO (tMCAO vs. pMCAO, infarct size [mm3], 43.3 ± 5.01 vs. 59.4 ± 2.4, P < 0.05; % swelling, 13.3 ± 2.6 vs. 37.8 ± 3.2, P < 0.001), and there was no difference between WT and CD36 KO mice (Figure 2C and D). The results suggest that pMCAO is associated with less inflammatory responses but results in larger injury, and CD36 does not play a role in ischemic injury without reperfusion.
Figure 2.
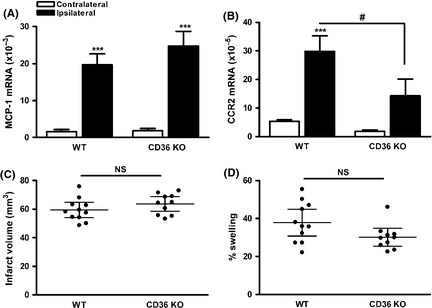
Absence of CD36 does not reduce inflammation and injury size in permanent focal ischemia. (A and B) MCP‐1 (A) and CCR2 (B) mRNA levels in the ischemic brain at 1 day postischemia. n = 5/group, ***P < 0.001 versus contralateral, # P < 0.05 versus wild‐type (WT), two‐way ANOVA. (C and D) Infarct volume (C) and % swelling (D) in WT and CD36 KO mice 3 days after pMCAO. n = 10–11/group, NS, nonsignificant.
SS‐31 Reduces Inflammation and Brain Injury in Transient Focal Ischemia
SS‐31 has been shown to attenuate stroke‐induced glutathione depletion and ischemic brain injury, and the attenuation was associated with attenuation of CD36 up‐regulation 22. As SS‐31 reduced infarct size in tMCAO 22, we determined the effect of SS‐31 on MCP‐1 and CCR2 expressions following tMCAO. Stroke increased CD36 mRNA levels at 24 h after tMCAO, and it was attenuated by SS‐31 treatment (Figure 3A). SS‐31 also significantly reduced MCP‐1 mRNA levels in the stroked hemisphere (Figure 3B). Unlike CD36 KO mice, SS‐31 did not have any effect on CCR2 mRNA levels (Figure 3C). Infarct volume and % hemispheric swelling were significantly smaller in the mice treated with SS‐31 compared to vehicle‐treated mice (Figure 3D and E), showing the efficacy of SS‐31 in suppressing MCP‐1 expression that is associated with neuroprotection in transient focal ischemia.
Figure 3.
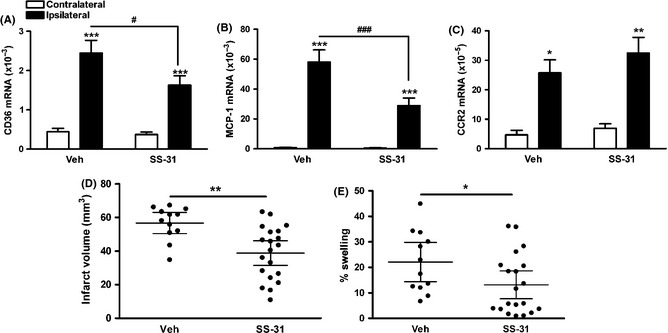
SS‐31 treatment attenuates CD36 and inflammatory response and ischemic/reperfusion injury. (A–C) Brain CD36 (A), MCP‐1 (B), and CCR2 (C) mRNA levels in mice treated with vehicle or SS‐31 24 h after tMCAO. n = 5–6/group, *P < 0.05, **P < 0.01, and ***P < 0.001 versus contralateral, # P < 0.05 and ### P < 0.001 versus veh, two‐way ANOVA, (D and E) stroke outcome measurement 3 days after tMCAO. (D and E) Infarct volume (D) and % swelling (E), n = 12–20/group, *P < 0.05 and **P < 0.01 versus veh, Student's t‐test.
SS‐31 has no Effect on Reducing Inflammation and Brain Injury in Permanent MCAO
We further investigated the effect of SS‐31 on permanent focal ischemia. While pMCAO also increased CD36, the increase was much smaller compared to tMCAO (tMCAO vs. pMCAO [×10−3], 2.4 ± 0.3 vs. 0.7 ± 0.1, P < 0.01), and it was not affected by SS‐31 treatment (Figure 4A). Likewise, the increase in MCP‐1 expression was much smaller compared to tMCAO (tMCAO vs. pMCAO [×10−3], 58.0 ± 8.1 vs. 15.6 ± 5.7, P < 0.01) and this was also not affected by SS‐31 (Figure 4B). In contrast, the increase in CCR2 mRNA levels at 1 day postischemia was comparable to tMCAO, and SS‐31 did not attenuate its expression (Figure 4C). Infarct size and % swelling were similar between vehicle‐ and SS‐31‐treated mice (Figure 4D and E).
Figure 4.
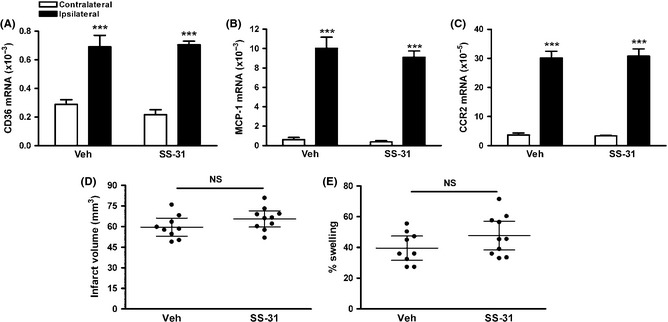
SS‐31 treatment does not reduce the expression of inflammatory markers and brain injury in permanent focal ischemia. (A–C) Brain CD36 (A), MCP‐1 (B), and CCR2 (C) mRNA levels in mice treated with saline (veh) or SS‐31 24 h after pMCAO, n = 4/group, ***P < 0.001 versus contralateral, Two‐way ANOVA. (D and E) Stroke outcome measurement 3 days after pMCAO. Infarct volume (D) and % swelling (E), n = 9–10/group, NS, nonsignificant.
Discussion
Stroke elicits multiple pathological processes including necrosis, apoptosis, oxidative stress, vascular dysfunction, and pro‐inflammatory responses 5, 6, 31, 32, 33. These events often temporally and spatially overlap, diverge, or cross talk, activating different sets of pathological pathways. The current study investigated stroke‐induced inflammation and acute outcome in two different ischemic stroke models (transient or permanent focal stroke), focusing on their responses to approaches directed at CD36. The results show that targeting CD36, either genetically or pharmacologically, demonstrated beneficial effects by reducing inflammation and injury in transient focal stroke, but failed to show benefit in permanent focal stroke.
CD36 expression occurs in a feed‐forward manner in the presence of ligands 34, 35, 36. With increased generation of CD36 ligands including long‐chain fatty acids, oxidized low‐density lipoprotein (oxLDL), and thrombospondins in stroke 37, 38, 39, 40, 41, CD36 elicits intracellular signaling and produces inflammatory cytokines and chemokines 17, 42, 43, 44. Among them, monocyte chemoattractant protein‐1 (MCP‐1) and CCR2, a receptor for MCP‐1, play an important role in attracting peripheral immune cells into the injury site 45, 46, 47, 48. Our previous study also suggested an involvement of CD36 in regulating the expression of MCP‐1 and CCR2 in focal ischemic stroke 9, 20. The current study addressed the influence of different types of stroke on MCP‐1 and CCR2 expressions by targeting CD36. We observed protection in CD36 KO mice subjected to tMCAO (Figure 1) but not pMCAO (Figure 2). Complementary to CD36 genetic approach, the pharmacological approach using SS‐31 similarly resulted in neuroprotection in tMCAO (Figure 3) but not in pMCAO (Figure 4).
Underlying mechanism that account for the difference in brain injury and swelling between the two models may derive from the extent of acute inflammatory responses associated with cerebral ischemia, with or without reperfusion. Acute inflammatory responses appear to be a major pathological event in ischemic stroke. Both tMCAO and pMCAO elicit the acute inflammatory response; however, this study shows that MCP‐1 expression was profoundly increased in tMCAO and much less in pMCAO. The down‐regulation of CD36 by genetic or pharmacological means reduced MCP‐1 up‐regulation in tMCAO (Figures 1A and 3B) but had no effect in pMCAO (Figures 2A and 4B). These results suggest that CD36 does not play a significant role in MCP‐1 induction in permanent focal stroke. Unlike MCP‐1, the modulation of CCR2 mRNA levels was different between genetic CD36 deficiency and pharmacological approaches. In both tMCAO and pMCAO, CCR2 expression was reduced in CD36 KO mice (Figures 1B and 2B), but the expression was not changed by SS‐31 treatment (Figures 3C and 4C). This may due to several reasons. The timing of infiltration of CCR2+ mononuclear phagocytes into the injured site, where up‐regulation of CCR2 in the postischemic brain, lags behind the up‐regulation of MCP‐1 20. Alternative explanations for the difference may include the regulation of CCR2 expression by other ligands in addition to MCP‐1 and a developmental compensation and/or a long‐term preventative effect in genetic CD36 deficiency, while SS‐31 treatment reflects the prevention of CD36 up‐regulation in acute stroke.
Although genes that are involved in stress responses, cell death, and metabolism are commonly induced in tMCAO and pMCAO, comparison of expression profiles between the models indicates that gene induction is unique to each model. Stroke with reperfusion induces genes encoding inflammation, apoptosis, and cell cycle, while genes associated with neurotransmitter receptors, ion channels, growth factors, and signaling molecules are linked to permanent occlusion 49. Despite the blunted inflammatory responses in pMCAO, larger injury size was observed in this study. Both infarct size and swelling were significantly greater in pMCAO compared to tMCAO and reperfusion reduced infarct size by ~30%, and swelling was almost threefold lower despite a larger increase in MCP‐1 and CCR2 expressions. These suggest that factors other than inflammation contribute to ischemic injury in the setting of stroke without reperfusion. A lack of ATP during ischemia results in apoptosis or necrosis, and rapid reperfusion minimizes cell death and infarct size. However, ischemia causes mitochondrial swelling, slows down the recovery of ATP synthesis upon reperfusion, and produces excess inflammatory mediators 50, 51, 52, 53, 54. Our study shows that CD36 plays a role in mediating this inflammatory response and benefits can be accomplished by attenuating the inflammatory process that occurs upon reperfusion.
SS‐31 is a cell permeable, mitochondria‐targeted tetrapeptide that has been shown to attenuate the up‐regulation of CD36 upon reperfusion 22. The SS peptides are highly water‐soluble, and their tissue distribution is largely determined by blood flow 55, thus, compared to tMCAO, SS‐31 would be delivered substantially less to the ischemic site in pMCAO. Because peripheral monocytes/macrophages contribute to stoke‐induced brain injury and SS‐31 modulates CD36 expression on these cells 22, it is possible to have indirect effects exerted by these immune cells to brain injury. However, pMCAO did not resulted in reduction of infarct in CD36 KO mice (Figure 2C). Therefore, the genetic evidence indicates that the reason for failure to reduce infarct in SS‐31‐treated pMCAO mice is likely due to differential pathophysiology associated with pMCAO model, which is distinct from CD36‐associated acute inflammatory pathology.
Early studies showed that SS‐31 selectively partitions to the inner mitochondrial membrane, and its dimethyltyrosine residue can scavenge excess electrons and reduce mitochondrial reactive oxygen species 56, 57. It was recently reported that SS‐31 interacts with cardiolipin on the inner mitochondrial membrane and modulates its interaction with cytochrome c 58, 59. As a result, SS‐31 inhibits cytochrome c peroxidase activity and promotes electron transport and ATP synthesis 58. SS‐31 can also reduce electron leak and reactive oxygen species production. By inhibiting cardiolipin peroxidation, the peptide protects mitochondrial structure, accelerates ATP recovery upon reperfusion, and reduces cell death and inflammation 59, 60. SS‐31 was effective in ameliorating reperfusion injury following myocardial ischemia in a preclinical study, which led to the EMBRACE‐STEMI trial with Bendavia (clinical formulation of SS‐31) for patients receiving percutaneous coronary reperfusion for ST‐segment elevation myocardial infarction (NCT01572909) 61. Additionally, its efficacy in improving renal outcome after revascularization in renal artery stenosis in pigs 23 led to a clinical trial for patients undergoing percutaneous angioplasty of the renal artery (NCT 01755858). Thus, our findings suggest that attenuating inflammatory responses directing CD36 is a promising strategy for acute transient ischemic stroke. Importantly, SS‐31/Bendavia may serve as a potential therapeutic for minimizing ischemia–reperfusion injury, especially in stroke patients who receive tPA.
Conflict of Interest
The SS peptides described in this article are licensed for commercial research and development to Stealth Peptides Inc, a clinical stage biopharmaceutical company, in which HHS, SC, and the Cornell Research Foundation have financial interests.
Acknowledgment
This work is supported by NIH Grants HL82511, NS07396 (SC) and the Burke Foundation.
References
- 1. Wardlaw JM, Murray V, Berge E, et al. Recombinant tissue plasminogen activator for acute ischaemic stroke: An updated systematic review and meta‐analysis. Lancet 2012;379:2364–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Carden DL, Granger DN. Pathophysiology of ischaemia‐reperfusion injury. J Pathol 2000;190:255–266. [DOI] [PubMed] [Google Scholar]
- 3. Pundik S, Xu K, Sundararajan S. Reperfusion brain injury: Focus on cellular bioenergetics. Neurology 2012;79:S44–S51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. White BC, Sullivan JM, DeGracia DJ, et al. Brain ischemia and reperfusion: Molecular mechanisms of neuronal injury. J Neurol Sci 2000;179:1–33. [DOI] [PubMed] [Google Scholar]
- 5. Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci 2003;4:399–415. [DOI] [PubMed] [Google Scholar]
- 6. Huang J, Upadhyay UM, Tamargo RJ. Inflammation in stroke and focal cerebral ischemia. Surg Neurol 2006;66:232–245. [DOI] [PubMed] [Google Scholar]
- 7. Amantea D, Nappi G, Bernardi G, Bagetta G, Corasaniti MT. Post‐ischemic brain damage: Pathophysiology and role of inflammatory mediators. FEBS J 2009;276:13–26. [DOI] [PubMed] [Google Scholar]
- 8. Cho S, Park EM, Febbraio M, et al. The class B scavenger receptor CD36 mediates free radical production and tissue injury in cerebral ischemia. J Neurosci 2005;25:2504–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kim E, Tolhurst AT, Qin LY, Chen XY, Febbraio M, Cho S. CD36/fatty acid translocase, an inflammatory mediator, is involved in hyperlipidemia‐induced exacerbation in ischemic brain injury. J Neurosci 2008;28:4661–4670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Febbraio M, Hajjar DP, Silverstein RL. CD36: A class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J Clin Invest 2001;108:785–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jimenez B, Volpert OV, Crawford SE, Febbraio M, Silverstein RL, Bouck N. Signals leading to apoptosis‐dependent inhibition of neovascularization by thrombospondin‐1. Nat Med 2000;6:41–48. [DOI] [PubMed] [Google Scholar]
- 12. Coraci IS, Husemann J, Berman JW, et al. CD36, a class B scavenger receptor, is expressed on microglia in Alzheimer's disease brains and can mediate production of reactive oxygen species in response to beta‐amyloid fibrils. Am J Pathol 2002;160:101–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Talle MA, Rao PE, Westberg E, et al. Patterns of antigenic expression on human monocytes as defined by monoclonal antibodies. Cell Immunol 1983;78:83–99. [DOI] [PubMed] [Google Scholar]
- 14. Bordessoule D, Jones M, Gatter KC, Mason DY. Immunohistological patterns of myeloid antigens: Tissue distribution of CD13, CD14, CD16, CD31, CD36, CD65, CD66 and CD67. Br J Haematol 1993;83:370–383. [DOI] [PubMed] [Google Scholar]
- 15. El Khoury JB, Moore KJ, Means TK, et al. CD36 mediates the innate host response to beta‐amyloid. J Exp Med 2003;197:1657–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Febbraio M, Abumrad NA, Hajjar DP, et al. A null mutation in murine CD36 reveals an important role in fatty acid and lipoprotein metabolism. J Biol Chem 1999;274:19055–19062. [DOI] [PubMed] [Google Scholar]
- 17. Rahaman SO, Lennon DJ, Febbraio M, Podrez EA, Hazen SL, Silverstein RL. A CD36‐dependent signaling cascade is necessary for macrophage foam cell formation. Cell Metab 2006;4:211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stewart CR, Stuart LM, Wilkinson K, et al. CD36 ligands promote sterile inflammation through assembly of a Toll‐like receptor 4 and 6 heterodimer. Nat Immunol 2010;11:155–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cho S. CD36 as a therapeutic target for endothelial dysfunction in stroke. Curr Pharm Des 2012;18:3721–3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim E, Febbraio M, Bao Y, Tolhurst AT, Epstein JM, Cho S. CD36 in the periphery and brain synergizes in stroke injury in hyperlipidemia. Ann Neurol 2012;71:753–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Moore KJ, El Khoury J, Medeiros LA, et al. A CD36‐initiated signaling cascade mediates inflammatory effects of beta‐amyloid. J Biol Chem 2002;277:47373–47379. [DOI] [PubMed] [Google Scholar]
- 22. Cho S, Szeto HH, Kim E, Kim H, Tolhurst AT, Pinto JT. A novel cell‐permeable antioxidant peptide, SS31, attenuates ischemic brain injury by down‐regulating CD36. J Biol Chem 2007;282:4634–4642. [DOI] [PubMed] [Google Scholar]
- 23. Eirin A, Li Z, Zhang X, et al. A mitochondrial permeability transition pore inhibitor improves renal outcomes after revascularization in experimental atherosclerotic renal artery stenosis. Hypertension 2012;60:1242–1249. [DOI] [PubMed] [Google Scholar]
- 24. Britton P, Lu XC, Laskosky MS, Tortella FC. Dextromethorphan protects against cerebral injury following transient, but not permanent, focal ischemia in rats. Life Sci 1997;60:1729–1740. [DOI] [PubMed] [Google Scholar]
- 25. Xiong L, Zhu Z, Dong H, Hu W, Hou L, Chen S. Hyperbaric oxygen preconditioning induces neuroprotection against ischemia in transient not permanent middle cerebral artery occlusion rat model. Chin Med J (Engl) 2000;113:836–839. [PubMed] [Google Scholar]
- 26. Zhang RL, Chopp M, Jiang N, et al. Anti‐intercellular adhesion molecule‐1 antibody reduces ischemic cell damage after transient but not permanent middle cerebral artery occlusion in the Wistar rat. Stroke 1995;26:1438–1442; discussion 1443. [DOI] [PubMed] [Google Scholar]
- 27. Febbraio M, Podrez EA, Smith JD, et al. Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J Clin Invest 2000;105:1049–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim E, Tolhurst AT, Cho S. Deregulation of inflammatory response in the diabetic condition is associated with increased ischemic brain injury. J Neuroinflammation 2014;11:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schiller PW, Nguyen TM, Berezowska I, et al. Synthesis and in vitro opioid activity profiles of DALDA analogues. Eur J Med Chem 2000;35:895–901. [DOI] [PubMed] [Google Scholar]
- 30. Lin TN, He YY, Wu G, Khan M, Hsu CY. Effect of brain edema on infarct volume in a focal cerebral ischemia model in rats. Stroke 1993;24:117–121. [DOI] [PubMed] [Google Scholar]
- 31. Iadecola C, Alexander M. Cerebral ischemia and inflammation. Curr Opin Neurol 2001;14:89–94. [DOI] [PubMed] [Google Scholar]
- 32. Choi DW. Ischemia‐induced neuronal apoptosis. Curr Opin Neurobiol 1996;6:667–672. [DOI] [PubMed] [Google Scholar]
- 33. Endres M, Dirnagl U. Ischemia and stroke. Adv Exp Med Biol 2002;513:455–473. [DOI] [PubMed] [Google Scholar]
- 34. Feng J, Han J, Pearce SF, et al. Induction of CD36 expression by oxidized LDL and IL‐4 by a common signaling pathway dependent on protein kinase C and PPAR‐gamma. J Lipid Res 2000;41:688–696. [PubMed] [Google Scholar]
- 35. Munteanu A, Taddei M, Tamburini I, Bergamini E, Azzi A, Zingg JM. Antagonistic effects of oxidized low density lipoprotein and alpha‐tocopherol on CD36 scavenger receptor expression in monocytes: Involvement of protein kinase B and peroxisome proliferator‐activated receptor‐gamma. J Biol Chem 2006;281:6489–6497. [DOI] [PubMed] [Google Scholar]
- 36. Tontonoz P, Nagy L, Alvarez JG, Thomazy VA, Evans RM. PPARgamma promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell 1998;93:241–252. [DOI] [PubMed] [Google Scholar]
- 37. Hayashi T, Noshita N, Sugawara T, Chan PH. Temporal profile of angiogenesis and expression of related genes in the brain after ischemia. J Cereb Blood Flow Metab 2003;23:166–180. [DOI] [PubMed] [Google Scholar]
- 38. Nihashi T, Inao S, Kajita Y, et al. Expression and distribution of beta amyloid precursor protein and beta amyloid peptide in reactive astrocytes after transient middle cerebral artery occlusion. Acta Neurochir (Wien) 2001;143:287–295. [DOI] [PubMed] [Google Scholar]
- 39. Pilitsis JG, Coplin WM, O'Regan MH, et al. Measurement of free fatty acids in cerebrospinal fluid from patients with hemorrhagic and ischemic stroke. Brain Res 2003;985:198–201. [DOI] [PubMed] [Google Scholar]
- 40. Shie FS, Neely MD, Maezawa I, et al. Oxidized low‐density lipoprotein is present in astrocytes surrounding cerebral infarcts and stimulates astrocyte interleukin‐6 secretion. Am J Pathol 2004;164:1173–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Uno M, Kitazato KT, Nishi K, Itabe H, Nagahiro S. Raised plasma oxidised LDL in acute cerebral infarction. J Neurol Neurosurg Psychiatry 2003;74:312–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Han CY, Park SY, Pak YK. Role of endocytosis in the transactivation of nuclear factor‐kappaB by oxidized low‐density lipoprotein. Biochem J 2000;350(Pt 3):829–837. [PMC free article] [PubMed] [Google Scholar]
- 43. Janabi M, Yamashita S, Hirano K, et al. Oxidized LDL‐induced NF‐kappa B activation and subsequent expression of proinflammatory genes are defective in monocyte‐derived macrophages from CD36‐deficient patients. Arterioscler Thromb Vasc Biol 2000;20:1953–1960. [DOI] [PubMed] [Google Scholar]
- 44. Lipsky RH, Eckert DM, Tang Y, Ockenhouse CF. The carboxyl‐terminal cytoplasmic domain of CD36 is required for oxidized low‐density lipoprotein modulation of NF‐kappaB activity by tumor necrosis factor‐alpha. Recept Signal Transduct 1997;7:1–11. [PubMed] [Google Scholar]
- 45. Babcock AA, Kuziel WA, Rivest S, Owens T. Chemokine expression by glial cells directs leukocytes to sites of axonal injury in the CNS. J Neurosci 2003;23:7922–7930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dimitrijevic OB, Stamatovic SM, Keep RF, Andjelkovic AV. Absence of the chemokine receptor CCR2 protects against cerebral ischemia/reperfusion injury in mice. Stroke 2007;38:1345–1353. [DOI] [PubMed] [Google Scholar]
- 47. Hughes PM, Allegrini PR, Rudin M, Perry VH, Mir AK, Wiessner C. Monocyte chemoattractant protein‐1 deficiency is protective in a murine stroke model. J Cereb Blood Flow Metab 2002;22:308–317. [DOI] [PubMed] [Google Scholar]
- 48. Chen Y, Hallenbeck JM, Ruetzler C, et al. Overexpression of monocyte chemoattractant protein 1 in the brain exacerbates ischemic brain injury and is associated with recruitment of inflammatory cells. J Cereb Blood Flow Metab 2003;23:748–755. [DOI] [PubMed] [Google Scholar]
- 49. Ford G, Xu Z, Gates A, Jiang J, Ford BD. Expression Analysis Systematic Explorer (EASE) analysis reveals differential gene expression in permanent and transient focal stroke rat models. Brain Res 2006;1071:226–236. [DOI] [PubMed] [Google Scholar]
- 50. Chen Q, Moghaddas S, Hoppel CL, Lesnefsky EJ. Ischemic defects in the electron transport chain increase the production of reactive oxygen species from isolated rat heart mitochondria. Am J Physiol Cell Physiol 2008;294:C460–C466. [DOI] [PubMed] [Google Scholar]
- 51. Iadecola C, Anrather J. The immunology of stroke: From mechanisms to translation. Nat Med 2011;17:796–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Levraut J, Iwase H, Shao ZH, Vanden HT, Schumacker PT. Cell death during ischemia: Relationship to mitochondrial depolarization and ROS generation. Am J Physiol Heart Circ Physiol 2003;284:H549–H558. [DOI] [PubMed] [Google Scholar]
- 53. Muir KW, Tyrrell P, Sattar N, Warburton E. Inflammation and ischaemic stroke. Curr Opin Neurol 2007;20:334–342. [DOI] [PubMed] [Google Scholar]
- 54. Wang Q, Tang XN, Yenari MA. The inflammatory response in stroke. J Neuroimmunol 2007;184:53–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kloner RA, Hale SL, Dai W, et al. Reduction of ischemia/reperfusion injury with bendavia, a mitochondria‐targeting cytoprotective Peptide. J Am Heart Assoc 2012;1:e001644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhao K, Luo G, Giannelli S, Szeto HH. Mitochondria‐targeted peptide prevents mitochondrial depolarization and apoptosis induced by tert‐butyl hydroperoxide in neuronal cell lines. Biochem Pharmacol 2005;70:1796–1806. [DOI] [PubMed] [Google Scholar]
- 57. Zhao K, Zhao GM, Wu D, et al. Cell‐permeable peptide antioxidants targeted to inner mitochondrial membrane inhibit mitochondrial swelling, oxidative cell death, and reperfusion injury. J Biol Chem 2004;279:34682–34690. [DOI] [PubMed] [Google Scholar]
- 58. Birk AV, Chao WM, Bracken C, Warren JD, Szeto HH. Targeting mitochondrial cardiolipin and the cytochrome c/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br J Pharmacol 2014;171:2017–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Birk AV, Liu S, Soong Y, et al. The mitochondrial‐targeted compound SS‐31 re‐energizes ischemic mitochondria by interacting with cardiolipin. J Am Soc Nephrol 2013;24:1250–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Szeto HH, Liu S, Soong Y, et al. Mitochondria‐targeted peptide accelerates ATP recovery and reduces ischemic kidney injury. J Am Soc Nephrol 2011;22:1041–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cruikshank SJ, Landisman CE, Mancilla JG, Connors BW. Connexon connexions in the thalamocortical system. Prog Brain Res 2005;149:41–57. [DOI] [PubMed] [Google Scholar]