

Abstract
This review covers advances in analytical technologies for high-throughput (HTP) glycomics. Our focus is on structural studies of glycoprotein glycosylation to support biopharmaceutical realization and the discovery of glycan biomarkers for human disease. For biopharmaceuticals, there is increasing use of glycomics in Quality by Design studies to help optimize glycan profiles of drugs with a view to improving their clinical performance. Glycomics is also used in comparability studies to ensure consistency of glycosylation both throughout product development and between biosimilars and innovator drugs. In clinical studies there is as well an expanding interest in the use of glycomics—for example in Genome Wide Association Studies—to follow changes in glycosylation patterns of biological tissues and fluids with the progress of certain diseases. These include cancers, neurodegenerative disorders and inflammatory conditions. Despite rising activity in this field, there are significant challenges in performing large scale glycomics studies. The requirement is accurate identification and quantitation of individual glycan structures. However, glycoconjugate samples are often very complex and heterogeneous and contain many diverse branched glycan structures. In this article we cover HTP sample preparation and derivatization methods, sample purification, robotization, optimized glycan profiling by UHPLC, MS and multiplexed CE, as well as hyphenated techniques and automated data analysis tools. Throughout, we summarize the advantages and challenges with each of these technologies. The issues considered include reliability of the methods for glycan identification and quantitation, sample throughput, labor intensity, and affordability for large sample numbers.
Keywords: Glycomics, High-throughput, Derivatization, Integration, Automation, Analysis
Introduction
This review focuses on advances in analytical technologies for high-throughput (HTP) glycomics (i.e. the large-scale study of glycoconjugate glycosylation patterns). Our emphasis will be on glycomics to support (1) realization of glycoprotein biopharmaceuticals and (2) development of glycan biomarkers of disease for clinical diagnostics.
Glycans play key roles in important biological processes such as protein folding, host-pathogen interaction and signal transduction [1]. They are present both in free form, or bound to proteins or lipids, and can modify the biological activities of the conjugate. The glycomics technologies covered in this review are mainly for analysis of protein glycosylation, the major types of which are N-linked glycans attached at Asn-X-Ser/Thr motifs on the peptide backbone and O-linked glycans attached to Ser/Thr [2]. Protein glycosylation is a co-translational and post-translational modification where glycans are attached to proteins during translation, assisting in the folding and quality control of the protein. Attached glycans are subsequently modified in the endoplasmic reticulum and Golgi to varying levels of complexity, causing glycosylation to be one of the most complex and diverse types of protein modification.
Glycomics is of interest to biopharma companies because glycans can greatly modify safety and efficacy profiles of the therapeutic protein to which they are attached. For example, IgG effector functions such as antibody-dependent cellular cytotoxicity and complement-dependent cytotoxicity of monoclonal antibodies are modulated by N-glycosylation of its fragment crystallizable (Fc) portion [3]. N-glycosylation modifications are also involved in determining the plasma half-life of glycoproteins by modulating the interaction with various receptors [4, 5]. Even small changes in glycosylation can lead to serious issues such as anaphylaxis in patients and the destruction of therapeutic activity. These effects have been found in drugs such as Cetuximab (a monoclonal antibody (mAb) for treatment of metastatic colorectal cancer), Herceptin (a mAb for treatment of breast cancer) and erythropoietin (EPO—a drug for increasing red blood cells in patients with renal failure and cancer patients undergoing chemotherapy). Given this, there are now increasing pressures from regulatory authorities for drug manufacturers to measure, optimize and control their drug’s glycosylation [6]. This task is very challenging given the structural complexity of biopharmaceutical glycosylation and achieving it has required adoption of a new approach to drug design—namely Quality by Design (QbD). This methodology has been actively promoted by the FDA and other drug regulators and is being adopted by leading biopharma companies as the system of best practice for the design, development, and manufacture of biologic drugs. QbD has the potential to deal with the complexities of drug glycans and could help simplify difficult tasks such as demonstrating comparability of biopharmaceutical glycosylation [7]. However, the glycoprofiling effort required for QbD is considerable. Studies of the QbD approach by the CMC Biotech Working Group (a consortium of experts from leading biopharma organizations including Genentech, Amgen, Eli Lilly, and Pfizer), indicate that definition of the QbD Design Space may require analysis of tens of thousands of drug samples [8]. Successful adoption of QbD for development of glycoprotein therapeutics requires glycoprofiling systems that can cope with this very high level of sample throughput—in effect it will need to be built on a glycomics framework [6].
In addition to its use in biopharmaceutical realization, there is also increasing interest in glycomics for development of new clinical diagnostics based on glycosylation markers of diseases. This follows from the key roles that glycans play in major biological processes from the fertilization of eggs by sperm to cell death and their involvement in a multitude of disease etiologies and progressions. The potential of glycans as sensitive disease biomarkers is particularly high, since they are synthesized by the concerted action of multiple proteins, and influenced by various disease-associated factors such as genetic variations, epigenetic signatures and metabolic distortions. Glycan patterns of secreted proteins often confer information on the pathophysiological status of the secreting cells, making glycosylation analysis of relevant glycoproteins a suitable diagnostic tool. Changes in glycosylation are associated with various diseases like cancer, neurodegenerative and inflammatory diseases [9–15]. These types of associated changes have resulted in an increased interest in studying the alterations in glycosylation patterns of biological fluids as disease biomarkers as well as for patient stratification and personalized medicine [16].
To date, two glycan biomarkers have been introduced for routine clinical diagnostics, namely carbohydrate-deficient transferrin for the detection of alcohol abuse [17] and fucosylated serum alpha-fetoprotein for the early diagnosis of hepatocellular carcinoma [18]. Importantly, glycomic changes are not limited to single conditions but are rather a hallmark of virtually any human disease, as stated recently in a whitepaper by Walt et al. [16] and diverse promising glycomic biomarkers have been described for MODY-type diabetes and immunoglobulin G myeloma [19, 20]. However, most of the glycan biomarkers discovered so far have been studied using glycoanalytical technologies that would not be suitable for use in routine clinical diagnostic labs—the shortfall being due mainly to limitations in sample throughput, resolution and affordability of the methods.
Given the above, it is clear that development of reliable, affordable, high-resolution HTP glycomics technologies would be essential in the biopharma industry for efficient optimization of the glycosylation patterns of glycoprotein therapeutics as well as advancement of practical clinical diagnostics based on glycosylation biomarkers of disease.
The rest of this review will cover a number of recent advances in glycomics technologies for these two applications. It highlights current challenges in analytical glycomics methods and presents some approaches of how the bottlenecks in protein glycosylation analysis may be overcome.
High-Throughput Sample Preparation
Many reviews describe glycan sample preparation steps such as N- and O-glycan release, de-glycosylated sample desalting and clean-up, derivatization of released glycans and analysis using different analytical platforms [21–25]. Here we describe a number of recent sample preparation methods for HTP glycosylation analysis. Next to their application in biomedical research, these methods are often applied for the characterization of biopharmaceuticals.
Liquid chromatography (LC) is a widely used glycoanalytical technique with good quantification, reproducibility, and separation of glycan isomers. Royle et al. [26] have demonstrated a HTP sample preparation for high performance LC (HPLC) using a polyvinylidene fluoride (PVDF) membrane and an in-gel block method for glycan release. The enzymatic release step is followed by 2-aminobenzamide (2-AB) labeling and purification using microplates, and subsequent HPLC analysis. Automation of the procedure was achieved to a large extent, as well as the automated analysis of the obtained results, but the resulting method is still labor intensive, particularly the in-gel block preparation and glycan release stage. Performing the whole process on 96 samples takes 3 days with an additional 2 days for the acquisition of the chromatographic data. Cook et al. [27] used the rapid deglycosylation kit and instant 2-AB kit from Prozyme to demonstrate an approach that only took 3.5 h of sample preparation time. A downside here is that the required kits are a considerable cost factor, which may limit the number of samples that can be studied. Burnina et al. [28], on the other hand, have described a cost effective HTP sample preparation method for the characterization of therapeutic antibody N-glycosylation taking approximately 90 min. The workflow included the following steps, denaturation, reduction, deglycosylation using a hydrophobic Immobilon-P PVDF membrane filter plate, fluorescent labeling and clean-up using a 96-well hydrophilic filter plate. They demonstrated the use of orthogonal assays and obtained LC and MS which were in agreement.
Callewaert et al. [29–31] reported capillary gel electrophoresis with laser-induced fluorescence detection (CGE-LIF) using a DNA sequencer for the analysis and profiling of total serum N-glycans labeled with the negatively charged, fluorescent label 8-aminopyrene-1,3,6-trisulfonic acid (APTS). Ruhaak et al. [32] have demonstrated the HTP application of this by optimizing sample preparation and employing a system with multiplexed capillaries. After protein denaturation and N-glycan release, the glycans were labeled with APTS in a 96 well plate and subjected to hydrophilic interaction chromatography (HILIC) purification. Using the multiplexed CGE-LIF system, 48 samples can be analyzed in parallel (scalable to 96 samples) allowing the analysis of 96 samples with a hands-on time of 2.5 h. A recent large-scale application making use of the multiplexed CGE-LIF system is the analysis of alpha1-antitrypsin and immunoglobulin A from over 2,400 human plasma samples [33]. Set up to discover novel biomarkers, the method revealed protein glycosylation to be associated with age and sex, as well as with cardiovascular and metabolic diseases.
Varadi et al. [34] described the use of magnetic beads for rapid, HTP sample preparation of N-glycans from therapeutic antibodies and indicate that this method is easily automatable. The carboxyl coated magnetic beads are capable of capturing released N-glycans due to the ionic interaction between the positively charged glycosylamines and the negatively charged carboxylated beads. The glycans were captured using the magnetic beads and were labeled with APTS for 2 h. Once again the magnetic beads were used for binding the labeled glycans and elution was accomplished by washing the beads with water. This eluent was suitable for direct analysis by CE-LIF.
Mass spectrometric analysis of glycans is difficult when dealing with isobaric structures and particularly difficult when dealing with sialylated species. Sialic acids are known to degrade under harsh sample preparation and ionization conditions, as well as showing a strong preference towards negative ionization. Sialylated glycans are often derivatized before mass spectrometric analysis, either by carboxylic acid-specific methods or permethylation [35].
Gil et al. [36] reported a one pot sialic acid amidation reaction using acetohydrazide, allowing neutralization of sialylated N-glycans still attached to proteins. Accidental Schiff base formation with the acetohydrazide is prevented because the glycans are still attached to the protein. N-glycan release could then be performed afterwards, as well as labeling with the negatively charged fluorescent label 2-aminobenzoic acid (2-AA). Clean-up was performed by 96-well filter plate, and analysis by MALDI-time of flight (TOF)-MS. The authors used recombinant human glycoproteins to determine changes in fucosylation and changes in sialylation and compared the MALDI–TOF–MS data with HILIC HPLC data proving that they were in good agreement. Another approach used by Jeong et al. [37] for HTP quantitative N-glycan analysis was solid phase permethylation. Sialylated N-glycans were protected with a methyl group at the carboxylic acid by the permethylation reaction, and glycan mixtures were subsequently analyzed by MALDI–TOF–MS in positive ion mode. Sample preparation steps were performed in a 96-well format, comprising glycoprotein binding on PVDF, deglycosylation, purification of N-glycans by porous graphitized carbon (PGC), and solid phase permethylation. By spiking a known concentration of internal standard mixture with ovalbumin and porcine thyroglobulin, the authors obtained absolute quantitation for these samples, as well as relative quantification of 49 glycans from human serum prostate specific antigen. The complete sample preparation utilizing this method takes 2 days, but is quite labor intensive due to multiple manual pipetting steps.
Miura et al. [38] have developed a protocol for the methyl esterification of sialic acids using 3-methyl-1-p-tolyltriazene (MTT) as sole reactant and methyl group donor. This derivatization was performed on both the free N-glycans, requiring a subsequent purification step, as well as after immobilization of the N-glycans on Affi-gel beads, allowing a much increased throughput. Glycans could be analyzed afterwards by MALDI–TOF–MS with significantly increased stability. Using this method, both the neutral glycans and the previously negatively charged sialylated glycans could be observed in the same positive mode spectrum. Liu et al. [39], have shown a similar increase in sialic acid stability after methylamidation of the carboxylic acid residues with a combination of methylamine and tripyrrolidinophosphonium hexafluorophosphate (PyAOP). They have shown this approach to be suitable for the derivatization of O-acetylated sialic acids, and were able to perform both MALDI and electrospray ionization (ESI)-MS (/MS) experiments on the glycans. Both the MTT and the PyAOP stabilization methods only require a few hours of preparation time (purification included), making them suitable for HTP glycomics.
Another variant of methods for sialic acid modification can make use of the chemical nature of α2,3-linked sialic acids to lactonize with the subterminal galactose resulting in a loss of water that can be observed in mass spectrometry, whilst α2,6-linked sialic acids show no such behavior. Published by Wheeler et al. [40], the use of 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium (DMT–MM) in combination with methanol showed good reaction selectivity for the sialic acid linkages, generating either lactones or methyl esters. Alley et al. [41] have used DMT-MM to generate amides instead of esters, performing the reaction in the presence of ammonium chloride. Subsequent permethylation still showed linkage-specific mass differences, with the added benefit of more informative fragmentation spectra. Another extension of the DMT-MM deriviatization method was shown by Tousi et al. [42] who performed nano-HILIC-Orbitrap-MS of the modified glycans, effectively increasing the number of individual resolvable peaks after HILIC separation, as well as allowing linkage-specific MS(/MS). Similar to MTT and PyAOP, DMT-MM-assisted modification can be performed in less than 3 h (with the reaction conditions and reaction time depending on the modification to be introduced), and is highly suitable for HTP sample preparation.
An alternative to the DMT-MM method for linkage-specific modification has recently been published by us using the combination of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) and 1-hydroxybenzotriazole (HOBt) as chemicals for derivatization [43]. Performed in ethanol, the reaction yields highly linkage-specific products when incubating at 37 °C for 1 h (lactones for α2,3-linked sialic acids and ethyl esters for α2,6-linked sialic acids), with more resistance to side reactivity (such as amidation) than with DMT-MM. As a result, the method could be performed directly on impure glycan-containing mixtures such as a peptide-N-glycosidase F (PNGase F)-released plasma N-glycome. The protocol has been validated for HTP application in 96-well plate format, and shows consistent results with ultra-performance liquid chromatography (UPLC)-based methods of analysis [Fig. 1]. An application has recently been published by Bondt et al. [44] who have studied glycosylation changes on IgG fragment antigen-binding (Fab) and Fc, at different time points during and after pregnancy. By analyzing a total of 576 glycan profile spectra, pregnancy-associated changes were revealed in IgG Fab glycan sialylation, galactosylation, bisection, fucosylation, and high mannose structures.
Fig. 1.
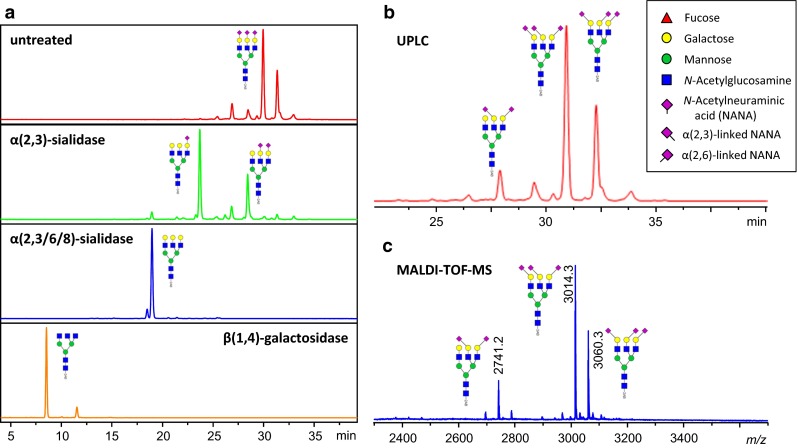
2-AB-labeled A3 glycan standard from Ludger (CAB-A3-01) was analyzed by UPLC and MALDI–TOF–MS after ethyl esterification. a Overnight incubation at 37 °C of Ludger 2-AB-labeled A3 standard with buffer (red), α(2,3)-sialidase (green), α(2,3/6/8)-sialidase (blue), α(2,3/6/8)-sialidase + β(1,4)-galactosidase (orange). Separation was performed on a Waters Acquity UPLC H-class system using an Acquity BEH 1.7 µm 2.1 × 150 mm glycan column. b Linkage-specific assignment of the undigested UPLC data [100]. c MALDI–TOF–MS spectrum of the 2-AB-labeled A3 standard ([M + Na]+) after 1 h of ethyl esterification with EDC and HOBt at 37 °C and subsequent HILIC purification according to Reiding et al. [43]. Profiles obtained from both methods are highly comparable and show similar ratios with regard to sialic acid occupancy and linkage. HILIC peak assignments are based on the exoglycosidase digests, as well as use of internal standards. Structural schemes of glycans are depicted following the CFG notation: N-acetylglucosamine (blue square), fucose (red triangle), mannose (green circle), galactose (yellow circle), N-acetylneuraminic acid (purple diamond). Known N-acetylneuraminic acid linkages are indicated by a left angle (α2,3) or right angle (α2,6), and otherwise unspecified
Modification and stabilization of glycoconjugates has recently been attempted by Nishikaze et al. [45], showing a PyAOP-based methylamidation of all carboxylic acids present on sialylated glycopeptides. Not only does this derivatization protect the sialylated glycan species from breakdown, MALDI-quadrupole ion trap-TOF–MS fragmentation shows increased structural information as a consequence. Nishikaze and coworkers observed predominantly peptide fragmentation in positive mode collision-induced dissociation MS/MS, while negative mode led to more information on glycan structural features. Using a different glycopeptide derivatization method, Amano et al. [46] modified glycopeptides with 1-pyrenyldiazomethane, also specifically targeting carboxylic acids. Whilst this method leads to enhanced ionization for MALDI–MSn analysis, the subsequent exploration of the fragmentation has shown fragment ions reflecting predominantly sialic acid linkage cleavage [47].
For the HTP analysis of underivatized Fc IgG tryptic glycopeptides, Selman et al. [48] have published a method employing MALDI-Fourier transform ion cyclotron resonance (FTICR)-MS. The advantage of this intermediate-pressure instrument is the tendency for labile substituents to remain intact without chemical stabilization, allowing directly for the analysis of sialylated glycopeptide species [49]. Using 96-well sample preparation, Selman et al. showed a highly repeatable analysis of 384 samples in less than 36 h, comprising all of the steps from capturing of the IgG to automatic processing of the spectra. Additionally, the same group established a HPLC–MS method for measuring IgG-Fc glycopeptides requiring 16 min analysis time per sample, employing a sheath-flow electrospray setup for enhanced sensitivity and robustness [44]. Large-scale applications of this method have been performed by Bondt et al. [50], analyzing over 1,500 samples within 4 weeks with an internal standard coefficient of variation (CV) below 4 %. The results indicated an association between IgG galactosylation and improvement of rheumatoid arthritis (RA) during pregnancy. Another application was performed by Rombouts et al. [51] who have used the method to show changes in anti-citrullinated protein antibody Fc glycosylation even preceding the onset of RA. Both the FTICR and HPLC methods separate IgG glycopeptides on the basis of single amino acid differences (phenylalanine or tyrosine) allowing subclass-specific glycosylation analysis. An overview of various HTP methods for IgG glycosylation analysis has been published by Huffman et al. [52], comparing UPLC with fluorescent detection, MALDI–TOF–MS, multiplexed CGE-LIF and LC–ESI–MS, as well as explaining the advantages and disadvantages of each method.
Another highly sensitive technique for analysis of protein glycosylation utilizes multiple reaction monitoring (MRM) which is an LC–MS/MS technique performed on a triple quadrupole (QqQ). MRM is known for its ability to quantify low abundant compounds in highly complex mixtures. For example, glycoproteins from biological fluids like serum or plasma are reduced, alkylated and digested with trypsin and analyzed without further fractionation [53]. In MRM glycans or glycopeptides are scanned in the first quadrupole (Q1) for user specified precursor ions and these precursors undergo CID in the second quadrupole (Q2) and fragments are produced. Finally the fragments of the precursor ions, known as transitions are scanned in the third quadrupole [54]. Song et al. [54] used MRM for quantifying glycopeptides from both model glycoproteins and from depleted human blood serum. Hong et al. [53, 55] also reported the use of MRM for absolute quantitation of IgG glycopeptides and also published the use of MRM to study site specific glycosylation alterations for identifying glycan biomarkers. The advantages of the technique are the very high sensitivity which together with the high selectivity allows for the direct analysis of the resulting glycopeptides on the QqQ without requiring prior protein enrichment or sample clean-up. A couple of drawbacks to this technique are that (1) MRM analyses are targeted, and analytes/glycopeptide species which are not included in the method set-up cannot be detected by data mining in hindsight. (2) A considerable amount of time and effort need to be invested for assay set-up and for establishing transitions for different analytes. These above mentioned MRM protocols were not adapted to HTP sample preparation, but can certainly be implemented for large sample numbers and MRM is envisaged to facilitate glycan biomarker discovery and protein characterization studies in the near future.
Use of Internal Standards
The analysis of glycosylation is made difficult by technical variation with respect to separation (retention time, m/z values) and intensity (fluorescent signal, ion detection). In particular, the analysis of complex samples is compromised by the presence of the biological matrix that can influence labeling as well as ionization efficacy and migration positions [56, 57]. The use of standards has been found to assist tremendously in identification and quantification of glycans, and many interesting approaches have been shown in the literature to internalize standardization within HTP sample analysis. When using a DNA analyzer for CGE-LIF analysis of labeled glycans, the fact can be exploited that the system can analyze fluorescence on different wavelength channels. Using multiplexed CGE-LIF, co-injection of fluorescently labeled glycans and a base-pair nucleotide ladder fluorescing at a different wavelength was performed allowing for the correction of glycan migration times on the basis of the nucleotide ladder [32].
Glycan standards labeled with heavy isotopes facilitate quantitation during mass spectrometric analysis of derivatized glycans. For example, Prien et al. [58] have quantitatively analyzed the N-glycosylation of several standard proteins by MALDI–TOF- and nanospray multistage-MS after labeling with normal 2-AA and a heavy 13C version of 2-AA. The molecular weight difference between the two labels gives an offset of +6 Da. Mixing various ratios of the differently labeled samples revealed consistent quantitation by mass spectrometry. Using a different labeling chemistry, Walker et al. [59] developed stable-isotope labeled hydrazide reagents for ESI–MS(/MS) analysis and showed quantitative detection after a sample preparation procedure that takes approximately 4 h. As another example, Zhang et al. [60] have presented stable isotopically labeled phenyl-3-methyl-5-pyrazolone at the N-glycan reducing end with quantitation achievable within a 10-fold dynamic range.
Another labeling strategy for quantification lies in the use of isobaric tags. Variants of these labels have the same mass, but differ in the distribution of heavy isotopes on opposite sides of a bond that readily undergoes fragmentation by CID. Reporter ions in MSn experiments can be used for relative quantification, making quantitation of highly complex samples intrinsically more robust than in MS mode quantitation, as the latter method can easily suffer from interfering, overlapping signals. An example of the use of isobaric tags in glycosylation analysis is provided by Ahn et al. [61] who show LC–MS/MS data on tryptic glycopeptides derivatized with isobaric tags for relative and absolute quantitation (iTRAQ) at the peptide primary amines. Another version of isobaric tags are tandem mass tags (TMT), which have been used by Gong et al. [62] to label released N-glycans still in their glycosylamine form. Glycosylamines are generated when PNGase F release is performed, but usually hydrolyze to the reducing sugar under most conditions. However, keeping the pH slightly alkaline preserved the glycosylamines and allowed for TMT labeling by N-hydroxysuccinimide ester chemistry [62]. Both iTRAQ and TMT not only facilitate internal standardization, but also allow the discrimination of multiple different samples during a single analysis, speeding up analysis time as a consequence.
Permethylation provides another opportunity to generate internal standards. For example, when a 13C permethylated glycan standard of known concentration is mixed with a 12C permethylated glycan sample and analyzed by MS, relative glycan quantitation data can be obtained. Manilla et al. [63] have published the use of 13C methyl iodide for permethylation, showing expected ratios at various concentrations when comparing to 12C permethylated human milk oligosaccharides using MALDI–TOF–MS. 12C and 13C permethylated human IgG N-glycan standards developed at Ludger are an example of system suitability standards used for relative quantitation [Fig. 2]. When in possession of mass spectrometric instruments with high resolving power, Atwood et al. have shown isobaric variants can also be used for permethylation. Using 13CH3I and 12CH2DI as methyl donors, a minimal mass difference (0.0029 Da) is imparted upon the glycan permethylation site. When analyzed with high resolution instruments such as FTICR-MS the variants can be separated, achieving relative quantitation over two orders of magnitude, while a single peak can be generated for the two masses at lower resolution.
Fig. 2.

MALDI–TOF–MS spectrum of the 12C and 13C permethylated human-IgG N-glycan standards from Ludger (Cat# Cpm13C-IgG-01 and Cpm12C-IgG-01). 13C was spiked with 12C on the same sample spot to showcase the comparison of relative quantities of the major IgG N-glycans. The mass values shown in the spectra are [M + Na]+ of permethylated glycans, with 13C permethylated masses in parentheses
Integration of Analysis Workflows
Various groups have attempted to integrate different parts of the work-up procedure and simplified sample preparation steps. The integration of workflows has the potential to expedite analyses of a wide range of glycans, and the approaches can provide significant advantages in productivity, sensitivity, speed, and efficiency. Methods focusing on the integration of workflows are discussed below.
BlotGlyco kits produced by Sumitomo Bakelite in Japan integrate and support an “all-in-one” solution for automated and HTP analysis [65]. Glycoproteins are cleaved with trypsin, glycans are released by PNGase F, captured chemoselectively on hydrazide-functionalized beads coupled to 2-AB, and modified at the sialic acids by MTT as described previously. Subsequently, the N-glycans are released from the beads by reduction of sulfide bonds, but by that process retain 2-AB as a label. Using this integration method up to 96 samples can be purified and labeled in about 8 h. A newer method of Sumitomo Bakelite uses hydrazide beads to capture glycans in a process called glycoblotting, the captured glycans are purified, released from the beads and then labelled with 2AB on a 96 well filter plate [66].
Another integrated system is the Agilent mAb-glyco chip kit which has been designed for automated characterization of N-glycans from monoclonal antibodies (mAb) [67]. The system integrates on-chip deglycosylation of the mAb, HPLC chromatographic separation, quadrupole time-of-flight (QTOF)-MS analysis of the released glycans, as well as data analysis by Molecular Feature Extractor which is a part of Agilent’s data analysis software MassHunter. A study comparing on-chip and in-solution deglycosylation methods showed that the analysis time could be reduced from 5 to 6 h to 12 min [68]. Another microchip-based integrated platform is the Caliper LabChip GXII Microchip-CE Platform, which uses the Caliper ProfilerPro glycan profiling kit to provide a HTP method for analyzing N-glycosylation patterns on mAbs and other glycosylated proteins [69]. The system contains plates for denaturation, deglycosylation, and glycan labeling. The analysis is performed on a LabChip GXII CE instrument together with an internal standard. By this method, the relative abundance of the major N-glycans found on mAbs can be determined in a short span of time, requiring only 90 min for the preparation of 96 samples, although the glycan peak separation is compromised.
Automation of Sample Preparation
While the development of HTP methods for analysis of glycosylation facilitates the glycomic study of large clinical cohorts, performing manual sample preparation is still a time-consuming process. Additionally, while performing manual work-up is suitable for small sample sets, scaling up to larger sizes may lead to errors and inconsistency, resulting in poor repeatability. This set of circumstances has led to an increasing demand for automation of workflows, with laboratories requiring a system that is simple to operate, has scalable sample preparation and a process that is both repeatable and reliable. Currently, high quality glycomics data can be obtained using liquid handling robots for sample processing, clean-up and sample preparation [70]. A few approaches for automated and HTP glycan analysis will be reviewed.
Stöckmann et al. published an automated workflow for IgG N-glycan release, hydrazide bead capture of glycans through glycoblotting adapted from the protocol of Furukawa et al. mentioned above, 2-AB labeling, solid phase extraction (SPE), LC separation, and quantification. This workflow was developed for the efficient glycoprofiling of bio-therapeutics and clinical samples on the Hamilton Star liquid handling robot [14]. This approach was tested for the analysis of protein A captured IgG derived from CHO cells, as well as for the analysis of 100 samples of protein G-captured human IgG. The automated glycan preparation was verified with conventional and manual in-gel block release for the human serum IgG N-glycans, with CVs below 10 % for all major glycan peaks, and with relative ratios in agreement with expectation.
A second integrated and automated technology developed specifically for biomolecule sample preparation is the Agilent AssayMAP platform, which is supported by the Bravo automated liquid-handling robot [71]. This workflow utilizes ProZyme GlykoPrep-plus chemistry kits, with an optional affinity purification step to capture glycoproteins from cell lysates, cell culture supernatants, or serum. Recoveries in glycoprotein purification approach 100 % for most applications, with CVs of ≤5 %. The purified glycoproteins are denatured and immobilized onto hydrophobic cartridges. Next, the immobilized glycoprotein samples are treated with PNGase F, the glycans fluorescently labeled, and cleaned on HILIC cartridges prior to analysis by HPLC, CE, or LC–MS [72]. The automation platform enables processing of complex samples, increases throughput, decreases variability and decreases assay time from a couple of days to 3–5 h. However, the cost of the kit could be a limiting factor when analyzing a large number of samples.
Another automated analytical workflow supported by the Hamilton STARlet liquid handling robot has been developed at Ludger for HTP glycomics for QbD studies. The automated workflow combines glycan release, 2-AB labeling, clean-up, and sample preparation for UPLC analysis. A validation study was performed, starting with 16 human IgG samples that were released by using PNGase F. Each sample was divided into four and the four subsets of samples were further divided equally into two 96 well PCR plates. Two operators used the liquid handling robot to label one plate each with 2-AB and cleaned-up using the liquid handling robot. The CVs for the major human IgG N-glycan peaks between the different operators were below 5 % [73]. In addition, a fully robotized analysis for 48 samples of human IgG was performed, starting from the N-glycan release up to UHPLC sample preparation, showed CVs less than 5 % for all peaks with average areas above 1 % [Fig. 3].
Fig. 3.

a Workflow depicting validation study of semi-automated sample preparation of 48 replicates of human IgG samples, comprising digestion by PNGase F, 2-AB-labeling, HILIC SPE enrichment and preparation of samples for injection onto UHPLC performed with Hamilton Starlet liquid handling robot [73, 101, 102]. b Typical UHPLC chromatogram of 2-AB-labeled human IgG glycans prepared using the robot. Analysis of the data from these 48 samples showed CVs less than 5 % for all major peaks. The data shows that the automated, high-throughput method is repeatable. The liquid handling robot allows for fast, reliable and robust analyses of glycans. Glycan peaks in the chromatogram were assigned by exoglycosidase digestion, standard inclusion, and literature [103]
For mass spectrometric analysis of glycopeptides, Reusch et al. [74] have developed a fully automated HTP sample preparation method employing the Hamilton Microlab STAR to monitor mAb IgG Fc glycosylation by Orbitrap ESI–MS. They studied IgG from fermentation supernatant, automating the protein A capturing, tryptic digest, and HILIC-SPE, showing that the automation produces highly similar results when compared to HILIC-HPLC analysis of 2-AB labeled glycans, MALDI–TOF–MS and high-pH anion-exchange chromatography-pulsed amperometric detection of released glycans, as well as ESI-QTOF–MS of reduced mAb Fc. The glycopeptide sample preparation performed in this protocol facilitates discrimination between Fc and Fab portions, allows subclass-specific analysis, and is usable for a wide range of readout methods.
Data Analysis
Innovations in sample preparation have increased the scope of glycomics considerably, allowing recently, for example, for large-scale GWAS [19, 75]. However, increased data production leads to an accompanying need for robust and HTP data analysis procedures. The techniques focused on in this review allow the detection of large numbers of samples either by being HTP, or by featuring automation leading to decreased hands-on time. Such methods can be used for the discovery of analytes (glycans), but are most helpful for the repeated detection of similar glycosylation features across large cohorts. As a consequence, there is need for software capable of repeated and robust HTP feature extraction and quality control. Most analysis platforms and integrated methods have their software solutions, several having been mentioned above, but usually these are closed source, require licensing and may not cater to the functionality required by a user.
We recently reported an automated data extraction method for MALDI–TOF–MS spectra of N-glycans after sialic acid ethyl esterification [76]. This script requires Python, a programming language to be installed (freely available from www.python.org), which is capable of repeatedly integrating a list of glycan compositions from any number of mass spectra. The script calculates the expected glycan masses, allows a variety of modifications, takes the resulting isotopic distributions into account, and performs local background subtraction. As a downside, the program needs already calibrated data, and requires the user to still manually perform quality control.
Next to software for repeated analysis of data, several groups have published tools for the interpretation and assignment of glycomic and glycoproteomic data. These tools have been comprehensively reviewed by Woodin et al. [77] and Dallas et al. [78]. Notable and freely available examples of de novo interpretation of mass spectrometric data include Glyco-Peakfinder [79], GlycoSpectrumScan [80], GlycoPep Grader [77], SysBioWare [81], GlycoMiner [82], and GlypID [83]. Interpretation by matching recorded data to a previously annotated database can be performed for mass spectrometric data by GlycoPeptide Search [84], GlycoSearchMS [85], and GlycoPep DB [86], while HPLC derived glycan traits can be interpreted by GlycoExtractor [87] and autoGU [88].
Glycomic and glycoproteomic databases have been reviewed by Hizal et al. [89] and Campbell et al. [90], of note being the metadatabases GlycomeDB (containing entries from CFG, KEGG, GLYCOSCIENCES.de, BCSDB and Carbbank) [91], and UniCarbKB (containing entries from GlycosuiteDB, EUROCarbDB, UniCarb-DB and GlycoBase) [92]. GlycomeDB and UniCarbKB contain a wealth of theoretical and experimentally observed glycan structures, site-specific information on protein glycosylation, alongside various tools for retrieving and displaying the information.
Helpful with the graphical annotation of spectra are the web-based GlycanBuilder and DrawRings, allowing the facile construction of CFG or Oxford type glycan cartoons [93, 94]. Particularly useful for glycan analysis is the desktop application GlycoWorkBench [95]. The program combines many aspects of glycan annotation and glycan spectral interpretation into a user-friendly package, incorporating GlycanBuilder for cartoon annotation, GlycoPeakfinder for theoretical structure prediction and interpretation, as well as several others for the automatic assignment of spectra, database searching, and many other functions.
All tools mentioned here are freely accessible web-based applets, or can be obtained without requiring payment.
Conclusion
Analysis of glycosylation is often a major challenge due to the vast macro-heterogeneity of glycoproteins, i.e., a single protein can have a varying number of occupied N- and O-glycosylation sites. Additionally, a single site can be occupied by a variety of glycans, which is known as micro-heterogeneity [2]. Furthermore, the analysis of glycans is complicated by variations in linkage and branching patterns which may lead to multiple isomers.
LC has long been the gold standard for analysis of fluorescently labeled glycans, providing good sensitivity, isomer separation, accurate glycan quantitation, repeatability, and in-depth characterization when supported by exoglycosidase sequencing. Downsides, however, are low sample throughput and relatively high cost per sample when comparing to, for example, MS-based methods of analysis [52]. MS on the other hand, provides good sensitivity, structural elucidation options via MSn experiments, good sensitivity, as well as very high throughput in case of MALDI ionization. Downsides here are the limitations in isomer separation and quantitation, and instability of labile groups when no derivatization is performed [96]. CGE-LIF supports high sensitivity, high throughput when multiplexed, and low running costs especially when comparing to LC. Unfortunately, databases for CGE-LIF annotation are small and could be a limiting factor.
Hyphenation of glycomic LC and CE methods with online MS detection, optionally in combination with fluorescence detection of labeled glycans, may add confidence in glycan structural assignment as both migration position and (tandem) mass spectrometric information can be combined [97, 98]. However, it should be noted that certainly CE separation of glycans is often compromised by the restrictions faced with MS coupling. Further limitations in LC and CE hyphenation for glycomics are the relatively high costs due to medium sample throughput.
As demonstrated in this review, many of the analysis methods are moving towards increasingly HTP sample preparation, making use of multiwell formats, employing derivatizations with brief and manageable reaction times, integration of time-consuming steps and providing solutions for rapid analysis of the acquired data. Additionally, this progress towards simpler protocols provides the opportunity for liquid handling robots like the Hamilton and Bravo types to take over many of the time-consuming and labor- as well as cost-intensive steps. These automated platforms often show similar performance to expert manual labor, while even outperforming manual preparation when large cohorts are measured.
A common motif for HTP methodology is the inclusion of a solid support glycan binding or adsorption step. This can be used for purification, often in the form of covalent binding. When using chemical methods for capturing such as beads containing reactive hydrazide groups, this provides the opportunity for efficient labeling. This kind of integration is an excellent example of development towards shorter and easier protocols. Labeling itself is moving towards the use of heavy isotopes, to allow for reliable quantitation with mass spectrometry. The derivatization steps required for proper analysis of sialylated glycan species, notably permethylation and sialic acid-specific strategies, and several methods have been successful to date [99].
While mass spectrometry is one of the most HTP techniques possible for the analysis of glycans and glycoconjugates, it is hampered by its inability to provide quantitative data. This makes the various efforts undertaken for internal standardization, and thereby allowing quantitation, an exciting field to watch. The two main strategies for internal standardization are the use of heavy isotopic variants of glycans as well as the use of isobaric tags with unequal mass distribution. In both cases, the most convenient way to introduce mass differences is by employing derivatization steps already part of an analysis protocol, such as labeling or stabilization. A second strategy would be the spiking of a sample with a known concentration of a standard modified with a heavy isotope, which would also allow for the use of heavy isotopic variants of underivatized glycans. Convenient mass differences for internal standards are a few milliDalton showing parallel isotopic distributions on high resolution instruments, and onward from a few Dalton to prevent the overlapping of the natural istotopic distribution of glycans and glycopeptides.
With increasingly large studies being performed, the bottleneck of glycomics is shifting towards the data analysis. Current software for automation of analysis mainly focuses on interpretation rather than repeatable analysis of already interpreted and annotated spectra. There is a clear need for software allowing proper feature extraction, denoising, background subtraction, and most importantly of all, quality control both on individual glycans and complete spectra. Automated interpretation is mostly performed assisted by a predefined set of possibilities, or by matching experimental data to a database. While several programs exist to perform true unassisted de novo annotation, this approach has only shown successful on data that is of high quality. Particularly for hyphenated analysis methods such as LC–MS, manual annotation is still the way to go, and is a highly time consuming process.
Altogether, the methodological improvements discussed in this article show a clear trend towards more sensitive approaches, with faster analysis, lower costs and less hands-on time. These improvements allow for the increasing translation of glycomic approaches to clinical research, with envisioned applications in patient stratification and personalized medicine.
Acknowledgments
We would like to thank Louise Royle for expert assistance with the exoglycosidase digestion. This work was supported by the European Union (Seventh Framework Programme HighGlycan project, grant number 278535).
Conflict of interest
The authors declare no competing financial interest.
Contributor Information
Karli R. Reiding, Email: K.R.Reiding@lumc.nl
Manfred Wuhrer, Phone: +31-20-598-7527, Email: m.wuhrer@vu.nl.
References
- 1.Esko Jeffrey AV, Hudson F, Gerald H, Jamey Marth RC. Essentials of glycobiology. Essent Glycobiol. 1999;1:653. [Google Scholar]
- 2.Roth Z, Yehezkel G, Khalaila I. Identification and quantification of protein glycosylation. Int J Carbohydr Chem. 2012;2012:1–10. doi: 10.1155/2012/640923. [DOI] [Google Scholar]
- 3.Abès R, Teillaud J-L. Impact of glycosylation on effector functions of therapeutic IgG. Pharmaceuticals. 2010;3:146–158. doi: 10.3390/ph3010146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moremen KW, Tiemeyer M, Nairn AV. Vertebrate protein glycosylation: diversity, synthesis and function. Nat Rev Mol Cell Biol. 2012;13:448–462. doi: 10.1038/nrm3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arey BJ (2012) The role of glycosylation in receptor signaling. http://cdn.intechopen.com/pdfs-wm/39461.pdf. Accessed 31 Oct 2014
- 6.Fernandes DL. Systematic approach to optimization and comparability of biopharmaceutical glycosylation throughout the drug life cycle. Biopharm Prod Technol. 2012;1–2:545–583. doi: 10.1002/9783527653096.ch17. [DOI] [Google Scholar]
- 7.Fernandes DL (2008) A QbD approach to biopharmaceutical glycosylation. In: Lyscom N, (ed) Quality for biologics—critical quality attributes, process and change control, product variation, characterisation, and regulatory concerns. Biopharm Knowledge Publishing, Hampshire, UK
- 8.CMC Biotech Working Group (2009) A-Mab: a case study in bioprocess development. http://www.ispe.org/pqli/a-mab-case-studyversion-2.1. Accessed 31 Oct 2014
- 9.Dube DH, Bertozzi CR. Glycans in cancer and inflammation–potential for therapeutics and diagnostics. Nat Rev Drug Discov. 2005;4:477–488. doi: 10.1038/nrd1751. [DOI] [PubMed] [Google Scholar]
- 10.Dennis JW, Granovsky M, Warren CE. Protein glycosylation in development and disease. BioEssays. 1999;21:412–421. doi: 10.1002/(SICI)1521-1878(199905)21:5<412::AID-BIES8>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 11.Theodoratou E, Campbell H, Ventham NT, et al. The role of glycosylation in IBD. Nat Rev Gastroenterol Hepatol. 2014 doi: 10.1038/nrgastro.2014.78. [DOI] [PubMed] [Google Scholar]
- 12.Kapur R, Della Valle L, Sonneveld M, et al. Low anti-RhD IgG-Fc-fucosylation in pregnancy: a new variable predicting severity in haemolytic disease of the fetus and newborn. Br J Haematol. 2014 doi: 10.1111/bjh.12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Albrecht S, Unwin L, Muniyappa M, Rudd PM. Glycosylation as a marker for inflammatory arthritis. Cancer Biomark. 2014;14:17–28. doi: 10.3233/CBM-130373. [DOI] [PubMed] [Google Scholar]
- 14.Glycopro HI, Stockmann H, Adamczyk B et al (2013) Automated, high-throughput IgG-antibody glycopro fi ling platform [DOI] [PubMed]
- 15.Adamczyk B, Tharmalingam T, Rudd PM. Glycans as cancer biomarkers. Biochim Biophys Acta Gen Subj. 2012;1820:1347–1353. doi: 10.1016/j.bbagen.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 16.David Walt (2012) Committee on assessing the importance and impact of glycomics and glycosciences board on chemical sciences and technology board on life sciences division on earth and life studies
- 17.Bortolotti F, De Paoli G, Tagliaro F. Carbohydrate-deficient transferrin (CDT) as a marker of alcohol abuse: a critical review of the literature 2001–2005. J Chromatogr B Analyt Technol Biomed Life Sci. 2006;841:96–109. doi: 10.1016/j.jchromb.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 18.Gish RG (2014) Early detection of hepatocellular carcinoma through surveillance using biomarkers. Gastroenterol Hepatol 10(2):121–123 [PMC free article] [PubMed]
- 19.Lauc G, Essafi A, Huffman JE, et al. Genomics meets glycomics-the first GWAS study of human N-Glycome identifies HNF1α as a master regulator of plasma protein fucosylation. PLoS Genet. 2010;6:e1001256. doi: 10.1371/journal.pgen.1001256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Novokmet M, Lukić E, Vučković F, et al. Changes in IgG and total plasma protein glycomes in acute systemic inflammation. Sci Rep. 2014;4:4347. doi: 10.1038/srep04347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carlson DM, et al. Structures and immunochemical properties of oligosaccharides isolated from pig submaxillary mucins. J Biol Chem. 1968;243:616. [PubMed] [Google Scholar]
- 22.O’Neill R a. Enzymatic release of oligosaccharides from glycoproteins for chromatographic and electrophoretic analysis. J Chromatogr A. 1996;720:201–215. doi: 10.1016/0021-9673(95)00502-1. [DOI] [PubMed] [Google Scholar]
- 23.Geyer H, Geyer R. Strategies for analysis of glycoprotein glycosylation. Biochim Biophys Acta Proteins Proteomics. 2006;1764:1853–1869. doi: 10.1016/j.bbapap.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 24.Wada Y, Dell A, Haslam SM, et al. Comparison of methods for profiling O-glycosylation: human proteome organisation human disease glycomics/proteome initiative multi-institutional study of IgA1. Mol Cell Proteomics. 2010;9:719–727. doi: 10.1074/mcp.M900450-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zauner G, Kozak RP, Gardner Ra, et al. Protein O-glycosylation analysis. Biol Chem. 2012;393:687–708. doi: 10.1515/hsz-2012-0144. [DOI] [PubMed] [Google Scholar]
- 26.Royle L, Campbell MP, Radcliffe CM, et al. HPLC-based analysis of serum N-glycans on a 96-well plate platform with dedicated database software. Anal Biochem. 2008;376:1–12. doi: 10.1016/j.ab.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 27.Cook KS, Bullock K, Sullivan T. Development and qualification of an antibody rapid deglycosylation method. Biologicals. 2012;40:109–117. doi: 10.1016/j.biologicals.2011.12.010. [DOI] [PubMed] [Google Scholar]
- 28.Burnina I, Hoyt E, Lynaugh H, et al. A cost-effective plate-based sample preparation for antibody N-glycan analysis. J Chromatogr A. 2013;1307:201–206. doi: 10.1016/j.chroma.2013.07.104. [DOI] [PubMed] [Google Scholar]
- 29.Callewaert N, Geysens S, Molemans F, Contreras R. Ultrasensitive profiling and sequencing of N-linked oligosaccharides using standard DNA-sequencing equipment. Glycobiology. 2001;11:275–281. doi: 10.1093/glycob/11.4.275. [DOI] [PubMed] [Google Scholar]
- 30.Callewaert N, Van Vlierberghe H, Van Hecke A, et al. Noninvasive diagnosis of liver cirrhosis using DNA sequencer–based total serum protein glycomics. Nat Med. 2004;10:429–434. doi: 10.1038/nm1006. [DOI] [PubMed] [Google Scholar]
- 31.Laroy W, Contreras R, Callewaert N. Glycome mapping on DNA sequencing equipment. Nat Protoc. 2006;1:397–405. doi: 10.1038/nprot.2006.60. [DOI] [PubMed] [Google Scholar]
- 32.Ruhaak LR, Huhn C, Borowiak M, et al. Optimized workflow for preparation of APTS-labeled N-glycans allowing high-throughput analysis of human plasma glycomes using 48-channel multiplexed CGE-LIF. J Proteome Res. 2010;12:6655–6664. doi: 10.1021/pr100802f. [DOI] [PubMed] [Google Scholar]
- 33.Ruhaak LR, Koeleman C a M, Uh H-W, et al. Targeted biomarker discovery by high throughput glycosylation profiling of human plasma alpha1-antitrypsin and immunoglobulin A. PLoS One. 2013;8:e73082. doi: 10.1371/journal.pone.0073082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Váradi C, Lew C, Guttman A. Rapid magnetic bead based sample preparation for automated and high throughput N-glycan analysis of therapeutic antibodies. Anal Chem. 2014;86:5682–5687. doi: 10.1021/ac501573g. [DOI] [PubMed] [Google Scholar]
- 35.Harvey DJ. Derivatization of carbohydrates for analysis by chromatography; electrophoresis and mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2011;879:1196–1225. doi: 10.1016/j.jchromb.2010.11.010. [DOI] [PubMed] [Google Scholar]
- 36.Gil G-C, Iliff B, Cerny R, et al. High throughput quantification of N-glycans using one-pot sialic acid modification and matrix assisted laser desorption ionization time-of-flight mass spectrometry. Anal Chem. 2010;82:6613–6620. doi: 10.1021/ac1011377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jeong H-J, Kim Y-G, Yang Y-H, Kim B-G. High-throughput quantitative analysis of total N-glycans by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Anal Chem. 2012;84:3453–3460. doi: 10.1021/ac203440c. [DOI] [PubMed] [Google Scholar]
- 38.Miura Y, Shinohara Y, Furukawa J, et al. Rapid and simple solid-phase esterification of sialic acid residues for quantitative glycomics by mass spectrometry. Chemistry. 2007;13:4797–4804. doi: 10.1002/chem.200601872. [DOI] [PubMed] [Google Scholar]
- 39.Liu X, Qiu H, Lee RK, et al. Methylamidation for sialoglycomics by MALDI–MS: a facile derivatization strategy for both α2,3- and α2,6-linked sialic acids. Anal Chem. 2010;82:8300–8306. doi: 10.1021/ac101831t. [DOI] [PubMed] [Google Scholar]
- 40.Wheeler SF, Domann P, Harvey DJ (2009) Derivatization of sialic acids for stabilization in matrix-assisted laser desorption/ionization mass spectrometry and concomitant differentiation of a (2⟶3)- and a (2⟶6)-isomers. Rapid Commun Mass Spectrom 23(2):303–312. doi:10.1002/rcm.3867 [DOI] [PubMed]
- 41.Alley WR, Novotny MV. Glycomic analysis of sialic acid linkages in glycans derived from blood serum glycoproteins research articles. J Proteome Res. 2010;9:3062–3072. doi: 10.1021/pr901210r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tousi F, Bones J, Hancock WS, Hincapie M. Differential chemical derivatization integrated with chromatographic separation for analysis of isomeric sialylated N-glycans: a nano-hydrophilic interaction liquid chromatography-MS platform. Anal Chem. 2013;85:8421–8428. doi: 10.1021/ac4018007. [DOI] [PubMed] [Google Scholar]
- 43.Reiding KR, Blank D, Kuijper DM et al (2014) High-throughput profiling of protein N-glycosylation by MALDI–TOF–MS employing linkage-specific sialic acid esterification. Anal Chem 86(12):5784–5793. doi:10.1021/ac500335t [DOI] [PubMed]
- 44.Selman MHJ, Derks RJE, Bondt A, et al. Fc specific IgG glycosylation profiling by robust nano-reverse phase HPLC–MS using a sheath-flow ESI sprayer interface. J Proteomics. 2012;75:1318–1329. doi: 10.1016/j.jprot.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 45.Nishikaze T, Kawabata S, Tanaka K. In-depth structural characterization of N-linked glycopeptides using complete derivatization for carboxyl groups followed by positive- and negative-ion tandem mass spectrometry. Anal Chem. 2014;86:5360–5369. doi: 10.1021/ac500340t. [DOI] [PubMed] [Google Scholar]
- 46.Amano J, Nishikaze T, Tougasaki F, et al. Derivatization with 1-pyrenyldiazomethane enhances ionization of glycopeptides but not peptides in matrix-assisted laser desorption/ionization mass spectrometry. Anal Chem. 2010;82:8738–8743. doi: 10.1021/ac101555a. [DOI] [PubMed] [Google Scholar]
- 47.Nishikaze T, Nakamura T, Jinmei H, Amano J. Negative-ion MALDI–MS2 for discrimination of α2,3- and α2,6-sialylation on glycopeptides labeled with a pyrene derivative. J Chromatogr B Analyt Technol Biomed Life Sci. 2011;879:1419–1428. doi: 10.1016/j.jchromb.2010.10.032. [DOI] [PubMed] [Google Scholar]
- 48.Selman MHJ, McDonnell La, Palmblad M, et al. Immunoglobulin G glycopeptide profiling by matrix-assisted laser desorption ionization Fourier transform ion cyclotron resonance mass spectrometry. Anal Chem. 2010;82:1073–1081. doi: 10.1021/ac9024413. [DOI] [PubMed] [Google Scholar]
- 49.O’Connor PB, Budnik B a, Ivleva VB, et al. A high pressure matrix-assisted laser desorption ion source for Fourier transform mass spectrometry designed to accommodate large targets with diverse surfaces. J Am Soc Mass Spectrom. 2004;15:128–132. doi: 10.1016/j.jasms.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 50.Bondt A, Selman MHJ, Deelder AM, et al. Association between galactosylation of immunoglobulin G and improvement of rheumatoid arthritis during pregnancy is independent of sialylation. J Proteome Res. 2013;12:4522–4531. doi: 10.1021/pr400589m. [DOI] [PubMed] [Google Scholar]
- 51.Rombouts Y, Ewing E, van de Stadt LA, et al. Anti-citrullinated protein antibodies acquire a pro-inflammatory Fc glycosylation phenotype prior to the onset of rheumatoid arthritis. Ann Rheum Dis. 2013 doi: 10.1136/annrheumdis-2013-203565. [DOI] [PubMed] [Google Scholar]
- 52.Huffman JE, Pučić-Baković M, Klarić L, et al. Comparative performance of four methods for high-throughput glycosylation analysis of immunoglobulin G in genetic and epidemiological research. Mol Cell Proteomics. 2014;13:1598–1610. doi: 10.1074/mcp.M113.037465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hong Q, Lebrilla CB, Miyamoto S, Ruhaak LR (2013) Absolute quantitation of immunoglobulin G and its glycoforms using multiple reaction monitoring [DOI] [PMC free article] [PubMed]
- 54.Song E, Pyreddy S, Mechref Y. Quantification of glycopeptides by multiple reaction monitoring liquid chromatography/tandem mass spectrometry. Rapid Commun Mass Spectrom. 2012;26:1941–1954. doi: 10.1002/rcm.6290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hong Q, Ruhaak LR, Totten SM et al (2015) Label-free absolute quantitation of oligosaccharides using multiple reaction monitoring [DOI] [PMC free article] [PubMed]
- 56.Mao W, Thanawiroon C, Linhardt RJ. Capillary electrophoresis for the analysis of glycosaminoglycans and glycosaminoglycan-derived oligosaccharides. Biomed Chromatogr. 2002;16:77–94. doi: 10.1002/bmc.153. [DOI] [PubMed] [Google Scholar]
- 57.Domann PJ, Pardos-Pardos AC, Fernandes DL, et al. Separation-based glycoprofiling approaches using fluorescent labels. Proteomics. 2007;7(Suppl 1):70–76. doi: 10.1002/pmic.200700640. [DOI] [PubMed] [Google Scholar]
- 58.Prien JM, Prater BD, Qin Q, Cockrill SL. Mass spectrometric-based stable isotopic 2-aminobenzoic acid glycan mapping for rapid glycan screening of biotherapeutics. Anal Chem. 2010;82:1498–1508. doi: 10.1021/ac902617t. [DOI] [PubMed] [Google Scholar]
- 59.Walker SH, Budhathoki-Uprety J, Novak BM, Muddiman DC. Stable-isotope labeled hydrophobic hydrazide reagents for the relative quantification of N-linked glycans by electrospray ionization mass spectrometry. Anal Chem. 2011;83:6738–6745. doi: 10.1021/ac201376q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang P, Zhang Y, Xue X, et al. Relative quantitation of glycans using stable isotopic labels 1-(d0/d5) phenyl-3-methyl-5-pyrazolone by mass spectrometry. Anal Biochem. 2011;418:1–9. doi: 10.1016/j.ab.2011.07.006. [DOI] [PubMed] [Google Scholar]
- 61.Ahn J-M, Sung H-J, Yoon Y-H, et al. Integrated glycoproteomics demonstrates fucosylated serum paraoxonase 1 alterations in small cell lung cancer. Mol Cell Proteomics. 2014;13:30–48. doi: 10.1074/mcp.M113.028621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gong B, Hoyt E, Lynaugh H, et al. N-glycosylamine-mediated isotope labeling for mass spectrometry-based quantitative analysis of N-linked glycans. Anal Bioanal Chem. 2013;405:5825–5831. doi: 10.1007/s00216-013-6988-9. [DOI] [PubMed] [Google Scholar]
- 63.Alvarez-Manilla G, Warren NL, Abney T, et al. Tools for glycomics: relative quantitation of glycans by isotopic permethylation using 13CH3I. Glycobiology. 2007;17:677–687. doi: 10.1093/glycob/cwm033. [DOI] [PubMed] [Google Scholar]
- 64.Iii JAA, Cheng L, Alvarez-manilla G et al (2008) Quantitation by isobaric labeling: applications to glycomics research articles. 367–374 [DOI] [PubMed]
- 65.Miura Y, Hato M, Shinohara Y, et al. BlotGlycoABCTM, an integrated glycoblotting technique for rapid and large scale clinical glycomics. Mol Cell Proteomics. 2008;7:370–377. doi: 10.1074/mcp.M700377-MCP200. [DOI] [PubMed] [Google Scholar]
- 66.Furukawa J, Shinohara Y, Kuramoto H, et al. Comprehensive approach to structural and functional glycomics based on chemoselective glycoblotting and sequential tag conversion. Anal Chem. 2008;80:1094–1101. doi: 10.1021/ac702124d. [DOI] [PubMed] [Google Scholar]
- 67.Bynum M a, Yin H, Felts K, et al. Characterization of IgG N-glycans employing a microfluidic chip that integrates glycan cleavage, sample purification, LC separation, and MS detection. Anal Chem. 2009;81:8818–8825. doi: 10.1021/ac901326u. [DOI] [PubMed] [Google Scholar]
- 68.Trojer L, Gromadski K, van de Goor T et al. (2011) The mAb-Glyco Chip Kit—a workflow solution for rapid and fully automated characterisation of N-linked glycans from monoclonal antibodies. Chromatography today 4(3). http://www.chromatographytoday.com/article_read/1066/. Accessed 31 Oct 2014
- 69.Caliper LifeSciences (2010) Rapid analysis of N-glycans on the LabChip GXII Microchip-CE platform. Application note 403. http://www.level.com.tw/html/ezcatfiles/vipweb20/img/img/52857/(4)LCGX-Nglycan.pdf. Accessed 31 Oct 2014
- 70.Doherty M, Bones J, McLoughlin N, et al. An automated robotic platform for rapid profiling oligosaccharide analysis of monoclonal antibodies directly from cell culture. Anal Biochem. 2013;442:10–18. doi: 10.1016/j.ab.2013.07.005. [DOI] [PubMed] [Google Scholar]
- 71.Russell JD, Van Den Heuvel Z, Bovee M et al (2013) Automation for LC/MS sample preparation: high throughput in-solution digestion and peptide cleanup enabled by the agilent AssayMAP Bravo Platform. Agilent Technologies, Inc., USA. http://www.chem.agilent.com/Library/applications/5991-2957EN.pdf. Accessed 31 Oct 2014
- 72.Murphy S, Bovee M, van den Hevel Z et al (2013) Characterize N-glycans using a new quantitative, automated sample preparation platform. Agilent Technologies poster. http://www.chem.agilent.com/Library/posters/Public/WCBP2013-GlykoPrep%20Poster%202013-01-26.pdf. Accessed 31 Oct 2014
- 73. Hamilton. N-glycan characterisation of biopharmaceuticals: simplifying sample preparation with the Microlab STARlet
- 74.Reusch D, Haberger M, Selman MHJ, et al. High-throughput work flow for IgG Fc-glycosylation analysis of biotechnological samples. Anal Biochem. 2013;432:82–89. doi: 10.1016/j.ab.2012.09.032. [DOI] [PubMed] [Google Scholar]
- 75.Ruhaak LR, Uh H-W, Beekman M, et al. Plasma protein N-glycan profiles are associated with calendar age, familial longevity and health. J Proteome Res. 2011;10:1667–1674. doi: 10.1021/pr1009959. [DOI] [PubMed] [Google Scholar]
- 76.Reiding KR, Blank D, Kuijper DM et al (2014) High-throughput profiling of protein N-glycosylation by MALDI–TOF–MS employing linkage-specific sialic acid esterification [DOI] [PubMed]
- 77.Woodin CL, Hua D, Maxon M, et al. GlycoPep grader: a web-based utility for assigning the composition of N-linked glycopeptides. Anal Chem. 2012;84:4821–4829. doi: 10.1021/ac300393t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dallas DC, Martin WF, Hua S, German JB. Automated glycopeptide analysis—review of current state and future directions. Brief Bioinform. 2013;14:361–374. doi: 10.1093/bib/bbs045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Maass K, Ranzinger R, Geyer H, et al. “Glyco-peakfinder”–de novo composition analysis of glycoconjugates. Proteomics. 2007;7:4435–4444. doi: 10.1002/pmic.200700253. [DOI] [PubMed] [Google Scholar]
- 80.Deshpande N, Jensen PH, Packer NH et al (2010) GlycoSpectrumScan: fishing glycopeptides from MS spectra of protease digests of human colostrum sIgA. J Proteome Res 9(2):1063–1075. doi:10.1021/pr900956x [DOI] [PubMed]
- 81.Vakhrushev SY, Dadimov D, Peter-Katalinić J. Software platform for high-throughput glycomics. Anal Chem. 2009;81:3252–3260. doi: 10.1021/ac802408f. [DOI] [PubMed] [Google Scholar]
- 82.Pollreisz F (2008) GlycoMiner: a new software tool to elucidate glycopeptide composition. 3245–3254. doi: 10.1002/rcm [DOI] [PubMed]
- 83.Wu Y, Mechref Y, Klouckova I et al (2010) Mapping site-specific protein N-glycosylations through liquid chromatography/mass spectrometry and targeted tandem mass spectrometry. Rapid Commun Mass Spectrom 24(7):965–972. doi: 10.1002/rcm.4474 [DOI] [PubMed]
- 84.Pompach P, Chandler KB, Lan R, et al. Semi-automated identification of N-Glycopeptides by hydrophilic interaction chromatography, nano-reverse-phase LC–MS/MS, and glycan database search. J Proteome Res. 2012;11:1728–1740. doi: 10.1021/pr201183w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lohmann KK, von der Lieth C-W. GlycoFragment and GlycoSearchMS: web tools to support the interpretation of mass spectra of complex carbohydrates. Nucleic Acids Res. 2004;32:W261–W266. doi: 10.1093/nar/gkh392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Go EP, Rebecchi KR, Dalpathado DS et al (2007) GlycoPep DB : a tool for glycopeptide analysis using a “Smart Search”. Anal Chem 79(4):1708–1713 [DOI] [PubMed]
- 87.Artemenko NV, Campbell MP, Rudd PM. GlycoExtractor: a web-based interface for high throughput processing of HPLC-glycan data. J Proteome Res. 2010;9:2037–2041. doi: 10.1021/pr901213u. [DOI] [PubMed] [Google Scholar]
- 88.Campbell MP, Royle L, Radcliffe CM, et al. GlycoBase and autoGU: tools for HPLC-based glycan analysis. Bioinformatics. 2008;24:1214–1216. doi: 10.1093/bioinformatics/btn090. [DOI] [PubMed] [Google Scholar]
- 89.Hizal DB, Wolozny D, Colao J, et al. Glycoproteomic and glycomic databases. Clin. Proteomics. 2014;11:1–10. doi: 10.1186/1559-0275-11-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Campbell MP, Ranzinger R, Lütteke T, et al. Toolboxes for a standardised and systematic study of glycans. BMC Bioinform. 2014;15(Suppl 1):S9. doi: 10.1186/1471-2105-15-S1-S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ranzinger R, Herget S, von der Lieth C-W, Frank M. GlycomeDB—a unified database for carbohydrate structures. Nucleic Acids Res. 2011;39:D373–D376. doi: 10.1093/nar/gkq1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Campbell MP, Peterson R, Mariethoz J, et al. UniCarbKB: building a knowledge platform for glycoproteomics. Nucleic Acids Res. 2014;42:D215–D221. doi: 10.1093/nar/gkt1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Damerell D, Ceroni A, Maass K, et al. The GlycanBuilder and GlycoWorkbench glycoinformatics tools: updates and new developments. Biol Chem. 2012;393:1357–1362. doi: 10.1515/hsz-2012-0135. [DOI] [PubMed] [Google Scholar]
- 94.Akune Y, Hosoda M, Kaiya S, et al. The RINGS resource for glycome informatics analysis and data mining on the Web. OMICS. 2010;14:475–486. doi: 10.1089/omi.2009.0129. [DOI] [PubMed] [Google Scholar]
- 95.Ceroni A, Maass K, Geyer H et al (2008) GlycoWorkbench: a tool for the computer-assisted annotation of mass spectra of glycans. J Proteome Res(4):1650–1659. doi:10.1021/pr7008252 [DOI] [PubMed]
- 96.Alley WR, Mann BF, Novotny MV (2013) High-sensitivity analytical approaches for the structural characterization of glycoproteins. Chem Rev 113(4):2668–2732. doi:10.1021/cr3003714 [DOI] [PMC free article] [PubMed]
- 97.Pabst M, Bondili JS, Stadlmann J, et al. Mass + retention time = structure: a strategy for the analysis of N-glycans by carbon LC-ESI-MS and its application to fibrin N-glycans. Anal Chem. 2007;79:5051–5057. doi: 10.1021/ac070363i. [DOI] [PubMed] [Google Scholar]
- 98.Zhao SS, Zhong X, Tie C, Chen DDY. Capillary electrophoresis-mass spectrometry for analysis of complex samples. Proteomics. 2012;12:2991–3012. doi: 10.1002/pmic.201200221. [DOI] [PubMed] [Google Scholar]
- 99.Alley WR, Novotny MV. Structural glycomic analyses at high sensitivity: a decade of progress. Annu Rev Anal Chem (Palo Alto Calif) 2013;6:237–265. doi: 10.1146/annurev-anchem-062012-092609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ahn J, Bones J, Yu YQ, et al. Separation of 2-aminobenzamide labeled glycans using hydrophilic interaction chromatography columns packed with 1.7 microm sorbent. J Chromatogr B Analyt Technol Biomed Life Sci. 2010;878:403–408. doi: 10.1016/j.jchromb.2009.12.013. [DOI] [PubMed] [Google Scholar]
- 101.Fernandes DL (2009) Turning to colour. Samedan Ltd. http://www.ludger.com/docs/articles/EBR-Glycan-Labelling-Summer-2009.pdf. Accessed 31 Oct 2014
- 102.Tharmalingam T, Adamczyk B, Doherty M a, et al. Strategies for the profiling, characterisation and detailed structural analysis of N-linked oligosaccharides. Glycoconj J. 2013;30:137–146. doi: 10.1007/s10719-012-9443-9. [DOI] [PubMed] [Google Scholar]
- 103.Lauc G, Huffman JE, Pučić M, et al. Loci associated with N-glycosylation of human immunoglobulin G show pleiotropy with autoimmune diseases and haematological cancers. PLoS Genet. 2013;9:e1003225. doi: 10.1371/journal.pgen.1003225. [DOI] [PMC free article] [PubMed] [Google Scholar]