

Abstract

Ferryl porphyrins, P−FeIV=O, are central reactive intermediates in the catalytic cycles of numerous heme proteins and a variety of model systems. There has been considerable interest in elucidating factors, such as terminal oxo basicity, that may control ferryl reactivity. Here, the sulfonated, water-soluble ferryl porphyrin complexes tetramesitylporphyrin, oxoFeIVTMPS (FeTMPS-II), its 2,6-dichlorophenyl analogue, oxoFeIVTDClPS (FeTDClPS-II), and two other analogues are shown to be protonated under turnover conditions to produce the corresponding bis-aqua-iron(III) porphyrin cation radicals. The results reveal a novel internal electromeric equilibrium, P−FeIV=O ⇆ P+−FeIII(OH2)2. Reversible pKa values in the range of 4–6.3 have been measured for this process by pH-jump, UV–vis spectroscopy. Ferryl protonation has important ramifications for C–H bond cleavage reactions mediated by oxoiron(IV) porphyrin cation radicals in protic media. Both solvent O–H and substrate C–H deuterium kinetic isotope effects are observed for these reactions, indicating that hydrocarbon oxidation by these oxoiron(IV) porphyrin cation radicals occurs via a solvent proton-coupled hydrogen atom transfer from the substrate that has not been previously described. The effective FeO–H bond dissociation energies for FeTMPS-II and FeTDClPS-II were estimated from similar kinetic reactivities of the corresponding oxoFeIVTMPS+ and oxoFeIVTDClPS+ species to be ~92–94 kcal/mol. Similar values were calculated from the two-proton P+−FeIII(OH2)2 pKaobs and the porphyrin oxidation potentials, despite a 230 mV range for the iron porphyrins examined. Thus, the iron porphyrin with the lower ring oxidation potential has a compensating higher basicity of the ferryl oxygen. The solvent-derived proton adds significantly to the driving force for C–H bond scission.
INTRODUCTION
Ferryl porphyrin species, P+−FeIV=O and P−FeIV=O, play pivotal roles in biological oxidations mediated by heme proteins.1 These two reactive intermediates, historically referred to as compound I and compound II,2 respectively, differ in the oxidation state of the porphyrin ring (P). Among the heme enzymes, only those bearing an axial thiolate ligand, as in the cytochromes P450 (CYP)3 and aromatic peroxygenases (APO),4 have the ability to perform the most challenging oxidation, the oxy-functionalization of unactivated sp3 C–H bonds. The consensus hydroxylation mechanism of CYP enzymes involves a hydrogen atom abstraction from a substrate C–H bond by P+−FeIV=O (P450-I) to afford a thiolateiron(IV) hydroxide5 (P450-II) and a substrate radical. The carbon-centered radical formed in this process then rebounds to the FeIV−OH hydroxyl, forming the product alcohol and reducing the heme center back to the ferric resting state. The strength of the FeO–H bond formed upon hydrogen atom abstraction is of considerable importance in determining the ability of a metal-oxo compound to abstract a hydrogen atom from a substrate,6 as is the electronic configuration.7 This bond strength, D(OH), can be dissected into a one-electron reduction potential of P+−FeIV=O (E0cpd I) and the pKa of P−FeIVO–H (eq 1).8For example, we have estimated the reduction potential of APO-I to be ~1.4 V and the FeO–H
| (1) |
bond dissociation energy (BDE) of APO-II to be ~100 kcal/mol.4b,c Likewise, a highly reactive synthetic iron porphyrin, oxoFeIVTM-4-PyP+, also has a 100 kcal/mol FeO–H bond.9 A more basic FeO–H pKa would lead to a stronger FeIVO–H bond. Green et al. have suggested that the axial thiolate ligand of P450 hydroxylases is an innovation used to engineer more basic ferryl states while sacrificing some redox potential.5a The net result is a stronger FeIVO–H bond and the ability to oxidize unactivated aliphatic C–H bonds while protecting the protein from oxidative damage.5,10 Support for this hypothesis derives from the recent determination that P450-II has a pKa ≈ 12, existing as an iron(IV) hydroxide under turnover conditions.
While P450-II is protonated, there is still spirited debate concerning the ferryl states of heme enzymes lacking thiolate axial ligands. For example, while X-ray data have suggested an elongation of the FeIV−O bond in cytochrome c peroxidase compound II (CcP-II) and ascorbate oxidase compound II,11 that effect has also been attributed to photoreduction of the sample in the X-ray beam.12 A recent neutron diffraction study of CcP-I has shown that the ferryl is not protonated.13 Likewise, ferryl myoglobin resists protonation even below pH 3.14 There are no reports of basic ferryls in model porphyrin systems, although basic oxomanganese and non-heme iron complexes have been discussed,15 and dimethoxyiron(IV) porphyrin complexes are known.16
In this work we provide a direct measure of the basicity of sulfonated ferryl porphyrins (Figure 1) and the effects of ferryl basicity on their reactivity toward C–H bonds. Protonation of P−FeIV=O species using rapid-mixing, pH-jump, stopped-flow spectrophotometry reveals an electromeric equilibrium with measurable pKa values for oxoFeIV porphyrins and the corresponding iron(III) porphyrin cation radicals, P+−FeIII(OH2)2. We also present evidence that the observed pKa influences C–H bond scission by compound I models through synchronous solvent proton and substrate hydrogen atom transfer (HAT).
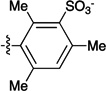
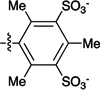
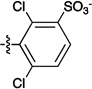
Figure 1.

Sulfonated ferryl porphyrins used in this study arranged, left to right in order of increasing electron-donation ability of the meso substituent, Ar. to right in order of increasing electron-donation ability of the meso substituent, Ar.
RESULTS
Iron tetramesitylporphyrin octasulfonate (FeTMPS) is a well-behaved, water-soluble iron porphyrin system. Previous work has shown that oxidation of FeIIITMPS with mCPBA or oxone above pH 7 affords oxoFeIVTMPS, λmax = 425–431 nm (pH dependent as shown in Figure S1); 1H NMR δ 3.1 (para-methyl), −1.2 (β-pyrrole).17 Similarly, when FeIIITMPS was treated with mCPBA at pH 5.0, a new intermediate was formed with a weak, blue-shifted λmax centered at 400 nm, a broad Q-band absorbance at ~650 nm, and strongly shifted 1H NMR resonances at δ 23, 25 (ortho-methyls) and δ −26 (β-pyrrole) (Figures S2 and S3). We have assigned this species as oxoiron(IV) TMPS porphyrin cation radical (oxoFeIVTMPS+) due to the closely analogous literature spectra of the tetramesityl derivative, oxoFeIVTMP+,18 and similar species.9,18,19 Electrochemical measurements with (H2O)2FeIII-TMPS showed a reversible, pH-independent oxidation wave at 940 mV vs Ag/AgCl between pH 2 and pH 4, while spectroelectrochemical oxidation at 1100 mV afforded the corresponding iron(III) porphyrin cation radical, FeIIITMPS+, with λmax = 390 nm and a broad Q-band feature (Figures S4 and S5).19d The UV–vis spectra of the various oxidation/protonation states of FeTMPS are compared in Figure 2. Oxidation of FeIIITDClPS with 1 equiv of mCPBA at pH 3.0 also produced a compound I analogue, oxoFeIVTDClPS+, with a similar, characteristic UV–vis spectrum (Figure S6).
Figure 2.

UV–vis spectra of various oxidation states of FeTMPS: FeIIITMPS, λmax = 397 nm (green); oxoFeIVTMPS, λmax = 425 nm at pH 8 (blue); FeIIITMPS+, λmax = 390 nm (red); and oxoFeIVTMPS+, λmax = 400 nm (black).
The interconversion of oxoFeIVTMPS and FeIIITMPS+ was investigated using pH-jump, stopped-flow experiments. OxoFeIVTMPS was formed via the stoichiometric addition of mCPBA to an unbuffered solution of FeIIITMPS adjusted to pH 11.5. The position of the Soret λmax shifted upon mixing from a broad absorbance at 416 nm for (HO)FeIIITMPS to a sharper, red-shifted band at 425 nm for oxoFeIVTMPS.17a The stability of oxoFeIVTMPS at high pH allowed for bulk generation of this species for use in single-mixing, pH-jump, stopped-flow experiments. Significantly, jumping the pH from 11.5 to <4 cleanly converted oxoFeIVTMPS to FeIIITMPS+, while jumping the pH to values >4 led to mixtures of oxoFeIVTMPS and FeIIITMPS+ (Figure 3A). A clear titration curve was revealed when the absorbance of oxoFeIVTMPS at 425 nm observed within 10 ms of mixing was plotted against the pH of the final mixed solution (Figure 3B, black line). Fitting these data yielded an apparent pKa = 5.5 ± 0.07. Over longer periods, the mixture of oxoFeIVTMPS and FeIIITMPS+ disproportionated to form oxoFeIVTMPS+ and FeIIITMPS. The identities of the species present after disproportionation were determined via generation of authentic oxoFeIVTMPS+, as shown in Figures 2 and S2. Similar measurements for oxoFeIVTDPS, oxo-FeIVTSMP,19c,d and oxoFeIVTDClPS (Figures S7 and S8) and the corresponding iron(III) porphyrin cation radicals gave a range of pKa values from 4.0 to 6.3 (Figures 3B and S9−S11). These systems were chosen for their resistance to forming µ-oxo FeIII dimers that could complicate the analyses.20 Reverse pH-jump experiments of acidic solutions of FeIIITMPS+ to higher pH produced oxoFeIVTMPS (Figure 4 inset). In this reaction sequence, FeIIITMPS at pH 2.5 was oxidized with 1 equiv of mCPBA to oxoFeIVTMPS+, which quickly converted to FeIIITMPS+ (λmax = 390 nm) under these conditions. Jumping the pH to ~13 produced oxo-hydroxoFeIVTMPS (λmax = 431 nm). The pH dependence of the UV–vis spectra of oxoFeIVTMPS under highly alkaline conditions indicates the presence of a second pKa = 12.5 ± 0.06 with the formation of HO−FeIV=O (Figure S1), as has been observed for other ferryl porphyrins.21
Figure 3.

(A) UV–vis spectra observed within 50 ms after rapid dilution of oxoFeIVTMPS (10 µM, pH 11.5) with 50 mM phosphate/acetate buffer at the target pH: oxoFeIVTMPS at pH 8.2 (red trace), FeIIITMPS+ at pH 4.7 (black trace), and both species at pH 5.4 (blue trace). (B) Plots of oxoFeIVporphyrin absorbance vs pH for oxoFeIVTDClPS (green trace, pKa = 4.0 ± 0.05), oxoFeIVTMPS (black trace, pKa = 5.5 ± 0.07), oxoFeIVTSMP (blue trace, pKa = 5.9 ± 0.04), and oxoFeIVTDPS (red trace, pKa = 6.3 ± 0.06).
Figure 4.

Reduction of oxoFeIVTMPS+ with TEMPO at various pH values (10 µM porphyrin in 50 mM phosphate/acetate buffer). Inset: Conversion of (H2O)2FeIIITMPS+ (10 µM) in pH 2.5 buffer (100 mM acetate) at 5 °C to oxoFeIVTMPS at pH 13 upon bulk addition of NaOH. All concentrations and pH measurements are final after mixing.
The same equilibrium mixtures of oxoFeIVTMPS and FeIIITMPS+ electromers could be generated via one-electron reduction of oxoFeIVTMPS+. As shown in Scheme 1, oxoFeIV-TMPS+ was generated via stoichiometric oxidation of FeIII-TMPS with mCPBA in the first push of a double-mixing, stopped-flow experiment. After an aging delay of 1 s, 1 equiv of TEMPO in buffer at the target pH was added as a reducing agent in the second push.22 The reduction of oxoFeIVTMPS+ by TEMPO was complete within the dead time of the measurement (~1 ms). By contrast, further reduction of oxoFeIVTMPS to FeIIITMPS under these conditions occurred much more slowly (k ≈ 104 M−1 s−1, Figure S12). Reduction of oxoFeIVTMPS+ below pH 6.5 afforded mixtures of oxoFeIV-TMPS and FeIIITMPS+, with greater amounts of the latter at lower pH, in accord with a prototropic equilibrium with a pKa = 5.5 (Figure 4A). A summary of spectral data, porphyrin electrochemistry, and measured pKa values for all porphyrins is presented in Table 1.
Scheme 1.

Design of a Double-Mixing, Stopped-Flow Experiment in Which OxoFeIVTMPS+ Was Generated Transiently at pH 5.0 (Unbuffered), pH-Jumped, and Reduced with TEMPO To Yield Mixtures of OxoFeIVTMPS and (H2O)2FeIIITMPS+
Table 1.
Collected Parameters for the Fe Porphyrin Model Systems Used in This Study
| FeIIITDPS | FeIIITSMP | FeIIITMPS | FeIIITDCIPS | |
|---|---|---|---|---|
| meso substituent Ar = | 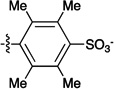 |
 |
 |
 |
| pKa P-FeIII(OH2)2 | 6.65 (phos/acetate) | 6.619d,19e (unbuffered) | 7.5 (phos/acetate)17 6.8 (HEPES, CHES, MES) | 7.85 (unbuffered)20,23 |
| pKa P+FeIII(OH2)2/P-FeIV=O | pKaobs= 6.3 | pKaobs= 5.9 | pKaobs= 5.5 | pKaobs= 4.0 |
| E1/2 (vs. Ag/AgCl) ZnP/ZnP+ | 558 mV (pH 4.0) | 630 mV (pH 4.0)19e | 596 mV (pH 4.0) | 790 mV (pH 4.0) |
| oxoFeIVP UV-vis (pH 11.5) | 423 nm | 424 nm19d | 425 nm17 | 420 nm20,23 |
| (H2O)2FeIIIP+UV-vis | 394 nm | 388 nm19d | 390 nm17b | 387 nm |
Both oxoFeIVTMPS+ and oxoFeIVTDClPS+ were reduced to the ferric form by dihydroanthracene (DHA). Bimolecular rate constants for DHA oxidation by oxoFeIVTMPS+ and oxoFeIV-TDClPS+, determined at pH 5 and pH 3, respectively, in single-turnover, stopped-flow experiments were 1023 ± 53 and 5015 ± 170 M−1 s−1 (Figures S13 and S14). OxoFeIVTDClPS+ was shown to oxidize xanthene with a bimolecular rate constant of (2.47 ± 0.15) × 104 M−1 s−1 (R2 = 0.99), while for oxoFeIVTMPS+ background decay obscured this reaction. Use of xanthene-d2 resulted in a slower rate constant of (1.37 ± 0.18) × 104 M−1 s−1 (R2 = 0.95) and, accordingly, a substrate kinetic isotope effect (KIE) of 1.80. This reaction was also shown to have a solvent KIE by oxidizing proteo xanthene in buffer made with D2O (pD 3.0). The bimolecular oxidation rate constant in this case was (1.50 ± 0.11) × 104 M−1 s−1 (R2 = 0.99), yielding a solvent KIE of 1.65. Under identical conditions, xanthene-d2 in D2O afforded a bimolecular rate of (1.12 ± 0.09) × 104 M−1 s−1 (R2 = 0.98), indicating a combined substrate−solvent isotope effect of 2.2.
DISCUSSION
A unique feature of heme iron enzymes and iron porphyrin model systems is the non-innocence of the porphyrin ring. Iron(III)/iron(IV) and porphyrin ring redox potentials are strongly intermingled, with both processes occurring at ~1 V. The rapid pH-jump experiments we have described here show that the terminal oxo ligands of oxoiron(IV) porphyrin complexes can be protonated in mildly acidic media, exhibiting electromeric equilibria between the ferryl species, P−FeIV=O, and the corresponding bis-aqua-iron(III) porphyrin cation radicals, P+−FeIII(OH2)2 (Figure 1). Apparent pKa values from 4.0 to 6.3 were observed that vary with the redox potential of the porphyrin. Re-formation of P−FeIV=O upon deprotonation of P+−FeIII(OH2)2 with base has shown that this equilibrium is reversible. Further, one-electron reduction of the corresponding compound I analogues, P+−FeIV=O, with TEMPO over the same range of pH produced the same mixtures of electromers and the same pKa values. In the following discussion we analyze these interconversions and their implications for C–H oxidations mediated by these P+−FeIV=O species.
Analysis of the P−FeIV=O ⇆ P+−FeIII−OH2 Equilibrium
An important aspect of this study was differentiating among the various species involved. Fortunately, the UV–vis spectrum of FeIIITMPS+ is distinct from those of FeIIITMPS, oxoFeIVTMPS+, and oxoFeIVTMPS (Figure 2). In particular, FeIIITMPS+ exhibits a strong blue-shifted Soret band (λmax = 390 nm) and broad, elevated Q-bands that are hallmarks of a porphyrin cation radical.19d By contrast, the Soret band of oxoFeIVTMPS+ is much weaker and centered at 400 nm, while the broad cation radical Q-bands were centered at longer wavelengths (Figure 2, black trace). These spectral features were particularly useful in observing FeIIITMPS+ before the appearance of oxoFeIVTMPS+ via disproportionation. Electrochemical generation of FeIIITMPS+ from FeIIITMPS was shown to occur through a one-electron, no proton process from the lack of a pH dependence of the oxidation wave below pH 4. This observation indicates that the axial ligation of FeIIITMPS+ is bis-aqua, (H2O)2FeIIITMPS+, as is the starting FeIIITMPS.17b Similar behavior has been described by Su et al. for a closely related porphyrin, FeIIITSMP.19d
Rapid pH-jumping of preformed basic solutions of oxoFeIV-TMPS to lower pH revealed the instantaneous formation of (H2O)2FeIIITMPS+. When the pH was jumped to intermediate values, both oxoFeIVTMPS and (H2O)2 FeIIITMPS+ were observed in solution, with the ratio of these two species favoring oxoFeIVTMPS as the pH increased (Figure S15). The isosbestic nature of this change and singular value decomposition (SVD) analysis of the data indicate that the only species present in solution just after mixing are oxoFeIVTMPS and (H2O)2FeIIITMPS+. The rapid establishment of these mixtures (~1 ms) indicates that a fast and reversible equilibrium exists between them. A clear, pKaobs = 5.5 was observed for (H2O)2FeIIITMPS+ by plotting the ratio of species present vs the final pH (Figure 3).24 For comparison, the first pKa of the ferric derivative, (H2O)2FeIIITMPS, is 7.5 under these conditions,17b thus providing a measure of the effect of porphyrin ring oxidation of the acidity of the ligated water protons. Interestingly, DFT computations have predicted six relatively low-lying electromers of HRP compound II, of which the hydroxoFeIII and aquaFeIII porphyrin cation radicals were indicated as the most stable isomers.25 Two other pertinent examples of electromeric equilibria in iron(IV) porphyrin systems are apparent in CcP-II26 and bis-azido-iron(IV) porphyrins.27
One-electron reduction of oxoFeIVTMPS+ near the pKaobs of (H2O)2FeIIITMPS+ was explored as an alternative approach to the equilibrium mixture of oxoFeIVTMPS and (H2O)2FeIII-TMPS+. We found that the nitroxyl radical, TEMPO (E0 = 760 mV), reacted rapidly with oxoFeIVTMPS+ to provide the ferryl species, oxoFeIVTMPS.22 Thus, stopped-flow mixing of oxoFeIVTMPS+ with 1 equiv of TEMPO over a range of pH values resulted in the transient formation of mixtures of oxoFeIVTMPS and (H2O)2FeIIITMPS+ (Figure 4), as was observed with the pH-jump experiments above (Figure 3). Further reduction of oxoFeIVTMPS with TEMPO was found to occur at a much slower rate, thus allowing for the observation of both (H2O)2FeIIITMPS+ and oxoFeIVTMPS without an appreciable accumulation of the ferric porphyrin. The apparently instantaneous establishment of the equilibrium mixture via TEMPO reduction is another indication that the interconversion of (H2O)2FeIIITMPS+ and oxoFeIVTMPS is fast and reversible and that the product of reduction (which electromer or mixture of electromers) depends upon the pH. This reversibility was further confirmed by pH-jumping an acidic solution of (H2O)2FeIIITMPS+ to high pH and observing the clean formation of oxoFeIVTMPS.
To elucidate the effect of ligand electronics on the observed aquaFeIIIP+/oxoFeIV porphyrin pKaobs, we examined several other sulfonated iron porphyrins: oxoFeIVTSMP, oxoFeIV-TDClPS, and oxoFeIVTDPS. Electrochemical analysis of the zinc derivatives showed that the porphyrin ring oxidation potentials of these ligands spanned 230 mV (Table 1), allowing for examination of this parameter on the ferryl pKaobs. As was observed with oxoFeIVTMPS, rapid pH-jump experiments with each of these oxoFeIV porphyrins revealed an apparent pKaobs (Figure 3B). The most electron-donating durenyl sulfonate meso substituent produced the most basic pKaobs = 6.3 for (H2O)2FeIIITDPS+, while the most electron-withdrawing 2,6-dichlorophenyl sulfonate substituent led to the lowest pKaobs = 4 for (H2O)2FeIIITDClPS+. SVD and evolving factor analyses for each case indicated that only two species were present in significant concentrations in these equilibrium mixtures (Figures S17−S30). Each of these FeIII porphyrin cation radical complexes exhibited a broader, blue-shifted Soret λmax than that of the oxoFeIV porphyrin upon jumping to acidic pH regimes. Additionally, the Q-band regions of these new species were all typical of porphyrin cation radical spectra. In the case of oxoFeIVTSMP, the UV–vis spectrum of the protonated species matched that of electrochemically generated bis-aqua-FeIIITSMP porphyrin cation radical, which has been previously characterized.19d These results, coupled with the evidence that protonation of oxoFeIVTMPS produces (H2O)2FeIIITMPS+, indicate that the protonation of oxoFeIV porphyrin systems to their corresponding FeIII porphyrin cation radicals is a general phenomenon.
Comparing the reduction potential of the corresponding Zn porphyrin cation radicals to the (H2O)2FeIII pKaobs revealed an inverse relationship between these two parameters. The lower the redox potential of the porphyrin ring, the higher the observed ferryl pKaobs. This trend is important for several reasons. First, the (H2O)2FeIIIP+/ferryl pKaobs appears to be very sensitive to changes in the oxidation potential of the porphyrin ring. When comparing the two extremes of the series, (H2O)2FeIIITDClPS+ is more than 500-fold more acidic than (H2O)2FeIIITDPS+, while 230 mV separates the oxidation potentials of their porphyrin rings. Thus, stabilizing the bisaqua-FeIII porphyrin cation radical through a lower porphyrin redox potential shifts the PFeO-H2 pKaobs to a higher value, creating an effectively more basic ferryl oxygen.
Discussion of ferryl heme basicity has generally focused on the role of the trans-axial ligand (L) as it affects the L−FeIV−OH ⇆ L−FeIV=O equilibrium. Specifically, the ability of a cysteine thiolate to allow for basic ferryl hemes as a way for heme-thiolate proteins such as cytochrome P450 and APO to oxidize strong C–H bonds in the presence of an oxidizable protein superstructure.4b,c,5a,c Spectroscopic data indicate that the iron centers of CPO-II and P450-II exist as iron(IV) hydroxides.5a,c The differences in structure and pKa between the protonated CPO-II or P450-II and the protonated model systems used here can be attributed to the thiolate axial ligand in the active site. The anionic, strongly donating thiolate ligand is able to stabilize the iron(IV) center upon oxo protonation to a greater extent than the analogous aqua ligand present in FeTMPS-II,10 although a recent case involving an amidate oxyanion ligand apparently does not.28 The relative importance of various parameters, such as the nature of the anionic ligand, Coulombic effects, σ- and π-trans-axial ligand effects, and field effects of other charges in the active sites of these proteins, has yet to be evaluated. The axial ligand is only one factor in determining the basicity of oxoFeIV porphyrins, however. As can be seen from the results, ligand electronics represent another way to modulate ferryl basicity, which in this case corresponds to a two-proton Brønsted acidity of P+−FeIII(OH2)2 and the accompanying electromeric equilibrium. As such, it appears likely that the compound II of some heme enzymes lacking a thiolate axial ligand could be protonated.11a
An important aspect of the current study is the fast establishment of the electromeric equilibria determining the observed pKaobs values before the subsequent disproportionations. Disproportionation of iron(IV) porphyrins to oxoiron-(IV) porphyrin cation radicals and iron(III) species has long been known.16,19f The tetrasulfonated iron porphyrin, oxo-FeIVTSMP, has been reported to disproportionate to a mixture of oxoFeIVTSMP+ and FeIIITSMP in 4:1 methanol−water.19c While a similar disproportionation reaction was observed in the current study in aqueous buffer over a longer time regime (>50 ms), the electromeric equilibrium was clearly established prior to this. One explanation for the difference in observed behavior between the tetra- and octasulfonated systems may be the different solvents. When FeTMPS was oxidized in basic methanol, oxidation to dimethoxyFeIVTMPS was observed, with its distinctive UV–vis spectrum (Q-bands at 550 and 580 nm), rather than oxoFeIVTMPS (Figure S16).17b DimethoxyFeIV porphyrins have been shown by us to disproportionate to oxoFeIV porphyrin cation radicals and FeIII porphyrins upon addition of acid.16
Analysis of Solvent Proton-Coupled Substrate Hydrogen Atom Transfer
Now we consider the reduction of P+−FeIV=O via HAT, which can be seen as an alternative way to access the P−FeIV = O/P+−FeIII−OH2 mixtures. Overall, the process is similar to the one-electron reduction of P+−FeIV=O with TEMPO described above, except that the electron and one proton derive from the same C–H bond. The observed equilibrium between P−FeIV=O and P+−FeIII−OH2 (pKaobs = 5.5 for FeTMPS) allows an estimate of the effective FeO–H BDE for hydrogen abstraction by P+−FeIV=O and the construction of an energy landscape for this process. We propose a mechanism for C–H bond scission that involves a simultaneous transfer of a proton from the medium to the ferryl oxygen (Scheme 2).
Scheme 2. Proposed Mechanism for Solvent Proton-Assisted C–H Bond Scissiona.

aThe solvent proton is shown approaching the ferryl oxygen lone pair, while the substrate proton is engaging the ferryl π* SOMO orbital.
A significant aspect of the behavior of this system is that pKaobs represents a two-proton equilibrium. As mentioned above, SVD analysis of the titration data (Figures S17−S30) indicates that the main two components, P−FeIV=O and P+−FeIII−OH2, account for >98% of all species present. A small amount of disproportionation during the time of the measurement is expected from the observed kinetics that could account for the residual. There could also be ~1% of a monoprotonated intermediate, although no such species was observed. Accordingly, we adopt a two-state model, P−FeIV=O ⇆ P+−FeIII−OH2, with no other intermediates accumulating at any pH over the range of the measurements (Figure S18), simplifying the analysis, although interactions of P+−FeIII−OH2 with the phosphate buffer are certainly also occurring.24 We estimate that pKa1 for TMPS+−FeIII−OH2 should be at least 7. A value of 6.5 or less would significantly increase the amount of monoprotonated intermediates predicted at pH 5.5, which is not supported by the data or SVD analysis. This value also follows from the measured first pKa for the ferric complex, P−FeIII−OH2, which is 7.5 for FeTMPS under these conditions.17b Thus, pKa1 for P+−FeIII−OH2 should be similar. With pKa1 estimated to be ~7, and pKaobs measured at 5.5, we can calculate pKa2 of P+−FeIII−OH2 to be ~4. The ferryl O-protonation event and the resulting electromeric equilibrium between P−FeIV and P+−FeIII are contained in pKa2. This unusual situation for a dibasic acid (pKa2 < pKa1) will be discussed further below. Finally, we estimate E′ for P+−FeIV=O/P−FeIV=O to be 1.06 V (vs NHE) from the measured porphyrin ring oxidation for FeTSMP of 1.1 V under these conditions and the very clear ring oxidations observed for the zinc porphyrin analogues (Table 1).19d,e Similar analyses were performed for each iron porphyrin.
The results show that the effective ferryl basicity reflected in the P−FeIV=O ⇆ P+−FeIII−OH2 equilibrium has a measurable effect on reactivity in this aqueous model system. OxoFeIVTMPS+ and oxoFeIVTDClPS+ react with DHA with similar rate constants, 1075 ± 52 and 5015 ± 170 M−1 s−1, respectively. Estimates of the effective FeO-H BDE formed during substrate HAT by these two oxidants were ~92 and ~94 kcal/mol, respectively, determined by comparing the rate constants for other oxidants using the Brønsted−Evans−Polyani relationship for the same substrate in the usual way (Figure 6).29
Figure 6.

Rate constants for reduction of oxoFeIVTMPS+ and oxoFeIVTDClPS+ by DHA mapped onto the Brønsted−Evans−Polyani relationship. Estimated FeO-H BDEs for FeTM-4-PyP and AaeAPO, determined by similar kinetic means using xanthene, are shown for comparison (cf. refs 4c and 9).
The accuracy of these BDE estimates could be confirmed electrochemically in this case. To account for the two-proton equibrium discussed above, we define a new FeO-H BDE parameter, D(OH2), in a modified Bordwell equation (eq 2) that reflects both the homolytic FeO-H BDE for the hydrogen atom in flight from the substrate and pKa1 for the proton derived from the medium.30
| (2) |
Thus, D(OH2), which is pH dependent, can be calculated to be 90–93 kcal/mol from the two measured quantities, E′ = 1.06 V and pKaobs = 5.5, over the pH range of the kinetic measurements (pH 3–5) (Scheme 3). It is satisfying that D(OH2) determined in this way is the same as the estimate determined kinetically in Figure 6. As can be seen in eq 2, there is a considerable increase in the driving force for C–H bond scission due to the large contribution from the two-proton pKaobs (15 kcal/mol for oxoFeIVTMPS+). Notably, a ferryl basicity corresponding to pKa = 11 for P−FeIV−OH would be required to obtain this much of a driving force enhancement in a single-proton event. Since the C–H BDE of DHA is 76 kcal/mol, the overall driving force for C–H bond homolysis is 16.5 kcal/mol if the solvent protonation is included, while it is only 11 kcal/mol without solvent assistance. The energetics for this concerted, solvent proton-assisted HAT are compared to a stepwise process in Figure 7.
Scheme 3. Two-Proton Equilibrium and HAT Driving Force for OxoFeIVTMPS+a.

a Simultaneous hydrogen atom abstraction and ferryl protonation from the medium increase the overall driving force for C–H bond scission.
Figure 7.

Energy landscape for C–H bond scission in dihydroanthracene by oxoFeIVTMPS+. A concerted process involving simultaneous hydrogen atom abstraction and ferryl protonation has a driving force of 16.5 kcal/mol and D(OH2) = 92.5 kcal/mol at pH 3, while a stepwise hydrogen atom transfer has only an 11 kcal/mol driving force and D(OH) = 87 kcal/mol.
It is of interest to consider the extent to which the equilibrium P−FeIV=O ⇆ P+−FeIII−OH2, as well as the solvent proton-coupled C–H bond scission process in Scheme 3, are concerted, two-proton events. Since the reactions are carried out in water, solvent hydrogen bonds are expected to be in place at the terminal oxo ligand. The electromeric equilibrium P−FeIV=O ⇆ P+−FeIII−OH2 creates an interesting spring-loaded situation wherein pKa2 is significantly more acidic than pKa1. Thus, the second ferryl protonation in the P−FeIV=O ⇆ P+−FeIII−OH2 equilibrium may be synchronous with the first because the unseen P+−FeIII−OH is more basic than P−FeIV=O. In this context, the trigger for this increased Fe−OH basicity is the electron transfer from the porphyrin ligand. Concerted two-proton transfers have been observed in other proton relays31 and in photoexcited hydrogen-bonded pairs.32 Winkler and Gray have shown that laser flash oxidation of ferric microperoxidase-8 affords an aquairon(III) porphyrin cation radical below pH 6 and the corresponding ferryl species at pH 7.33 Loss of the two protons in P+−FeIII−OH2 in that case was too fast to be resolved (>104 s−1). Simultaneous two-proton transfers, with an attendant increase in driving force, also seem likely for the one-electron reductions for typical enzymatic ferryl species, such as HRP-II, for which the corresponding ferric-aqua pKa values are known to be in the range of 7–11,34 and for non-heme ferryl species, for which the FeIII−OH2 pKa is in that same range.
Applied to the HAT event from the substrate mediated by P+−FeIV=O, a concerted ferryl protonation/hydrogen abstraction would have the advantage of the additional 5.5 kcal/mol driving force expressed in pKa1 for oxoFeIVTMPS+ (Figure 7). The experimental observation of both solvent and substrate isotope effects can be interpreted to result from this synchronicity, although a protonation pre-equilibrium leading to the HAT transition state could also give this result.
Interestingly, D(OH2) values for TDPS+FeIII−OH2, TSMP+FeIII−OH2, and TDClPS+FeIII−OH2 were determined to be 91, 91.5, and 90.5 kcal/mol, respectively (SI), almost identical to that of oxoFeIVTMPS+, despite a 230 mV range for the porphyrin ring oxidation potentials, corresponding to ~5 kcal/mol in expected driving force. Thus, the D(OH2) values for all four porphyrins studied are essentially the same and identical to the effective FeO–H BDE estimated from the rates of C–H bond scission. Clearly, the porphyrin redox potential difference is not reflected in the observed rates of DHA oxidation by FeTMPS-I and FeTDClPS-I. If the FeO-H BDE is indeed determined via eq 2, the ferryl heme basicity reflected in pKaobs values may be compensating for the lower redox potential to lead to such similar C–H cleavage rates. In this regard, it is interesting to compare the much higher rates of reaction (>100-fold) observed with oxoFeIVTM-4-PyP+.9 The ring oxidation potential for this cationic porphyrin has been measured at 1.22 V vs NHE, about 200 mV higher than that of FeTDClPS,35 about the same as the redox potential difference between FeTMPS and FeTDClPS. Why should this be? The results suggest that the relatively high pKaobs of (H2O)2FeIIITMPS+ (5.5, and the correspondingly high pKa1 ≈ 7) is sufficient to contribute to the overall driving force and lead to a higher reactivity than expected. Conversely, the pKa of (H2O)2FeIIITDClPS+ is only 4, and that of (H2O)2FeTM-4-PyP+, while unknown, is certainly lower. Thus, the hydrogen abstraction rates for these low-pKa iron porphyrins are largely determined by their relative oxidation potentials, with reduced contributions from the ferryl oxygen basicity. Obviously, it would be useful to have synthetic, biomimetic C–H oxygenation catalysts that have high C–H scission rates at relatively low oxidation potentials.15e
The idea that protonation or hydrogen-bonding of a ferryl oxo can influence the reactivity of such complexes has been suggested for non-heme oxoFeIV complexes. Ferryl basicity has been discussed recently in connection with differences between C–H and O–H or N−H bond scission.15b Also, addition of strong acids to the non-heme oxoFeIV complex [(N4Py)-FeIV(O)]2+ resulted in an increased reduction potential and increased rates for C–H oxidation.36 Concomitant with this increase in rate, however, was the loss of the substrate KIE and the appearance of an inverse solvent isotope effect, suggesting that the rate-determining step is electron transfer rather than H-atom abstraction. By contrast, oxoFeIVTDClPS+ displays both substrate and solvent KIEs, suggesting that the solvent proton is assisting in the HAT from the substrate as opposed to changing the mechanism from H-atom abstraction to electron transfer. Brønsted acid catalysis of substrate C–H bond cleavage may also be occurring in heme proteins such as cytochrome P450 and APO. The proton relay channel of cytochrome P450 has been well studied and is usually invoked to catalyze the O−O bond heterolysis in a ferric hydroperoxo precursor (compound 0) of the reactive oxoiron(IV) porphyrin cation radical and a water molecule.13,37 The same proton relay channel could use the newly formed water molecule to activate the ferryl oxygen through protonation in a similar manner as in these iron porphyrin model systems. More generally, ferryl protonation could play a significant role in the reactivity and selectivity of non-heme iron proteins such as the α-ketoglutarate-dependent halogenase SyrB238 and in (S)-2-hydroxypropyl-phosphonic acid epoxidase (HppE),39 which mediates the unusual epoxide formation in the biosynthesis of fosfomycin. In these cases, simultaneous ferryl protonation and substrate hydrogen abstraction could both facilitate C–H bond scission and disfavor hydroxylation of the incipient substrate radical.
SUMMARY AND CONCLUSIONS
We have shown that the oxo groups of sulfonated oxoFeIV porphyrin model systems can be protonated under mild conditions, with apparent pKa values for the P+−FeIII(OH2)2 ⇆h P−FeIV=O interconversion between 4.0 and 6.3, depending on the porphyrin meso substituent. The protonated form of these oxoFeIV porphyrins is a bis-aqua-FeIII porphyrin cation radical, or a buffer-bound equivalent. The reversible protonation of this family of ferryl porphyrins reveals a novel electromeric equilibrium, which, while predicted by theory,25 has not been previously described. Hydrogen abstraction by the compound I analogues, oxoFeIVTMPS+ and oxoFeIVTDClPS+, leads to protonated rebound intermediates when the pH of the medium is below the pKaobs of P+−FeIII(OH2)2. The rates of reaction of oxoFeIVTMPS+ and oxoFeIVTDClPS+ with dihydroanthracene are within a factor of 5 of each other, despite a 200 mV difference in the ring oxidation potential. The observed pKaobs values for (H2O)2FeIIITMPS+ and (H2O)2FeIIITDClPS+ differ by 1.5 pH units but in a compensating direction. Using kinetic and electrochemical estimations, the effective FeO–H BDEs of FeTMPS-II and FeTDClPS-II, D(OH2), have been estimated to be ~90–93 kcal/mol, involving both the hydrogen atom donated by the substrate and a proton delivered simultaneously from the medium. The presence of solvent O–H and substrate C–H deuterium KIEs indicate that hydrocarbon oxidation by these anionic oxoFeIV porphyrin cation radicals occurs via a novel solvent protoncoupled hydrogen atom transfer process. Thus, hydrogen atom abstraction from the substrate by the ferryl species produces a basic Fe−OH intermediate, the protonation of which can increase the overall driving force for C–H bond scission by at least 5 kcal/mol.
MATERIALS AND METHODS
H2TSMP, H2TMP, and H2TDClP were purchased from Frontier Scientific and used without further purification. Ferrous ammonium sulfate, Dowex AG-50W X8 ion-exchange resin (Na form), Sephadex LH-20 size exclusion gel, anhydrous sodium phosphate monobasic, acetic acid, perchloric acid, potassium hydroxide, TEMPO, and methanol were purchased from Sigma-Aldrich and used as received. mCPBA was obtained from Sigma-Aldrich and was purified by washing with pH 7.0 phosphate buffer (100 mM) to remove mCBA. D2O and methanol-d4 were purchased from Cambridge Isotope Laboratories, Inc. Water was distilled and deionized (Millipore, Milli-Q). Buffer solutions were prepared from a stock solution of 100 mM sodium phosphate/acetic acid and were corrected using concentrated KOH or perchloric acid solutions. pH values of the ACN/buffer mixtures were determined using the method of Bosch and co-workers.40 pH was measured using a sympHony meter that had been calibrated prior to use (pH 4.0, 7.0, and 10.0).
Porphyrin Synthesis
H2TMPS was prepared as previously reported from commercially available H2TMP (Frontier Scientific).41 H2TDClPS was prepared from commercially available H2TDClP (Frontier Scientific) using the method of Gonsalves et al.42 H2TDPS was prepared from H2TDP using the method of Badger et al.43

H2TDPS-Na4
The sulfonation of H2TDP was performed using the method reported by Gonsalves et al.42 An oven-dried 25 mL round-bottom flask equipped with a stir bar and a rubber septum was charged with H2TDP (0.12 mmol) and ~6 mL of neat chlorosulfonic acid. The mixture was allowed to stir for 1 h at room temperature, at which point it was quenched by slowly pouring onto 100 g of ground ice (Caution: this mixture will violently splatter!). The chlorosulfonyl porphyrin intermediate was extracted with chloroform (3 × 50 mL). The combined organic washings were then washed with saturated NaHCO3 (50 mL) and brine (50 mL) and dried over MgSO4. The purple solution was dried in vacuo to yield a shiny purple solid. The solid was then resuspended in water (50 mL) and refluxed overnight to yield the corresponding tetrasulfonic acid. The compound was purified via ion exchange (Dowex AG-50W X8, Na form) to yield H2TDPS-Na4 as a purple solid (101 mg, 69% of theoretical). UV–vis (λ, nm; ε, M−1 cm−1): 397 (27 000), 416 (140 000), 516 (6500), 553 (3300), 583 (3300), 591 (2700) (100 mM PBS, pH 7.4). 1H NMR (D2O, 500 MHz, δ): 8.47 (s, 8H, β-pyrrole), 2.77 (s, 24H, meta-methyl), 1.71 (s, 24H, ortho-methyl). 13C NMR (D2O, 500 MHz, ppm): 140.8, 139.1, 136.8, 131.5, 126.5, 120.0, 20.2, 19.0.
Iron Porphyrins
The insertion of iron into the porphyrin ligands was performed according to literature methods.19d Briefly, sulfonated porphyrin ligand (0.04 mmol) was dissolved in 15 mL of unbuffered water and pH corrected to pH 4.0 with sulfuric acid. The solution was added to a 25 mL round-bottom flask equipped with a reflux condenser and an argon inlet. The mixture was brought to and held at reflux for 30 min under argon. (NH4)2Fe(SO4)2 (100 mg, 0.26 mmol) was then quickly added to the refluxing solution under argon. The mixture was allowed to stir at reflux overnight, at which point the argon inlet was removed and the mixture refluxed open to air for 1 h. NaOH was added to the mixture to precipitate out the excess iron salts in solution. The mixture was then filtered through a bed of Celite and concentrated in vacuo. The resulting solid was purified via ion exchange (Dowex AG-50W X8, Na form) using water as the eluent. Finally, the mixture was desalted via size exclusion chromatography (Sephadex LH-20), eluting with 20% aqueous methanol, yielding the desired Fe complex. FeTMPS was prepared and characterized as we have previously described.17a
FeTDPS-HSO4-Na4
13.9 mg, 25% of theoretical. UV–vis (λ, nm; ε, M−1 cm−1): 328 (14 000), 416 (43 500), 505 (4000), 638 (1500). HR-ESI-MS (flying as a formate salt) for C61H58FeN4O14S43−: expected 418.073 m/z, observed 418.073 m/z.

Zinc Porphyrins
ZnTMPS-Na8
H2TMPS (100 mg, 0.063 mmol) was added to a 50 mL round-bottom flask equipped with a stir bar and a reflux condenser. A minimal amount of water was added to dissolve the solid. The solution was brought to reflux, followed by addition of ZnOAc (77.5 mg, 0.35 mmol). The reaction was monitored by UV–visible spectroscopy, and an increasing amount of Zn was added until the reaction was complete. Dowex AG 50W X8 (sodium form, 1 g) was then added and the mixture stirred for 1 h. The mixture was concentrated in vacuo and redissolved in a minimal amount of 50:50 methanol/water. This solution was then chromatographed on Sephadex LH-20, eluting with 50:50 methanol/water, to yield ZnTMPS as a shiny purple sodium salt (36.2 mg, 30%). UV–vis (λ, nm; ε, M−1 cm−1): 405 (47 000), 426 (578 000), 562 (19 500), 601 (7300) (100 mM PBS, pH 7.4). 1H NMR (D2O, 500 MHz, δ): 8.73 (s, 8H, β-pyrrole), 3.17 (s, 12H, para-methyl), 2.11 (s, 24H, ortho-methyl). HR-ESI-MS for C56H49N4O24S8Zn3−: expected 493.660 m/z, observed 493.659 m/z.
ZnTDClPS-Na4
Synthesized as above; 19.3 mg, 30%. UV–vis (λ, nm; ε, M−1 cm−1): 402 (46 500), 423 (535 000), 557 (20 000), 592 (22 000) (100 mM PBS, pH 7.4). 1H NMR (D2O, 500 MHz, δ): 8.74 (s, 8H, β-pyrrole), 8.32 (m, 4H), 7.89 (m, 4H). HR-ESI-MS for C44H16Cl8N4O12S4Zn4−: expected 316.910 m/z, observed 316.911 m/z.
ZnTDPS-Na4
Synthesized as above, starting with H2TDPS (0.038 mmol, 43.9 mg); 24.6 mg, 42%. UV–vis (λ, nm; ε, M−1cm−1): 404 (52,000), 423 (400,000), 560 (19,000), 599 (7500). 1H NMR (MeOD, 300 MHz, δ) 8.47 (s, 8H, β-pyrrole), 2.84 (s, 24H), 1.80 (s, 24H). HR-ESI-MS for C60H56ZnN4O12S44− expected 304.052 m/z, observed 304.051 m/z.


Instrumentation
UV–vis spectra were recorded using either a Hewlett-Packard 8453 diode array spectrophotometer or a Varian Cary Bio-300 double-beam spectrophotometer with temperature control. Rapid-mixing stopped-flow experiments were obtained using a thermostated Hi-Tech SF-61 DX2 double-mixing instrument with a 1 cm or 2 mm path length cuvette equipped with a diode array detector. NMR spectra were recorded using either a Bruker Avance (500 MHz) or a Bruker Ultra-Shield (300 MHz) spectrometer. HR-MS data were recorded using an Agilent 6220 accurate-mass LC-TOF mass spectrometer operating in negative mode. Electrochemical measurements were obtained using a BAS 100B/W electrochemical workstation.
Electrochemical Measurements
Cyclic voltammograms were obtained using a three-electrode cell: a glassy carbon electrode (3 mm diameter) as the working electrode, a platinum wire as the counter electrode, and a saturated Ag/AgCl reference electrode purchased from BASi. The electrochemical measurements were performed in 50 mM phosphate/acetate solution, which served as both buffer and supporting electrolyte.
Rapid-Mixing pH-Jump Experiments/pKa Determination
All kinetic data were obtained at 14.5 °C unless otherwise noted using a pH-jump technique similar to that reported by Yosca.5a Each experiment was carried out in runs composed of six individual tests, which were then averaged and duplicated at least twice. Reactions that were completed in under 1 min were measured using a thermostated Hi-Tech SF-61 DX2 double-mixing stopped-flow instrument with a 1 cm path length cuvette in single-mixing mode or in double-mixing mode with the reported aging time. Reactions taking longer than 1 min were measured using a Varian Carey Bio-300 double-beam spectrophotometer operating in single-wavelength mode. pH-jump experiments for oxoFeIVTMPS, TSMP, and TDClPS were performed in single-mixing mode by the 1:1 rapid mixing (and dilution) of a high-pH oxoFeIVporphyrin solution and 100 mM buffer (phosphate acetate). pH-jump experiments for oxoFeIVTDPS were performed in double-mixing mode, where the first push is used to generate oxoFeIVTDPS and the second push to jump the pH of the newly formed oxoFeIV species. Ionic strength was not compensated and varied between 0.055 and 0.175 M, depending upon the pH of the phosphate−acetate buffer after the pH jump. The pH of the resulting solutions was measured for each run. Spectrophotometric titrations were made using the first scan of the experiment after the instrumental dead time (~1 ms).
Formation of OxoFeIVporphyrin Complexes
OxoFeIVTSMP and oxoFeIVTMPS were formed in bulk by the addition of stoichiometric mCPBA (100 mM stock solution in ACN) to a solution of FeIIITSMP or FeIIITMPS (20 µM, pH 11.5, unbuffered), respectively. OxoFeIVTDClPS was formed by the bulk addition of 4 equiv of mCPBA (100 mM stock solution in ACN) to a solution of FeIIITDClPS (20 µM, pH 11.5 unbuffered). OxoFeIVTDPS was formed under rapid-mixing stopped-flow conditions by the 1:1 mixing of FeIIITDPS (40 mM, pH 11.5, unbuffered) and mCPBA (40 mM, pH 11.5, unbuffered).
Kinetic Measurements
All kinetic experiments were performed at 14.5 °C in 50 mM phosphate acetate buffer. Oxidations of hydrocarbon substrates were performed in 20% ACN buffer mixtures to increase solubility. Each experiment was carried out in runs composed of six individual shots, which were then averaged and duplicated at least twice. The concentrations presented are final after mixing. Time-resolved UV–vis spectra are recorded using Hi-Tech SF-61 DX2 double mixing instrument with a 1 cm path length cuvette equipped with diode array detector. Substrate reactions with xanthene and dihydroanthracene were performed in double-mixing mode where the oxoFeIV porphyrin cation radical species were generated by mixing FeIIIporphyrin complexes and mCPBA in the first push. The substrate was then introduced in the second push after the appropriate aging time. Values of kobs were obtained by fitting the kinetic profile to a single-exponential equation using Hi-Tech KinetAsyst 2.28 software. Bimolecular reaction rate constants were then obtained from a linear fit of kobs versus substrate concentration. Global spectral analyses, evolving factor analyses and singular value decomposition analyses44 were performed using the ReactLab Kinetics 1.1 software package.
Supplementary Material
Figure 5.

Oxidation of xanthene by oxoFeIVTDClPS+ under pseudofirst-order conditions. Reaction with proteo xanthene in proteo buffer (pH 3.0) yielded a bimolecular rate constant of (2.47 ± 0.15) × 104 M−1 s−1 (R2 = 0.99) (red). Xanthene-d2 showed a slower bimolecular rate constant of (1.37 ± 0.18) × 104 M−1 s−1 (R2 = 0.95), which yields a substrate KIE of 1.80 (blue). Reaction of oxoFeIVTDClPS+ with proteo xanthene in deutero buffer (pD 3.0) also showed a slower bimolecular rate constant than in the proteo buffer case, (1.50 ± 0.11) × 104 M−1 s−1 (R2 = 0.99), which yields a solvent KIE of 1.65 (black). Xanthene-d2 in D2O afforded a bimolecular rate of (1.12 ± 0.09) × 104 M−1 s−1 (R2 = 0.98), indicating a combined substrate−solvent isotope effect of 2.2 (purple).
ACKNOWLEDGMENTS
We are grateful for support of this research by the Center for Catalytic Hydrocarbon Functionalization, an Energy Frontier Research Center, U.S. Department of Energy, Office of Science, Basic Energy Sciences, under award no. DE-SC0001298. Partial support of this work was provided by the National Institutes of Health (2R37 GM036298).
Footnotes
ASSOCIATED CONTENT
S Supporting Information
UV–vis spectra and stopped-flow transients, kinetic traces, NMR spectra, and electrochemical data for compounds studied. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- 1.(a) Poulos TL. Chem. Rev. 2014;114:3919. doi: 10.1021/cr400415k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Groves JT. J. Inorg. Biochem. 2006;100:434. doi: 10.1016/j.jinorgbio.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 2.(a) Hasinoff BB, Dunford HB. Biochemistry. 1970;9:4930. doi: 10.1021/bi00827a015. [DOI] [PubMed] [Google Scholar]; (b) George P. Biochem. J. 1953;54:267. doi: 10.1042/bj0540267. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chance B. Arch. Biochem. Biophys. 1952;41:404. doi: 10.1016/0003-9861(52)90469-4. [DOI] [PubMed] [Google Scholar]; (d) Keilin D, Mann T. Proc. R. Soc. B-Biol. Sci. 1937;122:119. [Google Scholar]
- 3.(a) Ortiz de Montellano PR. Chem. Rev. 2010;110:932. doi: 10.1021/cr9002193. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shaik S, Cohen S, Wang Y, Chen H, Kumar D, Thiel W. Chem. Rev. 2010;110:949. doi: 10.1021/cr900121s. [DOI] [PubMed] [Google Scholar]; (c) Groves JT. In: Cytochrome P450: Structure Mechanism and Biochemistry. 3rd ed. Ortiz de Montellano PR, editor. New York: Kluwer Academic/Plenum; 2005. pp. 1–44. [Google Scholar]; (d) Denisov IG, Makris TM, Sligar SG, Schlichting I. Chem. Rev. 2005;105:2253. doi: 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]
- 4.(a) Hofrichter M, Ullrich R. Curr. Opin. Chem. Biol. 2014;19:116. doi: 10.1016/j.cbpa.2014.01.015. [DOI] [PubMed] [Google Scholar]; (b) Wang X, Peter S, Ullrich R, Hofrichter M, Groves JT. Angew. Chem. Int. Ed. 2013;52:9238. doi: 10.1002/anie.201302137. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wang X, Peter S, Kinne M, Hofrichter M, Groves JT. J. Am. Chem. Soc. 2012;134:12897. doi: 10.1021/ja3049223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Yosca TH, Rittle J, Krest CM, Onderko EL, Silakov Alexey, Calixto JC, Behan RK, Green MT. Science. 2013;342:825. doi: 10.1126/science.1244373. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rittle J, Green MT. Science. 2010;330:933. doi: 10.1126/science.1193478. [DOI] [PubMed] [Google Scholar]; (c) Green MT, Dawson JH, Gray HB. Science. 2004;304:1653. doi: 10.1126/science.1096897. [DOI] [PubMed] [Google Scholar]
- 6.Saouma CT, Mayer JM. Chem. Sci. 2014;5:21. doi: 10.1039/C3SC52664J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Usharani D, Lai W, Li C, Chen H, Danovicha D, Shaik S. Chem. Soc. Rev. 2014;43:4968. doi: 10.1039/c4cs00043a. [DOI] [PubMed] [Google Scholar]
- 8.(a) Warren JJ, Tronic TA, Mayer JM. Chem. Rev. 2010;110:6961. doi: 10.1021/cr100085k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mayer JM. Acc. Chem. Res. 1998;31:441. [Google Scholar]; (c) Lind J, Shen X, Eriksen TE, Merenyi GJ. Am. Chem. Soc. 1990;112:479. [Google Scholar]; (d) Bordwell FG. Acc. Chem. Res. 1988;21:456. [Google Scholar]; (e) Janousek BK, Reed KJ, Brauman JI. J. Am. Chem. Soc. 1980;102:3125. [Google Scholar]
- 9.Bell SR, Groves JT. J. Am. Chem. Soc. 2009;131:9640. doi: 10.1021/ja903394s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Groves JT. Nat. Chem. 2014;6:89. doi: 10.1038/nchem.1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Gumiero A, Metcalfe CL, Pearson AR, Raven EL, Moody PC. J. Biol. Chem. 2011;286:1260. doi: 10.1074/jbc.M110.183483. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Efimov I, Badyal SK, Metcalfe CL, Macdonald I, Gumiero A, Raven EL, Moody PCE. J. Am. Chem. Soc. 2011;133:15376. doi: 10.1021/ja2007017. [DOI] [PubMed] [Google Scholar]; (c) Alfonso-Prieto M, Oberhofer H, Klein ML, Rovira C, Blumberger J. J. Am. Chem. Soc. 2011;133:4285. doi: 10.1021/ja1110706. [DOI] [PubMed] [Google Scholar]
- 12.Meharenna YT, Doukov T, Li H, Soltis SM, Poulos TL. Biochemistry. 2010;49:2984. doi: 10.1021/bi100238r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Casadei CM, Gumiero A, Metcalfe CL, Murphy EJ, Basran J, Concilio MG, Teixeira SCM, Schrader TE, Fielding AJ, Ostermann A, Blakeley MP, Raven EL, Moody PCE. Science. 2014;345:193. doi: 10.1126/science.1254398. [DOI] [PubMed] [Google Scholar]
- 14.Yosca TH, Behan RK, Krest CM, Onderko EL, Langston MC, Green MT. J. Am. Chem. Soc. 2014;136:9124–9131. doi: 10.1021/ja503588n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Widger LR, Davies CG, Yang TH, Siegler MA, Troeppner O, Jameson GNL, Ivanovic-Burmazovic I, Goldberg DPJ. Am. Chem. Soc. 2014;136:2699. doi: 10.1021/ja410240c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Usharani D, Lacy DC, Borovik AS, Shaik SJ. Am. Chem. Soc. 2013;135:17090. doi: 10.1021/ja408073m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Latifi R, Sainna MA, Rybak-Akimova EV, de Visser SP. Chem.—Eur. J. 2013;19:4058. doi: 10.1002/chem.201202811. [DOI] [PubMed] [Google Scholar]; (d) Jaccob M, Ansari A, Pandey B, Rajaraman G. Dalton Trans. 2013;42:16518. doi: 10.1039/c3dt52290c. [DOI] [PubMed] [Google Scholar]; (e) Borovik AS. Chem. Soc. Rev. 2011;40:1870. doi: 10.1039/c0cs00165a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Lansky DE, Goldberg DP. Inorg. Chem. 2006;45:5119. doi: 10.1021/ic060491+. [DOI] [PubMed] [Google Scholar]
- 16.Groves JT, Quinn R, Mcmurry TJ, Nakamura M, Lang G, Boso B. J. Am. Chem. Soc. 1985;107:354. [Google Scholar]
- 17.(a) Shimanovich R, Groves JT. Arch. Biochem. Biophys. 2001;387:307. doi: 10.1006/abbi.2000.2247. [DOI] [PubMed] [Google Scholar]; (b) Bell SR. Ph.D. Thesis. Princeton University; 2010. [Google Scholar]
- 18.Groves JT, Haushalter RC, Nakamura M, Nemo TE, Evans BJ. J. Am. Chem. Soc. 1981;103:2884. [Google Scholar]
- 19.(a) Fujii H. Coord. Chem. Rev. 2002;226:51. [Google Scholar]; (b) Fujii H. J. Am. Chem. Soc. 1993;115:4641. [Google Scholar]; (c) Wolak M, van Eldik R. Chemistry. 2007;13:4873. doi: 10.1002/chem.200601148. [DOI] [PubMed] [Google Scholar]; (d) Liu MH, Su YO. J. Electroanal. Chem. 1998;452:113. [Google Scholar]; (e) Liu MH, Yeh CY, Su YO. Chem. Commun. 1996:1437. [Google Scholar]; (f) Balch AL, Latosgrazynski L, Renner MW. J. Am. Chem. Soc. 1985;107:2983. [Google Scholar]; (g) Takahashi A, Kurahashi T, Fujii H. Inorg. Chem. 2011;50:6922. doi: 10.1021/ic102564e. [DOI] [PubMed] [Google Scholar]; (h) Takahashi A, Yamaki D, Ikemura K, Kurahashi T, Ogura T, Hada M, Fujii H. Inorg. Chem. 2012;51:7296. doi: 10.1021/ic3006597. [DOI] [PubMed] [Google Scholar]
- 20.Panicucci R, Bruice TC. J. Am. Chem. Soc. 1990;112:6063. [Google Scholar]
- 21.Rodgers KR, Reed RA, Su YO, Spiro TG. Inorg. Chem. 1992;31:2688. [Google Scholar]
- 22.We thank Dr. Xiaoshi Wang for this suggestion.
- 23.Murata K, Panicucci R, Gopinath E, Bruice TC. J. Am. Chem. Soc. 1990;112:6072. [Google Scholar]
- 24.The shapes of the titration curves in Figure 3B are all more gradual than expected for an ideal two-proton event. The rates (dx values reported in the SI) are between 0.32 and 0.50, instead of the usual value of 0.22, as shown in Figure S18. We thank a reviewer for pointing this out. We attribute this effect to the obvious interaction of the buffer with these iron porphyrins (measured pKa values for P−Fe(III) are buffer dependent) and the fact that the ionic strength of the medium shifts with pH in these pH-jump, fast dilution experiments. For simplicity we have referred to the various iron(III) species as diaqua complexes, although the actual situation is certainly more complex, involving buffer-bound species that will have their own prototropy. Non-coordinating buffers interfered with the redox chemistry.
- 25.Derat E, Shaik S. J. Am. Chem. Soc. 2006;128:8185. doi: 10.1021/ja0600734. [DOI] [PubMed] [Google Scholar]
- 26.(a) Coulson AFW, Erman JE, Yonetani T. J. Biol. Chem. 1971;246:917. [PubMed] [Google Scholar]; (b) Hahm S, Miller MA, Geren L, Kraut J, Durham B, Millett F. Biochemistry. 1994;33:1473. doi: 10.1021/bi00172a025. [DOI] [PubMed] [Google Scholar]
- 27.Ikezaki A, Takahashi M, Nakamura M. Chem. Commun. 2013;49:3098. doi: 10.1039/c3cc40319j. [DOI] [PubMed] [Google Scholar]
- 28.England J, Bigelow JO, Heuvelen KMV, Farquhar ER, Martinho M, Meier KK, Frisch JR, Mück E, Lawrence Que J. Chem. Sci. 2014;5:1204. doi: 10.1039/C3SC52755G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.(a) Gardner KA, Kuehnert LL, Mayer JM. Inorg. Chem. 1997;36:2069. doi: 10.1021/ic961297y. [DOI] [PubMed] [Google Scholar]; (b) Arends IWCE, Mulder P, Clark KB, Wayner DDM. J. Phys. Chem. 1995;99:8182. [Google Scholar]; (c) Colussi AJ. Chemical Kinetics of Small Organic Radicals. Boca Raton: CRC Press; 1988. [Google Scholar]; (d) Howard JA, Scaiano JC. Oxyl-,Peroxyl- and Related Radicals. New York: Springer-Verlag; 1984. p. 13. [Google Scholar]
- 30.See Supporting Information for a derivation of eq 2. We thank a reviewer for insightful comments.
- 31.(a) Westlake BC, Brennaman MK, Concepcion JJ, Paul JJ, Bettis SE, Hampton SD, Miller SA, Lebedeva NV, Forbes MDE, Moran AM, Meyer TJ, Papanikolas JM. Proc. Natl. Acad. Sci. U.S.A. 2011;108:8554. doi: 10.1073/pnas.1104811108. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chen XH, Ma GC, Sun WC, Dai HJ, Xiao D, Zhang YF, Qin X, Liu YJ, Bu YX. J. Am. Chem. Soc. 2014;136:4515. doi: 10.1021/ja406340z. [DOI] [PubMed] [Google Scholar]
- 32.(a) Catalan J, del Valle JC, Kasha M. Proc. Natl. Acad. Sci. U.S.A. 1999;96:8338. doi: 10.1073/pnas.96.15.8338. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Catalan J, Perez P, del Valle JC, de Paz JLG, Kasha M. Proc. Natl. Acad. Sci. U.S.A. 2002;99:5793. doi: 10.1073/pnas.052703899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.(a) Berglund J, Pascher T, Winkler JR, Gray HB. J. Am. Chem. Soc. 1997;119:2464. [Google Scholar]; (b) Low DW, Winkler JR, Gray HB. J. Am. Chem. Soc. 1996;118:117. [Google Scholar]
- 34.Song Y, Mao J, Gunner MR. Biochemistry. 2006;45:7949. doi: 10.1021/bi052182l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lei JP, Ju HX, Ikeda O. J. Electroanal. Chem. 2004;567:331. [Google Scholar]
- 36.(a) Park J, Morimoto Y, Lee Y-M, Nam W, Fukuzumi SJ. Am. Chem. Soc. 2012;134:3903. doi: 10.1021/ja211641s. [DOI] [PubMed] [Google Scholar]; (b) Fukuzumi S. Coord. Chem. Rev. 2013;257:1564. doi: 10.1016/j.ccr.2012.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Park J, Lee Y-M, Nam W, Fukuzumi S. J. Am. Chem. Soc. 2013;135:5052. doi: 10.1021/ja311662w. [DOI] [PubMed] [Google Scholar]; (d) Park J, Morimoto Y, Lee YM, Nam W, Fukuzumi S. Inorg. Chem. 2014;53:3618. doi: 10.1021/ic403124u. [DOI] [PubMed] [Google Scholar]
- 37.(a) Batabyal D, Poulos TL. Biochemistry. 2013;52:8898. doi: 10.1021/bi401330c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gerber NC, Sligar SG. J. Am. Chem. Soc. 1992;114:8742. [Google Scholar]; (c) Groves JT, Boaz NC. Science. 2014;345:142. doi: 10.1126/science.1256754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.(a) Pratter SM, Light KM, Solomon EI, Straganz GD. J. Am. Chem. Soc. 2014;136:9385. doi: 10.1021/ja503179m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wong SD, Srnec M, Matthews ML, Liu LV, Kwak Y, Park K, Bell CB, Alp EE, Zhao JY, Yoda Y, Kitao S, Seto M, Krebs C, Bollinger JM, Solomon EI. Nature. 2013;499:320. doi: 10.1038/nature12304. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kulik HJ, Drennan CL. J. Biol. Chem. 2013;288:11233. doi: 10.1074/jbc.M112.415570. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Matthews ML, Neumann CS, Miles LA, Grove TL, Booker SJ, Krebs C, Walsh CT, Bollinger JM. Proc. Natl. Acad. Sci. U.S.A. 2009;106:17723. doi: 10.1073/pnas.0909649106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.(a) Huang H, Chang WC, Lin GM, Romo A, Pai PJ, Russell WK, Russell DH, Liu HW. J. Am. Chem. Soc. 2014;136:2944. doi: 10.1021/ja4100035. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Milaczewska A, Broclawik E, Borowski T. Chem.—Eur. J. 2013;19:770. doi: 10.1002/chem.201202825. [DOI] [PubMed] [Google Scholar]; (c) Liu PH, Mehn MP, Yan F, Zhao ZB, Que L, Liu HW. J. Am. Chem. Soc. 2004;126:10306. doi: 10.1021/ja0475050. [DOI] [PubMed] [Google Scholar]; (d) Wang C, Chang W-c, Guo Y, Huang H, Peck SC, Pandelia ME, Lin G-m, Liu H-w, Krebs C, Jr, B JM. Science. 2013;342:991. doi: 10.1126/science.1240373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gagliardi LG, Castells CB, Rafols C, Roses M, Bosch E. Anal. Chem. 2007;79:3180. doi: 10.1021/ac062372h. [DOI] [PubMed] [Google Scholar]
- 41.(a) Hoffmann P, Labat G, Robert A, Meunier B. Tetrahedron Lett. 1990;31:1991. [Google Scholar]; (b) Song R, Robert A, Bernadou J, Meunier B. Inorg. Chim. Acta. 1998;272:228. [Google Scholar]
- 42.Rocha Gonsalves AMdA, Johnstone RAW, Pereira MM, de SantAna AMP, Serra AC, Sobral AJFN, Stocks PA. Heterocycles. 1996;43:829. [Google Scholar]
- 43.Badger GM, Jones RA, Laslett RL. Aust. J. Chem. 1964;17:1028. [Google Scholar]
- 44.Rittle J, Younker JM, Green MT. Inorg. Chem. 2010;49:3610. doi: 10.1021/ic902062d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.