

Summary
Stable RNA maturation is a key process in the generation of functional RNAs and failure to correctly process these RNAs can lead to their elimination through quality control mechanisms. Studies of the maturation pathways of ribosomal RNA and transfer RNA in B. subtilis showed they were radically different from E. coli and led to the identification of new B. subtilis-specific enzymes. We noticed that, despite their important roles in translation, a number of B. subtilis small stable RNAs still did not have characterised maturation pathways, notably the tmRNA, involved in ribosome rescue, and the RNase P RNA, involved in tRNA maturation. Here, we show that tmRNA is matured by RNase P and RNase Z at its 5′ and 3′ extremities, respectively, while the RNase P RNA is matured on its 3′ side by RNase Y. Recent evidence that several RNases are not essential in B. subtilis prompted us to revisit maturation of the scRNA, a component of the signal recognition particle involved in co-translational insertion of specific proteins into the membrane. We show that RNase Y is also involved in 3′ processing of scRNA. Lastly, we identified some of the enzymes involved in the turnover of these three stable RNAs.
Keywords: stable RNA maturation, ssrA, 10Sa, SRP, 4.5S, ribonuclease
Introduction
Many stable RNAs (ribosomal RNAs, transfer RNAs and other RNAs typically with functions related to translation) are produced as precursor species that require maturation of either their 5′ and 3′ ends, or both. In some cases, 5′ extensions can be circumvented by initiating transcription at the first nucleotide of the mature RNA sequence (Randau et al., 2008); however, 3′ extensions usually consist of sequences necessary to terminate transcription and are generally unavoidable. Processing of these RNAs is often an important step towards ensuring their resistance to degradation by other ribonucleases. Indeed, in many cases, lack of accurate processing constitutes an active signal for a quality control pathway that leads to elimination of the RNA, typically via a polyadenylation step (Li et al., 1998b, Li et al., 2002, Maes et al., 2012). 5′ or 3′ exoribonucleases can latch onto unprocessed extensions and, through their processive activity, degrade the molecule in its entirety (Mathy et al., 2007, Gudipati et al., 2012, Guy et al., 2014).
The RNA degradation machineries of E. coli and B. subtilis, two of the best-studied model organisms, are very different, with only eight shared enzymes from a repertoire of over twenty RNases in each organism. B. subtilis possesses a 5′ exoribonuclease activity (Mathy et al., 2007) and has replaced the key endoribonuclease RNase E with a non-homologous enzyme called RNase Y, for example (Shahbabian et al., 2009). It is therefore highly likely that the maturation pathways of the stable RNAs are different in these two organisms and it is important that they be characterised in detail in both systems. While most of the enzymes involved in rRNA and tRNA maturation have been identified in B. subtilis (Condon et al., 2001, Pellegrini et al., 2003, Wen et al., 2005, Britton et al., 2007, Redko et al., 2008), and have indeed proven to be emphatically different from those in E. coli, the processing pathways of a number of B. subtilis small stable RNAs with important functions related to translation are not yet known or have only been partially determined.
Transfer-messenger (tm) RNA (also known as 10Sa RNA) is about 360 nucleotides (nts) in length and plays an important role in rescuing ribosomes that are trapped on fragments of mRNA lacking a stop codon. Encoded by the ssrA gene, the tmRNA consists of a tRNA-like structure, created by pairing between the 5′ and 3′ ends of the molecule, with a central portion that serves as an mRNA. The tmRNA is aminoacylated, enters the P-site of the stalled ribosome and provokes a conformational change that induces the ribosome to resume translation on the coding portion of the tmRNA (for recent review, see (Giudice & Gillet, 2013)). Upon reaching the stop codon, the ribosome is released and the truncated peptide, bearing a tag specified by the tmRNA, is degraded. The processing pathway for the E. coli tmRNA is known and is very similar to that of E. coli tRNAs. The mature 5′ end is generated by RNase P (Komine et al., 1994) and the 3′ end by RNase E cleavage and nibbling by a number of redundant 3′ exoribonucleases, but principally RNase T (Li et al., 1998a, Lin-Chao et al., 1999). RNase III has been proposed to generate an intermediate for these final processing reactions (Makarov & Apirion, 1992), an echo of its role in rRNA maturation.
RNase P was one of the first RNA molecules shown to have catalytic activity, catalysing the 5′ maturation of tRNA (Guerrier-Takada et al., 1983). It consists of a ~400 nt RNA, encoded by the rnpB gene and a small protein subunit, encoded by rnpA, both of which are essential in vivo. Until recently, 5′ maturation of tRNA by RNase P was considered a universally conserved reaction. However, some organisms (e.g. Nanoarchaeum equitans) forego the necessity for RNase P by initiating tRNA transcription at the first nucleotide of the mature sequence (Randau et al., 2008). Additionally, a protein-only form of RNase P (PRORP) has been discovered in animal mitochondria (Holzmann et al., 2008) and has completely replaced the RNA form of RNase P in plants (Gutmann et al., 2012). The mature 5′ end of E. coli RNase P RNA (also known as P-RNA or M1 RNA) corresponds to the principal site of transcription initiation. 3′ processing is catalysed by RNase E, which cleaves a few nucleotides downstream of the mature sequence and is followed by 3′ exonucleolytic trimming by RNases T, D, PH or BN/Z (Kim et al., 1996, Li et al., 1998a).
The signal recognition particle (SRP) RNA, known as 4.5S RNA in E. coli and scRNA in B. subtilis, is an integral part of a ribonucleoprotein complex whose role is to guide ribosomes to the membrane-bound FtsY/SecYEG assembly for the co-translational insertion of specific proteins into the cytoplasmic membrane (for recent review, see (Akopian et al., 2013)). The SRP RNA is essential, but its precise role in membrane protein targeting is still unclear. It is bound by a protein partner Ffh, whose role is to bind the large subunit of the ribosome and the signal peptide as it emerges from the exit tunnel. The E. coli 4.5S RNA (114 nts) is matured by RNase P at its 5′ end (Peck-Miller & Altman, 1991) and by 3′ exonucleolytic trimming at the 3′ end by RNases T, D, PH or BN/Z (Li et al., 1998a). The B. subtilis scRNA is more than twice the length (271 nts) of the E. coli SRP RNA. The major pathway for scRNA processing involves RNase III processing to yield the mature 5′ end and an intermediate with four extra nucleotides requiring 3′ exonucleolytic trimming (principally, but not exclusively by RNase PH) at the 3′ end (Oguro et al., 1998). A second 3′ processing pathway involves endonucleolytic cleavage at a site 12 nts downstream of the mature 3′ end (Yao et al., 2007). Here we revisit this second 3′ processing pathway of scRNA maturation and characterize the maturation pathways of B. subtilis tmRNA and RNase P RNA.
Results
Maturation of B. subtilis tmRNA is catalysed by RNase P and RNase Z
To understand the maturation pathway of tmRNA, we first mapped the 5′ ends of the precursor species by primer extension. We identified two possible Sigma A-dependent promoters, P1 and P2, respectively centred around 150 and 75 base pairs upstream of the annotated mature tmRNA sequence. Primer extension assays with appropriate oligonucleotides confirmed 5′ ends at nts −129 and −55 relative to the 5′ end of the mature tmRNA sequence (annotation confirmed), consistent with transcription initiation from these predicted promoter sequences (Fig. 1). A predicted Rho-independent transcription terminator (ter) ends 51 nts downstream of the tmRNA. Thus, complete maturation of tmRNA is likely to require removal of both 5′ leader and 3′ trailer sequences from two precursor transcripts, which we call P1-ter and P2-ter.
Figure 1.

Mapping of 5′ ends of tmRNA precursors. (A) Primer extension assay mapping the 5′ ends of transcripts from the putative P1 and P2 promoters and the 5′ end of the mature tmRNA (oligo CC1444). Reactions were performed on RNA isolated from wild-type (wt) and RNase P-depleted (P↓) strains, with the latter strain showing increased accumulation of P1 and P2-originating transcripts. Sequence lanes are labelled as their reverse complement to facilitate direct read-out. The origin of the species marked with an asterisk migrating just above the mature tmRNA is unknown. (B) Sequence of the ssrA gene, encoding tmRNA. The −35 and −10 regions of the putative P1 and P2 promoters are highlighted in grey and the 5′ ends of the corresponding transcripts labelled as +1 (underlined). The tmRNA is indicated in bold type, with the confirmed 5′ end of the mature sequence indicated by M. Mapped 5′ ends of degradation intermediates (DI) identified in cells depleted for RNase Z are indicated. The transcription terminator is underlined.
The 5′ and 3′ ends of the tmRNA fold into a tRNA-like structure that can be recognised by alanyl-tRNA synthetase and aminoacylated. We therefore anticipated that tmRNA maturation would be similar to tRNA processing. Transfer RNAs are almost universally processed at their 5′ ends by RNase P. Two pathways for tRNA 3′ maturation are known in B. subtilis, depending largely on whether or not the tRNA has an encoded CCA motif. Transfer RNAs lacking an encoded CCA motif are primarily matured at their 3′ ends by the endoribonuclease RNase Z, encoded by the essential rnz gene (Pellegrini et al., 2003), that cleaves just downstream of the unpaired discriminator nucleotide. Those tRNAs containing an encoded CCA motif are poor substrates for RNase Z cleavage and are instead matured by a 3′ to 5′ exoribonucleolytic pathway involving RNase PH, encoded by the rph gene (Wen et al., 2005). We performed Northern blots on total RNA isolated from an array of key endo- and exoribonuclease mutants to identify the enzymes involved in tmRNA processing (and possibly turnover).
In wild-type cells, a probe hybridising close to the 5′ end of the mature tmRNA sequence revealed four RNA species containing the tmRNA (Fig. 2A; lane 8). The major species corresponded in size to the mature tmRNA (363 nts), while the sizes of two of the larger minor species were consistent with transcripts extending from the P1 or P2 promoters to the transcription terminator. The P1-ter transcript has a predicted size of 540 nts, while P2-ter has a calculated length of 466 nts. The fourth minor species visible in the wild-type lane corresponded in size to a transcript extending from P1 to the 3′ end of the mature tmRNA (492 nts) i.e. lacking the transcription terminator. The identities of each of these transcripts were confirmed by probing Northern blots with oligonucleotides that hybridise close to the 5′ ends of transcripts emanating from P1 or P2, or to the transcription terminator (Fig. S1).
Figure 2.

Processing and turnover of tmRNA by different B. subtilis endo- and exoribonucleases. (A) Northern blot analysis of total RNA isolated from different B. subtilis RNase mutants and probed for the 5′ end of mature tmRNA (oligo CC1444). Gene symbols are as follows: rnjA (RNase J1), rnjB (RNase J2), rph (RNase PH), rnr (RNase R), yhaM (YhaM), pnp (polynucleotide phosphorylase), rny (RNase Y), rnc (RNase III), rnpA (RNase P protein subunit, rnpB (RNase P RNA subunit); rnz (RNase Z). The migration positions of RNA size markers are shown. The sizes of the intermediates are inferred from the sequence and confirmed with oligos specific for the 5′ ends of the P1, P2 transcripts and the transcription terminator (Fig. S1). For those species containing the mature 3′ end of the tmRNA, the 3 extra nts corresponding to the CCA motif have been added for size calculation. (B) Primer extension assay on RNase Z (Z) cleaved tmRNA P2-ter precursor. Position of RNase Z cleavage is indicated by a black arrowhead. Sequence lanes are labelled as their reverse complement to facilitate direct read-out. (C) Processing pathway of tmRNA. The mature sequence (before CCA addition) is shown in red and ends in the discriminator A-residue. The locations of the predicted transcription start sites from the P1 and P2 promoters are indicated. Three Mulfold-predicted stem loops (Zuker, 2003) are labelled SL1, SL2 and SL3 and the transcription terminator is labelled ter. Processing and degradation by RNases P, Z, J1 and the 3′ exoribonucleases (3′ exos) are indicated by larger bold-face type and arrows. A proposed cleavage by an unknown endoribonuclease is indicated by a question mark. The predicted sizes of the different fragments are indicated. (D) Sequence of the circular RT-PCR product corresponding to mature tmRNA. The junction between the 5′ and 3′ ends is shown and the non-coded CCA is underlined in red.
Processing of tmRNA was clearly defective in cells depleted for either the RNA (rnpB) or the protein (rnpA) subunit of RNase P (Fig. 2A; lanes 9 and 13). The genes encoding both RNase P subunits are essential and depletions were performed using constructs under the control of either xylose (Pxyl-rnpA) or IPTG-dependent promoters (Pspac-rnpB). In cells depleted for either subunit of RNase P, we observed an accumulation of precursor species consistent with transcripts extending from P1 and P2 to the mature 3′ end of the tmRNA (Fig. 2A, lanes 9 and 13; 492 nts and 418 nts, respectively). The identity of these two precursors was confirmed by hybridisation with the P1 and P2 probes (Fig. S1A, B; lanes 9 and 13). This experiment shows that RNase P is responsible for processing of the 5′ end of tmRNA, as anticipated for a tRNA-like structure.
In cells depleted for RNase Z (using the Pspac-rnz construct), the P1-ter and P2-ter species were visible (Fig. 2A; lane 11) and a precursor species corresponding in size to a transcript extending from the mature 5′ end of tmRNA to the transcription terminator accumulated (M-ter; 411 nts). This species also hybridised to a terminator specific probe (Fig. S1C; lane 11), confirming its identity and showing that 3′ maturation of tmRNA is performed by RNase Z. Paradoxically, the 411 nt M-ter precursor species was more intense in the presence of IPTG than its absence in the Pspac-rnz strain (Fig. 2A, lanes 11 and 12). Northern blot analysis of these RNA preparations probed for the rnz transcript showed that it was significantly reduced compared to wild-type even in the presence of IPTG, and undetectable as expected without IPTG (data not shown). We consider possible reasons for the accumulation of the M-ter species and other notable differences between the partially and fully depleted RNase Z strains in the Discussion section.
We confirmed the site of RNase Z cleavage by digesting the tmRNA P2-ter precursor species with purified RNase Z in vitro and subjecting the downstream fragment to a primer extension assay. We detected a new 5′ end in the sample incubated with RNase Z that mapped to 2 nts from the 3′ end of the primer (CC1445) (Fig. 2B). This confirms cleavage at the 3′ end of the tmRNA by RNase Z immediately downstream of the discriminator position, as expected from its behaviour in tRNA processing. A summary of the tmRNA processing steps is shown in Fig. 2C.
The annotated 3′ end of the tmRNA in the B. subtilis genome ends with a CCA sequence. We therefore initially anticipated that the tmRNA 3′ processing pathway would be similar to that of tRNAs with an encoded CCA motif and be dependent on RNase PH. This was clearly not the case, however (Fig. 2A; lane 7). An examination of the predicted secondary structure of the B. subtilis tmRNA showed that the two C-residues of the encoded CCA sequence are paired with G-residues from the 5′ end, and the 3′ terminal A-residue occupies the discriminator position (Fig. 2C). Thus the encoded CCA sequence is involved in tmRNA secondary structure and is likely not available for aminoacylation. Following RNase Z cleavage downstream of the encoded CCA sequence, a second CCA motif must therefore be added to create a functional tmRNA, presumably by nucleotidyl transferase. The addition of the non-coded CCA motif on mature tmRNA was confirmed by circular RT-PCR on total RNA from the wild-type strain using oligos CC1475 and CC1538. The sequence of the junction between the 5′ and 3′ ends of the mature tmRNA is shown in Fig. 2D.
Turnover of tmRNA and tmRNA processing fragments
The Northern blots shown in Fig. 2A and Fig. S1 also provided us with information on the enzymes responsible for the turnover of the upstream and downstream products of RNase P and Z cleavage and on the degradation of the tmRNA itself. A significant accumulation of the mature tmRNA species occurred in strains lacking the endoribonuclease RNase Y (Fig. 2A; lane 3) and to a lesser extent in strains lacking the four known 3′-exoribonucleases of B. subtilis, RNase PH, RNase R, PNPase and YhaM (Fig. 2A; lane 2). This suggests a role for these enzymes in tmRNA turnover or quality control. Interestingly a number of degradation intermediates (DI) were observed in the primer extension assay of total RNA isolated from the RNase Z-depleted strain (Fig. S2). The 5′ ends of these degradation intermediates mapped within the mature tmRNA sequence (Fig. 1B) and could correspond to sites of RNase Y cleavage (or another endonuclease) to initiate tmRNA turnover.
A fragment hybridising to the transcription terminator probe accumulated in strains lacking the 5′-exoribonuclease RNase J1 (Fig S1C; lanes 1 and 4), indicating that RNase J1 is responsible for degradation of the downstream product of RNase Z cleavage. Its size is smaller than the anticipated 51 nts, however, suggesting that some other enzyme (symbolized by a question mark in Fig. 2C) first cuts this fragment. Under conditions of strong RNase Z depletion, a new band appeared in the 475–480 nt range (marked with an asterisk in Fig. 2A, lane 11). This band also hybridised to the P1-probe (Fig. S1C; lane 11) and thus likely corresponds to a transcript extending from P1 to the site of cleavage by this unknown enzyme.
The P2-probe hybridised to bands of ~130 nts and ~50 nts in the strain lacking the four 3′-exoribonucleases (Fig. S1B, lane 2), corresponding in size to the upstream products of RNase P cleavage of the P1 and P2 transcripts (129 and 55 nts, respectively). The P1-specific probe hybridised to the ~130 nts species and to a number of P1-proximal fragments of smaller size (70–115 nts) in the same strain (Fig. S1A, lane 2). These experiments show that the 3′-exonucleases are responsible for turnover of the 5′ leader fragments. The 5′ proximal stem loop structure (SL1 in Fig. 2B) has a higher predicted ΔG than SL2, which is in turn higher than SL3, likely explaining the accumulation of 5′ proximal fragments containing SL1 and SL2 in the absence of the key 3′ exonucleases and suggesting the presence of at least one other enzyme of this polarity in B. subtilis.
3′ maturation of the RNase P RNA by RNase Y
We adopted a similar strategy to determine the processing pathway of the RNA subunit of the RNase P ribozyme, encoded by the rnpB gene. We first identified two potential sites of transcription initiation by primer extension assay (Fig. 3A). One is located just downstream of the Sigma-A promoter predicted to drive transcription of the gpsB gene (PgpsB), encoding a protein involved in cell division. The 5′ end of this transcript is located 382 nts upstream of the annotated RNase P RNA sequence, which was also confirmed. A second, much stronger promoter is predicted to drive rnpB expression itself (PrnpB). Although transcription typically starts from a purine (A or G) residue 6 nts downstream of the last residue of the consensus −10 sequence (TATAAT), in this case the 5′ end was mapped to a G residue 8 nts downstream. We confirmed that the same 5′ end was observed in an RNase J1 mutant, which could potentially have removed two nts from the primary transcript by 5′ exonucleolytic nibbling. Like the tmRNA, the RNase P RNA has a predicted Rho-independent transcription terminator ending 47 nts downstream of the mature rnpB sequence.
Figure 3.

Mapping of 5′ ends of RNase P precursors. (A) Primer extension assay mapping the 5′ ends of transcripts from the putative PgpsB (top panel; oligo CC1492) and PrnpB (lower panel; oligo CC1006) promoters. Reactions were performed on wild-type (WT) RNAs and on RNAs isolated from strains lacking RNase Y (ΔY) and RNase J1 (ΔJ1). Sequence lanes are labelled as their reverse complement to facilitate direct read-out. (B) Sequence of the gpsB and rnpB genes. The −35 and −10 regions of the putative PgpsB and PrnpB promoters are highlighted in grey and the 5′ ends of the corresponding transcripts labelled as +1 (underlined). The gpsB coding sequence and mature RNase P RNA are indicated in bold type. The transcription terminator is underlined.
Northern blots of total RNA isolated from the same series of endo- and exoribonuclease mutants were probed with an oligonucleotide that hybridised close to the 5′ end of the mature rnpB sequence. In wild type cells, we observed the mature RNase P species (401 nts) and two minor precursor transcripts (Fig. 4A; lane 8). Using oligonucleotides specific for the transcription terminator downstream of rnpB and for the 5′ end of the gpsB gene, we identified these minor species as PgpsB-ter (830 nts) and PrnpB-ter (448 nts) (Fig. S3B, C; lanes 8). In cells lacking RNase Y, we observed a strong accumulation of PrnpB-ter at the expense of the mature RNase P RNA (Fig. 4A; lane 3). The PgpsB-ter species also accumulated in cells lacking RNase Y, indicating that this enzyme also removes the rnpB terminator from the 3′ end of read-through transcripts from the upstream gpsB gene. 3′ processing is not completely abolished in the absence of RNase Y, suggesting an alternative maturation pathway exists. A previous paper has shown that RNase P can autocatalyze the maturation of both the 5′ and 3′ ends of its own RNA in vitro, although the 3′ cleavage reaction yielded an RNase P RNA lacking 4 terminal nts (Loria & Pan, 2000). Our data do not rule out the possibility that this is the alternative pathway in vivo; however, we did not see a relative accumulation of either the PrnpB-ter or PgpsB-ter precursors compared to the mature RNase P RNA in cells depleted for the RNase P protein subunit RnpA (Fig. 4A; lane 9).
Figure 4.
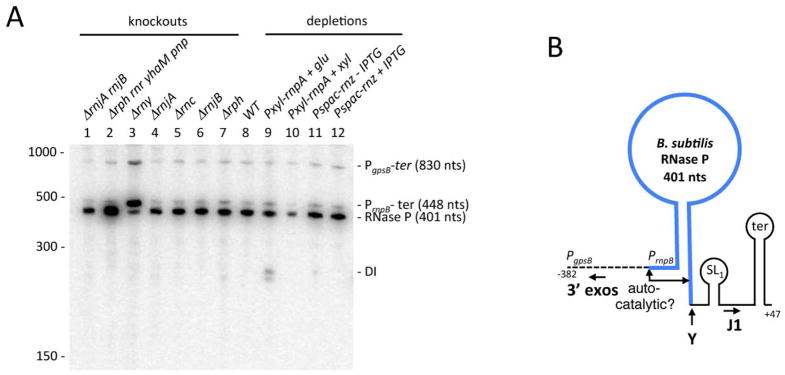
Processing and turnover of RNase P by different B. subtilis endo- and exoribonucleases. (A) Northern blot analysis of total RNA isolated from different B. subtilis RNase mutants probed for the 5′ end of the RNase P RNA (oligo CC1006). Gene symbols are as follows: rnjA (RNase J1), rnjB (RNase J2), rph (RNase PH), rnr (RNase R), yhaM (YhaM), pnp (polynucleotide phosphorylase), rny (RNase Y), rnc (RNase III), rnpA (RNase P protein subunit, rnpB (RNase P RNA subunit); rnz (RNase Z). The migration positions of RNA size markers are shown. The sizes of the intermediates are inferred from the sequence and confirmed with oligos specific for the 5′ ends of the PgpsB transcript and the transcription terminator (Fig. S3). (B) Processing pathway of RNase P. The mature sequence is shown in blue. The sites of autocatalytic cleavage observed in vitro in (Loria & Pan, 2000) are indicated. The locations of the transcription start sites from the PgpsB and PrnpB promoters are indicated. A Mulfold-predicted stem loop is labelled SL1 and the transcription terminator is labelled ter. Processing and degradation by RNases Y, J1 and the 3′ exoribonucleases (3′ exos) are indicated by larger bold-face type and arrows. The predicted sizes of the different fragments are indicated.
Similar to tmRNA, there was a significant accumulation of the mature RNase P in the strain lacking the four 3′-exoribonucleases (Fig. 4A; lane 2), suggesting that these enzymes may be involved in RNase P turnover or quality control. Interestingly, we also saw an accumulation of a degradation intermediate (DI) in a strain depleted for the RnpA protein subunit of RNase P (Fig. 4A; lane 9), suggesting that the protein subunit protects the RNA subunit from turnover, in line with previous findings (Wegscheid & Hartmann, 2007).
A fragment containing the transcription terminator accumulated in strains lacking the 5′ exoribonuclease RNase J1 (Fig. S3B; lanes 1 and 4). Thus, as for the tmRNA, RNase J1 is responsible for turnover of the downstream maturation product. Degradation intermediates corresponding to 5′ proximal fragments of the gpsB mRNA accumulated in the 3′-exoribonuclease deficient strain (Fig. S3C, lane 2), suggesting these enzymes are involved in the turnover of the gpsB portion of the transcript. A summary of the processing and degradation reactions for the RNase P RNA is shown in Fig. 4B.
3′ maturation of scRNA by RNase Y
Earlier studies on scRNA processing showed that the mature 5′ end is created directly by efficient RNase III cleavage at the “IIIA” site (Fig. 5A). The 3′ end can be created by less efficient RNase III cleavage 4 nts downstream of the mature 3′ end, at the “IIIb” site, followed by 3′-exonucleolytic processing (Oguro et al., 1998), or by an alternative pathway that was thought to be due to the endonuclease activity of RNase J1 cleaving 12 nts downstream of the mature 3′ end, followed by 3′-exonucleolytic processing (Yao et al., 2007). These experiments were done before it was discovered that RNase J1 is non-essential in B. subtilis (Figaro et al., 2013), and before the discovery of the endonuclease RNase Y (Commichau et al., 2009, Shahbabian et al., 2009), which has also been shown not to be essential (Figaro et al., 2013). These recent findings allowed us to revisit scRNA processing, using mutant strains that were completely deficient in the various endonuclease activities.
Figure 5.
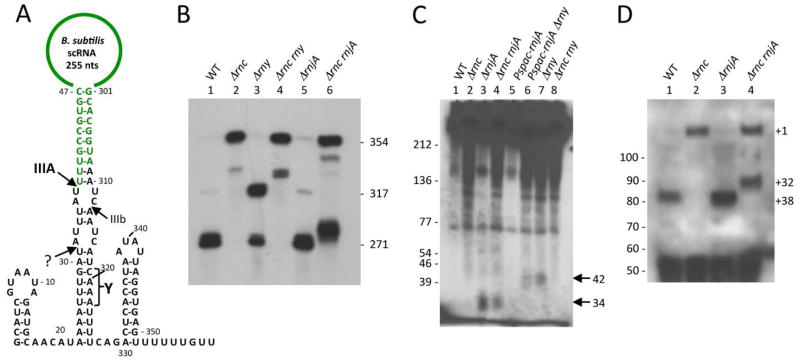
Processing and turnover of scRNA by different B. subtilis endoribonucleases. (A) Processing pathways of scRNA. The mature sequence (271 nts; in green) extends from the upstream site of RNase III cleavage (IIIA) at nt 38 to nt 308. The secondary cleavage site by RNase III is labelled ‘IIIb’. The approximate site of RNase Y cleavage site is labelled ‘Y’ and the proposed quality control processing event by an unknown ribonuclease is labelled with a question mark. (B) Northern blot of total RNA probed with the scRNA loop probe. Gene symbols are as follows: rnc (RNase III), rny (RNase Y), rnjA (RNase J1). The sizes of the intermediates are inferred from the sequence or taken from previous publications (Oguro et al., 1998, Yao et al., 2007) and confirmed with oligos specific for the 5′ and 3′ ends of the scRNA transcript (Fig. S4). (C) High resolution Northern blot showing accumulation of 3′ proximal fragments in strains lacking RNase J1. (D) Primer extension assay showing accumulation of an aberrant 5′ end at nt +32 in strains lacking RNase III and RNase J1. The primer used was DHB1299 (Table 2). The mature 5′ end of scRNA is labelled +38 and the transcription start site +1.
Total RNA was isolated from strains that were deleted for the rnc gene (encoding RNase III), and/or the rnjA and rny gene. An oligonucleotide probe (DHB1299) complementary to the internal loop portion of scRNA was used to detect scRNA species in the Northern blot analysis shown in Fig. 5B. (The percentages in the description below refer to the amount of signal in each band, relative to the total signal from all bands in that lane.) In the wild-type strain (lane 1), the major band (73%) represented the fully mature scRNA (271 nts). This band ran close to a 275-nt RNA (16%) which is believed to arise from RNase III cleavage at sites IIIA and IIIb (Yao et al., 2007). In addition to the mature scRNA, the probe detected small amounts of full-length precursor scRNA (354 nts; 4%) and a 317-nt intermediate (7%). We have shown previously that the 317-nt product is the result of RNase III cleavage at IIIA without additional endonuclease cleavage on the downstream side of the scRNA stem (Yao et al., 2007). The pattern was identical in the strain lacking RNase J1 (Fig. 5B, lane 5), indicating that RNase J1 is not involved in the processing of precursor scRNA. In the rnc deletion strain (lane 2), mostly full-length precursor scRNA was detected (90%), as well as a minor band (10%) that runs higher than the 317-nt band detected in lane 1. This minor band was detected by a 5′-terminal probe (Fig. S4B) but not by a 3′-terminal probe (Fig. S4C), and we hypothesize that it arises from single cleavage by RNase Y at the Y site (see below). No mature species was detected in the absence of RNase III. As depletion of scr expression results in cell death (Nakamura et al., 1992), these results indicate that unprocessed scRNA is functional.
In the strain that was deleted for RNase Y (lane 3), the 317-nt band representing cleavage at the IIIA site-only was much more prominent than in lane 1 (77% vs. 7%), and there was less of the mature scRNA (20% vs. 73%). These results indicated strongly that, in the wild-type strain, cleavage of the primary scRNA transcript occurs at the IIIA site to give the mature 5′ end, followed by RNase Y cleavage at or near site Y (Fig. 5A).
The location of the Y and IIIb cleavage sites was confirmed by probing with a 3′-terminal probe for small downstream products of cleavage (Fig. 5C). In the wild-type and rnc strains (lanes 1 and 2), no small products were detected. However, in the rnjA deletion strain (lane 3) a fragment running below the 39-nt marker was detected, and this was also detectable in the absence of RNase III (lane 4). We hypothesize that this small fragment is the expected 34-nt downstream product of RNase Y cleavage. The fragment is degraded from the 5′ end by RNase J1, such that it is undetectable in strains with wild-type levels of RNase J1 (lanes 1 and 2). In rny strains (lanes 6 and 7), the 34-nt 3′ product was no longer detectable and instead a larger product of ~42 nts was detected. This is the expected size for the 3′ product of cleavage by RNase III at IIIb. That this small fragment is detectable even when RNase J1 is present (lane 6) may be because it is harder for RNase J1 to attack if the 5′ and 3′ products of RNase III cleavage at IIIA and IIIb remain hybridized.
We constructed an rnc rny double mutant and examined scRNA processing in that strain (Fig. 5B, lane 4). Most of the scRNA was unprocessed (88%), except for a small amount of a doublet that ran between the bands representing cleavage at IIIA (317-nt) and cleavage at Y (lane 2). This doublet was detected by the 5′-terminal probe (Fig. S4B) but not by the 3′-terminal probe (Fig. S4C). Apparently, an additional endonuclease is capable of cleaving scRNA on the downstream side of the stem. As expected, a small 3′ fragment was not observed in the rnc rny strain (Fig. 5C, lane 8).
In an rnc rnjA strain (Fig. 5B, lane 6), the loop probe detected full-length precursor scRNA (52%), minor bands migrating between ~320–340 nts (13%) – the smaller of which likely arose from RNase Y-only cleavage as suggested for the lower band in lane 2 – and a surprisingly large amount (36%) of an RNA that appeared to be a few nucleotides larger than the mature RNA. Primer extension analysis of scRNA in rnc and rnjA mutant strains was performed using a primer (DHB1299) that was complementary to nts 98–116 of the scRNA precursor transcript (Fig. 5D). The predicted mature 5′ end at nt +38 was observed in strains containing RNase III (lanes 1 and 3), while the +1 transcription start site (TSS) was observed in the strain lacking RNase III but containing RNase J1 (lane 2). The data for the rnc rnjA strain (lane 4) showed a 5′ end that was a few nts upstream of the mature scRNA 5′ end at +38. This 5′ end was mapped precisely to nt +32 by an RNA-Seq method to be described elsewhere (JMD and DHB, unpublished). We hypothesize that this RNA is the result of an endonucleolytic cleavage at +32 that targets pre-scRNA that has not been cleaved by RNase III (nor, as a consequence, at the downstream Y site). This RNA is seen only when 1) RNase III is not present to initiate scRNA processing with cleavage at +38, and 2) RNase J1 is not present, which otherwise rapidly degrades the cleaved pre-scRNA from the 5′ end generated by the hypothesized quality control endonuclease cleavage. The minor band migrating at ~340 nts is likely the result of cleavage by this endonuclease without further cleavage at site Y. This band is detected by the 3′-terminal probe (Fig. S4C).
Discussion
In this paper we have identified the key endonucleases involved in the maturation of the B. subtilis tmRNA, scRNA and RNase P RNA. The enzymes involved vary; RNase P and RNase Z process the 5′ and 3′ ends of the tmRNA, while RNase Y processes the 3′ ends of both the RNase P RNA and scRNA. RNase III has previously been shown to perform the 5′ processing reaction for scRNA, while most of the RNase P RNA does not require 5′ maturation as it is generated directly through transcription initiation. In all cases, the 5′ exoribonuclease activity of RNase J1 is required for degradation of the downstream endonucleolytic cleavage product that includes the transcription terminator, while the upstream cleavage product is chiefly degraded by 3′ exoribonucleases. This is very similar to the strategy used by B. subtilis to degrade mRNAs (Durand et al., 2012a), the key difference being the long-lived persistence of the highly structured central portion of the primary transcript corresponding to the stable RNAs.
Our data shed some light on the preferred order of cleavages in the processing of the tmRNA. The intermediate extending from the P1 promoter to the mature 3′ end of the tmRNA (P1-Z) was visible in wild-type cells, but P2-Z was not detected. This suggests RNase P cleaves the P2 transcript more efficiently than P1, perhaps due to reduced accessibility of the P1 transcript. We also did not observe significant amounts of an intermediate extending from the site of RNase P cleavage to the terminator (M-ter) in wild-type cells, suggesting that, of the two enzymes, RNase Z is more efficient for pre-tmRNA processing. We were surprised by the observation that the 411 nt M-ter precursor species was more intense in the presence of IPTG than in its absence in the Pspac-rnz strain (Fig. 2A, lanes 11 and 12). While the reduction in RNase Z mRNA levels under the control of the fully induced Pspac promoter compared to wild-type levels explains why we see the M-ter precursor in the presence of IPTG, it does not explain why we see more than in the absence of inducer. We were also surprised to see significant amounts of P1-Z and detectable P2-Z transcripts in cells strongly depleted for RNase Z. Furthermore, we also saw a species that likely corresponds to a transcript extending from P1 to the site of cleavage by an unknown endonuclease (P1-?) downstream of the mature 3′ end of tmRNA that was specific to the strongly depleted RNase Z strain (asterisk in Fig. 2A, lane 11). Our data suggest that the unknown endonuclease cleaves more efficiently in the absence of RNase Z. Cleavage at this site would liberate a free 3′ end, that could be digested back to the mature 3′ end by 3′-exoribonucleases, thus accounting for the detection of ‘P1-Z’ and ‘P2-Z’ species under conditions of strong RNase Z depletion. This model would also account for the detection of less M-ter in strongly depleted vs. partially depleted RNase Z cells, since in the absence of RNase Z, this species would also be cut more efficiently by the unknown endonuclease and processed by 3′ exonucleases into mature sized tmRNA. Verification of this model and how cleavage at the downstream site is affected by the presence of RNase Z will require prior identification of the enzyme involved.
We have previously shown that 3′ processing of tRNA precursors by RNase Z is stimulated about 200-fold by the presence of a uracil immediately downstream of the cleavage site (Pellegrini et al., 2012). In harmony with this observation, the encoded CCA sequence of the tmRNA is also followed by a U-residue and likely to contribute to cleavage efficiency.
We identified RNase Y as the enzyme responsible for maturation of the 3′ end of the RNase P RNA. We cannot state definitively whether RNase Y processes precisely at the 3′ end of RNase P or whether it cleaves a few nucleotides downstream followed by 3′ exoribonuclease digestion. In vitro cleavage assays with purified RNase Y and PrnpB-ter precursor were inconclusive, and an attempt to map the 5′ end of the short terminator-containing fragment that accumulates in the strain lacking RNase J1 by primer extension did not yield a cDNA product (data not shown). In favour of direct processing by RNase Y, we did not see an accumulation of a larger RNase P RNA species in the strain lacking the four known 3′ exonucleases.
RNase Y was also identified as the principal endonuclease responsible for 3′ maturation of scRNA. In a strain lacking RNase Y, we observed a large accumulation of an intermediate that contained the mature 5′ end, generated directly by RNase III cleavage, and extended to the transcription terminator (Fig. 5B, lane 3). Thus, scRNA maturation requires RNase Y endonuclease cleavage on the downstream side of the stem shown in Fig. 5A, followed by 3′ exonuclease trimming to the mature 3′ end. Based on the size of an intermediate that was detected in a strain lacking RNase III (Fig. 5B, lane 2), and on the size of the 3′ terminal fragment that was detected in strains lacking the 5′ exonuclease RNase J1 that otherwise rapidly degrades this fragment (Fig. 5C), we tentatively map the RNase Y cleavage site to nt 320 of the scRNA transcript (Fig. 5A, Y site). Since 90% of the scRNA was unprocessed in the rnc mutant strain, we suggest that efficient cleavage at the Y site requires a preceding RNase III cleavage at site IIIA, and this may be due to the double-stranded nature of the sequence at site Y. However, RNase Y may be able to cleave somewhat at the double-stranded Y site even in the absence of prior RNase III cleavage (Fig. 5B, lane 2) due to an association between RNase Y and the CshA helicase (Lehnik-Habrink et al., 2010).
Cleavage by RNase Y is followed by efficient 3′ exonucleolytic degradation to give the mature 3′ end, such that an intermediate of 283 nts (from IIIA to Y) is not detected. In the previous detailed analysis using 3′ exonuclease mutant strains, we showed that this 3′ trimming is accomplished primarily by RNase PH, as a 283-nt intermediate accumulates in an rph deletion strain (Yao et al., 2007). The mature 3′ end of scRNA can also be created in the absence of RNase Y, by inefficient cleavage at site IIIb (yielding a 275-nt intermediate), followed by 3′ exonuclease trimming. We have shown previously that RNase III cleaves this side of the stem poorly, even in vitro (Yao et al., 2007), and thus disfavor the alternative hypothesis that prior cleavage by RNase Y increases cleavage by RNase III at site IIIb. We hypothesize that trimming after cleavage at the IIIb site, which leaves only a 2-nt single-stranded tail downstream of the upper stem shown in Fig. 5A, is less efficient than trimming after cleavage at the Y site, which leaves a 10-nt tail. Thus, the 275-nt band is detectable even in the wild-type strain (Fig. 5B, lane 1).
In the strain that had neither RNase III nor RNase J1 activity, we detected a novel scRNA species whose 5′ end mapped six nucleotides upstream of the mature 5′ end (Fig. 5B, lane 6; Fig. 5D). This may represent a pathway for degrading scRNA that has not been correctly cleaved by RNase III. We note that the Northern blot in Fig. 5C, which was overexposed to reveal small 3′-terminal fragments, shows evidence of extensive decay intermediates greater than 100 nts only in strains that are missing RNase III or RNase Y or both (lanes 2, 4, 6–8), suggesting difficulty in degrading improperly processed scRNA. Further work is needed to discover the identity of the endonuclease involved and whether it constitutes a quality control mechanism.
Experimental Procedures
Bacterial Strains
All of the bacterial strains used here are listed in Table 1 or have been described previously. Cells were grown in 2xYT medium (20 mL cultures) until mid-log phase (OD600 ≈ 0.5) before harvesting for RNA preparation by centrifugation and freezing of pellets.
Table 1.
Strains used in this study.
| Strain | Genotype | Source/Ref. |
|---|---|---|
| W168 | trp+ | Lab strain |
| CCB034 | W168 Pspac-rnjA (pMAP65) | (Britton et al., 2007) |
| CCB078 | W168 rnjB::spc | (Britton et al., 2007) |
| CCB322 | W168 rph::spc | This study/(Oussenko et al., 2005) |
| CCB364 | W168 ΔSkin ΔSPβ::PIID-sspB kan | (Durand et al., 2012b) |
| CCB396 | W168 rph::spc rnr::tc yhaM::pm pnp::kan | This study/(Oussenko et al., 2005) |
| CCB418 | W168 txpA -10Δ yonT::ery rnc::spc | (Durand et al., 2012b) |
| CCB434 | W168 rnjA::spc | (Figaro et al., 2013) |
| CCB441 | W168 rny::spc | (Figaro et al., 2013) |
| CCB422 | W168 ΔSkin ΔSPβ::PIID-sspB kan rnc::spc | (Durand et al., 2012b) |
| CCB501 | W168 rnjB::spc rnjA::kan | This study/(Figaro et al., 2013) |
| CCB504 | W168 Pxyl-rnpA cm | This study/ (Gossringer et al., 2006) |
| SSB318 | W168 Pspac-rnpB ery | (Wegscheid et al., 2006) |
| SSB321 | W168 Pspac-rnz ery pMAP65 kan | (Pellegrini et al., 2003) |
| BG322 | rnc::spc sup | (Herskovitz & Bechhofer, 2000) |
| BG877 | W168 ΔSkin ΔSPβ::PIID-sspB kan rnc::ery | This study |
| BG879 | W168 ΔSkin ΔSPβ::PIID-sspB kan rnc::ery rnjA::spc | This study |
| BG880 | W168 ΔSkin ΔSPβ::PIID-sspB kan rnjA::spc | This study |
| BG881 | W168 ΔSkin ΔSPβ::PIID-sspB kan rnc::ery rny::spc | This study |
| BG882 | W168 ΔSkin ΔSPβ::PIID-sspB kan rny::spc | This study |
| BG898 | W168 Pspac-rnjA rny::spc pMAP65 kan | This study |
Strain CC504 was made by transfering the Pxyl-rnpA construct from strain d7 (kind gift from R. Hartmann) to the W168 background. The genotype of the parent strain, CCB364 (Durand et al., 2012b), was W168ΔSkin SPβ::PIID-sspB kan, carrying a deletion of the Skin prophage and replacement of SPβ prophage with a spoIID-sspB fusion linked to a kanamycin-resistance marker. The rnc knockout strain, CB422, was a derivative of CCB364 with the rnc CDS replaced with a spectinomycin-resistance marker, as described (BG324; (Herskovitz & Bechhofer, 2000). The spectinomycin-resistance marker in CCB422 was converted to erythromycin resistance by transformation with ScaI-linearized pMDspcTOery (kindly provided by Jeanette Hahn), to give BG877. This allowed construction of BG879, which had, in addition to the replacement of the rnc CDS, a replacement of the rnjA coding sequence with a spectinomycin-resistance marker, as described (CCB433; (Figaro et al., 2013). BG880 was a control strain with wild-type rnc and deleted rnjA. BG881 was a derivative of BG877 with an additional deletion of the rny CDS, as described (CCB441; (Figaro et al., 2013)). BG882 was a control strain with wild-type rnc and deleted rny. BG898 was an rny deletion derivative of CCB034, in which rnjA is under Pspac control (Britton et al., 2007). Gene knockout constructs were confirmed by PCR. The wild-type sequence of the lac operator in strains with the Pspac-rnjA construct was also confirmed.
The following concentrations of antibiotics were used: chloramphenicol 4 μg/ml; erythromycin 5 μg/ml; kanamycin 5 μg/ml; neomycin 3 μg/ml; spectinomycin 200 μg/ml.
RNA isolation and analysis
RNA was isolated from frozen pellets by either the glass beads or hot phenol method (Bechhofer et al., 2008, Durand et al., 2012a) and analysed by Northern blot as previously described (Bechhofer et al., 2008, Durand et al., 2012a). Primer extension assays were performed as described in (Britton et al., 2007). Oligonucleotides used in this study are listed in Table 2.
Table 2.
Oligonucleotides used in this study.
| Oligo | Gene | Sequence |
|---|---|---|
| CC1006 | rnpB(mature) | TGCGAGCATGGACTTTCCTCTACAG |
| CC1012 | rnpB (3′) | CTCCTTATACGGGAGAGCTTG |
| CC1444 | tmRNA (mature) | CCATCCCTGTCGAATCCGTAACGTCC |
| CC1445 | tmRNA (3′) | CTTAAGAGGTTCTGCTTTATTGTCAGTATG |
| CC1463 | tmRNA (P1) | GTCAAATCCTTCAAATGAATGGCGTCGTGATC |
| CC1464 | tmRNA (P2) | GGGAAATACGGGAGAAAATATCAAGCTCTC |
| CC1475 | tmRNA (mature) | CGCAGCTCAAGCTCGATCCATCCC |
| CC1477 | tmRNA (3′) | CTGGGATTGAAATGGAAGTACGCAC |
| CC1479 | tmRNA (3′) bis | AAAGCAGAACCTCTTAAGA |
| CC1492 | gpsB | CGCAGAAAGCTTTACTTTATCAGC |
| CC1500 | tmRNA (T7) | GCTCTAATACGACTCACTATAGGGATAGCCAGTTTGAGTGAGAGCTTGATA |
| CC1538 | tmRNA | GCAGGCTACGCTCGTAGACGCTTAAG |
| DHB930 | scRNA (5′) | CAAATTTTATGTTGCATCTATTACGATGC |
| DHB944 | scRNA (3′) | CTAATATTAGGTGAGTCTGAAAATTTGATGATT |
| DHB1299 | scRNA (mature) | AACCTCGAGAAGGGATTCG |
For RNA isolation from normally-growing BG strains, a 5 ml starter culture with appropriate antibiotic was grown overnight in LB medium. Cells from the overnight culture were diluted 1:50 in fresh LB medium without antibiotic and were grown at 37°C, shaking at 300 rpm, until early log phase. Slower-growing strains (BG879, BG880, BG881, and BG882) were grown in 10 ml starter cultures with appropriate antibiotic for approximately 24 hours, and were then diluted as described above. For strains with rnjA under Pspac promoter control (CCB034 and BG898), mutations arising in the Pspac promoter were avoided by growing strains directly from frozen stocks in LB medium containing 0.125 mM IPTG and neomycin (selection for pMAP65 (Petit et al., 1998), a resident plasmid that provides extra copies of the LacI repressor). The CCB034 culture was started the same day as the RNA isolation with a 1:50 dilution from frozen stock, whereas the BG898 culture was started the day before RNA isolation with a 1:500 dilution from frozen stock.
For detection of scRNA, total RNA was separated on denaturing polyacrylamide gels (6% polyacrylamide/7M urea) and blotted onto positively-charged nylon membranes that are optimal for digoxigenin (DIG) detection (Roche). Hybridization conditions were as follows: 30 min pre-hybridization at 42°C with 10 ml of pre-warmed DIG Easy Hyb buffer (Roche), followed by addition of 5 pmols 5′-DIG-labeled probe in 10 ml of fresh DIG Easy Hyb buffer and overnight hybridization at 42°C. The membrane was washed twice for 20 min at 42°C with 50 ml of 5xSSC, 0.1% SDS. For DIG-tailed probes, washing was at 50°C. DIG detection was performed as per the manufacturer’s protocol (Roche). The scRNA 5′-terminal and 3′-terminal probes were DIG-tailed at the 3′ end, according to the Roche protocol (Version 6).
RNase Z cleavage assay
The P2-ter tmRNA precursor was produced by in vitro transcription using T7 RNA polymerase (Ambion) using a PCR fragment amplified with oligos CC1500 and CC1477 as a template. The tmRNA was denatured 5 mins at 80°C and renatured on ice for 20 mins before assay. One μg of tmRNA precursor was digested by 20 pmol RNase Z in a 10 μl reaction volume at 37°C for 30 mins as described previously (Pellegrini et al., 2012). The reactions were phenol extracted, ethanol precipitated and resuspended in 5 μl H2O. Primer extension assays with oligo CC1445 were performed as described in (Britton et al., 2007).
Circular RT-PCR
Circular RT-PCR (5′/3′ RACE) was performed as described previously (Britton et al., 2007) using oligonucleotides CC1475 and CC1538. The band corresponding to the expected amplicon size for mature tmRNA (~115 nts) was gel-purified and sequenced.
Supplementary Material
Figure S1. Processing and turnover of tmRNA by different B. subtilis endo- and exoribonucleases. Northern blot analysis of total RNA isolated from different B. subtilis RNase mutants probed with (A) the P1 probe (oligo CC1463), (B) the P2 probe (oligo CC1464) and (C) the terminator-specific probe (CC1445). The sizes of the intermediates are inferred from the sequence and confirmed with the different oligos used in each panel and the blot shown in Fig. 2. For those species containing the mature 3′ end of the tmRNA, the 3 extra nts corresponding to the CCA motif have been added for size calculation.
Figure S2. Mapping of degradation intermediates within the mature tmRNA sequence. Primer extension assay (oligo CC1445) identifying 5′ ends that accumulate in RNase Z-depleted strains. Sequence lanes are labelled as their reverse complement to facilitate direct read-out. Degradation intermediates are labelled DI and are reported on Fig. 1.
Figure S3. Processing and turnover of the RNase P RNA by different B. subtilis endo- and exoribonucleases. Northern blot analysis of total RNA isolated from different B. subtilis RNase mutants probed with (A) the mature probe (oligo CC1006), (B) the terminator-specific probe (oligo CC1012) and (C) the PgpsB-specific probe (CC1492). The sizes of the intermediates are inferred from the sequence and confirmed with the different oligos used in each panel and the blot shown in Fig. 4.
Figure S4. Processing and turnover of the scRNA by different B. subtilis endoribonucleases. Northern blot analysis of total RNA isolated from different B. subtilis RNase mutants probed with (A) the loop probe (B) the 5′ probe (C) the 3′ probe. The sizes of the intermediates are inferred from the sequence or taken from previous publications (Oguro et al., 1998, Yao et al., 2007) and confirmed with the different oligos used in each panel and the blot shown in Fig. 5.
Acknowledgments
This work was supported by funds from the CNRS (UPR 9073), Université Paris VII-Denis Diderot and the Agence Nationale de la Recherche (subtilRNA2 and asSUPYCO) to C.C., and by United States Public Health Service grant GM-100137 to D.H.B. We thank R. Hartmann for strain d7 (Pxyl-rnpA). This work has been published under the framework of Equipex (Cacsice) and LABEX programs (Dynamo) that benefit from a funding from the state managed by the French National Research Agency as part of the Investments for the Future program. We thank lab members for helpful discussion.
References
- Akopian D, Shen K, Zhang X, Shan SO. Signal recognition particle: an essential protein-targeting machine. Annu Rev Biochem. 2013;82:693–721. doi: 10.1146/annurev-biochem-072711-164732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechhofer DH, Oussenko IA, Deikus G, Yao S, Mathy N, Condon C. Analysis of mRNA decay in Bacillus subtilis. Methods Enzymol. 2008;447:259–276. doi: 10.1016/S0076-6879(08)02214-3. [DOI] [PubMed] [Google Scholar]
- Britton RA, Wen T, Schaefer L, Pellegrini O, Uicker WC, Mathy N, Tobin C, Daou R, Szyk J, Condon C. Maturation of the 5′ end of Bacillus subtilis 16S rRNA by the essential ribonuclease YkqC/RNase J1. Mol Microbiol. 2007;63:127–138. doi: 10.1111/j.1365-2958.2006.05499.x. [DOI] [PubMed] [Google Scholar]
- Commichau FM, Rothe FM, Herzberg C, Wagner E, Hellwig D, Lehnik-Habrink M, Hammer E, Volker U, Stulke J. Novel activities of glycolytic enzymes in Bacillus subtilis: interactions with essential proteins involved in mRNA processing. Mol Cell Proteomics. 2009;8:1350–1360. doi: 10.1074/mcp.M800546-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condon C, Brechemier-Baey D, Beltchev B, Grunberg-Manago M, Putzer H. Identification of the gene encoding the 5S ribosomal RNA maturase in Bacillus subtilis: Mature 5S rRNA is dispensable for ribosome function. RNA. 2001;7:242–253. doi: 10.1017/s1355838201002163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand S, Gilet L, Bessieres P, Nicolas P, Condon C. Three essential ribonucleases-RNase Y, J1, and III-control the abundance of a majority of Bacillus subtilis mRNAs. PLoS Genet. 2012a;8:e1002520. doi: 10.1371/journal.pgen.1002520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand S, Gilet L, Condon C. The essential function of B. subtilis RNase III is to silence foreign toxin genes. PLoS Genet. 2012b;8:e1003181. doi: 10.1371/journal.pgen.1003181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figaro S, Durand S, Gilet L, Cayet N, Sachse M, Condon C. Bacillus subtilis mutants with knockouts of the genes encoding ribonucleases RNase Y and RNase J1 are viable, with major defects in cell morphology, sporulation, and competence. J Bacteriol. 2013;195:2340–2348. doi: 10.1128/JB.00164-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giudice E, Gillet R. The task force that rescues stalled ribosomes in bacteria. Trends Biochem Sci. 2013;38:403–411. doi: 10.1016/j.tibs.2013.06.002. [DOI] [PubMed] [Google Scholar]
- Gossringer M, Kretschmer-Kazemi Far R, Hartmann RK. Analysis of RNase P protein (rnpA) expression in Bacillus subtilis utilizing strains with suppressible rnpA expression. J Bacteriol. 2006;188:6816–6823. doi: 10.1128/JB.00756-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudipati RK, Xu Z, Lebreton A, Seraphin B, Steinmetz LM, Jacquier A, Libri D. Extensive degradation of RNA precursors by the exosome in wild-type cells. Mol Cell. 2012;48:409–421. doi: 10.1016/j.molcel.2012.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrier-Takada C, Gardiner K, Marsh T, Pace N, Altman S. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell. 1983;35:849–857. doi: 10.1016/0092-8674(83)90117-4. [DOI] [PubMed] [Google Scholar]
- Gutmann B, Gobert A, Giege P. PRORP proteins support RNase P activity in both organelles and the nucleus in Arabidopsis. Genes Dev. 2012;26:1022–1027. doi: 10.1101/gad.189514.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy MP, Young DL, Payea MJ, Zhang X, Kon Y, Dean KM, Grayhack EJ, Mathews DH, Fields S, Phizicky EM. Identification of the determinants of tRNA function and susceptibility to rapid tRNA decay by high-throughput in vivo analysis. Genes Dev. 2014;28:1721–1732. doi: 10.1101/gad.245936.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herskovitz MA, Bechhofer DH. Endoribonuclease RNase III is essential in Bacillus subtilis. Mol Microbiol. 2000;38:1027–1033. doi: 10.1046/j.1365-2958.2000.02185.x. [DOI] [PubMed] [Google Scholar]
- Holzmann J, Frank P, Loffler E, Bennett KL, Gerner C, Rossmanith W. RNase P without RNA: identification and functional reconstitution of the human mitochondrial tRNA processing enzyme. Cell. 2008;135:462–474. doi: 10.1016/j.cell.2008.09.013. [DOI] [PubMed] [Google Scholar]
- Kim S, Kim H, Park I, Lee Y. Mutational analysis of RNA structures and sequences postulated to affect 3′ processing of M1 RNA, the RNA component of Escherichia coli RNase P. J Biol Chem. 1996;271:19330–19337. doi: 10.1074/jbc.271.32.19330. [DOI] [PubMed] [Google Scholar]
- Komine Y, Kitabatake M, Yokogawa T, Nishikawa K, Inokuchi H. A tRNA-like structure is present in 10Sa RNA, a small stable RNA from Escherichia coli. Proc Natl Acad Sci U S A. 1994;91:9223–9227. doi: 10.1073/pnas.91.20.9223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnik-Habrink M, Pfortner H, Rempeters L, Pietack N, Herzberg C, Stulke J. The RNA degradosome in Bacillus subtilis: identification of CshA as the major RNA helicase in the multiprotein complex. Mol Microbiol. 2010;77:958–971. doi: 10.1111/j.1365-2958.2010.07264.x. [DOI] [PubMed] [Google Scholar]
- Li Z, Pandit S, Deutscher MP. 3′ exoribonucleolytic trimming is a common feature of the maturation of small, stable RNAs in Escherichia coli. Proc Natl Acad Sci U S A. 1998a;95:2856–2861. doi: 10.1073/pnas.95.6.2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Pandit S, Deutscher MP. Polyadenylation of stable RNA precursors in vivo. Proc Natl Acad Sci U S A. 1998b;95:12158–12162. doi: 10.1073/pnas.95.21.12158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Reimers S, Pandit S, Deutscher MP. RNA quality control: degradation of defective transfer RNA. EMBO J. 2002;21:1132–1138. doi: 10.1093/emboj/21.5.1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin-Chao S, Wei CL, Lin YT. RNase E is required for the maturation of ssrA RNA and normal ssrA RNA peptide-tagging activity. Proc Natl Acad Sci U S A. 1999;96:12406–12411. doi: 10.1073/pnas.96.22.12406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loria A, Pan T. The 3′ substrate determinants for the catalytic efficiency of the Bacillus subtilis RNase P holoenzyme suggest autolytic processing of the RNase P RNA in vivo. RNA. 2000;6:1413–1422. doi: 10.1017/s1355838200000959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes A, Gracia C, Hajnsdorf E, Regnier P. Search for poly(A) polymerase targets in E. coli reveals its implication in surveillance of Glu tRNA processing and degradation of stable RNAs. Mol Microbiol. 2012;83:436–451. doi: 10.1111/j.1365-2958.2011.07943.x. [DOI] [PubMed] [Google Scholar]
- Makarov EM, Apirion D. 10Sa RNA: processing by and inhibition of RNase III. Biochemistry International. 1992;26:1115–1124. [PubMed] [Google Scholar]
- Mathy N, Benard L, Pellegrini O, Daou R, Wen T, Condon C. 5′-to-3′ exoribonuclease activity in bacteria: role of RNase J1 in rRNA maturation and 5′ stability of mRNA. Cell. 2007;129:681–692. doi: 10.1016/j.cell.2007.02.051. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Imai Y, Nakamura A, Yamane K. Small cytoplasmic RNA of Bacillus subtilis: functional relationship with human signal recognition particle 7S RNA and Escherichia coli 4.5S RNA. J Bacteriol. 1992;174:2185–2192. doi: 10.1128/jb.174.7.2185-2192.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oguro A, Kakeshita H, Nakamura K, Yamane K, Wang W, Bechhofer DH. Bacillus subtilis RNase III cleaves both 5′- and 3′-sites of the small cytoplasmic RNA precursor. J Biol Chem. 1998;273:19542–19547. doi: 10.1074/jbc.273.31.19542. [DOI] [PubMed] [Google Scholar]
- Oussenko IA, Abe T, Ujiie H, Muto A, Bechhofer DH. Participation of 3′-to-5′ exoribonucleases in the turnover of Bacillus subtilis mRNA. J Bacteriol. 2005;187:2758–2767. doi: 10.1128/JB.187.8.2758-2767.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peck-Miller KA, Altman S. Kinetics of the processing of the precursor to 4.5 S RNA, a naturally occurring substrate for RNase P from Escherichia coli. J Mol Biol. 1991;221:1–5. doi: 10.1016/0022-2836(91)80194-y. [DOI] [PubMed] [Google Scholar]
- Pellegrini O, Li de la Sierra-Gallay I, Piton J, Gilet L, Condon C. Activation of tRNA maturation by downstream uracil residues in B. subtilis. Structure. 2012;20:1769–1777. doi: 10.1016/j.str.2012.08.002. [DOI] [PubMed] [Google Scholar]
- Pellegrini O, Nezzar J, Marchfelder A, Putzer H, Condon C. Endonucleolytic processing of CCA-less tRNA precursors by RNase Z in Bacillus subtilis. EMBO J. 2003;22:4534–4543. doi: 10.1093/emboj/cdg435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit M-A, Dervyn E, Rose M, Entian K-D, McGovern S, Ehrlich DS, Bruand C. PcrA is an essential DNA helicase of Bacillus subtilis fulfilling functions both in repair and rolling-circle replication. Mol Micro. 1998;29:261–273. doi: 10.1046/j.1365-2958.1998.00927.x. [DOI] [PubMed] [Google Scholar]
- Randau L, Schroder I, Soll D. Life without RNase P. Nature. 2008;453:120–123. doi: 10.1038/nature06833. [DOI] [PubMed] [Google Scholar]
- Redko Y, Bechhofer DH, Condon C. Mini-III, an unusual member of the RNase III family of enzymes, catalyses 23S ribosomal RNA maturation in B. subtilis. Mol Microbiol. 2008;68:1096–1106. doi: 10.1111/j.1365-2958.2008.06207.x. [DOI] [PubMed] [Google Scholar]
- Shahbabian K, Jamalli A, Zig L, Putzer H. RNase Y, a novel endoribonuclease, initiates riboswitch turnover in Bacillus subtilis. EMBO J. 2009;28:3523–3533. doi: 10.1038/emboj.2009.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegscheid B, Condon C, Hartmann RK. Type A and B RNase P RNAs are interchangeable in vivo despite substantial biophysical differences. EMBO Rep. 2006;7:411–417. doi: 10.1038/sj.embor.7400641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegscheid B, Hartmann RK. In vivo and in vitro investigation of bacterial type B RNase P interaction with tRNA 3′-CCA. Nucleic Acids Res. 2007;35:2060–2073. doi: 10.1093/nar/gkm005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen T, Oussenko IA, Pellegrini O, Bechhofer DH, Condon C. Ribonuclease PH plays a major role in the exonucleolytic maturation of CCA-containing tRNA precursors in Bacillus subtilis. Nucleic Acids Res. 2005;33:3636–3643. doi: 10.1093/nar/gki675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao S, Blaustein JB, Bechhofer DH. Processing of Bacillus subtilis small cytoplasmic RNA: evidence for an additional endonuclease cleavage site. Nucleic Acids Res. 2007;35:4464–4473. doi: 10.1093/nar/gkm460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Processing and turnover of tmRNA by different B. subtilis endo- and exoribonucleases. Northern blot analysis of total RNA isolated from different B. subtilis RNase mutants probed with (A) the P1 probe (oligo CC1463), (B) the P2 probe (oligo CC1464) and (C) the terminator-specific probe (CC1445). The sizes of the intermediates are inferred from the sequence and confirmed with the different oligos used in each panel and the blot shown in Fig. 2. For those species containing the mature 3′ end of the tmRNA, the 3 extra nts corresponding to the CCA motif have been added for size calculation.
Figure S2. Mapping of degradation intermediates within the mature tmRNA sequence. Primer extension assay (oligo CC1445) identifying 5′ ends that accumulate in RNase Z-depleted strains. Sequence lanes are labelled as their reverse complement to facilitate direct read-out. Degradation intermediates are labelled DI and are reported on Fig. 1.
Figure S3. Processing and turnover of the RNase P RNA by different B. subtilis endo- and exoribonucleases. Northern blot analysis of total RNA isolated from different B. subtilis RNase mutants probed with (A) the mature probe (oligo CC1006), (B) the terminator-specific probe (oligo CC1012) and (C) the PgpsB-specific probe (CC1492). The sizes of the intermediates are inferred from the sequence and confirmed with the different oligos used in each panel and the blot shown in Fig. 4.
Figure S4. Processing and turnover of the scRNA by different B. subtilis endoribonucleases. Northern blot analysis of total RNA isolated from different B. subtilis RNase mutants probed with (A) the loop probe (B) the 5′ probe (C) the 3′ probe. The sizes of the intermediates are inferred from the sequence or taken from previous publications (Oguro et al., 1998, Yao et al., 2007) and confirmed with the different oligos used in each panel and the blot shown in Fig. 5.